

Summary
West Nile virus (WNV) infection is a mosquito‐borne zoonosis with increasing prevalence in the United States. WNV infection begins in the skin, and the virus replicates initially in keratinocytes and dendritic cells (DCs). In the skin and cutaneous lymph nodes, infected DCs are likely to interact with invariant natural killer T cells (iNKTs). Bidirectional interactions between DCs and iNKTs amplify the innate immune response to viral infections, thus controlling viral load and regulating adaptive immunity. iNKTs are stimulated by CD1d‐bound lipid antigens or activated indirectly by inflammatory cytokines. We exposed human monocyte‐derived DCs to WNV Kunjin and determined their ability to activate isolated blood iNKTs. DCs became infected as judged by synthesis of viral mRNA and Envelope and NS‐1 proteins, but did not undergo significant apoptosis. Infected DCs up‐regulated the co‐stimulatory molecules CD86 and CD40, but showed decreased expression of CD1d. WNV infection induced DC secretion of type I interferon (IFN), but no or minimal interleukin (IL)−12, IL‐23, IL‐18 or IL‐10. Unexpectedly, we found that the WNV‐infected DCs stimulated human iNKTs to up‐regulate CD69 and produce low amounts of IL‐10, but not proinflammatory cytokines such as IFN‐γ or tumour necrosis factor (TNF)‐α. Both CD1d and IFNAR blockade partially abrogated this iNKT response, suggesting involvement of a T cell receptor (TCR)–CD1d interaction and type I interferon receptor (IFNAR) signalling. Thus, WNV infection interferes with DC–iNKT interactions by preventing the production of proinflammatory cytokines. iNKTs may be a source of IL‐10 observed in human flavivirus infections and initiate an anti‐inflammatory innate response that limits adaptive immunity and immune pathology upon WNV infection.
Keywords: dendritic cell, flavivirus, human, invariant natural killer T cell
Introduction
West Nile virus (WNV) is an RNA genome flavivirus that is transmitted to humans via mosquito bites. While most infections are asymptomatic, some patients, especially elderly people and immunocompromised individuals, develop neuroinvasive disease, and 10% of these cases are lethal 1, 2. Despite this severe outcome, and the recent increase in the prevalence of WNV in the United States, the pathogenesis of WNV in humans remains ill defined, and there is no approved vaccine or therapy for human WNV infection.
WNV infection begins in the skin, and the virus replicates initially in keratinocytes and skin‐resident dendritic cells (DCs) 3. In the skin, or after migration to cutaneous lymph nodes, infected DCs are likely to interact with natural killer T cells (NKTs), an innate effector cell type important for control of pathogen infections 4, 5, 6. NKTs promote anti‐viral responses by activating DCs and contributing to innate immune responses controlling viral load and by regulating adaptive immunity [see 7, 8, 9, 10 for recent reviews]. NKTs recognize lipid antigens presented by the non‐classical major histocompatibility complex (MHC) molecule CD1d. Two categories of NKTs, types I and II, have been described. Type I or invariant NKTs (iNKTs) express a semi‐invariant T cell receptor (TCR) that uses a Vα24‐Jα18 rearrangement and Vβ11 TCR genes in humans. iNKTs respond to α‐galactosylceramide (αGalCer) and related lipids 5. This contrasts with type II NKTs (dNKTs), which do not react with αGalCer and express a much more diverse TCR repertoire 11. iNKTs are activated in response to pathogens either directly through their TCR or indirectly via cytokines. Direct activation is mediated by CD1d‐bound lipids derived from microbes (reviewed in 5), or can involve recognition of an altered complement of CD1d‐bound self‐lipids that arises during viral or bacterial infection as a result of changes in lipid metabolism or lipid loading onto CD1d 12, 13, 14, 15. Indeed, infection with hepatitis B virus leads to the synthesis of antigenic host lysophospholipids that are loaded on to CD1d, resulting in iNKT activation 16. Indirect activation of iNKTs is mediated by proinflammatory cytokines such as interleukin (IL)‐12, IL‐18 or type I interferons (IFNs) released by DCs in response to pathogen‐derived products. iNKTs are activated through this mechanism in response to the Toll‐like receptor (TLR)‐9 ligand cytosine–phosphate–guanosine (CpG) oligonucleotides 17, 18, 19, as well as cytomegalovirus (CMV) 18, 19, 20 and Dengue virus 21. Finally, iNKTs may be activated by a combination of weak responses to CD1d‐presented self‐antigens and inflammatory cytokines or type I IFN 13, 22, 23.
The nature of the DC response to virus infection influences immune regulatory pathways mediated by innate lymphocytes. Depending on the virus and the type of DC, infected DCs may undergo apoptosis or survive, modulate surface molecules recognized by lymphocytes and produce combinations of type I IFNs and pro‐ or anti‐inflammatory cytokines 24. Murine models of skin infection with the related Dengue flavivirus revealed two stages of DC infection – initial infection of resident dermal DCs within 12–24 h and a second wave of infection within 48 h of DCs derived from monocytes recruited to the inflamed dermis 25. Similarly, in a murine model of dermal WNV infection, bone marrow‐derived monocytes infiltrate the skin, cluster near infected fibroblasts and then differentiate into DCs 26. These reports suggest that human monocyte‐derived DCs are a relevant model for the study of DC‐intrinsic flavivirus infection. Indeed, prior reports showed that human monocyte‐derived DCs are permissive for WNV infection and produce type I IFN upon contact with the WNV dsRNA genome replication intermediate 27, 28, 29, 30. In human epithelial cells, WNV interferes with the host IFN response by inhibiting the type I IFN receptor (IFNAR) mediated activation of signal transducer and activator of transcription‐1 (STAT‐1) 31. This blockade of IFNAR signalling did not apparently occur in human DCs, indicating that DCs may have distinct mechanisms to control WNV infection 27.
While these prior studies have contributed to our understanding of human DC responses to WNV, the ability of WNV‐infected human DCs to activate innate lymphocytes has not been studied. Herein, we have determined the impact of WNV Kunjin (WNVKUN) infection on interactions between human DCs and iNKT cells. In WNV‐infected human monocyte‐derived DCs, we studied viral replication and protein expression, co‐stimulatory molecule and CD1d surface expression and production of type I IFNs, proinflammatory cytokines and IL‐10. We then determined the ability of WNV‐infected DCs to activate human blood iNKTs and investigated the role of CD1d and IFNAR in the iNKT response. Our data show that WNV‐infected DCs produce significant amounts of type I IFN but fail to activate the proinflammatory function of iNKTs.
Materials and methods
Generation of monocyte‐derived DCs
Heparinized peripheral blood was obtained from healthy volunteers with informed consent according to a venipuncture protocol approved by the OMRF Institutional Review Board. Leucocyte buffy coats from anonymous donors were purchased from the Oklahoma Blood Institute. Peripheral blood mononuclear cells (PBMCs) were isolated using lymphocyte separation medium gradients (Mediatech Inc., Manassas, VA, USA). CD14+ monocytes were isolated by negative selection using an EasySep human monocyte enrichment kit (Stem Cell Technologies, Vancouver, BC, Canada). Monocytes were cultured at 106/ml in RPMI, 10% fetal calf serum (FCS) with 30 ng/ml granulocyte–macrophage colony‐stimulating factor (GM‐CSF) and 20 ng/ml IL‐4 (recombinant cytokines from Peprotech, Rocky Hill, NJ, USA) for 6 days to promote DC differentiation as described previously 32. Differentiated DCs were CD14–CD11c+CD209+human leucocyte antigen D‐related (HLA‐DR)+.
WNV stocks
WNVKUN (a BSL2 isolate) and Aedes albopictus C6/36 mosquito cells were a kind gift from Dr M. Diamond (Washington University, Saint Louis, MO, USA). To generate virus stocks, exponentially growing C6/36 cells were infected with WNVKUN and grown at 28ºC for 4 days 33. Supernatants were collected, clarified by centrifugation, aliquoted and frozen at −80º C. One aliquot was used to titre the virus, using either plaque assays 33 or a flow cytometry‐based assay adapted from 34.
Assessment of DC activation
On day 6 after differentiation was initiated, DCs were harvested, replated at 5 × 105/ml and either left unstimulated, infected with WNVKUN [multiplicity of infection (MOI) 1, 5 or 50] or activated with polyIC (pIC, 50 μg/ml) or lipopolysaccharide (LPS) (100 ng/ml) + IFN‐γ (2000 IU/ml). After 24–48 h, both stimulated and unstimulated DCs were assessed for changes in cell surface markers using flow cytometry or placed in Trizol for later isolation of mRNA. DC supernatants were collected from duplicate wells, each containing 50 000 DCs in 200 μl, for measurement of secreted cytokines.
Monoclonal antibodies (mAbs) and flow cytometry
Cells were preincubated with human FcR‐binding inhibitor (eBioscience, San Diego, CA, USA) and 2% human serum, and labelled with optimally titred mAbs in fluorescence activated cell sorter (FACS) buffer [phosphate‐buffered saline (PBS), 5% newborn calf serum, 0.1% sodium azide]. DCs were stained with six to seven parameter combinations of fluorochrome‐labelled mAbs specific for CD14 (clone M5E2), CD11c (B‐LY6), CD209 (9E9A8), HLA‐DR (L243), CD40 (5C3), CD86 (IT2.2) and CD1d (51.1) [obtained from BD Biosciences, San Jose, CA, USA, eBioscience or Biolegend, San Diego, CA, USA]. iNKT cells were stained with mAbs specific for CD69 (FN50), OX‐40 (Ber‐ACT35) and CD107a/lysosomal‐associated membrane protein 1 (LAMP1) (H4A3), CD3 (OKT3) and CD1d‐PBS‐57 tetramer (obtained from the NIH tetramer facility). The following reagents were obtained through the National Institutes of Health (NIH) Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH: monoclonal anti‐West Nile virus Envelope (Env) protein, clone E34 (produced in vitro), NR‐10137 and clone E24 (produced in vitro), NR‐10136; and monoclonal anti‐WNV non‐structural protein 1, clone 22‐NS‐1 (produced in vitro), NR‐10145. Anti‐WNV Env and NS‐1 mAbs were labelled with an Alexa Fluor 647 monoclonal antibody labeling kit (Invitrogen, Carlsbad, CA, USA), and intracellular staining was accomplished using a BD cytofix/cytoperm kit. A live/dead fixable Aqua dye (Invitrogen) and a mAb to activated caspase 3 (BD Biosciences) were used to determine DC apoptosis and viability. Samples were run on an LSRII instrument (BD Biosciences) and the data were analysed with FlowJo software (TreeStar Inc., Ashland, OR, USA) software.
Analyses of gene expression
RNA was extracted using an RNeasy/Trizol hybrid protocol and cDNA was synthesized using high‐capacity cDNA reverse transcription with RNase inhibitor kit (Applied Biosystems). Relative expression of WNV ENV was determined using the 2–ΔCt method with normalization to actin, beta (ACTB) expression using TaqMan technology. A Ct of 40 for the WNV ENV gene was used in the calculations for the uninfected samples. Specific primer/probe sequences were ACTB forward: 5′‐ATCCTGGCCTCGCTGTCCAC‐3′, reverse: 5′‐GGGCCGGACTCGTCATAC‐3′ probe: 5′‐6FAM TCCAGCAGATGTGGATCAGCAAGCA tetramethylrhodamine (TAMRA)−3′; WNV ENV forward: 5′‐TTCTCGAAGGCGACAGCTG‐3′, reverse: 5′‐CCGCCTCCATATTCATCATc‐3′ probe: 5′‐6FAM ATGTCTAAGGACAAGCCTACCA TAMRA‐3′ 35. Relative expression of CD1D was determined using the ΔΔCt method with normalization to HPRT expression using Sybr green technology. Specific primer sequences were: HPRT forward 5′‐TTGGTCAGGCAGTATAATCC‐3′, reverse 5′‐GGGCATATCCTACAACAAAC‐3′ 36; CD1D forward 5′‐GTGGCCTCCTTGAGTCA‐3′, reverse 5′‐ACAGGCTTTGGGTAGAATC‐3′.
iNKT purification and expansion
PBMCs were purified from buffy coats by centrifugation on a Ficoll gradient, and resuspended in 10% FCS‐RPMI. The next day, the PBMCs were incubated with allophycocyanin (APC)‐labelled CD1d‐PBS‐57 tetramer (obtained from the NIH tetramer facility), incubated with anti‐APC beads (Miltenyi Biotec, San Diego, CA, USA). After washing again, the tetramer+ cells were selected positively using magnetic columns (Miltenyi Biotec). Enriched cells were stained with CD3 and CD1d‐PBS‐57 tetramer and sorted (Aria; BD). The sorted cells (>98% iNKT) were then cultured with irradiated heterologous PBMCs in 10% FCS‐RPMI containing IL‐2 (100 U/ml) and IL‐7 (100 U/ml). Cells were stimulated with αGalCer (100 ng/ml) (Funakoshi, Tokyo, Japan) or α‐CD3 + α‐CD28 (1 μg/ml each), and expanded in 96‐well round‐bottomed plates. Fresh medium with cytokines was added every 72 h. Before use in experiments, cells were rested in cytokine‐free medium for 24–36 h.
iNKT–DC co‐culture
Rested iNKTs were incubated with WNV‐infected or control DCs (5 : 1) in 10% RPMI in 96‐well round‐bottomed plates. After 24 h, the supernatant was collected for cytokine detection, and the cells washed and stained with CD3, CD1d‐PBS‐57 tetramer, CD69, CD107a and OX‐40 to assess activation. In some experiments, an α‐CD1d blocking mAb (clone 51.1, 10 μg/ml), an anti‐IFNAR chain 2 mAb (clone MMHAR‐2, 5 μg/ml) or an isotype control mAb, or purified IL‐12p70 (100 ng/ml) were added at the beginning of the culture.
Cytokine assays
Cytokines secreted by DCs and iNKTs were measured using Luminex assays in the OMRF Serum Analyte and Biomarker core facility (Oklahoma City, OK, USA). ProcartaPlex kits (eBioscience) for DC cytokines (IFN‐α, IFN‐β, IL‐12p70, IL‐10, IL‐23, IL‐18, IL‐1β) and NKT cell cytokines (TNF‐α, IL‐22, IFN‐γ, IL‐4, IL‐10) were used according to the manufacturer's instructions.
Statistics
Statistical analyses were performed using Prism GraphPad software. The data involving multiple donor DCs and NKTs each exposed to different (three or more) stimuli were analysed using repeated‐measure (RM) one‐way analyses of variance (ANOVAs), followed by multiple comparison tests as indicated in the figure legends. Reported significance values compare each stimulated value to the corresponding unstimulated value. In some cases, a comparison between unstimulated and stimulated cells was made with a paired t‐test. The significance of the changes in iNKT responses (parameter fold induction) in the presence or absence of α‐CD1d blocking mAb or the α‐IFNAR mAb was determined by ratio paired t‐tests.
Results
WNV replicates its RNA genome within DCs, leading to expression of viral Env and non‐structural (NS‐1) proteins without significant cell death
We used WNVKUN, a naturally attenuated (BSL2) strain isolated from an infected individual 37, which is 98% identical at the amino acid level to the highly pathogenic North American WNVNY99. While WNVKUN showed enhanced sensitivity to the host type I IFN response in mice, the virus limited the type I IFN response of human epithelial cells in vitro, indicating that at least some of the WNV host interference mechanisms are intact in WNVKUN 31, 37. Thus, WNVKUN is an informative model for understanding the innate response of human DCs to WNV infection.
To determine an optimal time‐frame for detection of viral RNA, human monocyte‐derived DCs were incubated with WNVKUN at a multiplicity of infection (MOI) of 5 for 4–44 h. These preliminary studies showed that RNA corresponding to the WNV Env protein peaked at ∼44 h (Fig. 1a). In subsequent experiments, DCs from multiple donors were exposed to WNV at MOI = 1 and MOI = 5 for 44 h. WNV‐exposed DCs contained significant amounts of RNA corresponding to the WNV Env protein at both MOI, although donor variability was observed (Fig. 1b). However, infection at MOI = 5 led to few DCs expressing the viral Env protein (detected using intracellular staining with a specific mAb) after 24 or 44 h (1‐7%), despite the robust production of viral RNA (Fig. 1c).
Figure 1.
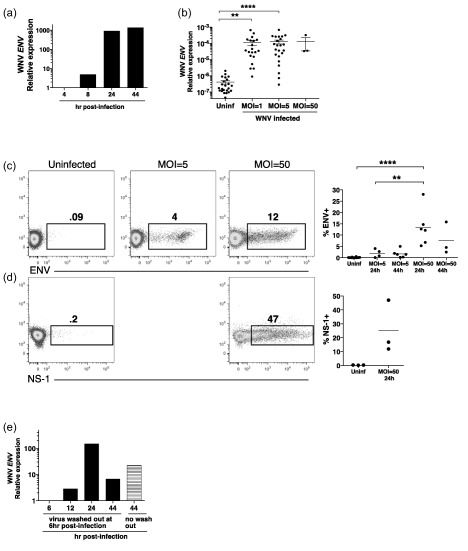
West Nile virus (WNV) infects human monocyte‐derived dendritic cells (DCs). (a) DCs were infected with multiplicity of infection (MOI) = 5 WNV Kunjin isolate (WNVKUN). RNA was collected at the indicated time‐points (4–44 h) post‐infection, and the cDNA assessed for expression of the ENV gene by quantitative polymerase chain reaction (qPCR). Shown is the expression of the WNV ENV gene relative to the host ACTB gene, with the data normalized to the values at the 4‐h time‐point. The data are representative of two independent experiments. (b) DCs were left uninfected (Uninf), or infected with MOI = 1 or MOI = 5 WNVKUN for 44 h. Symbols represent individual donors (n = 22). Three samples were infected at MOI = 50 for 24 h. The expression of the WNV ENV gene relative to the host ACTB gene is shown. Calculations were made using the 2–ΔCt method, as described in the Methods section, and a Ct of 40 for the WNV ENV gene was used in the calculations for the uninfected samples, as no signal for viral RNA was detected. The significance of the data was evaluated using a Friedman test followed by Dunn's multiple comparison test (comparing each stimulated value to the corresponding unstimulated value). **P < 0·01; ****P < 0·0001. (c,d) The presence of intracellular viral proteins envelope (Env) protein and non‐structural (NS‐1) protein in DCs (infected at the indicated MOIs) was determined by flow cytometry after 24 or 44 h. These data are representative of three to six similar experiments, and the results of all experiments varying MOIs and time post‐infection are compiled. (e) DCs were infected with WNV at MOI = 50, and the virus washed away after 6 h or left in for 44 h. RNA was collected at the indicated time‐points post‐infection, and the cDNA assessed for expression of the ENV gene by qPCR. Shown is the expression of the WNV ENV gene relative to the host ACTB gene, with the data normalized to the signal at the 6‐h time‐point. The data are representative of two independent experiments.
Because of the low percentage of DCs expressing viral protein, we next infected DCs at MOI = 50 and harvested the DCs 24 and 44 h post‐infection. The amount of viral Env RNA was comparable to that found in DCs incubated with WNV MOI = 5 (Fig. 1b). However, the fraction of DCs expressing Env protein (5–28%) was significantly higher upon exposure to MOI = 50 compared to MOI = 5 (Fig. 1c). Infected (MOI = 50 for 24 h) DCs also expressed the viral NS‐1 protein (12–47%) (Fig. 1d).
To determine if WNV replicated within DCs, the DCs were incubated with WNV at MOI = 50, and the virus was washed away after 6 h. RNA was collected at 6, 12, 24 and 44 h post‐infection. The amount of WNV Env RNA increased steadily between 6 and 24 h, after which it declined by 44 h to a level comparable to when the virus had not been washed out (Fig. 1e). This indicates that the WNV replicated its RNA genome within DCs. Taken together, the data shown in Fig. 1 suggest that virus infection was asynchronous at the population level, leading to detectable amounts of intracellular viral proteins in only 5–50% of DCs, depending on the MOI and time‐point post‐infection.
To determine if virus infection induced apoptosis, we determined levels of active caspase 3 in infected (MOI = 50) DCs at 24 h post‐infection. Fewer than 2% of the DCs contained active caspase 3, and most of those did not contain the Env protein (Fig. 2). Use of a viability stain showed very little cell death in the DC population. Similar results were obtained at 48 and 72 h post‐infection with MOI = 5 (not shown). Taken together, these experiments show that human monocyte‐derived DCs exposed to WNV were permissive to production of WNV RNA and protein without significant apoptosis or cell death.
Figure 2.
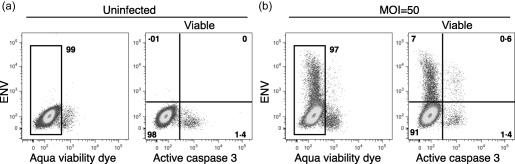
West Nile virus (WNV) infection did not induce significant dendritic cell (DC) apoptosis. DCs were (a) left uninfected or (b) infected with WNV Kunjin isolate (WNVKUN) at multiplicity of infection (MOI) = 50. After 24 h, cells were stained with monoclonal antibodies (mAbs) specific for envelope (Env) protein and activated caspase 3, and a fixable Aqua viability dye. The data are representative of five similar experiments.
WNV‐activated DCs produce type I IFN, but not significant amounts of other inflammatory cytokines
To determine DC production of soluble mediators post‐infection, we assayed DC culture supernatants for the presence of type I IFNs (IFN‐α and IFN‐β) and pro‐ and anti‐inflammatory cytokines, including IL‐12p70, IL‐18, IL‐1β, IL‐23 and IL‐10. We initially measured secreted cytokines at 24 and 48 h post‐infection with MOI = 5 and determined that the peak of cytokine production was at 48 h. At 24 h post‐infection, low amounts ( < 50 pg/ml) of IFN‐α were produced (Fig. 3a). At 48 h post‐infection, DCs infected with MOIs of 1 or 5 showed significant production of IFN‐α (300–700 pg/ml) and lesser amounts of IFN‐β (10–30 pg/ml) (Fig. 3a,b). This is consistent with a recent report that IFN‐α, more than IFN‐β, is a crucial mediator of the anti‐viral response to WNV in mice 38. Infection at MOI = 50 did not lead to increased amounts of secreted IFN‐α ( = 7.5 pg/ml at 24 h and = 198 pg/ml at 48 h) compared to the values at MOI = 5. Although donor variability was found and prior studies reported human sex differences in type I IFN production 39, 40, we did not identify a sex difference in the amounts of IFN‐α produced by WNV‐infected DCs; females, ± standard deviation (s.d.) = 700 ± 426 pg/ml and males, ± s.d. = 635 ± 478 pg/ml.
Figure 3.
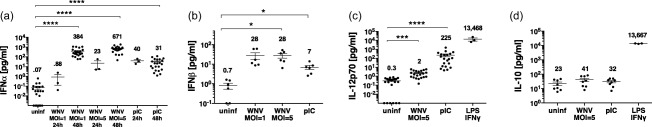
West Nile virus (WNV) infection induces dendritic cell (DC) production of interferon (IFN)‐α but not proinflammatory cytokines. DCs (50 000/well) were left unstimulated (uninf), infected with WNV Kunjin isolate (WNVKUN) at multiplicity of infection (MOI) = 1 or MOI = 5, or stimulated with poly‐inosine‐cytosine (pIC), and supernatants were harvested after 24 or 48 h. DC supernatants were assessed for amounts of (a) IFN‐α, (b) IFN‐β, (c) interleukin (IL)‐12p70 and (d) IL‐10 using Luminex assays. For IL‐12p70 and IL‐10, DCs were stimulated with lipopolysaccharide (LPS)/IFN‐γ as a positive control in some experiments. Symbols represent individual donors. Numbers within the graph are the mean amount of cytokine (pg/ml). Significance was evaluated using a repeated‐measure (RM) one‐way analysis of variance (anova) followed by a Dunnett's multiple comparison test (comparing each stimulated value to the corresponding unstimulated/uninfected value). *P < 0·05; ***P < 0·001; ****P < 0·0001.
At 48 h post‐infection with MOI = 1 or 5, WNV infected DCs produced minimal amounts of IL‐12p70 ( = 2 pg/ml) (Fig. 3c). Infected DCs also did not produce significant amounts of IL‐10 (Fig. 3d), IL‐18, IL‐1β or IL‐23 (not shown). DCs infected at MOI = 50 for 24 or 44 h did not secrete significant amounts of IL‐12p70 ( = 9 pg/ml) and IL‐10 ( = 23 pg/ml) relative to uninfected DCs. However, uninfected DCs stimulated via TLRs and other receptors with polyIC (pIC) and LPS/IFN‐γ were capable of robust production of IL‐12p70 and IL‐10 (Fig. 3c,d). In sum, WNV infection at MOI = 5 or MOI = 50 elicited DC production of type I IFN, but not IL‐12p70 or IL‐10. In subsequent experiments, we infected DCs at MOI = 5.
WNV infection led to increased expression of co‐stimulatory molecules by DCs
To determine if WNV infection induced DC up‐regulation of molecules important for T cell stimulation, we measured surface expression of the co‐stimulatory molecules CD40 and CD86. WNV infection induced the up‐regulation of CD40 and CD86 to a level comparable to that induced by pIC, a ligand for TLR‐3 and retinoic acid‐inducible gene 1 (RIG‐I) (Fig. 4). WNV exposure also led to increased expression of HLA‐DR, CD83, PDL1 and OX40L, but decreased expression of the IFN‐γR (not shown). The up‐regulation of co‐stimulatory molecules was uniform on the population, despite the fact that not all DCs showed expression of viral proteins. This suggests that type I IFNs or other mediators released from infected DCs activated most DCs in the population.
Figure 4.
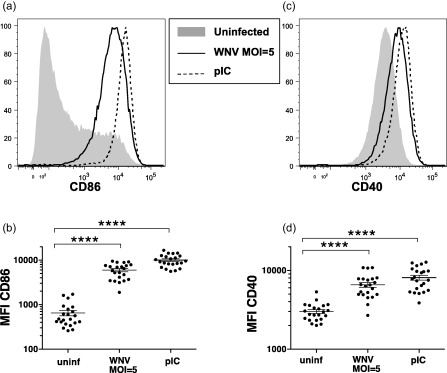
West Nile virus (WNV)‐infected dendritic cells (DCs) increase expression of T cell co‐stimulatory molecules. DCs were left unstimulated, infected with WNV Kunjin isolate (WNVKUN) at multiplicity of infection (MOI) = 1 or MOI = 5 or stimulated with poly‐inosine‐cytosine (pIC), and cells were harvested after 48 h. (a,b) CD86 and (c,d) CD40 surface expression (MFI = mean fluorescence intensity) on DCs was determined using flow cytometry. Symbols represent individual donors. Significance was evaluated using a repeated‐measure (RM) one‐way analysis of variance (anova) followed by a Dunnett's multiple comparison test (comparing each stimulated value to the corresponding unstimulated value). **P < 0·01; ***P < 0·001; ****P < 0·0001.
WNV‐activated DCs showed decreased levels of cell surface CD1d
We determined the impact of WNV infection on DC expression of CD1d, a lipid ligand‐bound molecule recognized by iNKTs. WNV infection of DCs induced increased levels of CD1D mRNA (Fig. 5a). However, display of cell surface CD1d on WNV exposed DCs actually decreased relative to uninfected cells (Fig. 5b,c). This effect was not unique to WNV exposure, as incubation of DCs with pIC and LPS/IFN‐γ also led to decreased surface CD1d (Fig. 5b,c). Incubation of DCs with the CD1d ligand αGalCer did not induce changes in CD1d surface expression (Fig. 5d) or elicit DC activation (not shown). This suggests that triggering of pattern recognition receptors leads to decreased surface expression of CD1d, and that the decrease in CD1d is unlikely to be due to a viral protein that blocks CD1d trafficking, as proposed for other viruses 41.
Figure 5.
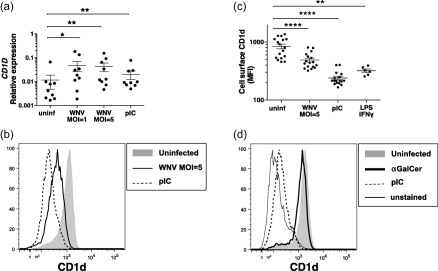
West Nile virus (WNV) infection decreases dendritic cell (DC) display of surface CD1d. DCs were left unstimulated, infected with WNV Kunjin isolate (WNVKUN) at multiplicity of infection (MOI) = 1 or MOI = 5 or stimulated with poly‐inosine‐cytosine (pIC), lipopolysaccharide/interferon‐gamma (LPS/IFN‐γ) or α‐galactosylceramide (αGalCer), and cells were harvested after 48 h. (a) DC cDNA was generated from RNA and assessed for expression of the CD1D gene by quantitative polymerase chain reaction (qPCR). Shown is the expression of the CD1D gene relative to the host ACTB gene. (b,d) CD1d surface expression (MFI) on DCs treated as indicated was determined using flow cytometry. (d) The signal of unstained DCs on the fluorescence channel used for anti‐CD1d. (a,c) Symbols represent individual donors. Significance was evaluated using a repeated‐measure (RM) one‐way analysis of variance (anova) followed by a Dunnett's multiple comparison test (comparing each stimulated value to the corresponding unstimulated value) or a paired t‐test (for unstimulated versus LPS/IFN‐γ). *P < 0·05; **P < 0·01; ****P < 0·0001.
WNV‐activated DCs activated iNKTs and stimulated their production of low levels of IL‐10 but not proinflammatory cytokines
iNKTs are activated optimally by CD1d, CD40 and IL‐12p70. WNV‐activated DCs produce type I IFN but not IL‐12p70, and express elevated CD40 but reduced CD1d. We determined if this DC profile led to activation of iNKTs. iNKTs were purified from blood of healthy donors based on their binding of CD1d‐PBS‐57 tetramers, expanded in vitro and rested prior to incubation with DCs. Because DC production of cytokines was optimal at MOI = 5, DCs were infected with WNV (MOI = 5) overnight and then cultured with iNKTs for 24 h. Unique combinations of DCs and iNKTs isolated from multiple donors were tested in this assay. iNKTs exposed to WNV‐infected DCs up‐regulated CD69 relative to those iNKTs exposed to unstimulated DCs (Fig. 6a,d,e). The CD69 increase was significant, although less pronounced than that induced by αGalCer‐pulsed DCs, indicating that WNV‐infected DCs were capable of activating iNKTs. In contrast, pIC activation of DCs led to minimal up‐regulation of CD69 by iNKTs (Fig. 6d,e). Other iNKTs activation markers such as OX‐40 and CD107a (LAMP1) were not up‐regulated significantly by WNV‐infected DCs (Fig. 6a–d).
Figure 6.
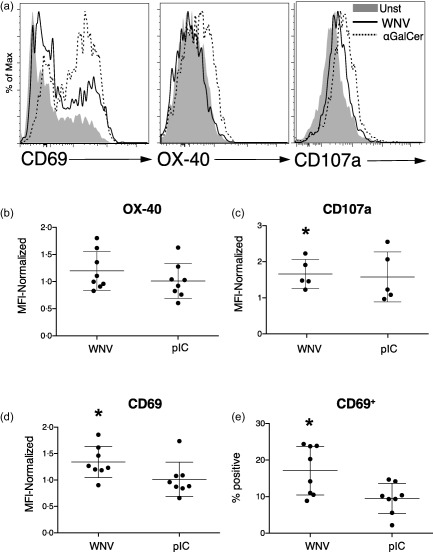
West Nile virus (WNV)‐infected dendritic cells (DCs) activate invariant natural killer T cells (iNKTs). DCs were left unstimulated, infected with WNVKUN at multiplicity of infection (MOI) = 5, incubated with the CD1d ligand α‐galactosylceramide (αGalCer) (100 ng/ml) or stimulated with poly‐inosine‐cytosine (pIC) (50 μg/ml) for 24 h. DCs (10 000/well) were then co‐cultured with iNKTs (50 000/well) that had been isolated from blood, expanded in vitro and rested. Purified iNKTs were CD3+ and bound the CD1d‐PBS−57 tetramer. (a) After 48 h, iNKT expression of CD69, OX‐40 and CD107a [lysosomal‐associated membrane protein 1 (LAMP1)] after incubation with unstimulated (Unst), WNV‐infected or αGalCer‐exposed DCs was determined by flow cytometry. (b,d) The surface expression of OX‐40, CD107a and CD69 on iNKTs in multiple experiments is compiled. In each experiment, mean fluorescence intensities (MFIs) on iNKTs incubated with stimulated (WNV or pIC) DCs were normalized to expression on iNKTs incubated with unstimulated DCs. (e) The percentage of CD69+ iNKTs is shown. The mean percentage of CD69+ iNKTs after exposure to unstimulated DCs was 10·1%. Symbols represent individual experiments with unique donor DC and iNKT combinations. Significance was evaluated using a repeated‐measure (RM) one‐way analysis of variance (anova) followed by a Dunnett's multiple comparison test (comparing each stimulated value to the corresponding unstimulated value). *P < 0·05.
To determine the iNKT production of cytokines elicited by WNV‐infected DCs, we measured amounts of secreted cytokines (IFN‐γ, IL‐10, TNF‐α, IL‐4, IL‐17, IL‐22) using Luminex assays. Because iNKTs from different donors varied in their baseline production of cytokines when incubated with unstimulated DCs, the iNKT production of cytokines in response to stimulated/infected DCs is expressed as fold induction over the response to unstimulated DCs. αGalCer‐exposed DCs elicited robust production of IFN‐γ, IL‐10 and TNF‐α from iNKTs, indicating that the iNKTs were functionally competent. In contrast, WNV‐infected DCs induced iNKTs to produce low amounts of IL‐10, but none of the other tested cytokines (Fig. 7a–c, and data not shown). To test whether the absence of IL‐12p70 explained the lack of IFN‐γ production by iNKTs we added purified IL‐12p70 to the cultures of infected DCs and iNKTs, but we did not observe any significant alteration in the production of IFN‐γ (Fig. 7d). Taken together, these data show that WNV‐exposed DCs fail to completely activate iNKTs.
Figure 7.
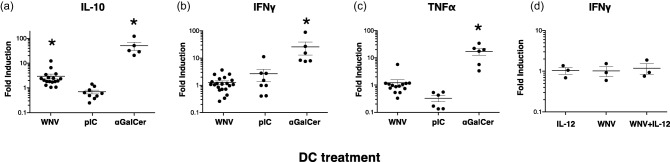
invariant natural killer T cells (iNKTs) produce interleukin (IL)−10 but not proinflammatory cytokines when exposed to WNV‐infected dendritic cells (DCs). DCs were left unstimulated, infected with West Nile virus Kunjin isolate (WNVKUN) at multiplicity of infection (MOI) = 5, incubated with the CD1d ligand α‐galactosylceramide (αGalCer) or stimulated with poly‐inosine‐cytosine (pIC) for 24 h. DCs (1 × 104/well) were then co‐cultured for 24 h with iNKTs (5 × 104/well) that had been isolated from blood, expanded in vitro and rested. Supernatants were assayed for the presence of (a) IL‐10, (b)interferon (IFN)‐γ and (c) tumour necrosis factor (TNF)‐α using Luminex assays. iNKT production of cytokines in response to stimulated/infected DCs is expressed as fold induction over iNKTs incubated with unstimulated DCs. The mean amount of IL‐10 in the cultures of WNV‐exposed DCs + iNKTs was 867 pg/ml, although the variability among donors was very high [95% confidence interval (CI) = 390–1345 pg/ml)]. DCs (1 × 104/well) incubated alone in the same assay produced very low amounts of IL‐10 ( = 4 pg/ml). WNV‐infected DCs did not elicit iNKT production of other inflammatory cytokines (IL‐4, IL‐17, IL‐22). (d) Addition of IL‐12p70 to the cultures did not alter IFN‐γ production significantly by iNKTs. Symbols represent individual experiments with unique donor DC and iNKT combinations. The significance of the fold induction for each condition was evaluated using a repeated‐measure (RM) one‐way analysis of variance (anova) followed by a Dunnett's multiple comparison test (comparing each stimulated (WNV or pIC) value to the corresponding unstimulated value) or a paired t‐test (for αGalCer versus unstimulated). *P < 0·05.
Blockade of CD1d–TCR interactions or IFNAR inhibits CD69 up‐regulation and IL‐10 production by iNKTs exposed to WNV‐infected DCs
iNKTs are activated in response to pathogens either directly through their TCR, or indirectly via cytokines such as IL‐12 or type I IFN 5. Other studies show that iNKTs may be activated by a combination of weak responses to CD1d‐presented self‐antigens and inflammatory cytokines or type I IFN 13, 22, 23.
Because WNV‐infected DCs secrete high amounts of IFN‐α, we tested if iNKT activation and IL‐10 production were mediated by IFNAR‐type I IFN interactions. WNV‐infected, αGalCer‐loaded or unstimulated DCs were incubated with iNKTs in the presence of an IFNAR‐blocking mAb or an isotype control mAb, and activation markers and cytokine production determined as described above. Blockade of IFNAR inhibited activation significantly (as judged by CD69 up‐regulation) and IL‐10 production of iNKTs exposed to WNV‐infected DCs (Fig. 8a,b), suggesting that a component of the activation process depends upon IFNAR signalling.
Figure 8.
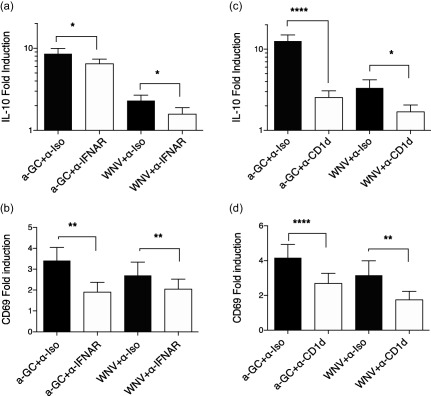
Invariant natural killer T cell (iNKT) up‐regulation of CD69 and interleukin (IL)−10 production after exposure to West Nile virus (WNV)‐infected dendritic cells (DCs) depends upon CD1d–T cell receptor (TCR) interactions and type I interferon receptor (IFNAR). (a,b) DCs were infected with WNV Kunjin isolate (WNVKUN) at MOI = 5 for 24 h, incubated with the CD1d ligand α‐galactosylceramide (αGalCer) or left unstimulated. DCs were then co‐cultured with iNKTs in the presence of a blocking anti‐IFNAR monoclonal antibody (mAb) (5 μg/ml) or a corresponding amount of isotype control mAb for 24 h. (a) Supernatants were assayed for the presence of interleukin (IL)−10 using Luminex assays. iNKT production of cytokines in response to stimulated DCs is expressed as fold induction over iNKTs incubated with uninfected DCs. (b) Expression of CD69 was determined by flow cytometry. Changes in CD69 mean fluorescence intensity (MFI) were normalized to the levels in NKT co‐cultured with uninfected DCs. (c,d) DCs were infected with WNVKUN at multiplicity of infection (MOI) = 5 for 24 h, incubated with the CD1d ligand αGalCer or left unstimulated. DCs were then co‐cultured with iNKTs in the presence of a blocking anti‐CD1d mAb (10 μg/ml) or an isotype control mAb for 24 h. (c) Supernatants were assayed for the presence of IL‐10 using Luminex assays. (d) Expression of CD69 was determined by flow cytometry. The data [ + standard error of the mean (s.e.m.)] summarize individual experiments with unique donor DC and iNKT combinations, n = 8 (IFNAR blockade) and n = 14 (CD1d blockade). The significance of the response differences in the presence or absence of blocking antibody was determined by ratio paired t‐tests. *P < 0·05; **P < 0·01; ****P < 0·0001.
However, as IFNAR blockade did not result in complete inhibition of the iNKT production of IL‐10, we also tested the role of the CD1d–TCR interaction using a CD1d‐blocking mAb. As shown in Fig. 8c,d, blockade of CD1d also partially inhibited CD69 up‐regulation and IL‐10 production in response to WNV‐infected DCs, suggesting that iNKT activation also depends upon TCR signalling.
Discussion
To understand more clearly early events in the immune response against WNV infections, we studied the ability of WNV‐infected human DCs to activate iNKTs, a population of innate lymphocytes that can amplify the adaptive immune response through secretion of different cytokines 7, 8, 9, 10. We studied the effect of WNV infection on DC molecules such as CD1d, CD40 and IL‐12p70 that are known to activate iNKTs optimally 42. Our results show that WNV‐exposed DCs synthesized viral RNA and proteins, produced type I IFN but not IL‐12p70 or IL‐10 and displayed elevated CD40 but reduced CD1d. This infected DC profile induced suboptimal iNKT activation, characterized by CD69 up‐regulation and limited production of IL‐10 but not proinflammatory cytokines. The iNKT response to infected DCs was dependent upon both CD1d–TCR and IFNAR‐type I IFN interactions.
It is well known that iNKTs are able to secrete copious amounts of many different cytokines in a very short time when stimulated in vitro with strong agonists, such as the CD1d ligand αGalCer or phorbol esters plus ionomycin. In fact, one of the original defining characteristics of iNKTs was that they could secrete IL‐4 and IFN‐γ simultaneously. However, iNKT responses in vivo are less promiscuous and depend upon their previous history of activation, the cytokine milieu and the activation state of the antigen‐presenting cells that interact with them. Our results show a very clear example of this. Interaction with WNV‐infected DCs results in iNKT up‐regulation of some, but not all, activation markers, and in secretion of IL‐10, but no other cytokines, either type 1 (IFN‐γ, TNF‐α) or types 2 or 3 (IL‐4, IL‐17, IL‐22).
WNV infection of the DCs altered their phenotype in ways that could influence the activation of iNKTs. Despite inducing increased CD1D mRNA, WNV infection reduced the surface expression of CD1d, which could influence the strength of TCR signalling in the iNKTs. Indeed, variable concentrations of αGalCer altered the pattern of cytokine production by iNKTs, suggesting that TCR signal strength is an important regulator of iNKT function 43. The effect of infection on CD1d surface residence is unlikely to be due to viral interference mechanisms that specifically target CD1d, as proposed for viruses such as HSV and HIV‐1 (reviewed in 41), because pattern recognition receptor ligands such as LPS and pIC also induced significant CD1d down‐regulation in our assays. As CD1d traffics through endocytic compartments with Ii chain, the profound changes in MHC‐II/Ii trafficking in activated DCs may alter CD1d trafficking and lead to reduced CD1d surface expression 44.
Alternatively, early studies of iNKT responses in the context of viral infections suggested that they were similar to those in the presence of TLR‐9 ligand (CpG ODN)‐stimulated DCs, which involved mainly IL‐12 and type I IFN, and were independent of TCR–CD1d interactions 18, 20. However, later studies have shown that some viruses, as well as innate stimuli, can alter the repertoire of lipids presented by CD1d in infected cells, and induce direct CD1d–TCR‐mediated responses of iNKTs 14, 16. These studies suggest that iNKT cell responses to viruses may be influenced both by changes in the lipid repertoire and by cytokine secretion of the infected DCs. Our experiments support this model. We show that the response of iNKTs to WNV‐infected human DCs can be blocked partially by α‐CD1d and α‐IFNAR, suggesting that both direct TCR triggering and IFNAR signalling contribute to the iNKT activation.
WNV‐exposed DCs produced IFN‐α but not IL‐12p70, due probably to IFNAR‐induced feedback mechanisms that dampen IL‐12 production 45, 46. Although IL‐12‐independent T cell IFN‐γ production promoted by type I IFN is observed in some virus infections 47, the absence of IL‐12p70 could explain why the iNKTs did not produce IFN‐γ. The presence of type I IFN in the absence of IL‐12p70 often leads to production of IL‐10 in T cells and myeloid cells 46, 48, 49, 50. However, in our experiments, supplementation of IL‐12p70 into cultures of infected DCs and iNKTs did not induce production of IFN‐γ or alter the production of IL‐10. Taken together, these results suggest that other cytokines and/or co‐stimulatory molecules influence the response pattern of iNKTs to WNV‐infected DCs.
The ability of freshly isolated iNKTs to produce IL‐10 has been controversial. In some experiments PMA and ionomycin stimulation did not induce IL‐10 production 51, 52, while others showed iNKT IL‐10 production after stimulation via anti‐CD3 + anti‐CD28 53. However, many experiments have demonstrated increased IL‐10 production of NKTs in vivo, especially after restimulation of αGalCer‐activated cells 54, 55, and a naturally existing subset of NKTs that produce primarily IL‐10 was identified recently 54. Furthermore, IL‐10 production by NKTs is important for their protective role in some autoimmune diseases such as type I diabetes 56 and experimental autoimmune encephalomyelitis 57. Reported populations of forkhead box protein 3 (FoxP3)+ regulatory iNKTs could be the source of this IL‐10 58, 59.
Clinical studies show that IL‐10 has an important role in regulating immune responses to flaviviruses in both humans and mice. There is a significant positive correlation between plasma IL‐10 levels and severity of disease in Dengue virus‐infected patients 60, and in‐vitro blocking of IL‐10 improves T cell responses from Dengue patients 61. In mice, IL‐10 modulates the morbidity and mortality of WNV infection and blockade of IL‐10 improves the immune response to WNV infection 62. Interestingly, in these experiments a CD4+CD3+ T cell population produced primarily the IL‐10 in vivo, a population that would include iNKTs. Although this work did not evaluate directly whether iNKTs produced IL‐10, our results suggest that iNKTs could be partly responsible for this in‐vivo effect, especially as IL‐10 production peaked by 72 h post‐infection. Consistent with this, evidence is emerging that iNKTs may be important in flavirus infections. iNKTs become activated in mouse models of Dengue 21, and iNKT activation correlates with disease severity in human Dengue patients 63. Taken together, our data suggest that upon interaction with WNV‐infected DCs, iNKTs could be responsible for IL‐10 production in the early stages of WNV infection.
Disclosure
The authors declare that no competing interests exist.
Author contributions
S. K. and J. A. designed experiments and wrote the manuscript; S. K., S. T., A. S. and J. A. performed experiments and analysed the data; T. P. and E. C. identified blood donors and performed phlebotomy.
Acknowledgements
This study was funded by NIH U19AI057234‐41000411348 (to S.K. and J. A.), U19AI057229‐60934766‐28291 (to S. K. and J. A.), U19AI62629 (to S. K.) and U54GM104938 (to E. C. and T. P.). We thank Dr Michael Diamond for the generous gift of WNV Kunjin and virus protocols, Dr Saghar Kaabinejadian for sharing advice and reagents and Dr Melissa Munroe in the OMRF Serum Analyte and Biomarker core facility.
References
- 1. Petersen LR, Brault AC, Nasci RS. West Nile virus: review of the literature. JAMA 2013; 310:308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wertheimer AM, Uhrlaub JL, Hirsch A et al Immune response to the West Nile virus in aged non‐human primates. PLOS ONE 2010; 5:e15514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Suthar MS, Diamond MS, Gale MJ. West Nile virus infection and immunity. Nat Rev Microbiol 2013; 11:115–28. [DOI] [PubMed] [Google Scholar]
- 4. Wu L, Van Kaer L. Natural killer T cells in health and disease. Front Biosci (Schol Edn) 2011; 3:236–51.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brigl M, Brenner MB. How invariant natural killer T cells respond to infection by recognizing microbial or endogenous lipid antigens. Semin Immunol 2010; 22:79–86. [DOI] [PubMed] [Google Scholar]
- 6. Tupin E, Kinjo Y, Kronenberg M. The unique role of natural killer T cells in the response to microorganisms. Nat Rev Microbiol 2007; 5:405–17. [DOI] [PubMed] [Google Scholar]
- 7. Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol 2013; 13:101–17. [DOI] [PubMed] [Google Scholar]
- 8. Tessmer MS, Fatima A, Paget C, Trottein F, Brossay L. NKT cell immune responses to viral infection. Expert Opin Ther Targets 2009; 13:153–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diana J, Lehuen A. NKT cells: friend or foe during viral infections? Eur J Immunol 2009; 39:3283–91. [DOI] [PubMed] [Google Scholar]
- 10. Juno JA, Keynan Y, Fowke KR. Invariant NKT cells: regulation and function during viral infection. PLOS Pathog 2012; 8:e1002838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kadri N, Blomqvist M, Cardell SL. Type II natural killer T cells: a new target for immunomodulation? Expert Rev Clin Immunol 2008; 4:615–27. [DOI] [PubMed] [Google Scholar]
- 12. Khovidhunkit W, Kim MS, Memon RA et al Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res 2004; 45:1169–96. [DOI] [PubMed] [Google Scholar]
- 13. Paget C, Mallevaey T, Speak AO et al Activation of invariant NKT cells by toll‐like receptor 9‐stimulated dendritic cells requires type I interferon and charged glycosphingolipids. Immunity 2007; 27:597–609. [DOI] [PubMed] [Google Scholar]
- 14. Salio M, Speak AO, Shepherd D et al Modulation of human natural killer T cell ligands on TLR‐mediated antigen‐presenting cell activation. Proc Natl Acad Sci USA 2007; 104:20490–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fox LM, Cox DG, Lockridge JL et al Recognition of lyso‐phospholipids by human natural killer T lymphocytes. PLOS Biol 2009; 7:e1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zeissig S, Murata K, Sweet L et al Hepatitis B virus‐induced lipid alterations contribute to natural killer T cell‐dependent protective immunity. Nat Med 2012; 18:1060–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Paget C, Bialecki E, Fontaine J et al Role of invariant NKT lymphocytes in immune responses to CpG oligodeoxynucleotides. J Immunol 2009; 182:1846–53. [DOI] [PubMed] [Google Scholar]
- 18. Tyznik AJ, Tupin E, Nagarajan NA, Her MJ, Benedict CA, Kronenberg M. Cutting edge: the mechanism of invariant NKT cell responses to viral danger signals. J Immunol 2008; 181:4452–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holzapfel KL, Tyznik AJ, Kronenberg M, Hogquist KA. Antigen‐dependent versus ‐independent activation of invariant NKT cells during infection. J Immunol 2014; 192:5490–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wesley JD, Tessmer MS, Chaukos D, Brossay L. NK cell‐like behavior of Valpha14i NKT cells during MCMV infection. PLOS Pathog 2008; 4:e1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Renneson J, Guabiraba R, Maillet I et al A detrimental role for invariant natural killer T cells in the pathogenesis of experimental dengue virus infection. Am J Pathol 2011; 179:1872–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hou R, Goloubeva O, Neuberg DS, Strominger JL, Wilson SB. Interleukin‐12 and interleukin‐2‐induced invariant natural killer T‐cell cytokine secretion and perforin expression independent of T‐cell receptor activation. Immunology 2003; 110:30–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leite‐De‐Moraes MC, Hameg A, Arnould A et al A distinct IL‐18‐induced pathway to fully activate NKT lymphocytes independently from TCR engagement. J Immunol 1999; 163:5871–6. [PubMed] [Google Scholar]
- 24. Olagnier D, Peri S, Steel C et al Cellular oxidative stress response controls the antiviral and apoptotic programs in dengue virus‐infected dendritic cells. PLOS Pathog 2014; 10:e1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schmid MA, Harris E. Monocyte recruitment to the dermis and differentiation to dendritic cells increases the targets for dengue virus replication. PLOS Pathog 2014; 10:e1004541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davison AM, King NJ. Accelerated dendritic cell differentiation from migrating Ly6C(lo) bone marrow monocytes in early dermal West Nile virus infection. J Immunol 2011; 186:2382–96. [DOI] [PubMed] [Google Scholar]
- 27. Qian F, Wang X, Zhang L et al Impaired interferon signaling in dendritic cells from older donors infected in vitro with West Nile virus. J Infect Dis 2011; 203:1415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martina BE, Koraka P, van den Doel P, Rimmelzwaan GF, Haagmans BL, Osterhaus AD. DC‐SIGN enhances infection of cells with glycosylated West Nile virus in vitro and virus replication in human dendritic cells induces production of IFN‐alpha and TNF‐alpha. Virus Res 2008; 135:64–71. [DOI] [PubMed] [Google Scholar]
- 29. Silva MC, Guerrero‐Plata A, Gilfoy FD, Garofalo RP, Mason PW. Differential activation of human monocyte‐derived and plasmacytoid dendritic cells by West Nile virus generated in different host cells. J Virol 2007; 81:13640–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rawle DJ, Setoh YX, Edmonds JH, Khromykh AA. Comparison of attenuated and virulent West Nile virus strains in human monocyte‐derived dendritic cells as a model of initial human infection. Virol J 2015; 12:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu WJ, Wang XJ, Mokhonov VV, Shi PY, Randall R, Khromykh AA. Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J Virol 2005; 79:1934–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O'Neill DW, Bhardwaj N. Differentiation of peripheral blood monocytes into dendritic cells. Curr Protoc Immunol 2005; Chapter 22:Unit 22F.4. [DOI] [PubMed] [Google Scholar]
- 33. Brien JD, Lazear HM, Diamond MS. Propagation, quantification, detection, and storage of West Nile virus. Curr Protoc Microbiol 2013; 31:15D.3.1–3.18. [DOI] [PubMed] [Google Scholar]
- 34. Lambeth CR, White LJ, Johnston RE, de Silva AM. Flow cytometry‐based assay for titrating dengue virus. J Clin Microbiol 2005; 43:3267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kong KF, Delroux K, Wang X et al Dysregulation of TLR3 impairs the innate immune response to West Nile virus in the elderly. J Virol 2008; 82:7613–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hua J, Kirou K, Lee C, Crow MK. Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti‐RNA binding protein autoantibodies. Arthritis Rheum 2006; 54:1906–16. [DOI] [PubMed] [Google Scholar]
- 37. Daffis S, Lazear HM, Liu WJ et al The naturally attenuated Kunjin strain of West Nile virus shows enhanced sensitivity to the host type I interferon response. J Virol 2011; 85:5664–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sheehan KC, Lazear HM, Diamond MS, Schreiber RD. Selective blockade of interferon‐α and ‐β reveals their non‐redundant functions in a mouse model of West Nile virus infection. PLOS One 2015; 10:e0128636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berghöfer B, Frommer T, Haley G, Fink L, Bein G, Hackstein H. TLR7 ligands induce higher IFN‐alpha production in females. J Immunol 2006; 177:2088–96. [DOI] [PubMed] [Google Scholar]
- 40. Meier A, Chang JJ, Chan ES et al Sex differences in the Toll‐like receptor‐mediated response of plasmacytoid dendritic cells to HIV‐1. Nat Med 2009; 15:955–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Opasawatchai A, Matangkasombut P. iNKT cells and their potential lipid ligands during viral infection. Front Immunol 2015; 6:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hegde S, Fox L, Wang X, Gumperz JE. Autoreactive natural killer T cells: promoting immune protection and immune tolerance through varied interactions with myeloid antigen‐presenting cells. Immunology 2010; 130:471–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang X, Chen X, Rodenkirch L et al Natural killer T‐cell autoreactivity leads to a specialized activation state. Blood 2008; 112:4128–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen X, Wang X, Keaton JM et al Distinct endosomal trafficking requirements for presentation of autoantigens and exogenous lipids by human CD1d molecules. J Immunol 2007; 178:6181–90. [DOI] [PubMed] [Google Scholar]
- 45. Trinchieri G. Type I interferon: friend or foe. J Exp Med 2010; 207:2053–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McRae BL, Semnani RT, Hayes MP, van Seventer GA. Type I IFNs inhibit human dendritic cell IL‐12 production and Th1 cell development. J Immunol 1998; 160:4298–304. [PubMed] [Google Scholar]
- 47. Cousens LP, Peterson R, Hsu S et al Two roads diverged: interferon alpha/beta‐ and interleukin 12‐mediated pathways in promoting T cell interferon gamma responses during viral infection. J Exp Med 1999; 189:1315–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stewart CA, Metheny H, Iida N et al Interferon‐dependent IL‐10 production by Tregs limits tumor Th17 inflammation. J Clin Invest 2013; 123:4859–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu BS, Janssen HL, Boonstra A. Type I and III interferons enhance IL‐10R expression on human monocytes and macrophages, resulting in IL‐10‐mediated suppression of TLR‐induced IL‐12. Eur J Immunol 2012; 42:2431–40. [DOI] [PubMed] [Google Scholar]
- 50. Zhang L, Yuan S, Cheng G, Guo B. Type I IFN promotes IL‐10 production from T cells to suppress Th17 cells and Th17‐associated autoimmune inflammation. PLOS ONE 2011; 6:e28432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Snyder‐Cappione JE, Tincati C, Eccles‐James IG et al A comprehensive ex vivo functional analysis of human NKT cells reveals production of MIP1‐alpha and MIP1‐beta, a lack of IL‐17, and a Th1‐bias in males. PLOS ONE 2010; 5:e15412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chan AC, Leeansyah E, Cochrane A et al Ex‐vivo analysis of human natural killer T cells demonstrates heterogeneity between tissues and within established CD4(+) and CD4(‐) subsets. Clin Exp Immunol 2013; 172:129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Coquet JM, Chakravarti S, Kyparissoudis K et al Diverse cytokine production by NKT cell subsets and identification of an IL‐17‐producing CD4‐NK1.1‐ NKT cell population. Proc Natl Acad Sci USA 2008; 105:11287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sag D, Krause P, Hedrick CC, Kronenberg M, Wingender G. IL‐10‐producing NKT10 cells are a distinct regulatory invariant NKT cell subset. J Clin Invest 2014; 124:3725–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Birkholz AM, Girardi E, Wingender G et al A novel glycolipid antigen for NKT cells that preferentially induces IFN‐γ production. J Immunol 2015; 195:924–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hammond KJ, Poulton LD, Palmisano LJ, Silveira PA, Godfrey DI, Baxter AG. Alpha/beta‐T cell receptor (TCR)+CD4‐CD8‐ (NKT) thymocytes prevent insulin‐dependent diabetes mellitus in nonobese diabetic (NOD)/Lt mice by the influence of interleukin (IL)−4 and/or IL‐10. J Exp Med 1998; 187:1047–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Singh AK, Wilson MT, Hong S et al Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J Exp Med 2001; 194:1801–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Huijts CM, Schneiders FL, Garcia‐Vallejo JJ, Verheul HM, de Gruijl TD, van der Vliet HJ. mTOR inhibition per se induces nuclear localization of FOXP3 and conversion of invariant NKT (iNKT) cells into immunosuppressive regulatory iNKT cells. J Immunol 2015; 195:2038–45. [DOI] [PubMed] [Google Scholar]
- 59. Monteiro M, Almeida CF, Caridade M et al Identification of regulatory Foxp3+ invariant NKT cells induced by TGF‐beta. J Immunol 2010; 185:2157–63. [DOI] [PubMed] [Google Scholar]
- 60. Butthep P, Chunhakan S, Yoksan S, Tangnararatchakit K, Chuansumrit A. Alteration of cytokines and chemokines during febrile episodes associated with endothelial cell damage and plasma leakage in dengue hemorrhagic fever. Pediatr Infect Dis J 2012; 31:e232–8. [DOI] [PubMed] [Google Scholar]
- 61. Malavige GN, Jeewandara C, Alles KM et al Suppression of virus specific immune responses by IL‐10 in acute dengue infection. PLOS Negl Trop Dis 2013; 7:e2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bai F, Town T, Qian F et al IL‐10 signaling blockade controls murine West Nile virus infection. PLOS Pathog 2009; 5:e1000610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Matangkasombut P, Chan‐In W, Opasawaschai A et al Invariant NKT cell response to dengue virus infection in human. PLOS Negl Trop Dis 2014; 8:e2955. [DOI] [PMC free article] [PubMed] [Google Scholar]