

Abstract
Constitutive Ca2+/calmodulin (CaM)-activation of adenylyl cyclases (ACs) types 1 and 8 in sinoatrial nodal cells (SANC) generates cAMP within lipid-raft-rich microdomains to initiate cAMP–protein kinase A (PKA) signaling, that regulates basal state rhythmic action potential firing of these cells. Mounting evidence in other cell types points to a balance between Ca2+-activated counteracting enzymes, ACs and phosphodiesterases (PDEs) within these cells. We hypothesized that the expression and activity of Ca2+/CaM-activated PDE Type 1A is higher in SANC than in other cardiac cell types.
We found that PDE1A protein expression was 5-fold higher in sinoatrial nodal tissue than in left ventricle, and its mRNA expression was 12-fold greater in the corresponding isolated cells. PDE1 activity (nimodipine-sensitive) accounted for 39% of the total PDE activity in SANC lysates, compared to only 4% in left ventricular cardiomyocytes (LVC). Additionally, total PDE activity in SANC lysates was lowest (10%) in lipid-raft-rich and highest (76%) in lipid-raft-poor fractions (equilibrium sedimentation on a sucrose density gradient). In intact cells PDE1A immunolabeling was not localized to the cell surface membrane (structured illumination microscopy imaging), but located approximately within about 150 nm inside of immunolabeling of hyperpolarization-activated cyclic nucleotide-gated potassium channels (HCN4), which reside within lipid-raft-rich microenvironments. In permeabilized SANC, in which surface membrane ion channels are not functional, nimodipine increased spontaneous SR Ca2+ cycling. PDE1A mRNA silencing in HL-1 cells increased the spontaneous beating rate, reduced the cAMP, and increased cGMP levels in response to IBMX, a broad spectrum PDE inhibitor (detected via fluorescence resonance energy transfer microscopy).
We conclude that signaling via cAMP generated by Ca2+/CaM-activated AC in SANC lipid raft domains is limited by cAMP degradation by Ca2+/CaM-activated PDE1A in non-lipid raft domains. This suggests that local gradients of [Ca2+]–CaM or different AC and PDE1A affinity regulate both cAMP production and its degradation, and this balance determines the intensity of Ca2+-AC-cAMP-PKA signaling that drives SANC pacemaker function.
Keywords: Heart, Sinoatrial node pacemaker cells, Phosphodiesterase Type 1A, Calcium, cAMP
1. Introduction
Spontaneous, local Ca2+ releases (LCRs) generated by the sarcoplasmic reticulum (SR) via spontaneous local diastolic ryanodine receptor (RyR) activation within cardiac sinoatrial nodal cells (SANC) is crucial for their normal automaticity. In the absence of β-adrenergic receptor stimulation, constitutive Ca2+/calmodulin (CaM) activation of neuronal type adenylyl cyclases (ACs) within SANC lipid raft microdomains generates cAMP [1,2], which directly activates hyperpolarization-activated cyclic nucleotide-gated potassium channels (HCNs) leading to If, “funny” current activation, and mediates protein kinase A (PKA)-dependent phosphorylation of SR, mitochondrial and ion channel proteins to regulate Ca2+ cycling that drives generation of spontaneous rhythmic action potentials (APs), i.e., normal automaticity [3,4] and ATP production [5–7] of these cells. This basal, Ca2+/CaM-activated, AC-cAMP-PKA-Ca2+ signaling in SANC greatly exceeds that in left ventricular cardiomyocytes (LVC) [4], and is intrinsically feed-forward in nature, because PKA-dependent protein phosphorylation leads to an increase in intracellular Ca2+ release, which further activates AC, leading to further increases in protein phosphorylation signaling, and so forth. Negative feedback mechanisms, therefore, which limit the extent of basal Ca2+-AC-cAMP- and cAMP-dependent protein phosphorylation are necessary to maintain the AP firing rate and stimulation of ATP production near mid-range set points [8].
It has been clearly established that cyclic nucleotide phosphodiesterase (PDE) inhibition in rabbit SANC markedly increases cAMP, augments Ca2+ cycling protein phosphorylation and accelerates the AP firing rate [9,10]. Thus, basal PDE activity in SANC can be a potent, negative modulator of the cAMP level [11] and therefore an important “brake” of feed-forward basal Ca2+-AC-cAMP-PKA-Ca2+- signaling. It is well known that expression patterns of PDEs and PDE activity vary substantially among different tissues, cells and species [12,13]. The importance of PDEs, their precise activity and cell localization for functioning is reflected in a wide variety of isoforms and post-translational modifications. Evidence in other cell types demonstrates that Ca2+/CaM-activated PDE1A activity is a key regulator of cross-talk between cAMP and Ca2+ messenger molecules [14] and that a balance within cells between Ca2+-activated counteracting enzymes, ACs and PDEs, maintains optimal levels of function of these cells. For example, PDE1A (a Ca2+/CaM-activated enzyme) expression in retinal cells in which Ca2+/CaM-activated AC types drive cAMP production, modulates cAMP levels [15,16]. Similarly, in transgenic mice overexpressing Ca2+/CaM-activated neuronal AC Type 8 in LVC, the PDE1A affinity for cAMP becomes increased to limit the exaggerated cAMP production [17], shielding L-type Ca2+ channels to prevent Ca2+ overload [17,18]. We hypothesized that, because Ca2+/CaM-activated AC Types 1 and 8 are expressed and active in SANC [1,2], the expression and activity of Ca2+/CaM-activated PDE Type 1A might also be higher in SANC than in other cardiac cells, and that PDE1A makes a substantial contribution to regulation of Ca2+-AC-cAMP-PKA-Ca2+ signaling and thus to basal pacemaker function.
2. Methods
We: (1) measured the transcript abundance of PDE1 subtypes in isolated SANC, and compared it to that of isolated LVC and right atrial cardiomyocytes (RAC); (2) designed a novel antibody to detect PDE1A protein levels in rabbit sinoatrial node (SAN) and other cardiac tissues, and to detect localization of PDE1A via immunolabeling in SANC by Structured Illumination Microscopy (SIM); 3) measured basal cAMP-hydrolyzing PDE activity in SANC and LVC lysates including nimodipine sensitive PDE1 contribution and its Ca2+ dependence, (4) assessed distribution of PDE activity in sucrose density gradient (SDG) fractions of SANC lysates; (5) quantified the effect of PDE1 inhibition on SR-generated cAMP-dependent spontaneous LCRs which are crucial regulators of normal SANC automaticity; (6) silenced PDE1A mRNA in a cardiac cell line (HL-1 cells), to determine the impact of its signaling on real time cAMP levels (via fluorescence resonance energy transfer (FRET) microscopy) in these cells and on their spontaneous beating rate.
A detailed description of the methods and a complete listing of the sources of reagents and techniques utilized in our study are provided in the Online data supplement (ODS).
3. Results
3.1. PDE1 subtype expression in cardiac cells
Fig. 1A illustrates representative examples of RT-qPCR amplification plots in SANC and LVC. PDE1 transcript abundance patterns, normalized to the expression of a housekeeping gene, Tubb2a (tubulin, beta 2A class IIa) in SANC, RAC and LVC are illustrated in Fig. 1B. Average abundance of mRNA for different PDE1 isoforms was strikingly different in SANC, LVC and RAC: PDE1A transcript abundance in SANC greatly exceeded that in LVC and exceeded that in RAC by 4-fold; the abundance of PDE1B transcript was equivalent in all three cell types; interestingly, abundance of PDE1C transcript in LVC greatly exceeded that in either SANC or RAC (Fig. 1B). Table II in ODS quantifies the proportion of PDE1 subtypes abundance of LVC and RAC to that of SANC.
Fig. 1.
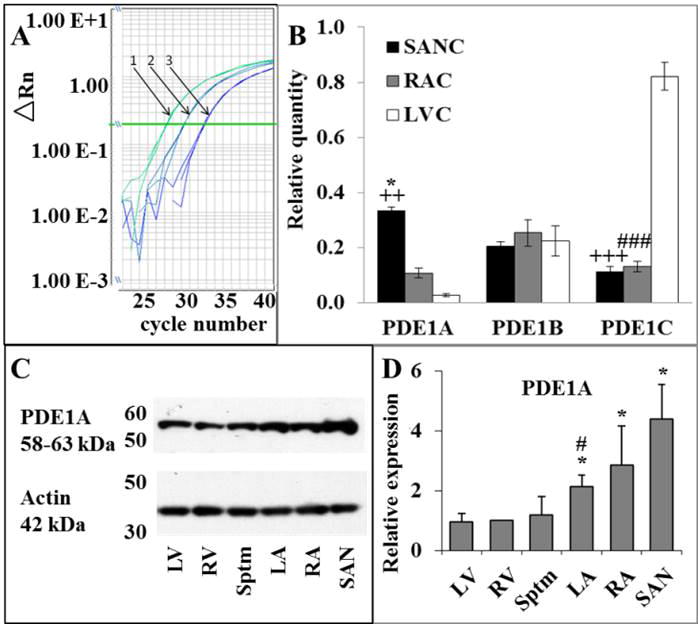
A, Example of amplification plot. 1 – SANC, 2 – RAC and 3 – LVC samples; B, expression profile of PDE1 subtype transcripts in SANC, RAC and LVC normalized to the expression of β-tubulin housekeeping gene, n = 6; * p < 0.05 for SANC versus RAC; ++ p < 0.01 for SANC versus LVC; +++ p < 0.001 for SANC versus LVC; ### p < 0.001 for RAC versus LVC; C, representative Western Blot of PDE1A immunolabeling of rabbit heart tissue lysates (left ventricle (LV), right ventricle (RV), Septum (Sptm), left atria (LA), right atria (RA), sinoatrial node (SAN)); D, averaged expression levels of PDE1A in rabbit heart tissues, normalized to the expression of actin, n = 3. * – differences between RA/SAN vs. ventricles/Sptm and LA, p < 0.05 ANOVA; *# – differences between LA and ventricles/Sptm, p < 0.05 ANOVA.
To ensure that major differences in PDE1 isoforms expressed among cell types (Fig. 1B) were not artifacts of slight differences in Tubb2a expression among cell types (ODS Fig. 1), we performed additional experiments to compare the relative transcript abundance of selected PDE types to another housekeeping gene, rabbit hypoxanthine guanine phosphoribosyl transferase, which was equally expressed in all cell types (ODS Fig. 1). Cell type differences in relative quantitation of PDE1A isoform expression normalized to hypoxanthine guanine phosphoribosyl transferase (ODS Fig. 2) were similar to those normalized to Tubb2a (Fig. 1B).
3.2. PDE1A protein expression in different cardiac cell types
Fig. 1C illustrates representative blot of different cardiac tissue lysates labelled with a monoclonal antibody to PDE1A (see ODS methods). Average expression of PDE1A normalized to actin is shown in Fig. 1D. Note that the relative protein levels of PDE1A in SAN > RA > LV is similar to the difference of expression of PDE1A transcripts in the corresponding cardiac myocytes.
3.3. Imaging of PDE1A immunolabeling
We employed SIM imaging of immunolabeled SANC in order to visualize the intracellular location of PDE1A. Fig. 2 illustrates an example of a representative SIM image of SANC with dual immunolabeling for PDE1A (Panel A) and HCN4 (Panel B). The merged images (Panels C, D) show that PDE1A (green) is localized near, but internal to, the cell surface membrane, as indicated by its position relative to HCN4 (red), which is known to be located within the cell membrane and in caveoli [19]. Panel C inset shows SIM maximum intensity projection Z-stack image of HCN4 and PDE1A immunolabeling of the same cell as in panels A–D. Distinct punctate immunolabeling of both proteins, observed in the merged SIM Z-section image, indicates that the PDE1A is likely localized within approximately 150 nm internal to the surface membrane localized HCN4 (i.e., based on the lateral resolution of N-SIM).
Fig. 2.
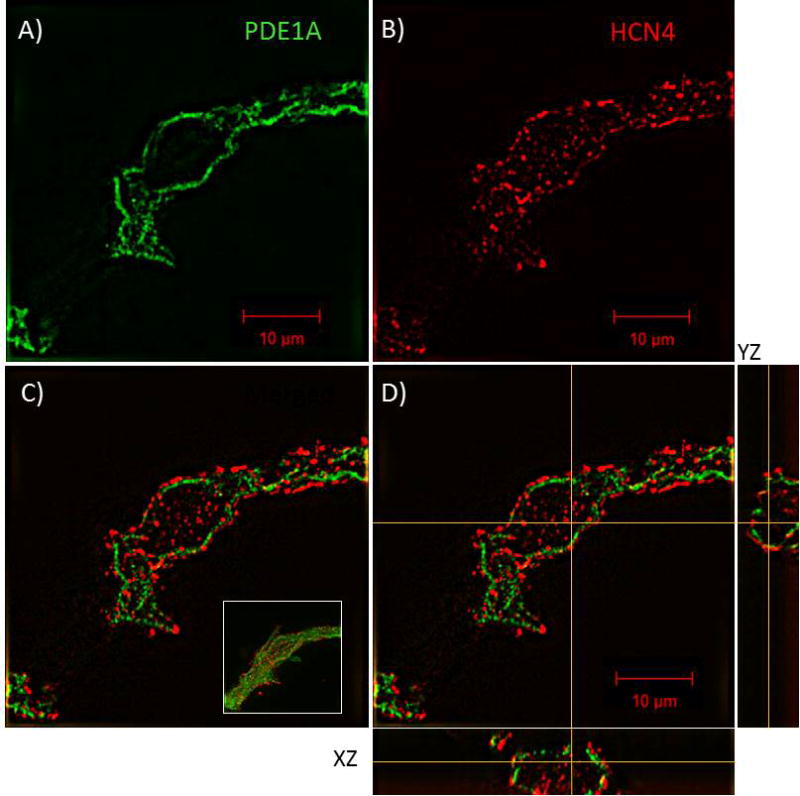
PDE1A localizes to the submembrane region of SANC. A–C, N-SIM acquired 0.5 μm thick Z-stack composite image of an immunolabeled SANC identifying the subcellular localization of PDE1A (green) as being in close proximity, but interior, to the cell membrane as indicated by its relative position to the cell membrane HCN4 (red); C, inset, maximum intensity projection Z-stack composite image showing PDE1A/HCN4 staining of the entire SA Node Cell. D, orthogonal sections (XZ and YZ) through the perinuclear region of the cell confirming PDE1A localization is primarily restricted to the submembrane region of the cell and that the majority of the PDE1A signal does not overlap that of HCN4.
3.4. PDE activity in SANC SDG fractions
We fractionated SANC lysates to determine the relative proportion of PDE activity in lipid-raft-rich and lipid-raft-poor fractions. A representative gel of SDG fractions and the average protein concentration in each SDG fraction are shown in ODS Fig. 7. Note that fraction I had negligible protein and lipid raft labeling [1]; this fraction was not used in further enzymatic analysis. Fig. 3A shows the average immunolabeling densities of caveolin-3 (Cav-3) and ganglioside M1 (GM-1) in SDG II–IV fractions. Note that because of day to day variability in the separation of lipid rafts versus non-lipid rafts, when averaged across nine different experiments in different pooled SANC, lipid raft densities do not completely segregate into lipid-raft-rich (high Cav-3 immunolabeling) and lipid-raft-poor gradient fractions. Still, on average, the highest Cav-3 immunolabeling density was in fraction II, and is 4- to 6-fold lower in fraction IV and this relative degree of separation is sufficient to roughly assess the relative presence of PDE activity residing within each lipid-raft-rich and lipid-raft-poor fractions. While the specific location of lipid-raft-rich microdomains within cells cannot be ascertained from SDG fractions, immunolabeling of caveolin in intact SANC is high in lipid rich lysate fractions (Fig. 3B). This suggests that, as expected, our lipid raft rich fraction contains proteins that are highly (but not necessarily uniquely [20]) localized to the cell surface membrane. The distribution of GM-1 across fractions is highly correlated with that of Cav-3 (ODS Fig. 8).
Fig. 3.
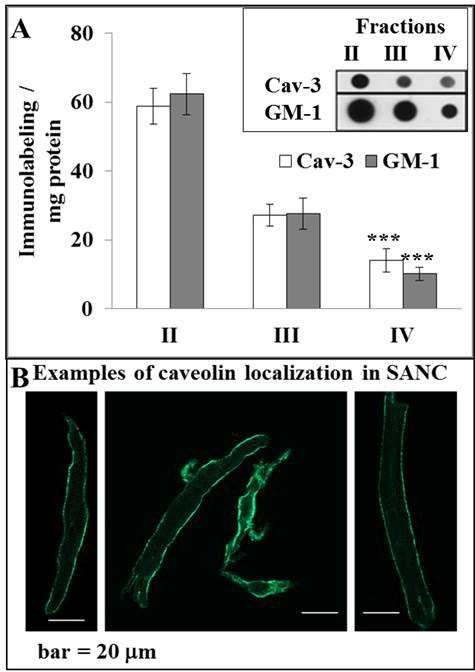
A, Average density of Cav-3 and GM1 immunolabeling measured in SANC fractions, n = 9, ***p < 0.001 versus fraction II, inset – representative dot blots of Cav-3 and GM1 immunolabeling in SANC fractions; B, representative examples of caveolin immunolabeling in SANC.
Fig. 4A illustrates a representative example of Cav-3 immunolabeling and PDE activity in the three SDG fractions of a representative SANC lysate. Note the reciprocal relationship of Cav-3 immunolabeling and PDE activity, with PDE activity progressively increasing from fraction II to IV while the Cav-3 immunolabeling progressively decreasing from fraction II to IV. Fig. 4B shows the average data. Even in the presence of less than perfect total separation of the lipid raft marker into a single fraction the figure clearly shows relatively high PDE activity content of lipid-raft-poor versus lipid-raft-rich fraction.
Fig. 4.
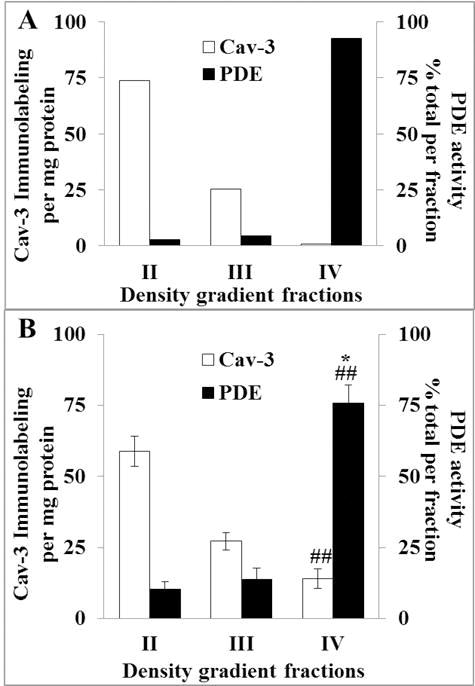
A, Cav-3 immunolabeling and PDE activity of a representative SANC lysate fractions; B, average Cav-3 immunolabeling (n = 9) and PDE activity (n = 7) in sucrose density gradient fractions. ##p < 0.01, versus fraction II, *p < 0.05, versus fraction III. Combined total PDE activity of all fractions was 258 ± 43.5 pmol cAMP/min/mg protein.
3.5. Total PDE activity in SANC and LVC lysates and effects of PDE inhibition on total PDE activity
Total c-AMP-specific PDE activity measured in SANC and LVC lysates is illustrated in Fig. 5. The total PDE activity was comparable in SANC and LVC lysates, and Fig. 5A illustrates PDE activity in the presence or absence of the broad spectrum PDE inhibitor, isobutylmethylxanthine (IBMX), in a representative SANC lysate. On average, IBMX decreased PDE activity by approximately 90% and 75% in SANC and LVC, respectively (Fig. 5B). Fig. 5C illustrates the average effect of PDE1 inhibition by nimodipine (a PDE1 inhibitor [21]) on SANC and LVC lysate total PDE activity. Suppression of total PDE activity in SANC lysates by nimodipine averaged 39 ± 1.8%. Nimodipine however, did not significantly inhibit PDE activity in LVC lysates (4 ± 2.2%). We also applied PF-04471141 to inhibit PDE1. We found that at 2 μM it significantly decreased total PDE activity in SANC lysates and this effect did not differ from the nimodipine effect (data not shown). Since PDE1 activity is Ca2+ regulated, the effect of nimodipine was further analyzed in cell lysates in the presence and absence of Ca2+ (Fig. 5C). In LVC PDE activities did not significantly differ in the presence or absence of Ca2+. In SANC, however, the effect of the nimodipine to inhibit PDE activity was robust, both in the presence or absence of Ca2+, and was slightly, but significantly, lower in the absence of Ca2+.
Fig. 5.
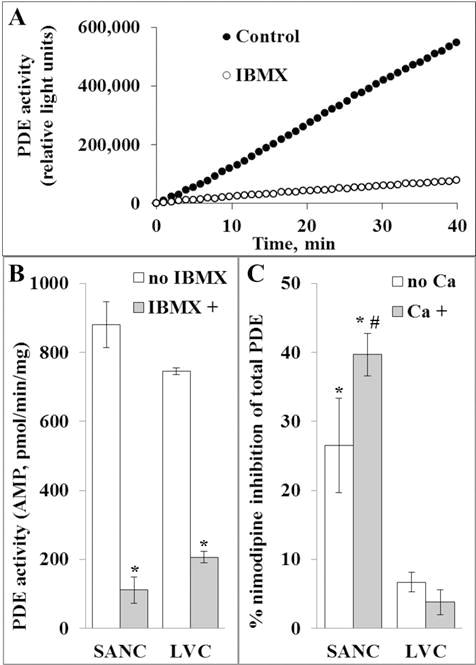
A, a representative example of IBMX inhibitory effect on PDE activity in SANC lysate; B, PDE activity of SANC and LVC lysates in the presence and absence of IBMX; *p < 0.05, versus –IBMX, n = 4 to 7; C, Inhibition of total PDE activity in SANC and LVC by nimodipine (specific for PDE1), Ca2+-dependence of relative nimodipine-sensitive inhibition of total PDE activity; *p < 0.05, #p = 0.0343, n = 3 to 10.
3.6. Functional effects of PDE1A inhibition in permeabilized SANC on local Ca2+ releases generated by spontaneous RyR activation
To determine the functional significance of PDE1 activity we assessed the effects of nimodipine on SANC LCRs, we used permeabilized SANC to avoid the effects on sarcolemmal L-type Ca2+ channels and to localize its effect to SR [Ca2+] cycling [22]. Further, free cytosolic [Ca2+] can be controlled at any desired level in single permeabilized SANC by adjusting the bathing [Ca2+]. To prove the requirement of cAMP-PDE signaling for LCRs in permeabilized SANC we first inhibited ACs with 20 μM of MDL-12330A (MDL), and then inhibited PDEs with 20 μM of IBMX in the presence of AC inhibition (Fig. 6A). The AC inhibitor, MDL, markedly suppressed LCRs; PDE inhibition (IBMX) in the continuous presence of MDL restored LCRs to control. Fig. 6B (top, representative examples and average data in Panels C–F) shows that in permeabilized SANC bathed in a physiological [Ca2+], the PDE1 inhibitor nimodipine increases spontaneous RyR activation manifested by increases in LCR size and duration, an increase in the Ca2+ signal of individual LCRs, and an increase of the ensemble LCR Ca2+ signal.
Fig. 6.
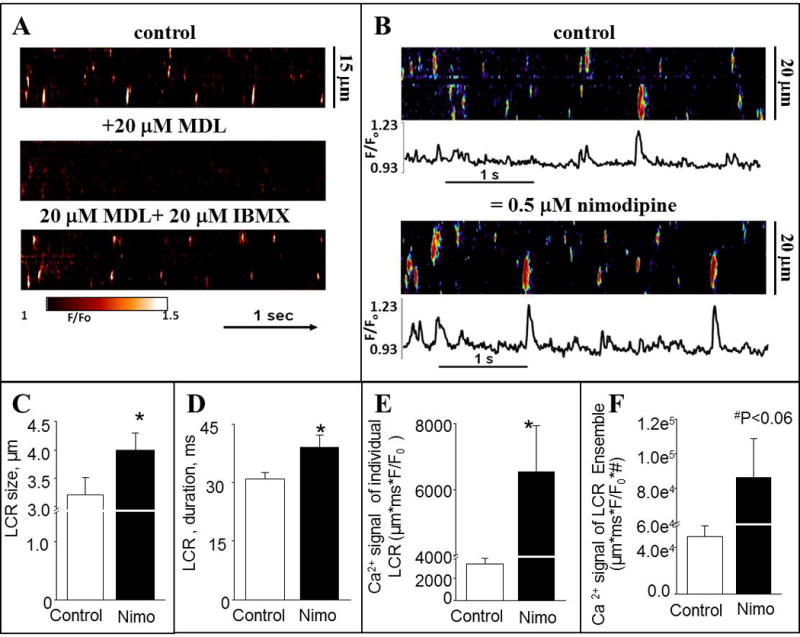
A, Confocal line-scan images of a representative permeabilized SANC prior (top), and during superfusion with 20 μM MDL, an AC inhibitor, alone (middle), and during the continued superfusion with MDL plus the addition of 20 μM IBMX (bottom). Free [Ca2+] was 100 nM. B, representative confocal line-scan images and Ca2+ transients of SANC bathed in 100 nM [Ca2+] in control conditions and after application of 0.5 μM nimodipine (Nimo). C–F, average characteristics of local Ca2+ releases (LCRs) in permeabilized SANC. C, average LCR size (μm). D, average LCR duration (ms). E, average Ca2+ signal of individual LCRs (μm ∗ ms ∗ nM Ca2+, see the Methods section). F, Ca2+ signals LCR Ensemble (sum of μm ∗ ms ∗ nMCa2+, see the Methods section). N = 6; *p < 0.05 vs control, paired t-test.
3.7. cAMP and spontaneous beating rate in cardiac HL-1 cells
HL-1 cells are an immortal cell line with a phenotype that is similar to adult cardiomyocytes and they represent somewhat of a hybrid between embryonic and adult myocytes rather than an intermediate stage of myocyte maturation [23]. An impressive literature demonstrates the similarity of peacemaking in HL-1 cells and SAN cells. HL-1 cells express HCN 1–4 channels, predominantly with 1, 2 and 4 isoforms [24]. Properties of If current in HL-1 cells are similar to those in SANC; inhibition of If current in HL-1 cells as in SANC significantly suppresses diastolic depolarization and reduces automaticity. L-type and T-type Ca channels, inward rectifier K+ channels, and Stim1 are also expressed in HL-1 cells [25–28]. The effects of application of thapsigargin and ryanodine to HL-1 cells as well as effects of PKA activation are the same as in SANC. Block of inward Na+ currents reduces beating rate in HL-1 cells [26]. Finally, NCLX gene, (mitochondrial NCXmit) also regulates automaticity in the HL-1 cardiomyocytes, possibly via modulation of SR CA2+ handling [29] so it does in rabbit SANC [30].
We employed genetic manipulation (PDE1A siRNA) in HL-1 cells to determine its effect on the spontaneous beating rate and real time cAMP levels. Fig. 7A illustrates representative beating rate recordings in HL-1 cells transfected with scrambled siRNA and in HL-1 cells with PDE1A silencing. On average (n = 4) PDE1A expression level was reduced to 30% of control in response to its mRNA silencing. Note that the spontaneous beating rate is significantly increased by 80% in cells with PDE1A knockdown (n = 18), on average (Fig. 7B).
Fig. 7.
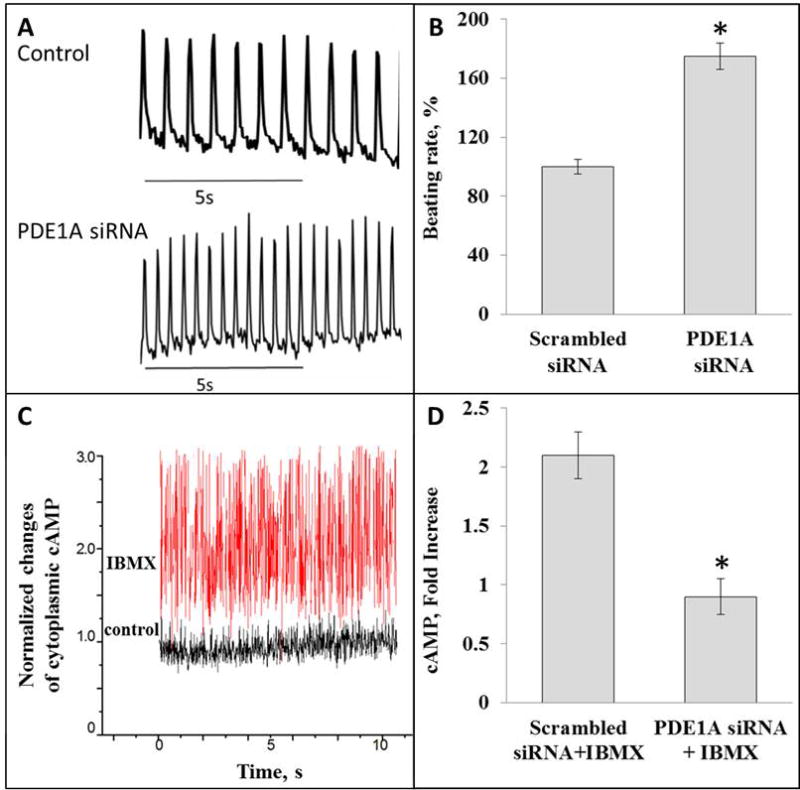
A, optically recorded contractions of HL-1 cells transfected with scrambled siRNA (control) or siRNA targeting PDE1A (bottom graph). B, increase of beating rate of HL-1 cells in response to inhibition of PDE1A mRNA expression with siRNA, *p < 0.05; C, example of FRET detection of cAMP level in live HL-1 cell transfected with scrambled siRNA. Note change of basal level of cAMP in cell in response to IBMX treatment. D. Average data of IBMX-induced increase of cAMP in HL-1 cells transfected with scrambled siRNA (2.1±0.22, n = 8) and cells transfected with siRNA targeting PDE1A (0.90±0.15, n = 8); *p < 0.05.
We employed fluorescence resonance energy transfer (FRET) microscopy to detect cAMP levels in HL-1 cells in order to investigate the impact of PDE1A on intracellular cAMP. Fig. 7C illustrates an example of FRET detection of cAMP from a single cell, which increases two fold in response to IBMX. Fig. 7D shows that in control cells (transfected with a scrambled siRNA), the broad spectrum PDE inhibitor, IBMX, on average, increase cAMP by about 2 fold. But in cells with PDE1A knockout, IBMX, on average, does not increase cAMP, indicating that PDE1A has a major contribution to the total PDE activity in HL-1 cells. Thus PDE1A activity in HL-1 cells suppresses both cAMP and the spontaneous beating rate. Of note, PDE1A knockdown also suppressed the relative ability of IBMX to increase cGMP to the same extent as cAMP (ODS, Fig. 9).
4. Discussion
Constitutive basal Ca2+/CaM activation of neuronal types AC types 1 and 8 in SANC leads to increased basal PKA-dependent protein phosphorylation of Ca2+ cycling and surface electrogenic proteins. This protein phosphorylation augments SR Ca2+ release, both AP induced and spontaneous LCRs during diastole, required for SANC to generate spontaneous APs at a normal rate. Spontaneous, rhythmic Ca2+ releases within local microdomains also lead to feed-forward local activation of AC/PKA signaling; degradation of cAMP by PDE activity indirectly limits the both A cyclase cAMP signaling initiation and downstream cAMP-mediated PKA-dependent phosphorylation. In addition to PKA-dependent phosphorylation, local increases in [Ca2+] activate Ca2+/CaM-dependent CaMKII, which leads to the basal phosphorylation of sarcolemmal and SR Ca2+ cycling proteins not only at PKA but also at CaMKII sites. Basal phosphorylation of both sites is prerequisite to the generation of spontaneous, rhythmic APs at a normal, basal rate, i.e., SANC normal automaticity [31]. Basal cAMP, PKA and CaMKII also regulate mitochondrial ATP production, ensuring it to be commensurate with ATP utilization to generate spontaneous APs at given rates [5–7].
The present study is the first (1) to report PDE1A protein levels in rabbit cardiac tissues, PDE1 isoform transcripts’ expression levels in isolated SANC, LVC and RAC, (2) to demonstrate PDE1A localization within SANC, and (3) to demonstrate its relevance to pacemaker cell activity. A most interesting finding is that although the mRNA expression of all of the major PDE types have previously been detected in cardiac muscle cells [11,13,32,33] the relative abundance of PDE1A isoform in SANC exceeds that in LVC by an order of magnitude (Fig.1A, B), as does PDE1A protein expression (Fig. 1C, D). As in human ventricular myocardium [34] in rabbit LVC in the present study PDE Type 1C transcripts and protein expression were substantially greater than in SANC.
While all three PDE1 subtypes are Ca2+/CaM-activated, some differences among subtypes have been observed in previous studies [35,36]. Ca2+/CaM binding activates PDE1A and PDE1C, and this activation is inhibited by PKA-dependent phosphorylation of the PDE itself. The activity of PDE1B, in contrast, is inhibited by CaMKII-dependent phosphorylation [37,38]. Almost 40% of total PDE activity in SANC lysates was inhibited by the PDE1 inhibitor, nimodipine. In contrast, nimodipine had a negligible effect on PDE activity in LVC. The high activity of PDE1A in SANC may be due, in part, not only to high PDE1A protein expression, but also to the high Vmax of that enzyme (10 to 450 μmol/min/mg for PDE1 compared to other PDEs, e.q., 0.03 to 8.5 μmol/min/mg for PDE3 or PDE4) [39].
The balance of synthesis–degradation of cAMP determines the set point for the local cAMP level. Specific functional coupling of individual PDE families to Gs-coupled receptors was identified in rat ventricular myocytes as a major mechanism enabling cardiac cells to generate heterogeneous cAMP signals in response to different hormones [40]. Spatially and temporally distinct cAMP signals can coexist within the same cell [41,42]. Elegant prior studies in rat ventricular myocytes provide evidence that localized PDE activity creates a functional cAMP diffusion barrier [40,43], that effectively restricts high cAMP levels to local AC domains where high cAMP is generated. With the exception of the one soluble AC species, all other ACs have twelve transmembrane domains and reside only within membranes [44,45]. A Ca2+-induced increase in cAMP generation would occur if the balance within a micro-environment differentially increases AC rather than PDE activation. Differential targeting of PDE1 isoforms is likely to result from different targeting mechanisms that control their spatial distribution (cf. cartoon in Fig. 8). The high level of coupled expression of Ca2+/CaM-activated AC8 and PDE1A in SANC is consistent with their coupled expression in the other cell types [15–17]. Thus, in SANC, as in the retina, the AC/PDE system is poised within microdomains to sense changes in Ca2+ or CaM and respond by cAMP production and degradation. cAMP signaling leads to changes in PKA and local Ca2+ releases, which feedback on the initial Ca2+ signal thus leads to an increase in Ca2+ for AC activation. Both positive and negative feedback can occur. PKA-dependent phosphorylation of PDE1A reduces its activity, leading to a feed forward effect to increase cAMP [14]. Ca2+ and CaM inhibit PDE1A phosphorylation. Interestingly, PP2B (calcineurin), a Ca2+/CaM-dependent phosphatase, dephosphorylates PDE1A, limiting cAMP-mediated, PKA-dependent phosphorylation of PDE1A [46]. Thus PP2B may have a role in balancing the Ca2+-CaM-AC-cAMP-PKA-signaling.
Fig. 8.
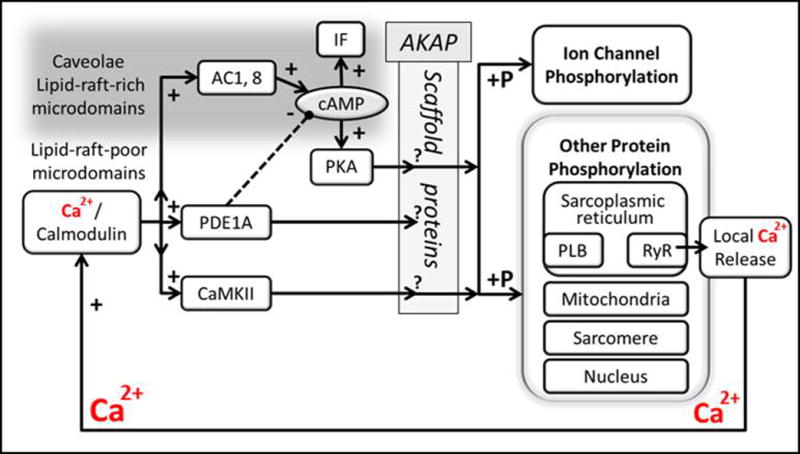
A speculative schematic depicting Ca2+/calmodulin activation of AC and PDE1 signaling in SANC lipid-raft-rich and lipid-raft-poor microdomains, respectively, and possible role of PDE in regulating cAMP, and downstream effects on phosphorylation of PKA-dependent proteins that regulate Ca2+ cycling in sarcoplasmic reticulum and activation of surface membrane ionic currents (solid lines indicate positive and dashed – negative regulation). Future experiments are required to determine the accuracy of this cartoon. Note also that SANC also express AC5 and 6 (not depicted in scheme), which are activated by β-adrenergic receptor ligands via activation of Gσa.
The present study (Fig. 4) is also the first to demonstrate that PDE activity is low in SANC lipid-raft-rich fractions, but is high in SANC fractions that are lipid-raft-poor (i.e. soluble fraction of density gradient separation of SANC lysates). Since our prior study had demonstrated that Ca2+-activated AC activity within SANC is mainly localized in lipid-raft-rich membrane microenvironments [1], the present results support the idea that PDE activity hovering in lipid-raft-poor domains outside but adjacent to lipid-raft-rich microdomains of SANC modulates cAMP diffusion from these domains to its targets (cf. cartoon in Fig. 8). HCN4 localized in caveoli is a target of cAMP [47,48]. A comparison of confocal and SIM images of PDE1A and HCN4 coimmunolabeling in SANC demonstrates that PDE1A is localized approximately 150 nm internal to the surface membrane where HCN4 is localized (Fig. 2). PDE1A location near to the cell membrane may limit cAMP diffusion from sarcolemmal lipid rafts where it is produced [1] to its numerous targets, including HCN4, thereby modulating HCN4 activity. Such PDE diffusion barriers, while limiting, do not completely prevent the spread of cAMP from lipid-raft-rich domains to potential targets outside lipid rafts. This idea does not preclude PDE mobilization into or out of a lipid raft environments.
Because PDE1 isoforms, in contrast to AC, have no membrane spanning regions any potential association with the plasma membrane would likely result from lipid modification or binding to scaffold proteins, e.g. A-kinase-anchoring protein (AKAP). While AKAPs were originally classified on their ability to compartmentalize PKA it is now recognized that this family of regulatory proteins also acts as scaffolding of receptors, protein kinase C, ACs, PDEs and phosphatases [49]. In cardiac myocytes AKAP6 localizes to the junctional membrane of the SR and to the perinuclear region of cardiac myocytes [50–52], and can form multienzyme complexes. In this region PDE type 3A is highly enriched within SR of LVC. Due to an incomplete understanding of Ca2+ regulation of PDE1, however, it is possible to only speculate on the role of the CaM-binding domain in the intracellular targeting and binding of PDEs along with PKA and other kinases to AKAPs [53]. It therefore is possible that the differences in PDE1 isoform distribution could be due to the cytoskeleton targeting the enzyme to discrete regions within the cell.
In addition to compartmentalization of AC types 1 and 8 and PDE1A, competition between these enzymes for a limited pool of CaM, may also be a determinant of the balance of their activities within SANC. Experiments on the kinetics of PDE activity using fluorescent cAMP sensors revealed that PDE activity in a physiological system is much faster than the speed of induced cAMP synthesis in the cells [54]. Thus, different kinetic properties of AC1, 8 and PDE1A also may play a major role in determining the cAMP balance resulting from an increase in Ca2+ [55].
Our experiments of PDE1 inhibition on SR-generated spontaneous LCRs provide some evidence to implicate PDE1 involvement in SR Ca2+ cycling. As noted, c-AMP-mediated-PKA dependent phosphorylation of SR Ca2+ cycling proteins, phospholamban and RyR is required for SANC normal automaticity [8]. The validity of the concept that AC-PDE balance regulates spontaneous SR-generated LCRs is established by our experiments in permeabilized SANC, an experimental preparation in which SR competes with EGTA for a fixed amount of free Ca2+ (Fig. 6). AC inhibition abolishes LCRs; PDE inhibition in the continual presence of AC inhibition is able to restore the spontaneous LCRs (Fig. 6). Type PDE1A, at least in part, accounts for local PDE activity effects on SR Ca2+ cycling causes the PDE1A inhibitor nimodipine augments LCRs signals. The increase in cAMP following PDE1A mRNA knockdown in HL-1 cardiac cells may increase the spontaneous beating rate via effects on If activation and/or SR Ca2+ cycling and also on other potential targets not measured in the present study.
The activity of other PDEs, however, is also critical for SANC normal automaticity. The basal spontaneous AP firing in intact SANC increases dramatically in response to PDE3 inhibition and is accompanied by a marked increase in PLB phosphorylation [10]. PDE4 is also expressed in SANC and has been implicated in their action potential firing rate [56]. A recent report on mouse neonatal cardiac myocytes provides evidence that PDE4D is also activated by Ca2+ via CaMKII [57]. Co-immunoprecipitation of PDEs and Ca2+ cycling proteins in ventricular cells indicate that PDE3-4 are linked to SR SERCA-2 and RyR, two of the most important Ca2+ cycling proteins in regulation of SANC automaticity [58–60]. The importance of PDE3A in heart rate regulation is demonstrated in PDE3A and PDE3B gene knockout mice [61].
5. Conclusions
Because of variable enzymatic microenvironments within cells in vivo, and AC, PDE, kinase, and phosphatase concentrations and activities, and [Ca2+] likely vary among microenvironments, and because of differences in PDE enzyme Km and Vmax for cGMP and cAMP, it is virtually impossible, in the absence of the additional measurements that go beyond those of the present study, to define the precise cellular microenvironment cAMP concentrations in SANC. Newly engineered methods based on sensitivity of different agents such as PKA or cyclic nucleotide-gated ion channels to cAMP permit detection of different pools of cAMP within cell compartments [41,62–65]. Still, it is tempting to speculate (Fig. 8) that local increases in Ca2+ within lipid-raft-rich microdomains activate AC in SANC, and in lipid-raft-poor microdomains activate PDE1A. The source of Ca2+ to activate AC1 and AC8, CaMKII, and PDE1A in SANC is likely Ca2+ influx into the 15 nm gap between the sarcolemma and SR proteins via L-type Ca2+ channels, or that via spontaneous rhythmic local, diastolic Ca2+ releases resulting from spontaneous local RyR activation, or from the global Ca2+ release initiated by the AP [66–69]. In mouse SANC recent evidence suggests that capacitative Ca2+ entry via transient receptor potential store operated channels may be a Ca2+ source [70,71].
Supplementary Material
Acknowledgments
We thank Dr. Willliam C. Claycomb (Department of Biochemistry and Molecular Biology, LSU Health Sciences Center, New Orleans, LA, USA) for providing the HL-1 cell line and Dr. Kees Jalink, PhD (Netherlands Cancer Institute) for providing the cAMP FRET sensors.
Sources of funding
This work was supported by the National Institute on Aging, National Institutes of Health Intramural Research Program, NIA and NIH contract number TAS::75 0872::TAS.
Abbreviations
- AC
adenylyl cyclase
- AKAP
A-kinase-anchoring protein
- AP
action potential
- CaM
calmodulin
- CaMKII
calmodulin-dependent protein kinase II
- Cav-3
caveolin-3
- FRET
fluorescence resonance energy transfer
- GM-1
ganglioside M1
- HCN
hyperpolarization-activated cyclic nucleotide-gated potassium channel
- IBMX
isobutylmethylxanthine
- If
“funny” current
- LCR
local Ca2+ release
- LVC
left ventricular cardiomyocytes
- MDL
MDL-12330A (cis-N-(2-phenylcyclopentyl)-azacyclotridec-1-en-2-aminehydrochloride)
- ODS
online data supplement
- PDE
phosphodiesterase
- PKA
protein kinase A
- RAC
right atrial cardiomyocytes
- RyR
ryanodine receptor
- SAN
sinoatrial node
- SANC
sinoatrial nodal cells
- SDG
sucrose density gradient
- SIM
structured illumination microscopy
- SR
sarcoplasmic reticulum
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.yjmcc.2016.06.064.
Footnotes
Disclosures
None.
Contributor Information
Antoine Younes, Email: Younesa@nia.nih.gov.
Alexey E. Lyashkov, Email: Alexey.lyashkov@nih.gov.
Kirill V. Tarasov, Email: Tarasovkv@mail.nih.gov.
Daniel R. Riordon, Email: RiordonD@grc.nia.nih.gov.
Joonho Lee, Email: Joon.lee@nih.gov.
Syevda G. Sirenko, Email: Sirenkos@grc.nia.nih.gov.
Evgeny Kobrinsky, Email: KobrinskiEv@grc.nia.nih.gov.
Bruce Ziman, Email: ZimanB@grc.nia.nih.gov.
Yelena S. Tarasova, Email: Tarasovay@grc.nia.nih.gov.
Magdalena Juhaszova, Email: Juhaszovam@grc.nia.nih.gov.
Steven J. Sollott, Email: SollottS@grc.nia.nih.gov.
David R. Graham, Email: Dgraham@jhmi.edu.
References
- 1.Younes A, Lyashkov AE, Graham D, Sheydina A, Volkova MV, Mitsak M, et al. Ca(2+)-stimulated basal adenylyl cyclase activity localization in membrane lipid microdomains of cardiac sinoatrial nodal pacemaker cells. J Biol Chem. 2008;283:14461–14468. doi: 10.1074/jbc.M707540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mattick P, Parrington J, Odia E, Simpson A, Collins T, Terrar D. Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea-pig sino-atrial node cells and modulates the I(f) pacemaker current. J Physiol. 2007;582:1195–1203. doi: 10.1113/jphysiol.2007.133439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vinogradova TM, Lakatta EG. Regulation of basal and reserve cardiac pacemaker function by interactions of cAMP-mediated PKA-dependent Ca2+ cycling with surface membrane channels. J Mol Cell Cardiol. 2009;47:456–474. doi: 10.1016/j.yjmcc.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vinogradova TM, Lyashkov AE, Zhu W, Ruknudin AM, Sirenko S, Yang D, et al. High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ Res. 2006;98:505–514. doi: 10.1161/01.RES.0000204575.94040.d1. [DOI] [PubMed] [Google Scholar]
- 5.Yaniv Y, Sirenko S, Ziman BD, Spurgeon HA, Maltsev VA, Lakatta EG. New evidence for coupled clock regulation of the normal automaticity of sinoatrial nodal pacemaker cells: bradycardic effects of ivabradine are linked to suppression of intracellular Ca cycling. J Mol Cell Cardiol. 2013 doi: 10.1016/j.yjmcc.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yaniv Y, Spurgeon HA, Ziman BD, Lakatta EG. Ca(2)(+)/calmodulin-dependent protein kinase II (CaMKII) activity and sinoatrial nodal pacemaker cell energetics. PLoS One. 2013;8:e57079. doi: 10.1371/journal.pone.0057079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yaniv Y, Spurgeon HA, Ziman BD, Lyashkov AE, Lakatta EG. Mechanisms that match ATP supply to demand in cardiac pacemaker cells during high ATP demand. Am J Physiol. 2013;304:H1428–H1438. doi: 10.1152/ajpheart.00969.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lakatta EG, Maltsev VA, Vinogradova TM. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ Res. 2010;106:659–673. doi: 10.1161/CIRCRESAHA.109.206078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J, Sirenko S, Juhaszova M, Ziman B, Shetty V, Rain S, et al. A full range of mouse sinoatrial node AP firing rates requires protein kinase A-dependent calcium signaling. J Mol Cell Cardiol. 2011;51:730–739. doi: 10.1016/j.yjmcc.2011.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vinogradova TM, Sirenko S, Lyashkov AE, Younes A, Li Y, Zhu W, et al. Constitutive phosphodiesterase activity restricts spontaneous beating rate of cardiac pacemaker cells by suppressing local Ca2+ releases. Circ Res. 2008;102:761–769. doi: 10.1161/CIRCRESAHA.107.161679. [DOI] [PubMed] [Google Scholar]
- 11.Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007;100:309–327. doi: 10.1161/01.RES.0000256354.95791.f1. [DOI] [PubMed] [Google Scholar]
- 12.Johnson WB, Katugampola S, Able S, Napier C, Harding SE. Profiling of cAMP and cGMP phosphodiesterases in isolated ventricular cardiomyocytes from human hearts: comparison with rat and guinea pig. Life Sci. 2012;90:328–336. doi: 10.1016/j.lfs.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 13.Hua R, Adamczyk A, Robbins C, Ray G, Rose RA. Distinct patterns of constitutive phosphodiesterase activity in mouse sinoatrial node and atrial myocardium. PLoS One. 2012;7:e47652. doi: 10.1371/journal.pone.0047652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma RK, Das SB, Lakshmikuttyamma A, Selvakumar P, Shrivastav A. Regulation of calmodulin-stimulated cyclic nucleotide phosphodiesterase (PDE1): review. Int J Mol Med. 2006;18:95–105. [PubMed] [Google Scholar]
- 15.Deplano S, Giorgi M, Maccarone R, Santone R, Nuccetelli V, Basso M, et al. Gene expression and protein localization of calmodulin-dependent phosphodiesterase during ontogenesis of chick retina. J Neurosci Res. 2008;86:1017–1023. doi: 10.1002/jnr.21570. [DOI] [PubMed] [Google Scholar]
- 16.Santone R, Giorgi M, Maccarone R, Basso M, Deplano S, Bisti S. Gene expression and protein localization of calmodulin-dependent phosphodiesterase in adult rat retina. J Neurosci Res. 2006;84:1020–1026. doi: 10.1002/jnr.21009. [DOI] [PubMed] [Google Scholar]
- 17.Georget M, Mateo P, Vandecasteele G, Lipskaia L, Defer N, Hanoune J, et al. Cyclic AMP compartmentation due to increased cAMP-phosphodiesterase activity in transgenic mice with a cardiac-directed expression of the human adenylyl cyclase type 8 (AC8) FASEB J. 2003;17:1380–1391. doi: 10.1096/fj.02-0784com. [DOI] [PubMed] [Google Scholar]
- 18.Georget M, Mateo P, Vandecasteele G, Jurevicius J, Lipskaia L, Defer N, et al. Augmentation of cardiac contractility with no change in L-type Ca2+ current in transgenic mice with a cardiac-directed expression of the human adenylyl cyclase type 8 (AC8) FASEB J. 2002;16:1636–1638. doi: 10.1096/fj.02-0292fje. [DOI] [PubMed] [Google Scholar]
- 19.S A, Barbuti Andrea, Mazzocchi Nausicaa, Terragni Benedetta, Baruscotti Mirko, DiFrancesco Dario. A caveolin-binding domain in the HCN4 channels mediates functional interaction with caveolin proteins. J Mol Cell Cardiol. 2012;53:187–195. doi: 10.1016/j.yjmcc.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 20.Fridolfsson HN, Roth DM, Insel PA, Patel HH. Regulation of intracellular signaling and function by caveolin. FASEB J. 2014;28:3823–3831. doi: 10.1096/fj.14-252320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lugnier C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: a new target for the development of specific therapeutic agents. Pharmacol Ther. 2006;109:366–398. doi: 10.1016/j.pharmthera.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 22.Higo K, Saito H, Matsuki N. Characteristics of [3H]nimodipine binding to sarcolemmal membranes from rat vas deferens and its regulation by guanine nucleotide. Jpn J Pharmacol. 1988;48:213–221. doi: 10.1254/jjp.48.213. [DOI] [PubMed] [Google Scholar]
- 23.Claycomb WC, Lanson NA, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gunther A, Baumann A. Distinct expression patterns of HCN channels in HL-1 cardiomyocytes. BMC Cell Biol. 2015;16:18. doi: 10.1186/s12860-015-0065-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stolting G, de Oliveira RC, Guzman RE, Miranda-Laferte E, Conrad R, Jordan N, et al. Direct interaction of CaVbeta with actin up-regulates L-type calcium currents in HL-1 cardiomyocytes. J Biol Chem. 2015;290:4561–4572. doi: 10.1074/jbc.M114.573956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Z, Murray KT. Ionic mechanisms of pacemaker activity in spontaneously contracting atrial HL-1 cells. J Cardiovasc Pharmacol. 2011;57:28–36. doi: 10.1097/FJC.0b013e3181fda7c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goldoni D, Zhao Y, Green BD, McDermott BJ, Collins A. Inward rectifier potassium channels in the HL-1 cardiomyocyte-derived cell line. J Cell Physiol. 2010;225:751–756. doi: 10.1002/jcp.22278. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen N, Biet M, Simard E, Beliveau E, Francoeur N, Guillemette G, et al. STIM1 participates in the contractile rhythmicity of HL-1 cells by moderating T-type Ca(2+) channel activity. Biochim Biophys Acta. 2013;1833:1294–1303. doi: 10.1016/j.bbamcr.2013.02.027. [DOI] [PubMed] [Google Scholar]
- 29.Takeuchi A, Kim B, Matsuoka S. The mitochondrial Na+–Ca2+ exchanger, NCLX, regulates automaticity of HL-1 cardiomyocytes. Sci Rep. 2013;3:2766. doi: 10.1038/srep02766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maltsev AV, Yaniv Y, Stern MD, Lakatta EG, Maltsev VA. RyR-NCX-SERCA local cross-talk ensures pacemaker cell function at rest and during the fight-or-flight reflex. Circ Res. 2013;113:e94–e100. doi: 10.1161/CIRCRESAHA.113.302465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sirenko S, Yang D, Li Y, Lyashkov AE, Lukyanenko YO, Lakatta EG, et al. Ca(2)(+)-dependent phosphorylation of Ca(2)(+) cycling proteins generates robust rhythmic local Ca(2)(+) releases in cardiac pacemaker cells. Sci Signal. 2013;6:ra6. doi: 10.1126/scisignal.2003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fischmeister R, Castro LR, Abi-Gerges A, Rochais F, Jurevicius J, Leroy J, et al. Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res. 2006;99:816–828. doi: 10.1161/01.RES.0000246118.98832.04. [DOI] [PubMed] [Google Scholar]
- 33.Mongillo M, Zaccolo M. A complex phosphodiesterase system controls beta-adrenoceptor signalling in cardiomyocytes. Biochem Soc Trans. 2006;34:510–511. doi: 10.1042/BST0340510. [DOI] [PubMed] [Google Scholar]
- 34.Vandeput F, Wolda SL, Krall J, Hambleton R, Uher L, McCaw KN, et al. Cyclic nucleotide phosphodiesterase PDE1C1 in human cardiac myocytes. J Biol Chem. 2007;282:32749–32757. doi: 10.1074/jbc.M703173200. [DOI] [PubMed] [Google Scholar]
- 35.Michibata H, Yanaka N, Kanoh Y, Okumura K, Omori K. Human Ca2+/calmodulin-dependent phosphodiesterase PDE1A: novel splice variants, their specific expression, genomic organization, and chromosomal localization. Biochim Biophys Acta. 2001;1517:278–287. doi: 10.1016/s0167-4781(00)00293-1. [DOI] [PubMed] [Google Scholar]
- 36.Fidock M, Miller M, Lanfear J. Isolation and differential tissue distribution of two human cDNAs encoding PDE1 splice variants. Cell Signal. 2002;14:53–60. doi: 10.1016/s0898-6568(01)00207-8. [DOI] [PubMed] [Google Scholar]
- 37.Hashimoto Y, Sharma RK, Soderling TR. Regulation of Ca2+/calmodulin-dependent cyclic nucleotide phosphodiesterase by the autophosphorylated form of Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 1989;264:10884–10887. [PubMed] [Google Scholar]
- 38.Florio VA, Sonnenburg WK, Johnson R, Kwak KS, Jensen GS, Walsh KA, et al. Phosphorylation of the 61-kDa calmodulin-stimulated cyclic nucleotide phosphodiesterase at serine 120 reduces its affinity for calmodulin. Biochemistry. 1994;33:8948–8954. doi: 10.1021/bi00196a012. [DOI] [PubMed] [Google Scholar]
- 39.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 40.Rochais F, Abi-Gerges A, Horner K, Lefebvre F, Cooper DM, Conti M, et al. A specific pattern of phosphodiesterases controls the cAMP signals generated by different Gs-coupled receptors in adult rat ventricular myocytes. Circ Res. 2006;98:1081–1088. doi: 10.1161/01.RES.0000218493.09370.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rich TC, Fagan KA, Tse TE, Schaack J, Cooper DM, Karpen JW. A uniform extracellular stimulus triggers distinct cAMP signals in different compartments of a simple cell. Proc Natl Acad Sci U S A. 2001;98:13049–13054. doi: 10.1073/pnas.221381398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jurevicius J, Fischmeister R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by beta-adrenergic agonists. Proc Natl Acad Sci U S A. 1996;93:295–299. doi: 10.1073/pnas.93.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leroy J, Abi-Gerges A, Nikolaev VO, Richter W, Lechene P, Mazet JL, et al. Spatiotemporal dynamics of beta-adrenergic cAMP signals and L-type Ca2+ channel regulation in adult rat ventricular myocytes: role of phosphodiesterases. Circ Res. 2008;102:1091–1100. doi: 10.1161/CIRCRESAHA.107.167817. [DOI] [PubMed] [Google Scholar]
- 44.Sunahara RK, Taussig R. Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol Interv. 2002;2:168–184. doi: 10.1124/mi.2.3.168. [DOI] [PubMed] [Google Scholar]
- 45.Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, et al. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 2009;7:e1000172. doi: 10.1371/journal.pbio.1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharma RK, Wang JH. Calmodulin and Ca2+-dependent phosphorylation and dephosphorylation of 63-kDa subunit-containing bovine brain calmodulin-stimulated cyclic nucleotide phosphodiesterase isozyme. J Biol Chem. 1986;261:1322–1328. [PubMed] [Google Scholar]
- 47.Barbuti A, Scavone A, Mazzocchi N, Terragni B, Baruscotti M, Difrancesco D. A caveolin-binding domain in the HCN4 channels mediates functional interaction with caveolin proteins. J Mol Cell Cardiol. 2012;53:187–195. doi: 10.1016/j.yjmcc.2012.05.013. [DOI] [PubMed] [Google Scholar]
- 48.Barbuti A, Gravante B, Riolfo M, Milanesi R, Terragni B, DiFrancesco D. Localization of pacemaker channels in lipid rafts regulates channel kinetics. Circ Res. 2004;94:1325–1331. doi: 10.1161/01.RES.0000127621.54132.AE. [DOI] [PubMed] [Google Scholar]
- 49.Smith FD, Scott JD. Scaffolding proteins: not such innocent bystanders. Curr Biol. 2013;23:R515–R7. doi: 10.1016/j.cub.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kapiloff MS, Jackson N, Airhart N. mAKAP and the ryanodine receptor are part of a multi-component signaling complex on the cardiomyocyte nuclear envelope. J Cell Sci. 2001;114:3167–3176. doi: 10.1242/jcs.114.17.3167. [DOI] [PubMed] [Google Scholar]
- 51.McCartney S, Little BM, Langeberg LK, Scott JD. Cloning and characterization of A-kinase anchor protein 100 (AKAP100). A protein that targets A-kinase to the sarcoplasmic reticulum. J Biol Chem. 1995;270:9327–9333. doi: 10.1074/jbc.270.16.9327. [DOI] [PubMed] [Google Scholar]
- 52.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 53.Welch EJ, Jones BW, Scott JD. Networking with AKAPs: context-dependent regulation of anchored enzymes. Mol Interv. 2010;10:86–97. doi: 10.1124/mi.10.2.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nikolaev VO, Gambaryan S, Engelhardt S, Walter U, Lohse MJ. Real-time monitoring of the PDE2 activity of live cells: hormone-stimulated cAMP hydrolysis is faster than hormone-stimulated cAMP synthesis. J Biol Chem. 2005;280:1716–1719. doi: 10.1074/jbc.C400505200. [DOI] [PubMed] [Google Scholar]
- 55.Goraya TA, Masada N, Ciruela A, Willoughby D, Clynes MA, Cooper DM. Kinetic properties of Ca2+/calmodulin-dependent phosphodiesterase isoforms dictate intracellular cAMP dynamics in response to elevation of cytosolic Ca2+ Cell Signal. 2008;20:359–374. doi: 10.1016/j.cellsig.2007.10.024. [DOI] [PubMed] [Google Scholar]
- 56.Vinogradova TM, Lyashkov AE, Li Y, Lukyanenko YO, Lakatta EG. Synergism of PDE3 and PDE4 activity regulates cAMP-mediated PKA-dependent local Ca2+ releases to modulate basal spontaneous firing of cardiac pacemaker cells. Circulation. 2012;126:A13555. [Google Scholar]
- 57.Mika D, Richter W, Conti M. A CaMKII/PDE4D negative feedback regulates cAMP signaling. Proc Natl Acad Sci U S A. 2015;112:2023–2028. doi: 10.1073/pnas.1419992112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 59.Lehnart SE, Wehrens XH, Reiken S, Warrier S, Belevych AE, Harvey RD, et al. Phosphodiesterase 4D deficiency in the ryanodine–receptor complex promotes heart failure and arrhythmias. Cell. 2005;123:25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Richter W, Xie M, Scheitrum C, Krall J, Movsesian MA, Conti M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res Cardiol. 2011;106:249–262. doi: 10.1007/s00395-010-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun B, Li H, Shakur Y, Hensley J, Hockman S, Kambayashi J, et al. Role of phosphodiesterase type 3A and 3B in regulating platelet and cardiac function using subtype-selective knockout mice. Cell Signal. 2007;19:1765–1771. doi: 10.1016/j.cellsig.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 62.Matulef K, Zagotta WN. Cyclic nucleotide-gated ion channels. Annu Rev Cell Dev Biol. 2003;19:23–44. doi: 10.1146/annurev.cellbio.19.110701.154854. [DOI] [PubMed] [Google Scholar]
- 63.Rich TC, Fagan KA, Nakata H, Schaack J, Cooper DM, Karpen JW. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J Gen Physiol. 2000;116:147–161. doi: 10.1085/jgp.116.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rich TC, Tse TE, Rohan JG, Schaack J, Karpen JW. In vivo assessment of local phosphodiesterase activity using tailored cyclic nucleotide-gated channels as cAMP sensors. J Gen Physiol. 2001;118:63–78. doi: 10.1085/jgp.118.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nikolaev VO, Bunemann M, Hein L, Hannawacker A, Lohse MJ. Novel single chain cAMP sensors for receptor-induced signal propagation. J Biol Chem. 2004;279:37215–37218. doi: 10.1074/jbc.C400302200. [DOI] [PubMed] [Google Scholar]
- 66.Vinogradova TM, Bogdanov KY, Lakatta EG. Novel perspectives on the beating rate of the heart. Circ Res. 2002;91:e3. doi: 10.1161/01.res.0000031164.28289.55. [DOI] [PubMed] [Google Scholar]
- 67.Bogdanov KY, Vinogradova TM, Lakatta EG. Sinoatrial nodal cell ryanodine receptor and Na(+)–Ca(2+) exchanger: molecular partners in pacemaker regulation. Circ Res. 2001;88:1254–1258. doi: 10.1161/hh1201.092095. [DOI] [PubMed] [Google Scholar]
- 68.Rigg L, Terrar DA. Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Exp Physiol. 1996;81:877–880. doi: 10.1113/expphysiol.1996.sp003983. [DOI] [PubMed] [Google Scholar]
- 69.Huser J, Blatter LA, Lipsius SL. Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J Physiol. 2000;524(Pt 2):415–422. doi: 10.1111/j.1469-7793.2000.00415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ju YK, Allen DG. Store-operated Ca2+ entry and TRPC expression; possible roles in cardiac pacemaker tissue. Heart Lung Circ. 2007;16:349–355. doi: 10.1016/j.hlc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 71.Ju YK, Chu Y, Chaulet H, Lai D, Gervasio OL, Graham RM, et al. Store-operated Ca2+ influx and expression of TRPC genes in mouse sinoatrial node. Circ Res. 2007;100:1605–1614. doi: 10.1161/CIRCRESAHA.107.152181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.