

Summary
The assessment of Toll‐like receptor (TLR) agonists as candidate adjuvants for induction of effective T helper type 1 (Th1) immunity continues to rely on the use of mice. However, the genetic variation among inbred mice may influence the efficacy of adjuvants and bias a study's conclusions. Here, we evaluated the differences in cellular and humoral responses of genetically non‐identical mouse strains immunized with ovalbumin (OVA) plus alum, TLR‐3, TLR‐4, TLR‐7/8 or TLR‐9 agonists. We found that all the tested TLR agonists recruited dendritic cells (DCs) and natural killer (NK) cells significantly into the lymph nodes, promoted DC–NK cross‐talk and enhanced the cellular responses in B6 strain. In contrast, TLR‐3 and TLR‐7/8 were the only two agonists that showed the cellular adjuvanticity in the BALB/c strain. Compared with other TLR agonists, TLR‐3 and TLR‐7/8 were demonstrated to be the most effective adjuvants to generate interferon (IFN)‐γ‐producing effector NK, CD4, and CD8 T cells in B6 and BALB/c strains, respectively. We also found that compared with alum, all adjuvants induced the recruitment of B cells and production of OVA‐specific immunoglobulin (Ig)G2a more effectively in both strains. In addition, the B6 strain recruited more B cells, but surprisingly produced significantly lower amounts of OVA‐specific IgG2a in response to all adjuvants. However, consistent with the frequency of IFN‐γ‐producing effector cells observed in individual strains following immunizations, we detected more OVA‐specific IgG2a in serum of B6 and BALB/c strains in response to TLR‐3 and TLR‐7/8, respectively. Our data suggest that genetic background should be taken into consideration when evaluating the activities of TLR agonists for the development of prophylactic and therapeutic vaccines.
Keywords: dendritic cells, natural killer cells, Toll‐like receptors (TLRs), vaccination
Introduction
Advances in genomics and proteomics have accelerated the identification of recombinant and synthetic molecules for the development of new vaccines. Unfortunately, these candidate proteins are often poorly immunogenic compared with the traditional live, attenuated or killed pathogens 1 which has surged the development and selection of improved and more powerful adjuvants to enhance their action. Although there has been a flurry of research on adjuvants for use in vaccines during the last decade very few vaccine adjuvants have been licensed for human vaccines, which include alum, MF59, AS03 and AS04 1. While more effective in inducing humoral immunity, none of the currently approved adjuvants enhance uniformly or sufficiently both the humoral and cellular immunity that is essential for the elimination of microorganisms, particularly intracellular pathogens and tumour cells 2. Toll‐like receptors (TLRs) are pathogen‐associated molecular pattern (PAMP) recognition receptors that are an important link between innate and adaptive immunity 3. In most instances, signalling through TLRs favours the development of a T helper cell type 1 (Th1) response, which has triggered interest in exploiting TLR agonists as potential adjuvants for prophylactic and therapeutic vaccines 4, 5. TLRs are expressed on most innate immune cells, including dendritic cells (DC), natural killer cells (NK), monocytes and granulocytes 6. Two classes of TLRs can be defined based on their cellular localization: TLRs‐1, ‐2 and ‐4–6 are expressed on the cell surface and recognize pathogenic components, while TLRs‐3, ‐7, ‐8 and ‐9 are expressed in intracellular endosome/lysosome membranes and are nucleic acid sensors 7. All TLRs trigger an intracellular signal transduction pathway initiated by the adaptor protein myeloid differentiation primary response gene (MyD88), with the exception of TLR‐3, which utilizes an alternative adaptor called TIR domain‐containing adaptor‐inducing interferon (IFN) β (TRIF). TLR‐4, however, activates both MyD88 and TRIF 8.
PAMPs or TLR agonists mediate the interaction between DCs and NK cells, which is a critical step in initiating an adaptive immunity. Indeed, following TLR engagement, DCs migrate from peripheral tissues to lymphoid organs while up‐regulating major histocompatibility complex and co‐stimulatory molecules, and acquiring the unique capacity to prime naive T cells 9. In draining lymph nodes, DCs determine the character of the ensuing immune response by secreting cytokines [e.g. interleukin (IL)‐12] that drive the development of pathogen‐specific naive T cells into Th1 cell type. DCs also potentiate their efficiency by interacting with NK cells recruited to the lymphoid tissues in response to PAMPs. IL‐12 released by mature DCs can activate NK cells. In turn, NK cells that are activated by IL‐12 and PAMPs through their own TLRs provide IFN‐γ, necessary for enhancing stable IL‐12 production by DCs and maintaining Th1 cell polarization 10. Th1, as well as Th2 polarization and their resultant cytokines, can, in turn, promote further differentiation of antigen‐specific naive B cells and affect the class of antibodies produced by plasma cells. IFN‐γ produced by Th1 cells induces B cells to class‐switch to immunoglobulin (Ig)G2a while IL‐4 and TGF‐β, released typically by Th2 cells, promote them to switch to IgG1 and IgG2b, respectively 11. Therefore, the ability to recruit NK cells and establish DC–NK cross‐talk has been considered as a criterion for selection of Th1 adjuvants.
There are no predictive in‐vitro systems to evaluate the safety and effectiveness of vaccines formulated with TLR agonists owing to the complexity of the immune system, which is difficult to mimic in cell culture systems. However, animal models have been very useful in the efficient translation of basic vaccine research. Indeed, inbred mice such as BALB/c and C57BL/6 (B6), with non‐identical genetic background, have been used extensively in preclinical research. However, one of the common drawbacks to many vaccine studies aimed to examine the protective effect of a candidate adjuvant is the use of a single mouse strain, which may potentially bias the study conclusion. For example, Rajagopl et al. showed that R848 has a limited effect in inducing an ovalbumin (OVA)‐specific CD4+ T cell immune response in B6 mice 12. Conversely, using BALB/c mice, Martin‐Fontecha et al. found that R848 but not cytosine–phosphate–guanosine (CpG) or complete Freund's adjuvant (CFA) was the robust adjuvant in boosting the OVA‐specific CD4+ T cell immune response 13, while Sun et al. showed that CpG but not CFA potentiated clonal expansion of an α‐globulin‐specific T cell response markedly in B6 mice 14. The difference in vaccine‐mediated protection between mouse strains was also demonstrated clearly in a report where prophylactic immunization with recombinant Helicobacter pylori adhesin A (HpaA) induced a reduction in H. pylori colonization in BALB/c but was ineffective in B6 mice 15. Hence, in this study we immunized two genetically non‐identical mouse strains with a protein‐based vaccine formulated with TLR agonists and analysed the recruitment and phenotypes of DCs and the generation of effector NK and T cells and antibodies in their lymphoid tissues and sera. Our study indicates that the genetic background of a strain biases significantly the interpretation of adjuvant effect of TLR agonists.
Materials and method
Mice
Wild‐type C57BL/6 (B6, H‐2b) and BALB/c (Ba, H‐2d) male mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). They were bred and maintained under specific pathogen‐free conditions in the animal facility of the Charles E. Schmidt College of Medicine at Florida Atlantic University. Mice were used at 6–8 weeks of age and treated in accordance with the National Institutes of Health guide for the care and use of laboratory animals in experiments approved by the Florida Atlantic University IACUC committee.
Immunization
Mice were injected on day 0 (NK recruitment, cell‐mediated response) or days 0 and 14 (humoral response) subcutaneously at the nape of the neck with 2 mg of OVA protein (Sigma, St Louis, MO, USA) mixed with 25 μg of TLR agonists [polyinosinic–polycytidylic acid (poly I:C)], MPLA, R848 or CpG‐C) or 50μl of aluminum hydroxide gel (alum; Invivogen, San Diego, CA, USA).
Animal preparation
For solid organ collection, animals were euthanized by overdose of CO2 by placing them into a chamber that contains CO2 and oxygen controlled by the CO2 flow regulator. Overdose CO2 treatment was followed by cervical dislocation after the animal was determined to be non‐responsive to noxious stimuli. For blood collection, mice were first anaesthetized by intraperitoneal injection of mixture of ketamine/xylazine (100/10 mg/kg body weight). Then, a midline incision was made through the skin and musculature and peritoneum from xiphoid to pubis. Up to 1 ml blood samples were collected from the abdominal aorta. Mice were euthanized following the blood collection.
Cell preparation
Axillary, inguinal and popliteal lymph nodes from immunized mice were harvested on days 2–3 following immunization. Single‐cell suspensions were obtained by grinding lymph nodes with two frosted glass slides. Cells were washed with phosphate‐buffered saline (PBS) buffer and then treated with ammonium–chloride–potassium (ACK) buffer [0·15 M ammonium chloride (NH4Cl)/1 mM potassium bicarbonate (KHCO3)/0·1 mM Na2 ethylenediamine tetraacetic acid (EDTA)] to remove erythrocytes before counting and staining with indicated fluorochrome‐labelled monoclonal antibodies.
Fluorescence activated cell sorter (FACS) analysis
Single‐cell suspensions from lymph nodes were stained with antibodies against B220 (RA3‐6B2), CD80 (16‐10A1), CD86 (GL1), CD19 (1D3), TLR‐3 (11F8), TLR‐4 (UT41), TLR‐9 (M9.D6), TLR‐7 (Polyclone, Mountain View, CA, USA), CXCR3 (CXCR3‐173), CD27 (LG7F9), CD69 (H1.2F3), CD4 (RM4‐5), major histocompatibility complex (MHC) II (M5/114.15.2), CD212 (114), CD62L (MEL‐14), CD11b (M1/70), CD122 (5H4), IFN‐γ (XMG1.2), CD3 (17A2), CD8 (53‐6.7), CD11c (N418), CD49b (DX5), Thy1.2 (53‐2.1), CCR7 (4B12) and CD19 (1D3). All the antibodies except anti‐TLR‐3 and anti‐TLR‐7 were purchased from either eBioscience (San Diego, CA, USA) or BD Biosciences (San Jose, CA, USA). Anti‐TLR‐3 and Anti‐TLR‐7 were purchased from BioLegend (San Diego, CA, USA) and Imgenex (San Diego, CA, USA), respectively. To stain cell‐surface molecules, cells were incubated with optimally diluted fluorochrome‐labeled monoclonal antibodies for 30 min on ice. For intracellular staining, cells were first stimulated with 2 μl of leucocyte activation cocktail (BD Biosciences) for 5 h and then stained for the cell‐surface markers. After washing, the cells were then fixed and permeabilized with Cytofix/Cytoperm solution (BD Biosciences) before staining with anti‐IFN‐γ antibodies, as per the manufacturer's instructions. Samples were acquired on Calibur or Aria (BD Biosciences) flow cytometers using CellQuest Pro and FACS Diva softwares, respectively. Data were analysed using FlowJo analysis software (Tree Star, Ashland, OR, USA).
Serum anti‐OVA IgG1, anti‐OVA IgG2a and anti‐OVA IgG2b levels
For measurement of anti‐OVA antibodies in the mice serum, enzyme immunoassay plates (Costar, Milpitas, CA, USA) were coated with 15 μg/ml OVA protein (Sigma‐Aldrich, St Louis, CA, USA) in PBS overnight at 4°C, blocked with 10% fetal calf serum (FCS)/PBS, washed and incubated for 2 h with serum samples or standards (AntibodyShop, Utrecht, the Netherlands). Plates were then washed, incubated with 2 mg/ml biotin‐conjugated anti‐mouse IgG1 (A85‐1), IgG2a (R19‐15) or IgG2b (R12‐3) antibodies for 2 h, followed by incubation with 1/2000 streptavidin–horseradish peroxidase (HRP) (BD Biosciences) for 1 h at room temperature. After the final washes, a one‐to‐one mixture of BD OptEIA substrate reagents A and B (BD Biosciences) was added as a chromogen. The reaction was stopped with 2 N H2SO4. The optical densities at 490 nm were measured, and the antibody titres were determined for each sample.
Statistical analysis
All experiments were performed in replicates. Statistical significance was evaluated using Student's t‐test with a 95% confidence interval. Results are expressed as means ± standard deviation. In the figures, P‐values of 0·05 are labelled with an asterisk. The specific values are shown when the P‐value is greater but close to 0·05. Analysis was performed with a Prism program (GraphPad, San Diego, CA, USA).
Results
TLR agonists induced DC recruitment in lymph nodes of B6 and BALB/c mice
DCs express the broadest repertoire of TLRs sensing a plethora of microbial compounds. After engagement of TLRs, immature DCs undergo a complex process of maturation, resulting in up‐regulation of their co‐stimulatory molecules and their migration from tissues to secondary lymphoid organs, where DCs interact with T cells, initiate and shape the adaptive immune response 16. Thus, the capability to activate DCs is an important consideration in the adjuvant research. We first compared the changes in the number and phenotype of DCs in lymph nodes of two different strains of mice immunized with OVA protein mixed with TLR‐3 (poly I:C), TLR‐4 [monophosphoryl lipid A (MPLA)], TLR‐7/8 (R848) and TLR‐9 (CpG) agonists, which are now in licensed products or in late clinical development 17.
Prior to immunization, the frequency of total DCs in the lymph nodes of B6 mice was almost twofold higher than in BALB/c mice (Fig. 1b). However, the absolute number of DCs between the two strains was comparable owning to a higher number of total cells in the lymph nodes of BALB/c mice (Fig. 1b). While both DCs expressed the same levels of TLR‐4 and TLR‐9, except for BALB/c DCs, which expressed lower levels of TLR‐7 and to a lesser extent TLR‐3, the expression levels of co‐stimulatory molecules CD80 and CD86 were slightly higher on B6 than BALB/c DCs (Fig. 1a,c). Interestingly, although the two strains had similar number of DCs in lymph nodes, the proportions of DC subsets were significantly different. Indeed, myeloid DCs constituted the largest population (46.08%), followed by CD8+ (26·46%) and plasmacytoid (23·14%) DCs in B6 mice (Fig. 1d). In contrast, the predominant DC subset in BALB/c mice was the plasmacytoid (58%), which was significantly higher than myeloid (21%) and CD8+ (17·8%) DC subsets (Fig. 1d). Of note, same results were observed when other markers used commonly to define DC subsets, including CD8/CD11b and B220/CD8, were used (data not shown).
Figure 1.
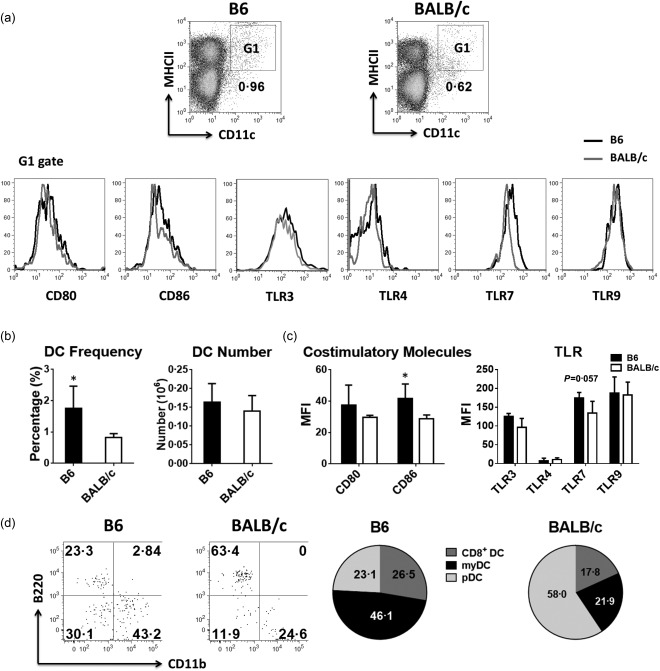
Number, frequency and phenotype of dendritic cells (DCs) in naive B6 and BALB/c mice. Single cell suspensions of lymph nodes were prepared from untreated or phosphate‐buffered saline (PBS)‐injected mice. (a–c) The frequency, absolute number, expression level of co‐stimulatory molecules and Toll‐like receptors (TLRs) on DC [major histocompatibility complex (MHC) II+CD11c+, G1 gate] were analysed by flow cytometry (a) and the differences between the two strains are shown in bar plots (b,c). (d) DC subsets were identified by flow cytometry as CD8+ lymphoid (lyDC, MHC II+CD11c + B220–CD11b–), myeloid (myDC, MHC II+CD11c+B220–CD11b+) and plasmacytoid (pDC, MHC II+CD11c+B220+CD11b–) DCs in the CD19– fraction. Pie charts shows the difference in the proportions of these three DC subsets. One representative flow cytometry data is shown. The bar graphs and pie charts represent the mean ± standard deviation (s.d.); n = 4∼8 (n denotes the number of individual animals).
Immunization with any of the TLR agonists tested induced a significant increase in the total number of DCs in lymph nodes of B6 mice (Fig. 2a). In contrast, TLR‐3 (poly (I:C) and TLR‐7/8 (R848) agonists were the only two adjuvants that could recruit and increase the number of DCs efficiently in the lymph nodes of BALB/c mice (Fig. 2a). However, the expression levels of the co‐stimulatory molecules CD80 and CD86 on DCs were up‐regulated variably in both strains after immunization. Interestingly, CD86 was more up‐regulated on DCs of immunized BALB/c mice while CD80 up‐regulation was more pronounced on DCs of immunized B6 mice, except for the TLR‐4 (MPLA) agonist (Fig. 2a). Finally, we observed that in both B6 and BALB/c mice, all TLR agonists tested mobilized the myeloid DC subset preferentially (Fig. 2b). However, the initial predominant myeloid DC subset in B6 and plasmacytoid DC subset in BALB/c remained high after immunization with any of the TLR agonists (Fig. 2b). Of note, the control adjuvant, alum, had a limited effect on DC recruitment and maturation.
Figure 2.
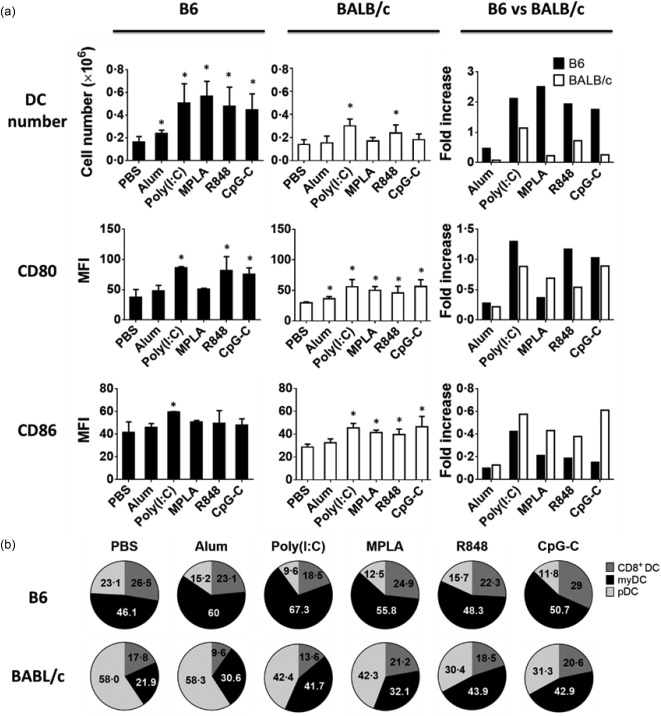
Recruitment of dendritic cells (DCs) in the lymph nodes of immunized mice. Mice were injected subcutaneously with ovalbumin (OVA) protein mixed with indicated Toll‐like receptor (TLR) agonists. (a, top panels) Bar graphs show the absolute numbers and fold increase over unimmunized mice of major histocompatibility complex (MHC) II+CD11C+ dendritic cells (DCs) in the lymph nodes of immunized B6 (filled black) and BALB/c (open bars) mice 2 days after immunization. (a, mid and bottom panels) Bar graphs show the expression level and fold change over unimmunized mice of CD80 and CD86 molecules on DCs from immunized B6 (filled black) and BALB/c (open bars) mice. (b) Pie charts display the changes in the distribution of the DCs subsets in the lymph nodes of the two strains after immunization. Data are expressed as mean ± standard deviation (s.d.); n = 2∼8 (n denotes number of individual animals).
TLR agonists induced NK recruitment in lymph nodes of B6 and BALB/c mice
NK cells express TLR‐1–9 and 11 18 and their recruitment to the lymph nodes promote Th1 immune response 13. Therefore, the ability to recruit NK cells has been considered as a criterion for selection of Th1 adjuvants. To gain insight into the influence of genetic background on the recruitment and activation of NK cells, we next monitored the changes in number, subsets and phenotype of NK cells in the lymph nodes of B6 and BALB/c mice 2 days after immunization with OVA protein mixed with the aforementioned adjuvants.
Prior to immunization, the absolute numbers of NK cells and NK subsets were significantly lower in lymph nodes of B6 compared to BALB/c mice (Fig. 3a,b). In addition, NK cells from B6 but not BALB/c mice expressed lower levels of CD69 (NK activation marker) and CD122 (the IL‐2/IL‐15 receptor β‐chain) and higher levels of CXCR3 and CD62L, both essential for the recruitment of NK cells to the lymph nodes 13 (Fig. 3c). Furthermore, we observed that NK cells from B6 mice expressed similar levels of TLR‐3 and TLR‐4 but significantly higher levels of TLR‐7 and TLR‐9 compared with their counterpart in BALB/c mice (Fig. 3c).
Figure 3.
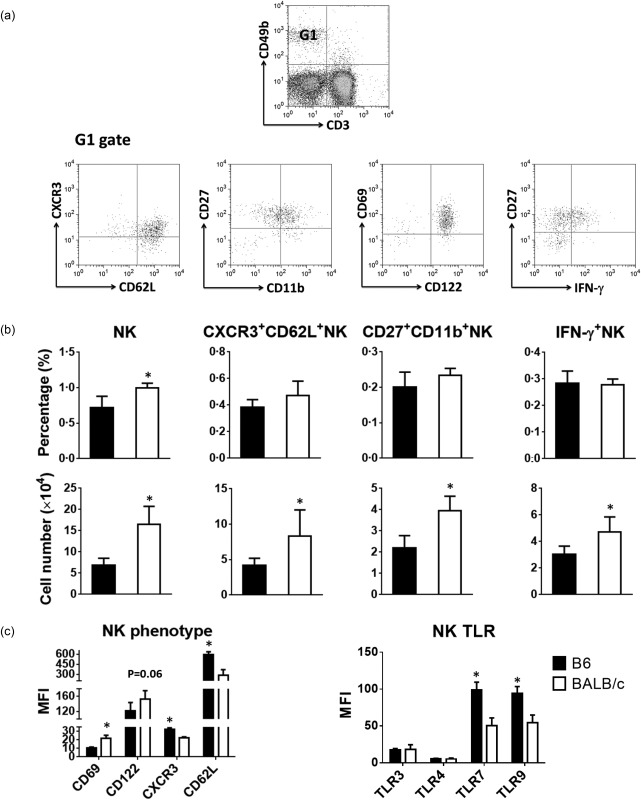
Number, frequency, and phenotype of natural killer (NK) cells in naive B6 and BALB/c mice. Cells from lymph nodes of untreated or phosphate‐buffered saline (PBS)‐injected mice were counted and analysed by flow cytometry. (a) Flow cytometry plots show the gating strategy to measure the frequency of CD3–CD49b+ natural killer (NK) cells and their subsets in the lymph nodes of B6 and BALB/c mice. One representative flow cytometry data is shown. (b) Bar graphs show the percentages and absolute numbers of NK cells and their subsets. (c) Bar graphs show the expression level of activation and migratory molecules and Toll‐like receptors (TLRs) on NK cells from B6 (filled black bars) and BALB/c (open bars). Data are expressed as mean ± standard deviation (s.d.); n = 4∼8 (n denotes number of individual animals).
After immunization, the number of NK cells was increased considerably in the lymph nodes of B6 mice by all TLR agonists, in particular TLR‐3 (poly I:C) (Fig. 4). In contrast, only TLR‐3 (poly I:C) or TLR‐7/8 (R848) agonists increased the NK cells significantly in lymph nodes of BALB/c mice (Fig. 4). Interestingly, poly I:C was more effective in recruiting NK cells in B6 than BALB/c mice, while R848 mobilized NK cells with similar effectiveness in both mouse strains (Fig. 4). We also observed that, in both B6 and BALB/c mice, the majority of the NK cells recruited to the draining lymph nodes were CXCR3‐ and CD62L‐expressing NK cells (Fig. 4) and displayed an activated phenotype, as evidenced by the up‐regulation of the activation marker CD69 (Fig. 4). Hayakawa et al. showed that activated NK cells expressing CD27+CD11b+ have a predominant role in cross‐talk with DCs and display effector functions and secrete high levels of IFN‐γ 19. We found that TLR‐3 (poly I:C) and TLR‐7/8 (R848) agonists were very effective in recruiting CD27+ NK cells producing IFN‐γ in B6 and BALB/c mice, respectively (Fig. 4). In contrast, TLR‐4 (MPLA) and TLR‐9 (CpG‐C) had little to no effect on the induction of these effector NK cells (Fig. 4) in BALB/c mice. Finally, we observed that alum had almost no effect on the recruitment and activation of NK cells in both strains (Fig. 4).
Figure 4.
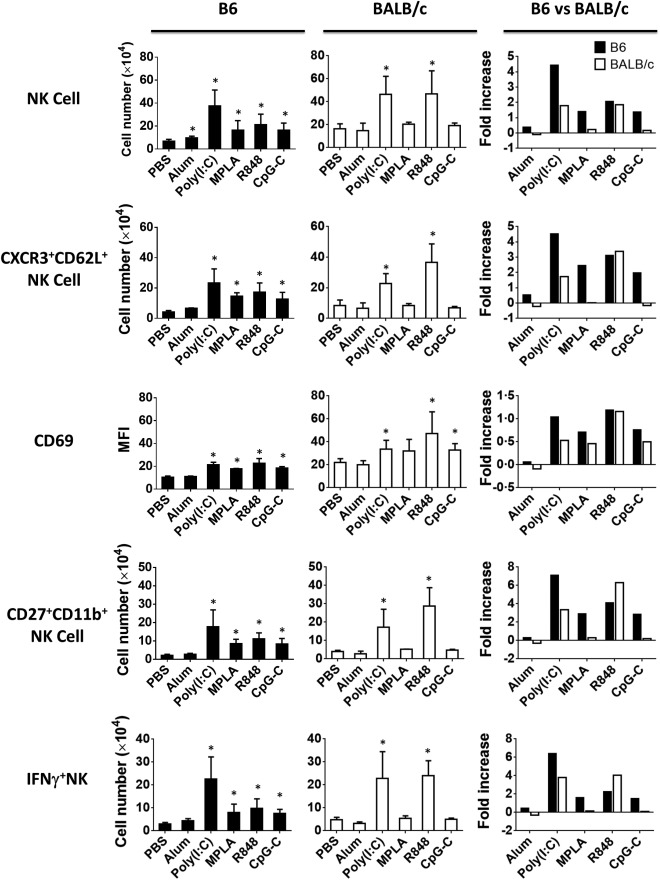
Recruitment and activation status of natural killer (NK) cells in the lymph nodes of immunized mice. Mice were injected subcutaneously with ovalbumin (OVA) protein mixed with indicated Toll‐like receptor (TLR) agonists. Two days after immunization, single cell suspensions from the collected lymph nodes were analysed by flow cytometry. Bar graphs (left and mid panels) show the difference in the absolute number of CD3–CD49b+ NK, CXCR3+CD62+ NK, CD27+CD11b+ NK, interferon (IFN)‐γ‐producing CD27+ NK cells, their expression of activation marker CD69 and fold increase (right panels) over unimmunized mice in the lymph nodes of immunized B6 (filled black) versus BALB/c (open bars) mice. Data are expressed as mean ± standard deviation (s.d.); n = 2∼8 (n denotes number of individual animals).
TLR agonists induced cellular and humoral‐mediated immunity in lymph nodes of B6 and BALB/c mice
The magnitude of immune response to vaccines can be measured by the production of effector CD4+ helper and CD8+ cytotoxic T cells and Igs. To evaluate the impact of genetic background on induction of immune response by the TLR agonists, we analysed the changes in number of CD4+ and CD8+ IFN‐γ‐secreting T cells and B cells producing Igs after immunization.
Prior to immunization, the percentage and absolute number of T cells, in particular CD4+ T cells in the lymph nodes of BALB/c mice, were significantly higher than those in B6 mice (Fig. 5a,b). Following immunization, the absolute numbers of IFN‐γ‐producing CD4+ and CD8+ T cells were increased variably from the baseline in the lymph nodes of both strains in response to all TLR agonists, except for CD4+ T cells in BALB/c mice immunized with alum and TLR‐4 (MPLA) agonist (Fig. 5c). Interestingly, TLR‐3 (poly I:C) agonist was the most effective adjuvant to induce both CD4+ and CD8+ effector T cells in B6 and CD4+ effector T cells in BALB/c mice (Fig. 5c). In contrast, TLR‐7/8 (R848) agonist was the most effective adjuvant to induce both CD4+ and CD8+ effector T cells in BALB/c mice and CD8+ effector T cells in B6 mice (Fig. 5c).
Figure 5.
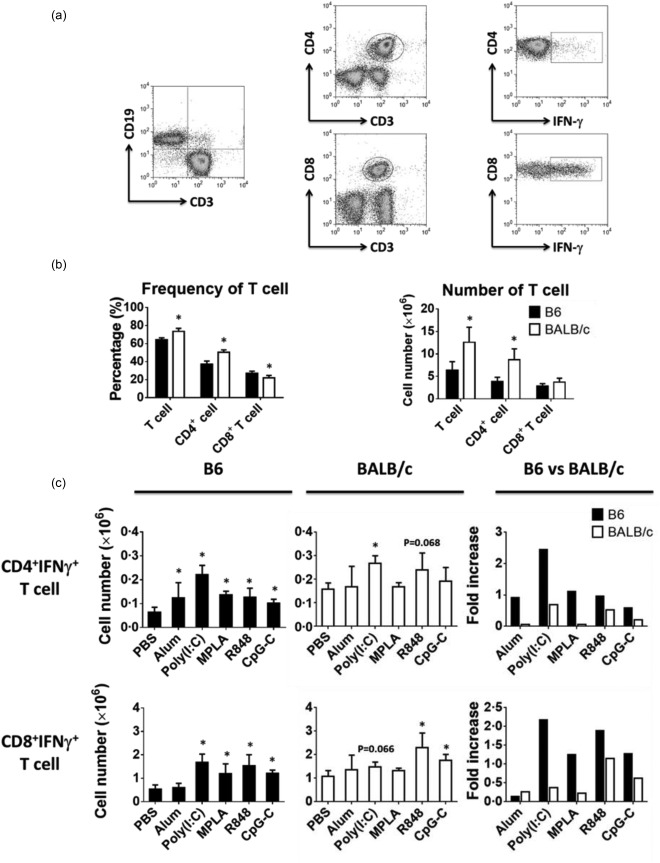
Difference in the number of T cells in naive B6 and BALB/c mice and development of interferon (IFN)‐γ‐producing effector T cell in response to immunization. (a) Flow cytometry plots show the gating strategy to identify B and T cells subsets from lymph nodes of untreated or phosphate‐buffered saline (PBS)‐injected mice. One representative flow cytometry data is shown. (b) Bar graph show the difference in the percentages and absolute numbers of T cells between the two strains. (c) Mice were injected subcutaneously with ovalbumin (OVA) protein mixed with indicated adjuvants. Bar graphs (left and mid panels) show the absolute number of IFN‐γ‐producing effector T cells developed 3 days after immunization and the fold increase (right panels) over PBS immunization in B6 (filled black) and BALB/c (open bars) mice. Data are expressed as mean ± standard deviation (s.d.); n = 2∼8 (n denotes number of individual animals).
In naive mice, the percentage and absolute number of B cells in both strains were almost identical (Fig. 6a,b). After immunization, however, the number of B cells increased in both strains variably, but with the same trend, in response to the TLR agonists (Fig. 7a). In addition, the fold increase in B cells was higher in the lymph nodes of B6 compared to BALB/c mice (Fig. 7a). Following immunization, both strains showed similar amounts of OVA‐specific Igs in their serum except for the TLR‐7/8 (R848) agonist, that produced higher but not significant amount of Igs in BALB/c mice (Fig. 7b). Our data also reveal the differences in the amounts of OVA‐specific Ig subsets in sera of these mice in response to various immunizations. Indeed, B6 mice produced significantly lower amounts of IgG2a in response to any of the TLR agonists. However, consistent with the frequency of IFN‐γ‐producing effector cells observed in individual strains following immunization, we detected more OVA‐specific IgG2a in serum of B6 and BALB/c strains in response to TLR‐3 (poly I:C) and TLR‐7/8 (R848) agonists, respectively. Finally, the BALB/c mice produced more IgG1 in response to TLR‐7/8 (R848) while B6 mice produced more IgG2b in response to the TLR‐4 (MPLA) agonist (Fig. 7b). Overall, our data also show that when considering the fold increase in absolute numbers of CD4+ and CD8+ effector T cells and B cells, B6 mice were more responsive to immunization by any of the TLR agonists than BALB/c mice. However, BALB/c mice produced more Igs, in particular IgG2a, than B6 mice in response to all agonists.
Figure 6.
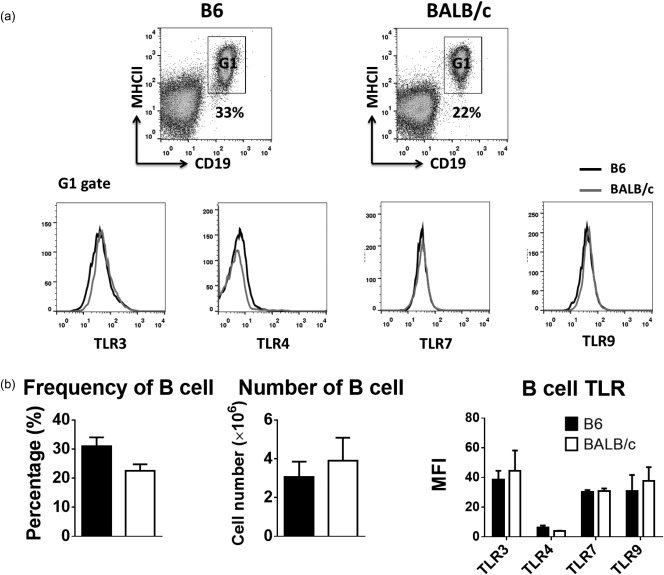
Number, frequency and Toll‐like receptor (TLR) expression of B cells in naive B6 and BALB/c mice. (a) Flow cytometry plots show the gating strategy to identify B cell subsets from lymph nodes of untreated or phosphate‐buffered saline (PBS)‐injected mice and the difference in expression levels of TLRs on B cells between two strains. One representative flow cytometry data is shown. (b) Bar graph show the difference in the percentages, absolute numbers and TLR expression of B cells between the two strains. Data are expressed as mean ± standard deviation (s.d.); n = 4∼8 (n denotes number of individual animals).
Figure 7.
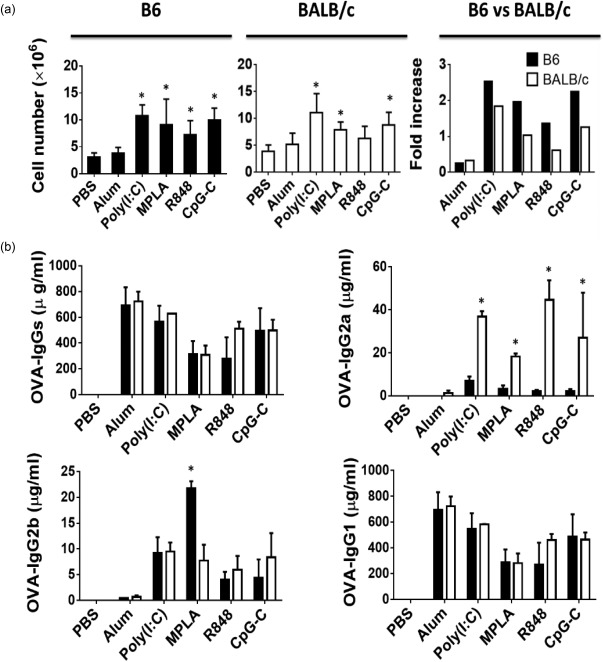
Absolute number of B cells and production of anti‐ovalbumin (OVA) antibodies in immunized mice. (a) Bar graphs show the percentages of B cells 3 days after immunization (left and mid panels) and the fold increase (right panels) over phosphate‐buffered saline (PBS) immunization in B6 (filled black) and BALB/c (open bars) mice. (b) Bar graphs show the differences in the amounts of OVA‐specific immunoglobulin (Ig) subsets in sera between the two strains a week after the second immunization. Data are expressed as mean ± standard deviation (s.d.); n = 2∼8 (n denotes number of individual animals).
Discussion
In the present study, we compared the cellular and humoral responses of two most commonly used inbred mouse strains to a protein vaccine formulated with different TLR agonists. Our data show that B6 mice overall recruited more DCs (Fig. 2) and NK cells (Fig. 4) to the lymph nodes and mounted strong cellular responses (Fig. 5). Among the TLR agonists tested, TLR‐3 (poly I:C) and TLR‐7/8 (R848) agonists were the most effective adjuvants in inducing IFN‐γ‐producing effector cells in both strains. However, poly I:C generated more effector cells in B6 mice, whereas R848 induced a comparable fold increase in effector cells in both strains. In addition, TLR‐4 (MPLA) and TLR‐9 (CpG‐C) agonists recruited DCs and NK cells and induced cellular responses moderately in B6 mice, although their cellular adjuvanticity in BALB/c mice were almost minimal. Finally, B6 mice recruited more B cells into the lymphoid tissues in response to all TLR agonists but overall produced fewer total OVA‐specific Igs, particularly IgG2a, with the exception of IgG2b in response to TLR‐4 (MPLA) (Fig. 7). These results could be interpreted by some of the pre‐existing factors that might contribute to the differences in innate and adaptive immune responses observed in two genetically disparate mouse strains.
First, prior to immunization, B6 mice had a higher percentage of DCs in the draining lymph nodes (Fig. 1b), the majority of which were conventional DCs (myeloid DCs and lymphoid CD8+ DCs). In contrast, the majority of DCs in BALB/c mice were plasmacytoid DCs (Fig. 1d), which was consistent with a previous report where plasmacytoid DC antigen 1 (PDCA‐1) was used to identify pDCs 20. It is known that myeloid DCs have a predominant role in MHC‐II presentation, and CD8+ lymphoid DCs are the source of IL‐12 21, 22, 23, 24, 25 and IFN‐γ at an early stage of infection 26 and have superior ability in cross‐presentation and activation of cytotoxic T cell 27, 28. Conversely, pDCs express lower levels of MHC II and co‐stimulatory molecules 29 and are incapable of stimulating proliferation and polarization of T cells unless activated by TLR‐7 or TLR‐9 agonists 30, 31. Immunization with TLR agonists increased the frequency of DCs variably in both strains (Fig. 2). However, we observed more recruitment of DCs expressing a higher level of migratory molecule, CCR7 (data not shown) to the lymph nodes of B6 mice, which consistently maintained higher percentages of myeloid and CD8+ lymphoid DCs in response to TLR agonists (Fig. 2). It is reported that co‐stimulatory signalling through CD80 may favour Th1, while CD86 favours Th2 polarization 32. We found that DCs in the draining lymph nodes of pre‐immunized B6 mice, similar to what has been reported for B6 splenic DCs 33, expressed higher levels of co‐stimulatory molecules, CD80 and CD86 (Fig. 1c). When compared to BALB/c DCs, they also showed a higher fold increase in CD80 expression after immunization. Overall, the differences in frequency, subtypes, recruitment and differential expression of co‐stimulatory markers observed between DCs of these two strains could contribute partially to stronger cellular responses observed in immunized B6 mice.
In addition to DCs, the number of NK cells recruited to the lymph nodes was shown to be higher in B6 mice following immunization (Fig. 4). Previous studies have indicated that the spleen is the source of NK cells recruited to the lymph nodes during the inflammation and NK expression of CD62L and CXCR3 are indispensable for their recruitment 13, 34, 35. Our finding that B6 splenic NK cells displayed higher levels of CD62L and CXCR3 may have favoured their recruitment potentially in response to CXCR3 chemokines, CXCR9 and CXCR10 36, 37, 38, which are shown to be produced in more quantities by activated B6 compared to BALB/c DCs 38. Naive BALB/c mice harbour a higher percentage and a greater number of NK cells with more a mature and pre‐activated phenotype (Fig. 3). Interestingly, however, they recruited further NK cells poorly in response to TLR‐4 and TLR‐9 compared to other TLR agonists (Fig. 4). This might be due to variable requirements for recruitment of NK cells by these agonists. Investigation by Uchida et al. indicated that depletion of CD11c+ conventional DC blocked TLR‐4‐mediated NK cell recruitment to the draining lymph nodes completely and had a mild to moderate effect on TLR‐9‐mediated NK cell recruitment 39. Therefore, the lower percentage of conventional DCs in BALB/c mice (39 versus 72·6%) could influence further the very low changes in NK recruitment seen in response the TLR‐4 and TLR‐9 agonists.
Our data show that recruited NK cells in B6 mice have a higher fold change in the expression levels of activation and effector markers CD69 and IFN‐γ, respectively, except for TLR‐7/8 (R848) (Fig. 4). The observed difference in the NK cell activation between two strains after immunization could be interpreted by the levels of cytokines produced by DCs. IL‐15 carries out a pivotal role in homeostasis and function of NK cells 40. Two research groups have shown that IL‐15 trans‐presented by DCs is necessary and sufficient for priming NK cells to perform cytotoxicity or to produce IFN‐γ in vitro and in vivo 41, 42. IL‐12, produced mainly by mature DCs, is another crucial cytokine involved in the DC–NK cross‐talk. In vivo, IL‐12 is produced at an early stage of infection and enhances the IFN‐γ production by NK cells 43 which, in turn, strengthen the IL‐12 secretion by DCs 44 and promote the differentiation of monocytes into DCs 45. Liu et al. reported that compared to BALB/c, DCs from B6 mice express higher levels of IL‐15 and IL‐12 after Listeria infection 46. Interestingly, we have observed that B6 DCs produce more IL‐12 in response to all TLR agonists except for TLR‐7/8 (R848), which induced a similar level of activation in both strains (data not shown). Altogether, our results suggest that B6 mice recruit DC and NK cells more effectively and promote DC–NK cross‐talk in response to vaccines formulated with various TLR agonists.
We observed that TLR‐3 (poly I:C) induces more recruitment of NK cells in B6 compared to BALB/c mice (Fig. 4), due possibly to the role of type I IFN in NK recruitment. A study by Uchida et al. showed that blockage of type I IFN abolished the TLR‐7‐mediated NK cell recruitment 39. Differing from plasmacytoid DCs, which is the exclusive source of TLR‐7 agonist‐induced type I IFN 12, the origin of type I IFN in response to TLR‐3 (poly I:C) is from multiple cell types, including haematopoietic cells (CD8+DEC205+ DCs and monocytes) and non‐haematopoietic stromal cells 47. Using a mouse cytomegalovirus (MCMV) infection model, a time–course analysis demonstrated that the production of type I IFN was biphasic. The initial peak occurred at 8 h post‐infection and stromal cells were the major contributor. The second wave appeared between 36 and 72 h and plasmacytoid DCs were the major supplier of IFN. Although both B6 and BALB/c strains responded in the same biphasic fashion, B6 produced a much higher level of type I IFN, especially at initial phase 48. Because the depletion of CD11c+ conventional DCs has a moderate effect on TLR‐3‐mediated NK recruitment 39 and TLR‐3 is absent on plasmacytoid DCs 49, TLR‐3 (poly I:C) may have induced only the initial phase of type I IFN production in both strains, which is reported to be higher in B6 mice 48. Interestingly, TLR‐7, with high expression on plasmacytoid DCs 49, induced a similar fold increase of NK cell recruitment in both B6 and BALB/c strains (Fig. 4) which could be facilitated by the abundant plasmacytoid DCs, the source of IFN‐α, found in lymph nodes of BALB/c mice (Fig. 1).
Our data show that TLR‐3 (poly I:C) and TLR‐7/8 (R848) were superior to other adjuvants to induce more Th1 effector cells in both strains (Fig. 5). However, TLR‐3 (poly I:C) was far more effective in inducing IFN‐γ‐producing effector cells in B6 mice because of at least two reasons. First, TLR‐3 (poly I:C) recruits more DCs and NK cells in B6 mice (Figs 2, 4). Secondly, B6 mice produce a higher level of IFN‐α, which is shown to be essential for poly I:C‐mediated development of Th1 effector cells 47. Conversely, TLR‐7/8 (R848) induced IFN‐γ‐producing effector cells more effectively in BALB/c mice. One of the reasons could be the observation that TLR‐7/8 (R848) recruits more plasmacytoid DCs in BALB/c mice that could respond to TLR‐7/8 (R848) by producing IFN‐α, which is known to be important in augmenting Th1 immunity 50. Also, we have found that ex‐vivo‐generated bone marrow‐derived BALB/c DCs produce more IL‐12 in response to TLR‐7/8 (R848) when compared to B6 DCs (data not shown), which could support further the development of IFN‐γ‐producing effector cells in BALB/c mice (Fig. 5). Indeed, besides type I IFN, IL‐12 was also shown to be indispensable for maximal production of Th1 effector cells in response to TLR‐7/8 signalling 12, 50. Collectively, our data suggest that, compared to poly I:C, the difference in IFN‐γ‐producing effector cells induced by TLR‐7/8 (R848) is less pronounced between the strains (Fig. 5).
A recent study, in which different TLR agonists (including TLR‐2/6, TLR‐1/2, TLR‐3, TLR‐4, TLR‐7/8, TLR‐9) were compared in the CxB6 F1 mouse model, identified TLR‐3 agonist followed by the TLR‐7/8 agonist as the most robust adjuvants in inducing CD4+ Th1 immunity 47. In line with this study, we also showed that TLR‐3 (poly I:C) ranked first, followed by TLR‐7/8 (R848) among five adjuvants in inducing Th1 effector cells in the B6 strain. However, this order was reversed when the same adjuvants were tested in BALB/c strain. Finally, although we observed a correlation between TLR‐3 (poly I:C) and TLR‐7/8 (R848)‐induced IFN‐γ‐producing effector cells and IgG2a production in these mice strains, it is surprising that B6 mice, with the marked overall propensity towards the development of Th1 cells, produced significantly lower amounts of OVA‐specific serum IgG2a. In summary, our study reveals the differences in TLR agonists‐induced engagement of innate and adaptive immune components for effective cellular and humoral immunity in genetically non‐identical mouse strains. It also provides a useful reference when the selection of adjuvants and interpretation of results are based on criteria such as DC subsets and NK recruitment and/or the magnitude of B and T cell responses.
Disclosure
The authors have no financial conflicts of interest.
Author contributions
M. N. S. designed and supervised the study. M. Z. and E. N. S. carried out the experiments. M. N. S., M. Z. and E. G. wrote the manuscript. All authors have read and approved the final manuscript.
Acknowledgements
This work was supported by the National Institute of Allergy and Infectious Diseases, National Institute of Health Grant 1R03AI103750‐01A1 (to M. N. S). We thank the staff in the animal facility for excellent animal care. We also thank Miss Saba Tamjidi and Miss Brittany Bible for their assistance in collecting the tissues and Ms Carrie Perez for editing the manuscript.
References
- 1. O'Hagan DT, De Gregorio E. The path to a successful vaccine adjuvant – ‘the long and winding road’. Drug Discov Today 2009; 14:541–51. [DOI] [PubMed] [Google Scholar]
- 2. Vasilakos JP, Tomai MA. The use of Toll‐like receptor 7/8 agonists as vaccine adjuvants. Expert Rev Vaccines 2013; 12:809–19. [DOI] [PubMed] [Google Scholar]
- 3. Akira S, Takeda K, Kaisho T. Toll‐like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2001; 2:675–80. [DOI] [PubMed] [Google Scholar]
- 4. Coffman RL, Sher A, Seder RA. Vaccine adjuvants: putting innate immunity to work. Immunity 33:492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Duin D, Medzhitov R, Shaw AC. Triggering TLR signaling in vaccination. Trends Immunol 2006; 27:49–55. [DOI] [PubMed] [Google Scholar]
- 6. Sandor F, Buc M. Toll‐like receptors. II. Distribution and pathways involved in TLR signalling. Folia Biol (Praha) 2005; 51:188–97. [DOI] [PubMed] [Google Scholar]
- 7. Takeda K, Akira S. Toll‐like receptors in innate immunity. Int Immunol 2005; 17:1–14. [DOI] [PubMed] [Google Scholar]
- 8. Akira S, Takeda K. Toll‐like receptor signalling. Nat Rev Immunol 2004; 4:499–511. [DOI] [PubMed] [Google Scholar]
- 9. Iwasaki A, Medzhitov R. Toll‐like receptor control of the adaptive immune responses. Nat Immunol 2004; 5:987–95. [DOI] [PubMed] [Google Scholar]
- 10. Cooper MA, Fehniger TA, Fuchs A, Colonna M, Caligiuri MA. NK cell and DC interactions. Trends Immunol 2004; 25:47–52. [DOI] [PubMed] [Google Scholar]
- 11. Deenick EK, Hasbold J, Hodgkin PD. Decision criteria for resolving isotype switching conflicts by B cells. Eur J Immunol 2005; 35:2949–55. [DOI] [PubMed] [Google Scholar]
- 12. Rajagopal D, Paturel C, Morel Y, Uematsu S, Akira S, Diebold SS. Plasmacytoid dendritic cell‐derived type I interferon is crucial for the adjuvant activity of Toll‐like receptor 7 agonists. Blood 2010; 115:1949–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martin‐Fontecha A, Thomsen LL, Brett S et al Induced recruitment of NK cells to lymph nodes provides IFN‐gamma for T(H)1 priming. Nat Immunol 2004; 5:1260–5. [DOI] [PubMed] [Google Scholar]
- 14. Sun S, Kishimoto H, Sprent J. DNA as an adjuvant: capacity of insect DNA and synthetic oligodeoxynucleotides to augment T cell responses to specific antigen. J Exp Med 1998; 187:1145–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sutton P, Doidge C, Pinczower G et al Effectiveness of vaccination with recombinant HpaA from Helicobacter pylori is influenced by host genetic background. FEMS Immunol Med Microbiol 2007; 50:213–9. [DOI] [PubMed] [Google Scholar]
- 16. Banchereau J, Briere F, Caux C et al Immunobiology of dendritic cells. Annu Rev Immunol 2000; 18:767–811. [DOI] [PubMed] [Google Scholar]
- 17. Reed SG, Bertholet S, Coler RN, Friede M. New horizons in adjuvants for vaccine development. Trends Immunol 2009; 30:23–32. [DOI] [PubMed] [Google Scholar]
- 18. Lauzon NM, Mian F, Ashkar AA. Toll‐like receptors, natural killer cells and innate immunity. Adv Exp Med Biol 2007; 598:1–11. [DOI] [PubMed] [Google Scholar]
- 19. Hayakawa Y, Smyth MJ. CD27 dissects mature NK cells into two subsets with distinct responsiveness and migratory capacity. J Immunol 2006; 176:1517–24. [DOI] [PubMed] [Google Scholar]
- 20. Asselin‐Paturel C, Brizard G, Pin JJ, Briere F, Trinchieri G. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J Immunol 2003; 171:6466–77. [DOI] [PubMed] [Google Scholar]
- 21. Maldonado‐Lopez R, De Smedt T, Michel P et al CD8alpha+ and CD8alpha‐ subclasses of dendritic cells direct the development of distinct T helper cells in vivo . J Exp Med 1999; 189:587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hochrein H, Shortman K, Vremec D, Scott B, Hertzog P, O'Keeffe M. Differential production of IL‐12, IFN‐alpha, and IFN‐gamma by mouse dendritic cell subsets. J Immunol 2001; 166:5448–55. [DOI] [PubMed] [Google Scholar]
- 23. Mashayekhi M, Sandau MM, Dunay IR et al CD8alpha(+) dendritic cells are the critical source of interleukin‐12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity 2011; 35:249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pulendran B, Smith JL, Caspary G et al Distinct dendritic cell subsets differentially regulate the class of immune response in vivo . Proc Natl Acad Sci USA 1999; 96:1036–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pulendran B, Lingappa J, Kennedy MK et al Developmental pathways of dendritic cells in vivo: distinct function, phenotype, and localization of dendritic cell subsets in FLT3 ligand‐treated mice. J Immunol 1997; 159:2222–31. [PubMed] [Google Scholar]
- 26. Ohteki T, Fukao T, Suzue K et al Interleukin 12‐dependent interferon gamma production by CD8alpha+ lymphoid dendritic cells. J Exp Med 1999; 189:1981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(–) dendritic cells cross‐prime cytotoxic T cells in vivo . J Exp Med 2000; 192:1685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dudziak D, Kamphorst AO, Heidkamp GF et al Differential antigen processing by dendritic cell subsets in vivo . Science 2007; 315:107–11. [DOI] [PubMed] [Google Scholar]
- 29. Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol 2011; 29:163–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Boonstra A, Asselin‐Paturel C, Gilliet M et al Flexibility of mouse classical and plasmacytoid‐derived dendritic cells in directing T helper type 1 and 2 cell development: dependency on antigen dose and differential toll‐like receptor ligation. J Exp Med 2003; 197:101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mouries J, Moron G, Schlecht G, Escriou N, Dadaglio G, Leclerc C. Plasmacytoid dendritic cells efficiently cross‐prime naive T cells in vivo after TLR activation. Blood 2008; 112:3713–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thompson CB. Distinct roles for the costimulatory ligands B7‐1 and B7‐2 in T helper cell differentiation? Cell 1995; 81:979–82. [DOI] [PubMed] [Google Scholar]
- 33. Liu T, Matsuguchi T, Tsuboi N, Yajima T, Yoshikai Y. Differences in expression of toll‐like receptors and their reactivities in dendritic cells in BALB/c and C57BL/6 mice. Infect Immun 2002; 70:6638–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pak‐Wittel MA, Yang L, Sojka DK, Rivenbark JG, Yokoyama WM. Interferon‐gamma mediates chemokine‐dependent recruitment of natural killer cells during viral infection. Proc Natl Acad Sci USA 2013; 110:E50–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen S, Kawashima H, Lowe JB, Lanier LL, Fukuda M. Suppression of tumor formation in lymph nodes by L‐selectin‐mediated natural killer cell recruitment. J Exp Med 2005; 202:1679–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sozzani S. Dendritic cell trafficking: more than just chemokines. Cytokine Growth Factor Rev 2005; 16:581–92. [DOI] [PubMed] [Google Scholar]
- 37. Yoneyama H, Narumi S, Zhang Y et al Pivotal role of dendritic cell‐derived CXCL10 in the retention of T helper cell 1 lymphocytes in secondary lymph nodes. J Exp Med 2002; 195:1257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zanoni I, Foti M, Ricciardi‐Castagnoli P, Granucci F. TLR‐dependent activation stimuli associated with Th1 responses confer NK cell stimulatory capacity to mouse dendritic cells. J Immunol 2005; 175:286–92. [DOI] [PubMed] [Google Scholar]
- 39. Uchida T, Scumpia PO, Murasko DM et al Variable requirement of dendritic cells for recruitment of NK and T cells to different TLR agonists. J Immunol 2007; 178:3886–92. [DOI] [PubMed] [Google Scholar]
- 40. Marcais A, Viel S, Grau M, Henry T, Marvel J, Walzer T. Regulation of mouse NK cell development and function by cytokines. Front Immunol 2014; 4:450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koka R, Burkett P, Chien M, Chai S, Boone DL, Ma A. Cutting edge: murine dendritic cells require IL‐15R alpha to prime NK cells. J Immunol 2004; 173:3594–8. [DOI] [PubMed] [Google Scholar]
- 42. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans‐presenting interleukin 15. Immunity 2007; 26:503–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Trinchieri G. Interleukin‐12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 2003; 3:133–46. [DOI] [PubMed] [Google Scholar]
- 44. Gerosa F, Baldani‐Guerra B, Nisii C, Marchesini V, Carra G, Trinchieri G. Reciprocal activating interaction between natural killer cells and dendritic cells. J Exp Med 2002; 195:327–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Goldszmid RS, Caspar P, Rivollier A et al NK cell‐derived interferon‐gamma orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity 2012; 36:1047–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu T, Nishimura H, Matsuguchi T, Yoshikai Y. Differences in interleukin‐12 and −15 production by dendritic cells at the early stage of Listeria monocytogenes infection between BALB/c and C57 BL/6 mice. Cell Immunol 2000; 202:31–40. [DOI] [PubMed] [Google Scholar]
- 47. Longhi MP, Trumpfheller C, Idoyaga J et al Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med 2009; 206:1589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schneider K, Loewendorf A, De Trez C et al Lymphotoxin‐mediated crosstalk between B cells and splenic stroma promotes the initial type I interferon response to cytomegalovirus. Cell Host Microbe 2008; 3:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Edwards AD, Diebold SS, Slack EM et al Toll‐like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol 2003; 33:827–33. [DOI] [PubMed] [Google Scholar]
- 50. Kastenmuller K, Wille‐Reece U, Lindsay RW et al Protective T cell immunity in mice following protein‐TLR7/8 agonist‐conjugate immunization requires aggregation, type I IFN, and multiple DC subsets. J Clin Invest 2011; 121:1782–96. [DOI] [PMC free article] [PubMed] [Google Scholar]