

Abstract
The genetic code serves as one of the natural links for life’s two conceptual frameworks—the informational and operational tracks—bridging the nucleotide sequence of DNA and RNA to the amino acid sequence of protein and thus its structure and function. On the informational track, DNA and its four building blocks have four basic variables: order, length, GC and purine contents; the latter two exhibit unique characteristics in prokaryotic genomes where protein-coding sequences dominate. Bridging the two tracks, tRNAs and their aminoacyl tRNA synthases that interpret each codon—nucleotide triplet, together with ribosomes, form a complex machinery that translates genetic information encoded on the messenger RNAs into proteins. On the operational track, proteins are selected in a context of cellular and organismal functions constantly. The principle of such a functional selection is to minimize the damage caused by sequence alteration in a seemingly random fashion at the nucleotide level and its function-altering consequence at the protein level; the principle also suggests that there must be complex yet sophisticated mechanisms to protect molecular interactions and cellular processes for cells and organisms from the damage in addition to both immediate or short-term eliminations and long-term selections. The two-century study of selection at species and population levels has been leading a way to understand rules of inheritance and evolution at molecular levels along the informational track, while ribogenomics, epigenomics and other operationally-defined omics (such as the metabolite-centric metabolomics) have been ushering biologists into the new millennium along the operational track.
Keywords: Eubacteria, Genome sequence, Compositional dynamics, Genetic code
Prologue
The first bacterial genome my research team sequenced in collaboration with colleagues at the Institute of Microbiology, Chinese Academy of Sciences, is a thermophile named Thermoanaerobacter tengcongensis [1], which was isolated from one of the fresh water hot springs in Tengchong County, Yunnan, China. Two interesting results in this study stand out among others. One is that the T. tengcongensis genome has 86.7% of its genes encoded on the leading strand and the other is that a strong correlation between the GC content of tDNA and rDNA genes and the optimal growth temperature is found among the sequenced thermophiles at the time. Although the reasoning for the latter is obvious but that of the former is not. Consequently, a research direction was set for investigating the former and as a result we have published more than a dozen papers over the past 10 years toward both understanding conceptual basics and developing related bioinformatic tools [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12], [13], [14], [15], [16].
Along the way, we are forced to synthesize new frameworks and to propose new concepts largely due to the complexity of the genomic view on biology [17], [18], [19], albeit beginning with the very basics (Figure 1). Life “plays” its four “cards” in a context of gene and genome sequences, i.e., nucleotide compositional dynamics of genes and genomes, leading to four basic variables: order, size, GC content and purine (GA or R) content. However, the variables beyond the basics seem very complex, including numerous informational (such as regulatory elements and duplication of genes) and operational definitions (such as chemical modifications of nucleotides and non-genic chromosomal elements) [18], [19]. Therefore, our investigations have to begin with prokaryotic genomes and their basic variables (Table 1).
Figure 1.

The four nucleotides and their variables in DNA sequence A. Life’s informational track has only four “cards”—four nucleotides A, T, G, and C—to “play” but a highly variable “deck” size. For instance, human has a deck of 3 billion “cards”. Although modified nucleotides do exist in genomes, their functional roles are often operational, such as 5-methylcytocine (5-mC) and 5-hydroxylcytocine (5-hmC). B. A “deck of cards” for all life forms has a limited number of basic variables.
Table 1.
Mechanisms associated with compositional dynamics of eubacterial genomes
| Mechanism | In-Track/Op-Track | GC-content/R-content | Mutation/selection | Selected Ref. |
|---|---|---|---|---|
| Global repair and replicationa | +++/++ | +++/++ | +++/++ | [2], [3], [7], [8] |
| Transcription-coupled DNA repair and transcriptiona | +/++ | ++/+ | ++/+ | [16], [20], [21], [22], [23] |
| Strand-biased nucleotide compositiona | ++/+ | +/++ | ++/+ | [4], [5], [6], [7], [8], [9], [10] |
| Strand-biased gene distributionb | +/++ | +/++ | +/++ | [4], [6], [8] |
| Horizontal gene transfer/transpositionb | +/++ | +/+ | +/++ | [8] |
| Genome size expansionb | +/++ | ++/+ | +/++ | [4], [8] |
| Environmental biotic and abiotic factors | +/+ | +/+ | +/+ | [7] |
Note: Some relevant molecular mechanisms and their impacts on the informational (In-Track) or (/) operational (Op-Track) tracks, GC- or R (purine)-contents, and mutation or selection are scored qualitatively (weakly positive, +; moderately positive, ++; and strongly positive, +++). A limited number of examples are provided as references (parentheses). Mechanisms related to composition alteration and those related to gene alteration are indicated by a and b, respectively.
First, it is obvious that to change a sequence in a prokaryotic genome, various DNA polymerases, which are responsible for both replication and repair, hold the keys, although DNA repair mechanisms are not limited to DNA polymerization. By classifying the alpha subunit of the DNA polymerase polII complex that catalyzes the replication of eubacterial genomes, we learnt that the enzyme dimeric combination and associated mutator genes dictate the GC content of the prokaryotic genomes [2], [3], [7], [8]. There is also another molecular mechanism found to alter GC content in a transcript-centric fashion and in a significant way [20], [21], which was first described by us for the rice transcripts [16] and validated subsequently by others in mammalian genomes [22], [23]. Since it is primarily related to the transcription-coupled DNA repair mechanism that is universal to all unicellular and multicellular organisms [24], we went on further to demonstrate its existence in eubacteria [4], [6], as well as most of unicellular [14] and multicellular eukaryotes, if not all (Zhang and Yu, unpublished data). In addition, we also demonstrated that alterations in GC content were associated with both mutation and selection [18], [25], not limited to substitution but also indel [25], [26], [27], [28], and involved in not only sequence variation but also horizontal gene transfer [5], [8].
Second, the alterations in purine content are always weaker (within a range of 20%), compared to those in GC content (within a range of 60%; also see the Model). The less pronounced difference in purine content explains why it was not noticed by Erwin Chargaff who discovered the base pairing rules of isolated DNA in several organisms [29], [30], [31]. However, our theoretical work and algorithms have both validated their importance [9], [10], [11], [12], [13]. We and others have pointed out that purine content is associated with strand-biased purine asymmetry (SPA), strand-biased gene distribution (SGD) and transcript-centric nucleotide composition biases [4], [5], [6], [7], [8], [9], [10], [11], [12], [13], [14], [20], [21], [22], [23], [28].
Third, as far as the dominant mutations (such as substitutions and indels) are concerned, the chances that mutations at the nucleotide level is linked to the function of proteins at amino acid level are rather high in prokaryotes and in the coding sequences of eukaryotes. The relationship between DNA and protein sequences is primarily governed by the organization of the genetic code [32], [33], [34], the canonical in most cases. As demonstrated in Figure 2, the three dnaE groups of eubacteria chose different strategies to allow alterations in GC content: high GC (the dnaE2 group that uses a dnaE1 homodimeric alpha subunit and dnaE2 as a major mutator gene), low GC (the dnaE3-polC group that uses a dnaE3 and polC heterodimeric alpha subunit), and varying in between high and low (the dnaE1 group that uses a dnaE1 homodimeric alpha subunit) [7], [8]. Not only are these choices exhausted, but also each has advantages over others. For instance, the dnaE1 group bacteria are able to alter their GC content in a broader range as opposed to the other two groups, such that they can enjoy the broader and more diverse codon usage while both the dnaE2 and dnaE3 groups are rather limited to higher or lower GC content, respectively. However, the low GC dnaE3 group bacteria have a more chemically-diverse codon set (the codons in the purine-sensitive quarter) [32], [33], [34] to exploit and the high GC dnaE2 group bacteria tend to have larger genomes [8]—just to mention a couple of the advantages—as we certainly have much more examples in mind.
Figure 2.
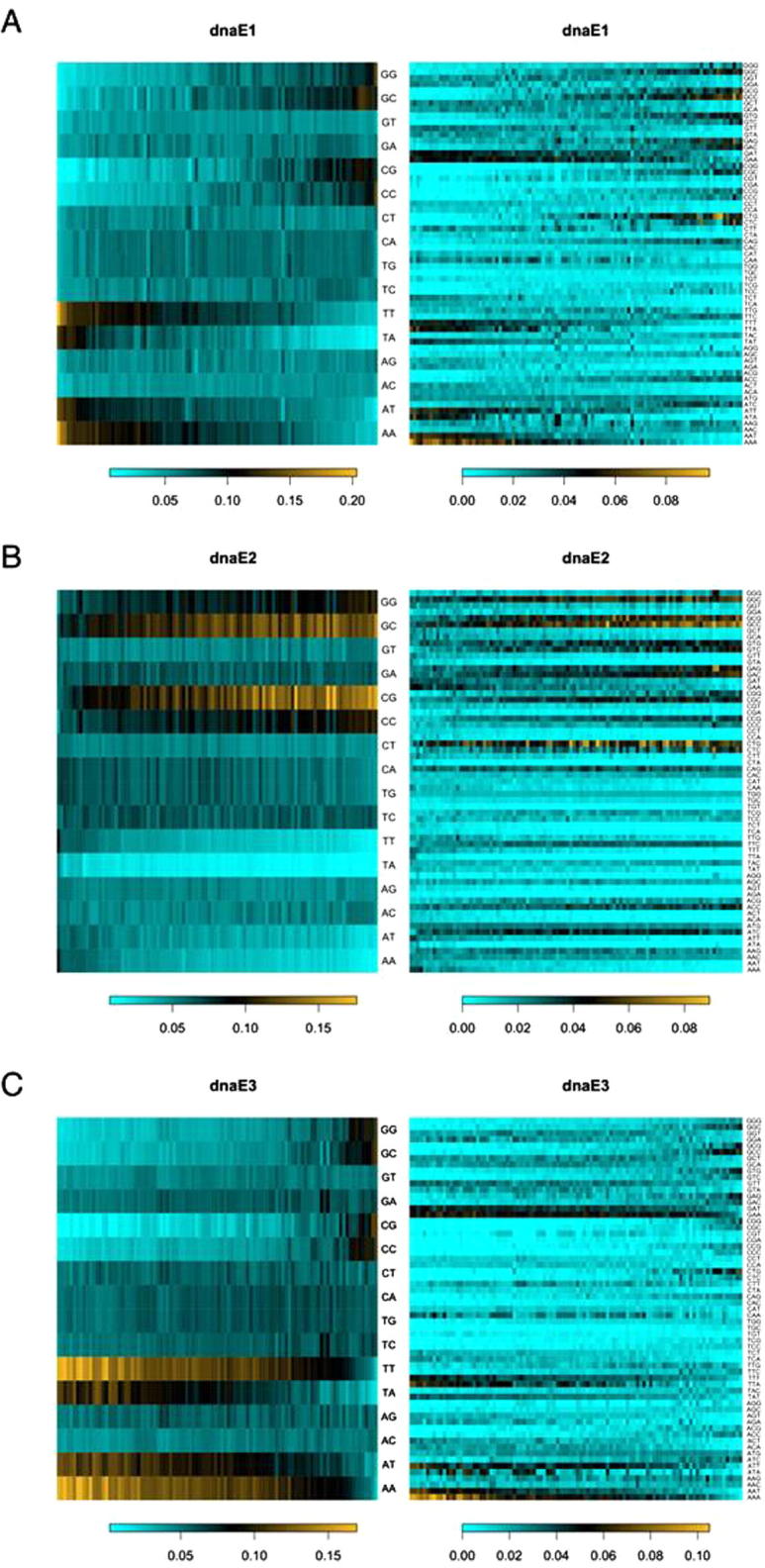
Dinucleotide and codon contents of prokaryotic genomes We used 300 genomes, 100 each, from the three dnaE-based groups including the dnaE1 (dnaE1–dnaE1) group (A), the dnaE2 (dnaE1–dnaE1 and dnaE2) group (B) and the dnaE3 (dnaE3–polC) group (C). Di-nucleotide contents (left panels) are sorted based on GC content increase (left to right; scale bars). Codons (right panels) are also sorted based on GC content changes (left to right; scale bars). The six-fold codons are separated into their corresponding two and four codon sets. Note that frequencies of dinucleotides are essentially equivalent to those of codons and that GC-rich and GC-poor codons are over-utilized in the dnaE2 and dnaE3 group bacteria.
We thus present a mechanistic model, detailed below, to explain the relationship among GC and purine contents, codon usage, and sequence variation of genes and genomes, as well as that between mutation and selection (Figure 3, Figure 4).
Figure 3.

The Pendulum Model Pendulum models were drawn for both GC and purine contents in the same figure. On the horizontal scale, the GC content variation is shown where the equilibrium position (dashed yellow massless rob and massive bob) points at 50%, although there are ample genomes whose GC contents deviate significantly from this position. The amplitude of GC variation is rather broad, leading to a 60% difference (from 20% to 80%, horizontal double-arrowed blue line). The dashed blue curve indicates bob’s trajectory. Other dashed massless robs and massive bobs (red and blue indicate GC-rich and GC-poor, respectively) and their connected arrowed dials connect different GC-content to amino acids; the arrowed dials (dashed arrowed lines indicate transient positions) are aligned linearly with the bobs. On the vertical scale, variation of purine content is shown, which has smaller amplitude than that of GC content, (40–60%, green massive bob and massless rob), a third of the amplitude for GC content variation. The vertical double-arrowed green line indicates the amplitude and the green dashed curve shows the bob’s trajectory. The equilibrium position is indicated with yellow dashed massless rob and massive bob, pointing at 50% that is roughly the average purine content for most of the genomes and genes. The connected arrowed dials are perpendicular in this part of the model to the pendulums and such connection has no particular meaning but to demonstrate the link between nucleotide to amino acid sequences. The face of the “pendulum clock” has two components, the 64-codon genetic code and the 20 amino acid set. The frictionless pivot (dark grey toothed button) is fixed in the genetic code to indicate the fact that the information flow is translated into protein sequences through the code and that the code has both evolved step-wise to fix the coding capacity in the operational track and selected to minimize the damage in the operational track when DNA sequence varies to change the amino acid sequence. Among prokaryotic genomes, GC content variation is the major force dominating composition dynamics, while purine content variation only becomes pronounced when GC-content becomes relatively low. Lower GC-content forces the genomes to select more G for protein coding diversity and more genes on the leading strand to achieve transcription efficiency and transcript stability (see the main text for details).
Figure 4.

A detailed illustration of the Pendulum Model Three pendulums are positioned in such a way where the equilibrium position is shown in color and the other two positions are shown in grey to indicate their transient nature. The bob’s trajectory is indicated with a dashed blue line. When the pendulum moves toward either GC increase (red) or AT increase (blue), the GC or AT quarters expand (gray pendulums) as indicated with three schematic circular representations of the clock faces at the three positions. We only filled in the lower half of the clock’s face here, since half of the codon table is not GC-content sensitive. The concentric circles (from the center) are the first (A and U in blue; G and C in red), second (A and U in blue; G and C in red) and the third (only R or purine and Y or pyrimidine are indicated in the AU quarter; N in the GC quarter is omitted) codon positions. The outermost circle displays the corresponding amino acids in a single letter code. The model demonstrates that alterations in GC content lead to the reshuffling of the codon composition in protein through the organization of the genetic code or the codon table. Although the GC-sensitive quarters of the genetic code is directly affected, other codons are not all standing still since the six-codon members of the genetic code including Arg (R), Leu (L) and Ser (S), balance both the purine-sensitive and purine-insensitive halves and GC-sensitive and GC-insensitive quarters [32], [33], [34].
The pendulum model
The mechanics: the genetic code is organized in such a way that the damage of extreme nucleotide variations is effectively minimized yet the potential change in amino acid sequences is also maintained for further natural selection at the population level
Sequence alterations at nucleotide level are assumed random only when replication and global repair mechanisms are concerned for non-protein-coding sequences, while they are mostly selected when transcripts (as a major effect) and other functional sequence elements (such as minimal introns as minor effect in eukaryotes) [25], [26], [27] are considered.
The larger amplitude: GC content variation represents the dominant change in nucleotide composition dynamics
The GC contents of eubacteria genomes vary from 20% to 80% and such a variation is also contributed by both global and transcript-centric mutation mechanisms, representing a major mutation force. The GC-rich codon encoded amino acids are relatively small and less diverse in terms of their physiochemical properties as opposed to the GC-poor codons that are mostly large and more diverse (Figure 4).
The smaller amplitude: purine content is often strongly selected when GC content goes extremity
The purine contents (R-contents) of some prokaryotic genomes, especially their genes, are extremely biased, although the genomic variation range of purine contents is three-fold narrower than that of GC contents (Figure 3). The selection on purine content is essentially due to the fact that purines at the second codon position control the physicochemical properties of the majority of the 20 amino acids [9], [10], [32], [33], [34].
The clock face: the genetic code links mutation to selection
Despite the fact that there are possibilities that other sequence elements and structures are also selected evolutionarily, the selection for protein-coding sequences is dominant across all superkingdoms. The GC content is also dominant over purine content and they are inversely related: purine content decreases when GC content increases [4]. Furthermore, selection should be more obvious but not necessarily maximized when GC content goes to the extremes. Therefore, the effect of selection may not be figured out easily since it is exceedingly context-dependent. However, mutation is often obvious as long as it is partitioned into global and transcript-centric portions, both of which are at the same order of magnitude, although the latter is weaker [20], [21].
Epilogue
On the one hand, composition dynamics as DNA sequence mutation is thought to be random, neutral, and free to evolve (drifting). On the other hand, however, a greater majority of the changes in coding sequence can lead to amino acid changes that have potentials to alter the structure of a protein and thus to affect its function. Natural selection then comes into play. Whenever there is a mutation, selection will come sooner or later, directly or indirectly. By working on selective mutations and populations or even case-control studies, we will never be able to fully understand the relationship between mutation and selection; a bigger, larger and broader picture is of essence. One example is gene clustering between the vertebrate and the arthropod lineages [18], [19], [35], [36], [37]. As one of the major mechanisms in the operational track, gene clustering contributes largely to negative selection. Another example is antisense regulation [38], [39], where transcript-centric mutation and selection from the 3’ end is inevitable. One additional example is genome duplication and its associated massive gene loss and fast accumulation of mutations in rice and mammals [40], [41]. A similar phenomenon has been reported recently in the yeast Saccharomyces cerevisiae [42].
Acknowledgements
We would like to thank Drs. Hao Wu and Dawei Huang for providing materials for the figures. National Basic Research Program (973 Program; Grant No. 2011CB944100 and 2011CB944101) and National Natural Science Foundation of China (Grant No. 90919024). This work was supported by grants from the “100-Talent Program” of Chinese Academy of Sciences (Grant No. Y1SLXb1365), National Programs for High Technology Research and Development (863 Program; Grant No. 2012AA020409).
References
- 1.Bao Q., Tian Y., Li W., Xu Z., Xuan Z., Hu S. A complete sequence of the T. tengcongensis genome. Genome Res. 2002;12:689–700. doi: 10.1101/gr.219302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao X., Hu J., Yu J. Comparative analysis of eubacterial DNA polymerase III alpha subunits. Genomics Proteomics Bioinformatics. 2006;4:203–211. doi: 10.1016/S1672-0229(07)60001-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao X., Zhang Z., Yan J., Yu J. GC content variability of eubacteria is governed by the pol III alpha subunit. Biochem Biophys Res Commun. 2007;356:20–25. doi: 10.1016/j.bbrc.2007.02.109. [DOI] [PubMed] [Google Scholar]
- 4.Hu J., Zhao X., Zhang Z., Yu J. Compositional dynamics of guanine and cytosine content in prokaryotic genomes. Res Microbiol. 2007;158:363–370. doi: 10.1016/j.resmic.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Hu J., Zhao X., Yu J. Replication-associated purine asymmetry (PAS) may contribute to strand-biased gene distribution (SGD) Genomics. 2007;90:186–194. doi: 10.1016/j.ygeno.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Qu H., Wu H., Zhang T., Zhang Z., Hu S., Yu J. Nucleotide compositional asymmetry between the leading and lagging strands of eubacterial genomes. Res Microbiol. 2010;161:838–846. doi: 10.1016/j.resmic.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 7.Wu H., Zhang Z., Hu S., Yu J. On the molecular mechanism of GC content variation among eubacterial genomes. Biol Direct. 2012;7:2. doi: 10.1186/1745-6150-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu H., Qu H., Zhang Z., Hu S., Yu J. Strand-biased gene distribution (SGD) and nucleotide composition (SNC) in Eubacteria. Genomics Proteomics Bioinformatics. 2012;10:186–196. doi: 10.1016/j.gpb.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Z., Yu J. Modeling compositional dynamics based on GC and purine contents of protein-coding sequences. Biol Direct. 2010;5:63. doi: 10.1186/1745-6150-5-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Z., Li J., Cui P., Ding F., Li A., Townsend J.P. Codon Deviation Coefficient: a novel measure for estimating codon usage bias and its statistical significance. BMC Bioinformatics. 2012;13:43. doi: 10.1186/1471-2105-13-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z., Li J., Wang J., Wong G.K., Yu J. KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinformatics. 2006;4:259–263. doi: 10.1016/S1672-0229(07)60007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang D., Zhang Y., Zhang Z., Zhu J., Yu J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics. 2010;8:77–80. doi: 10.1016/S1672-0229(10)60008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Z., Yu J. Evaluation of six methods for estimation synonymous and nonsynonymous substitution rates. Genomics Proteomics Bioinformatics. 2006;4:173–181. doi: 10.1016/S1672-0229(06)60030-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen K., Meng Q., Ma L., Liu Q., Tang P., Chiu C. A novel DNA sequence periodicity decodes nucleosome positioning. Nucleic Acids Res. 2008;36:6228–6236. doi: 10.1093/nar/gkn626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen K., Wang L., Yang M., Liu J., Xin C., Hu S. Sequence signatures of nucleosome positioning in Caenorhabditis elegans. Genomics Proteomics Bioinformatics. 2010;8:92–102. doi: 10.1016/S1672-0229(10)60010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong G.K., Wang J., Tao L., Tan J., Zhang J., Passey D.A. Compositional gradients in Gramineae genes. Genome Res. 2002;12:851–856. doi: 10.1101/gr.189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu J., Wong G.K. Genome biology: the second modern synthesis. Genomics Proteomics Bioinformatics. 2005;3:3–4. doi: 10.1016/S1672-0229(05)03002-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu J. Challenges to the common dogma. Genomics Proteomics Bioinformatics. 2012;10:55–57. doi: 10.1016/j.gpb.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu J. Life on two tracks. Genomics Proteomics Bioinformatics. 2012;10:123–126. doi: 10.1016/j.gpb.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui P., Ding F., Lin Q., Zhang L., Li A., Zhang Z. Distinct contributions of replication and transcription to mutation rate variation of human genomes. Genomics Proteomics Bioinformatics. 2012;10:4–10. doi: 10.1016/S1672-0229(11)60028-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui P., Lin Q., Ding F., Hu S., Yu J. The transcript-centric mutations in human genomes. Genomics Proteomics Bioinformatics. 2012;10:11–22. doi: 10.1016/S1672-0229(11)60029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green P., Ewing B., Miller W., Thomas P.J., Green E.D. Transcription-associated mutational asymmetry in mammalian evolution. Nat Genet. 2003;33:514–517. doi: 10.1038/ng1103. [DOI] [PubMed] [Google Scholar]
- 23.Majewski J. Dependence of mutational asymmetry on gene-expression levels in the human genome. Am J Hum Genet. 2003;73:688–692. doi: 10.1086/378134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanawalt P.C., Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 25.Wang D., Yu J. Both size and GC-content of minimal introns are selected in human population. PLoS One. 2011;6:e17945. doi: 10.1371/journal.pone.0017945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu J., Yang Z., Kibukawa M., Paddock M., Passey D.A., Wong G.K. Minimal introns are not “junk”. Genome Res. 2002;12:1185–1189. doi: 10.1101/gr.224602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu J., He F., Wang D., Liu K., Huang D., Xiao J. A novel role for minimal introns: routing mRNAs to the cytosol. PLoS One. 2010;5:e10144. doi: 10.1371/journal.pone.0010144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lobry L.J. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol. 1996;13:660–665. doi: 10.1093/oxfordjournals.molbev.a025626. [DOI] [PubMed] [Google Scholar]
- 29.Chargaff E. Chemical specificity of nucleic acids and mechanism of their enzymatic degradation. Experientia. 1950;6:201–209. doi: 10.1007/BF02173653. [DOI] [PubMed] [Google Scholar]
- 30.Magasanik B., Vischer E., Doniger R., Elson D., Chargaff E. The separation and estimation of ribonucleotides in minute quantities. J Biol Chem. 1950;186:37–50. [PubMed] [Google Scholar]
- 31.Rudner R., Karkas J.D., Chargaff E. Separation of B. Subtilis DNA into complementary strands. 3. Direct analysis. Proc Natl Acad Sci U S A. 1968;60:921–922. doi: 10.1073/pnas.60.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu J. A content-centric organization of the genetic code. Genomics Proteomics Bioinformatics. 2007;5:1–6. doi: 10.1016/S1672-0229(07)60008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao J., Yu J. A scenario on the stepwise evolution of the genetic code. Genomics Proteomics Bioinformatics. 2007;5:143–151. doi: 10.1016/S1672-0229(08)60001-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Z., Yu J. On the organizational dynamics of the genetic code. Genomics Proteomics Bioinformatics. 2011;9:1–9. doi: 10.1016/S1672-0229(11)60004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu J., Wong G.K., Wang J., Yang H. 2nd ed. Wiley-VCH; 2005. Shotgun sequencing (SGS) encyclopedia of molecular cell biology and molecular medicine. vol. 13. pp. 71–114. [Google Scholar]
- 36.Yang L., Yu J. A comparative analysis of divergently-paired genes (DPGs) of Drosophila and vertebrate genomes. BMC Evol Biol. 2009;9:55. doi: 10.1186/1471-2148-9-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cui P., Lin Q., Zhang L., Ding F., Xin C., Zhang D. The disequilibrium of nucleosomes distribution along chromosomes plays a functional and evolutionarily role in regulating gene expression. PLoS One. 2011;6:e23219. doi: 10.1371/journal.pone.0023219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui P., Liu W., Zhao Y., Lin Q., Zhang D., Ding F. Comparative analyses of H3K4 and H3K27 trimethylations between the mouse cerebrum and testis. Genomics Proteomics Bioinformatics. 2012;10:82–93. doi: 10.1016/j.gpb.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cui P., Liu W., Zhao Y., Lin Q., Ding F., Xin C. The association between H3K4me3 and antisense transcription. Genomics Proteomics Bioinformatics. 2012;10:74–81. doi: 10.1016/j.gpb.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J., Zhang J., Li R., Zheng H., Li J., Zhang Y. Evolutionary transients in the rice transcriptome. Genomics Proteomics Bioinformatics. 2010;8:223–228. doi: 10.1016/S1672-0229(10)60023-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu W., Zhao Y., Cui P., Lin Q., Ding F., Xin C. Thousands of novel transcripts identified in mouse cerebrum, testis, and ES cells based on ribo-minus RNA sequencing. Front Genet. 2011;2:93. doi: 10.3389/fgene.2011.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carvunis A.R., Rolland T., Wapinski I., Calderwood M.A., Yildirim M.A., Simonis N. Proto-genes and de novo gene birth. Nature. 2012;487:370–374. doi: 10.1038/nature11184. [DOI] [PMC free article] [PubMed] [Google Scholar]