

SUMMARY
B cell CLL/lymphoma 11A (BCL11A) is a transcription factor and regulator of hemoglobin switching that has emerged as a promising therapeutic target for sickle cell disease and thalassemia. In the hematopoietic system, BCL11A is required for B lymphopoiesis, yet its role in other hematopoietic cells, especially hematopoietic stem cells (HSCs) remains elusive. The extensive expression of BCL11A in hematopoiesis implicates context-dependent roles, highlighting the importance of fully characterizing its function as part of ongoing efforts for stem cell therapy and regenerative medicine. Here, we demonstrate that BCL11A is indispensable for normal HSC function. Bcl11a-deficiency results in HSC defects, typically observed in the aging hematopoietic system. We find that downregulation of cyclin-dependent kinase 6 (Cdk6), and the ensuing cell cycle delay, correlate with HSC dysfunction. Our studies define a mechanism for BCL11A in regulation of HSC function and have important implications for design of therapeutic approaches to targeting BCL11A.
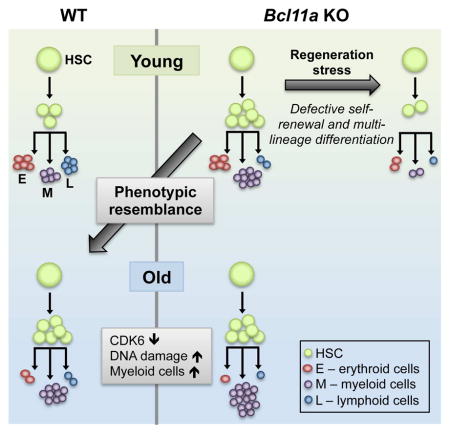
Graphical Abstract
INTRODUCTION
Hematopoietic development in mammals emerges in sequential waves that include the primitive and definitive waves. Primitive hematopoiesis gives rise to transient macrophages, megakaryocytes and large nucleated erythroid progenitors characterized by the production of embryonic globins. The definitive wave of hematopoiesis gives rise to hematopoietic stem cells (HSCs), defined by their ability to self-renew to sustain the HSC population, and differentiate to regenerate the entire hematopoietic system from the embryo into adult life (Palis, 2014; Seita and Weissman, 2010).
B cell CLL/lymphoma 11A (BCL11A) is a C2H2 zinc-finger transcription factor expressed in the hematopoietic system and brain (Liu et al., 2003). Bcl11a-null mice are perinatal lethal and have impaired lymphopoiesis, particularly in the B cell lineage (Liu et al., 2003; Sankaran et al., 2009). During embryonic development, the expression of BCL11A and other transcriptional regulators such as SOX6 and MYB, coincides with definitive hematopoiesis in both human and mouse (Palis, 2014; Sankaran et al., 2009; Xu et al., 2010), although a more recent report suggests that BCL11A may be expressed even earlier at the pre-HSC stage (Zhou et al., 2016).
Genome-wide association studies (GWAS) has identified BCL11A as a major fetal hemoglobin (HbF)-associated locus (Lettre et al., 2008; Menzel et al., 2007; Uda et al., 2008). Subsequent studies demonstrated that BCL11A is expressed in adult definitive erythroid cells and acts as a transcriptional repressor of human fetal and mouse embryonic β-like globin genes (Bauer et al., 2013; Sankaran et al., 2009; 2008; Xu et al., 2011). Given its critical role in hemoglobin switching, BCL11A has emerged as a promising therapeutic target for the major β-globin disorders. However, its essential role in normal B lymphopoiesis underscores the importance of delineating the full extent of BCL11A’s function in other cellular contexts within the hematopoietic system to address target-related toxicities in therapy. In fact, Bcl11a is expressed in multiple hematopoietic lineages besides B lymphoid and erythroid cells, including bone marrow (BM) progenitor cells and HSCs (Yu et al., 2012). Furthermore, its temporal expression in embryonic development coincides with the emergence of definitive hematopoiesis, warranting exploration of its role in establishing the identity and function of definitive HSCs. This is especially relevant considering current efforts to generate bona fide HSCs through directed differentiation of pluripotent embryonic stem cells (ESCs) and reprogramming of induced pluripotent stem cells (iPSCs) for disease-modeling and clinical applications. Although it is possible to make cells that phenotypically resemble definitive HSCs, it remains challenging to generate transplantable long-term definitive HSCs. The limited success of current strategies is due in part to the embryonic-like nature of the ESC/iPSC-derived hematopoietic cells that are developmentally restricted from becoming competent definitive HSCs. Hence, elucidating the role of transcription factors such as BCL11A in definitive hematopoiesis may provide insights into developing improved strategies to overcome these obstacles (Daniel et al., 2016).
Here, we use an inducible, conditional Bcl11a knockout (KO) mouse strain (Ippolito et al., 2014; Sankaran et al., 2009) to examine the role of Bcl11a in definitive hematopoiesis. We demonstrate that Bcl11a is indispensable for normal HSC function. Bcl11a-deficient HSCs exhibit cell cycle defects resembling a premature aging phenotype that is associated with impaired HSC multilineage differentiation and self-renewal. Our studies suggest a mechanism for BCL11A in regulating definitive hematopoiesis and have important implications for the therapeutic targeting of BCL11A.
RESULTS
Bcl11a is required for hematopoietic stem/progenitor cells in embryonic development
Bcl11a is widely expressed in the definitive hematopoietic system, including hematopoietic stem cells (HSCs) and downstream myeloid and lymphoid progenitors (Figure S1A) (Yu et al., 2012). To evaluate the role of BCL11A in steady-state hematopoiesis, we used a conditional Bcl11a mouse strain (Ippolito et al., 2014; Sankaran et al., 2009). crossed with the Gata1-Cre transgenic mice to achieve germline deletion (Jasinski et al., 2001) (Figure S1B). BCL11A is a critical repressor of human fetal hemoglobin and mouse embryonic β-like globin genes (εy and βh1) (Sankaran et al., 2009). Consistently, we observed a marked increase in mouse εy- and βh1-globin mRNA in embryonic day 18.5 (E18.5) Bcl11a-deficient fetal liver cells (Figures S1C and S1D).
Similar to the conventional Bcl11a KO mouse, Bcl11afl/fl × Gata1-Cre mice were perinatal lethal (Sankaran et al., 2009). B lymphopoiesis was significantly impaired in Bcl11a-null E18.5 fetal livers and spleens (Figures 1A and 1B; Figures S1E and S1F). In parallel, there was a marked decrease in the frequency of lymphoid-primed multipotent progenitor (LMPP; Lin−Sca-1+c-Kit+(LSK)Flt3+CD150−) and common lymphoid progenitor (CLP; Lin−Flt3+IL-7Ra+c-KitlowSca-1low), both considered as precursors of B cells, in Bcl11a-null E14.5 and E17.5 fetal livers (Figure 1C; Figures S1G and S1H). Although Bcl11a-deficiency minimally impacted mature myeloid cells (Liu et al., 2003), analysis of the myeloid progenitors, granulocyte-monocyte progenitor (GMP; Lin−Sca-1−c-Kit+CD41−CD150−CD16/32+) and megakaryocyte progenitor (MkP; Lin−Sca-1−c-Kit+CD150+CD41+) demonstrated a modest decrease in E17.5 Bcl11a-null embryos (Figure 1D; Figure S1I). Moreover, phenotypic HSCs were reduced by 3.8-fold and 1.9-fold in Bcl11a-null E14.5 and E17.5 embryos, respectively (Figure 1E; Figure S1J). These refined analyses demonstrate that Bcl11a is required not only for B lymphopoiesis but also for hematopoietic stem/progenitor cells during mouse embryonic development.
Figure 1. Decreases in HSCs and lymphoid progenitors in Bcl11a-deficient embryos.

(A) PreProB cell and ProB cell/PreB cell (ProB/PreB) frequency in E18.5 fetal liver of Bcl11afl/Δ × Gata1-Cre embryos.
(B) ProB/PreB and IgM+ B cell frequency in E18.5 fetal spleen of Bcl11afl/Δ × Gata1-Cre embryos.
(C) Lymphoid-primed multipotent (LMPP) and common lymphoid progenitor (CLP) frequency in E14.5 and E17.5 fetal liver of Bcl11afl/Δ × Gata1-Cre embryos.
(D) Granulocyte-monocyte progenitor (GMP) and megakaryocyte progenitor (MkP) frequency in E14.5 and E17.5 fetal liver of Bcl11afl/Δ × Gata1-Cre embryos.
(E) Hematopoietic stem cell (LSKCD48−Flt3−CD150+) frequency in E14.5 and E17.5 fetal liver of Bcl11afl/Δ × Gata1-Cre embryos.
Error bars represent mean ± SD. In (A–B) n = 5–6 WT, 4–8 Het, 2–4 KO from E18.5 embryos. In (C–E) n = 2 WT, 9 Het, 10 KO from E14.5 embryos and n = 2 WT, 2 Het, 4 KO from E17.5 embryos. *p<0.05; **p<0.01; ***p<0.001.
See also Figure S1.
Acute loss of Bcl11a in steady-state hematopoiesis impairs lymphopoiesis
Given the perinatal lethality following germline deletion of Bcl11a, we then crossed the Bcl11a floxed strain to the interferon-inducible myxovirus resistance-1 (Mx1)-Cre mouse strain (Kühn et al., 1995) to evaluate the role of BCL11A in postnatal hematopoiesis. We obtained non-deleted (wildtype, WT; Bcl11afl/fl × Mx1-Cre−), heterozygously (Het; Bcl11afl/wt × Mx1-Cre+) and homozygously deleted (KO; Bcl11afl/fl × Mx1-Cre+) animals by administration of Polyinosinic:polycytidylic acid, p(I:C) (Figure 2A). Both Het and KO mice were viable following p(I:C) treatment and the BCL11A protein was completely ablated from the BM of Bcl11a KO mice (Figure S2A). Although the Mx1 promoter is active in BM stromal cells, there was no evidence of BCL11A expression in the BM stromal cell compartment (Figures S2B and S2C). To facilitate the assessment and tracking of deleted cells, Bcl11afl/fl × Mx1-Cre mice were also crossed to the Rosa26-stop-EYFP (R26-eYFP) reporter strain (Srinivas et al., 2001).
Figure 2. Acute deletion of Bcl11a in steady-state hematopoiesis results in loss of lymphoid progenitors and B cells.

(A) Experimental design for characterization of the hematopoietic system following acute deletion of Bcl11a.
(B) Frequency of peripheral blood B, T and myeloid cells 3 and 10 weeks following Bcl11a gene deletion.
(C) Frequency of BM myeloid (M), erythroid (E), B and T cells 13 weeks following Bcl11a gene deletion.
(D) Frequency of BM B cell progenitors 13 weeks following Bcl11a gene deletion.
(E) Frequency of BM GMP, MkP and LMPP 13 weeks following Bcl11a gene deletion.
(F) Frequency of BM HSC 13 weeks following Bcl11a gene deletion (right) and representative FACS profiles (left). Mean frequencies of total HSCs from kit-enriched BM is shown in the FACS plots.
Error bars represent mean ± SD. In (B) n = 8 WT, 5 Het, 8 KO, in (C–F) n = 7 WT, 4 Het, 7 KO. *p<0.05; **p<0.01; ***p<0.001.
See also Figures S2 and S3.
Upon p(I:C)-induced gene deletion, B cells (B220+CD19+) was the only blood lineage negatively affected, while the frequency of T cells (CD3ε+Thy1.2+) remained largely unchanged and the frequency of myeloid cells (Mac-1+Gr-1+) was increased by 10 weeks in the peripheral blood (Figure 2B). R26-eYFP reporter expression showed high deletion efficiency in all lineages, which was also confirmed by mRNA expression of Bcl11a and embryonic β-like globin genes (Figures S2D–G). Consistently, among the mature blood lineages in the BM, only B cells were negatively affected by Bcl11a loss (Figure 2C; Figure S3A). Furthermore, Pro B cells (B220+CD43+IgM−CD19+CD24lowCD93+) and Pre B cells (B220+CD43+IgM−CD19+CD24hiCD93+) were absent and PrePro B cells (B220+CD43+IgM−CD19−CD24−CD93+) were markedly reduced in Bcl11a KO BM, suggesting a block in B cell development at the PrePro B and Pro B cell stages, consistent with a previous report (Yu et al., 2012) (Figure 2D; Figures S3B and S3C). Although both mature B cells and B cell progenitors were present in the heterozygous animals, they were decreased to levels intermediate of WT and KO mice (Figures 2C and 2D). Notably, the frequency of myeloid cells (Mac-1+Gr-1+) and T cells (CD3ε+) was increased in the Bcl11a-null BM, whereas erythroid cells (Ter119+CD71+) remained unaltered (Figure 2C; Figure S3A). However, early thymic progenitors (ETP; Lin−CD4−CD8a−c-Kit+CD25−) were significantly decreased in Bcl11a-deficient thymus, whereas double-negative (DN) 2 (Lin−CD4−CD8a−c-Kit+CD25+), DN3 (Lin−CD4−CD8a−c-Kit−CD25+) and DN4 (Lin−CD4−CD8a−c-Kit−CD25−) T cells were not significantly affected (Figure S3D). These findings suggest that Bcl11a is indispensable for both B and early T cell progenitors.
To assess whether Bcl11a is required upstream in the hematopoietic hierarchy, we next analyzed the primitive BM progenitor compartment. Consistent with a decrease in the B cell lineage and ETPs, LMPPs (LSKFlt3+CD150−) were nearly absent in Bcl11a KO mice, whereas the myeloid progenitor compartment including GMPs (Lin−Sca-1−c-Kit+CD41−CD150−CD16/32+) and MkPs (Lin−Sca-1−c-Kit+CD150+CD41+) (Figure 2E; Figures S3E and S3F) was modestly increased. Intriguingly, there was an increase in the frequency and number of phenotypic HSCs (LSKCD48−Flt3−CD150+) in Bcl11a KO mice (Figure 2F; Figure S3G), which was not due to inefficient gene deletion (Figures S3H and S3I). These data demonstrate that acute loss of Bcl11a in the BM resulted in impaired normal lymphopoiesis, a parallel increase in the myeloid compartment and alterations in the frequency of steady-state HSCs.
Bcl11a is required for normal HSCs function
Given the increased HSC frequency in Bcl11a-deficient mice, we next performed a non-competitive transplantation using 1 × 106 whole BM cells from Bcl11a KO mice to functionally evaluate the HSC activity. The donor contribution, measured as the frequency of R26-eYFP+ cells, in peripheral blood rapidly declined after transplantation. Donor chimerism (R26-eYFP +) in the BM was barely detectable after 13 weeks post-transplantation (Figure S4A). To assess whether the impaired repopulation capacity of the Bcl11a KO BM cells was due to an intrinsic effect, we purified phenotypic HSCs (LSKCD48−Flt3−CD150+) from Bcl11a WT, Het and KO and performed a competitive transplantation. To ascertain that Bcl11a-deficiency did not impede homing of the transplanted cells to the BM niche, gene deletion was induced only after confirming reconstitution of the injected cells (Figures 3A and 3B). Following Bcl11a deletion, donor contribution (CD45.2+) in the peripheral blood drastically decreased in the KO transplanted animals (Figure 3B). All donor-derived blood lineages (CD45.2+) in the peripheral blood, including B, T and myeloid cells were markedly reduced in both Het and KO transplanted mice. Remarkably, the myeloid predominance noted in steady-state hematopoiesis was more pronounced (Figure S4C). Moreover, Bcl11a Het HSCs reconstituted myeloid cells to intermediate levels compared to WT and KO HSCs, but were not as efficient in contributing to the lymphoid lineages, in particular B cells (Figure S4B). Consistently, analysis of the BM of transplanted mice at 18 weeks demonstrated that repopulation from Bcl11a KO cells to mature blood lineages (Figures 3C and 3D) and progenitor populations (GMP, MkP and LMPP) (Figure 3E) was severely impaired. Notably, the myeloid lineage appeared to be the least affected.
Figure 3. HSCs from Bcl11a-deficient mice have impaired repopulation ability.

(A) Experimental design to evaluate HSC activity from Bcl11a-deficient mice.
(B) Donor-chimerism in peripheral blood following transplantation of HSCs from Bcl11a WT, Het and KO mice before and after Bcl11a gene deletion.
(C) Frequency of donor-derived BM myeloid (M), B, NK and T cells 18 weeks following Bcl11a gene deletion.
(D) Frequency of donor-derived BM B cell progenitors 18 weeks following Bcl11a gene deletion.
(E) Frequency of donor-derived BM GMP, MkP and LMPP 18 weeks following Bcl11a gene deletion.
(F) Frequency of donor-derived BM HSC 18 weeks following Bcl11a gene deletion (right) and representative FACS profiles (left). Mean frequencies of total HSCs from kit-enriched BM is shown in the FACS plots.
Error bars represent mean ± SD. In (B) n = 7 WT, 8 Het, 7–8 KO, in (C–F) n = 6 WT, 7 Het, 6 KO. *p<0.05; **p<0.01; ***p<0.001.
See also Figure S4.
HSCs from both Bcl11a Het and KO mice failed to reconstitute HSCs to the same level as WT controls (Figure 3F; Figure S4D). The reconstitution ability of Bcl11a KO HSCs did not improve even under lower proliferation pressure by transplantation into sublethally irradiated recipients (Figure S4E). Upon transplantation of equal numbers of Bcl11a WT and KO HSCs into the same irradiated recipient, Bcl11a-deficient HSCs were rapidly outcompeted by WT cells (Figure S4F). Even five times more Bcl11a KO HSCs was not sufficient to maintain long-term repopulation (Figure S4G). Collectively, these results demonstrate that Bcl11a-deficient HSCs are at a competitive disadvantage and Bcl11a is indispensable for normal HSC repopulation capacity.
The inability of Bcl11a-deficient cells to reconstitute HSCs indicates that there might be a self-renewal defect in addition to impaired repopulation. Thus, we purified donor-derived phenotypic HSCs (LSKCD48−Flt3−CD150+CD45.2+ WT, or LSKCD48−Flt3− CD150+CD45.2+eYFP+ Het and KO) from primary transplanted recipients and performed secondary transplantation. Consistent with a self-renewal defect, Bcl11a-deficient HSCs completely failed to reconstitute secondary transplanted animals (Figure 4A). Although Bcl11a Het HSCs were successful in reconstituting secondary recipients, albeit to a lower degree compared to WT HSCs, they were ultimately unable to produce B and T lymphoid cells (Figure 4B; Figure S5).
Figure 4. Bcl11a-deficiency results in impaired HSC self-renewal capacity.

(A) Donor-chimerism in peripheral blood following secondary transplantation of Bcl11a WT, Het and KO HSCs isolated from primary transplanted mice.
(B) Donor-derived B, T and myeloid cells in peripheral blood from (A).
(C) Donor-chimerism in BM 21 weeks following secondary transplantation of Bcl11a WT, Het and KO HSCs.
(D) Frequency of donor-derived BM myeloid (M), B, NK and T cells 21 weeks following secondary transplantation.
(E) Frequency of donor-derived BM B cell progenitors 21 weeks following secondary transplantation.
(F) Frequency of donor-derived BM GMP, MkP and LMPP 21 weeks following secondary transplantation.
(G) Frequency of donor-derived BM HSC 21 weeks following secondary transplantation (right) and representative FACS profiles (left). Mean frequencies of total HSCs from kit-enriched BM is shown in the FACS plots.
Data and error bars represent mean ± SD. In (A–B) n = 9 WT, 9 Het, 7 KO, in (C–G) n = 9 WT, 9 Het, 7 KO. *p<0.05; **p<0.01; ***p<0.001.
See also Figure S5.
Analysis of the BM confirmed that Bcl11a KO HSCs were unable to repopulate the secondary recipients including reconstituting HSCs (Figure 4C–G). Bcl11a Het HSCs demonstrated only modest BM reconstitution (Figure 4C) and almost exclusively produced myeloid cells but failed to contribute to the lymphoid lineages (Figures 4D–E). Moreover, the myeloid progenitors GMP and MkP were present at a lower frequency in Bcl11a Het transplants compared to WT controls (Figure 4F). Unexpectedly, Bcl11a Het HSCs partially reconstituted LMPPs, albeit at low frequency, despite absence of downstream lymphoid cells (Figure 4F). Analysis of the Bcl11a heterozygous HSCs suggests that the observed lymphoid defect is exacerbated with serial transplantation. In summary, these findings demonstrate that Bcl11a is required not only for multi-lineage repopulation but also for HSC self-renewal.
Gene expression profiling of Bcl11a-deficient HSCs show cell cycle defects
To determine the molecular basis of the observed defects in Bcl11a-deficient HSCs, we performed global gene expression analysis. Comparison of transcriptomic profiles between Bcl11a WT and KO HSCs identified 620 genes to be differentially expressed (419 upregulated and 201 downregulated; fold change ≥2, p<0.01) (Figure 5A). To identify candidate cellular processes affected by Bcl11a loss, we performed pathway analyses (Experimental Procedures). Genes significantly upregulated in Bcl11a KO relative to WT HSCs were highly enriched in pathways involved in development, immune response and cell adhesion processes (Figure 5B; Table S1). In contrast, genes downregulated in Bcl11a KO HSCs reflected pathways required for cell cycle, apoptosis and survival, and DNA damage repair (Figure 5C; Table S2).
Figure 5. Bcl11a-deficient HSCs have downregulated Cdk6 expression and a quiescence signature.

(A) Heatmap representation of differentially regulated genes (≥2 fold; p<0.01) between Bcl11a WT and KO HSCs ranked according to log-fold change.
(B) Enriched pathways of genes upregulated in Bcl11a-deficient HSCs.
(C) Enriched pathways of genes downregulated in Bcl11a-deficient HSCs.
(D) Relative mRNA expression of Bcl11a and Cdk6 in sorted Bcl11a WT and KO HSCs.
(E) Relative mRNA expression of typical cell cycle promoting genes in sorted Bcl11a WT and KO HSCs.
(F) Relative mRNA expression of typical cell cycle inhibitor genes in sorted Bcl11a WT and KO HSCs.
(G) Gene set enrichment analysis of two quiescence gene signatures in Bcl11a WT and KO HSCs.
In (A–C, G) n = 3/genotype, in (D–F) n = 4–7/genotype. Data in (D–F) represent mean ± SD. *p<0.05; **p<0.01; ***p<0.001.
See also Figure S6 and Tables S1 and S2.
HSCs are normally quiescent and re-enter cell cycle as a response to various stresses (Pietras et al., 2011). Given the increased HSC frequency but impaired HSC multilineage differentiation and self-renewal in Bcl11a-null animals, we hypothesized that these defects are due to defects in cell cycle. We identified the cyclin-dependent kinase 6 (Cdk6) gene among the top 50 genes that were significantly downregulated in Bcl11a KO HSCs compared to the WT controls (Figure S6). qRT-PCR analysis validated the downregulation of Cdk6 in Bcl11a KO HSCs (P-value = 0.03; Figure 5D). CDK6 protein is expressed in the cell cycle stage gap 1 (G1) and plays an important role in G1 cell cycle progression (Laurenti et al., 2015). Because cell cycle is a highly controlled process regulated by stage-specific regulators, we also examined the expression of common cell cycle regulators (Moore, 2013; Passegue, 2005). In addition to Cdk6 (Figure 5D), we also found significant decreases in expression of the cell cycle promoting genes cyclin A2 (Ccna2) and cyclin B2 (Ccnb2), active in the synthesis (S) and mitosis (M) stages of cell cycle, respectively (Figure 5E) in Bcl11a KO HSCs. In contrast, expression of the cell cycle inhibitors p16, p21, p27 and p57 was comparable between WT and KO groups (Figure 5F). In addition to its role in cell cycle progression, CDK6 is also involved in regulating quiescence exit in human HSCs (Laurenti et al., 2015). To further explore whether decreased Cdk6 expression may affect the quiescence status of Bcl11a KO HSCs, we performed gene set enrichment analysis (GSEA) (Subramanian et al., 2005). Compared to the WT control, Bcl11a KO HSCs were significantly enriched in two independent quiescence signature gene sets (Oki et al., 2014; Venezia et al., 2004) (Figure 5G), indicating that Bcl11a KO HSCs are more quiescent. Collectively, these findings suggest that Bcl11a-deficiency in steady-state hematopoiesis alters cell cycle progression, resulting in increased quiescence in HSCs.
Bcl11a-deficient HSCs have delayed cell cycle kinetics
Given the decreased expression of several cell cycle regulators in Bcl11a KO HSCs, we next determined the functional impact on the cell cycle process. In contrast to the gene expression analysis, all cell cycle stages were comparable between Bcl11a WT and KO groups (Figure 6A). To further distinguish between cells in G0 and G1 cell cycle stages, we measured the expression of the cell proliferation marker Ki-67 in HSCs. Cells lacking Ki-67 protein are exclusively in the quiescent G0 stage. Similarly, there was no significant difference between Bcl11a WT and KO groups (Figure 6B). Since the S-phase regulator Ccna2 was significantly downregulated in Bcl11a-deficient HSCs (Figure 5E), we next examined the frequency of proliferating HSCs in vivo by measuring 5-bromo-2-deoxyuridine (BrdU) incorporation. Consistent with the cell cycle analysis, the frequency of proliferating cells from Bcl11a KO and WT mice were comparable at all time-points (Figure 6C).
Figure 6. Bcl11a-deficient HSCs have delayed cell cycle kinetics.

(A) Cell cycle analysis by DAPI staining of Bcl11a WT and KO HSCs (left) and the cell cycle profiles (right) 7 weeks post-p(I:C). n = 4 WT, 6 KO.
(B) Cell cycle analysis by Ki-67 and DAPI staining showing frequency of HSCs in G0 and G1 (left) and representative FACS profiles (right) 7 weeks post-p(I:C). n = 4 WT, 6 KO.
(C) Frequency of Bcl11a WT and KO HSCs with BrdU incorporation at 12, 24 and 60 hours after BrdU injection in vivo. n = 3–4 WT, 3 KO.
(D) Cell cycle analysis by DAPI staining (left) and the cell cycle profiles (right) following transplantation of Bcl11a WT and KO HSCs, 4–7 weeks post-p(I:C). n = 5 WT, 4 KO.
(E) Cell cycle analysis by Ki-67 and DAPI staining showing frequency of HSCs in G0 and G1 (left) and the representative FACS profiles (right) following transplantation of Bcl11a WT and KO HSCs, 4–7 weeks post-p(I:C). n = 9 WT, 8 KO.
(F) Cell division analysis 24 and 48 hours (hrs) after plating of clonal HSCs from Bcl11a WT and KO HSCs. n = 120 WT cells, 99 KO cells (24 hrs) and 152 WT cells, 125 KO cells (48 hrs).
(G) Flow cytometry analysis of cell division kinetics 6 days after plating HSCs from Bcl11a WT and KO HSCs. Data from 2 independent experiments.
(H) Quantification of cell expansion from cultured single HSCs at day 10. n = 153 WT cells, 98 KO cells.
(I) Frequency of Annexin V+ apoptotic HSCs in Bcl11a WT and KO mice. n = 7 WT, 6 KO.
Data and error bars represent mean ± SD. *p<0.05; **p<0.01; ***p<0.001.
The lack of functional defects in cell cycle following acute deletion of Bcl11a was not altogether unexpected given the impairment in HSC function was not evident until cells had experienced proliferation pressure, such as in transplantation. Consequently, we transplanted purified HSCs and analyzed the cell cycle status following Bcl11a gene deletion. The frequency of KO HSCs in G0/G1 was significantly increased and cells in G2/M was markedly decreased (Figure 6D). However, Ki-67 staining analysis revealed a significantly lower frequency of KO HSCs in G0 and a higher frequency of cells in G1 (Figure 6E). Given that no cell cycle stages other than G1 had increased frequency of KO HSCs, we hypothesized that the Bcl11a-deficent HSCs are arrested or delayed in G1 due to decreased Cdk6 expression. To test this, we purified HSCs and cultured single cells in vitro to assess their cell division (Figure 6F). After 24 hours, most single HSCs had gone through only one cell division or none. Nearly twice as many WT single HSCs (40%) had already gone through one cell division compared to KO HSCs (21%), whereas the majority of KO cells (69%) had still not yet divided compared to WT control cells (54%). At 48 hours, the majority of WT cells (67%) had divided more than once, compared to only 44% of the Bcl11a KO HSCs (P-value = 0.04). In contrast, significantly more Bcl11a KO cells (44%) had still only gone through one cell division compared to WT HSCs (21%) (P-value = 0.04). These findings strongly suggest that Bcl11a-deficient HSCs have a delayed cell cycle. Specifically, Bcl11a-deficient HSCs divide more slowly than WT HSCs despite being recruited into cell cycle to a higher degree (Figures 6D–F). To further validate these results, we used an alternative approach to evaluate cell cycle kinetics. Purified HSCs were incubated with CellTrace Far Red, a cell trace dye, which dilutes with every cell division. Consistent with previous findings, Bcl11a KO HSCs had gone through significantly fewer cell divisions compared to WT HSCs (Figure 6G).
It is conceivable that a delay in cell cycle kinetics would result in longer duration in cell cycle and ultimately to a reduction in total cell number. To test this hypothesis, we cultured purified single HSCs in vitro and measured their cellular expansion. After 10 days of culture, WT control HSCs had expanded on average 3,000-fold compared to only 1,900-fold from single Bcl11a KO HSCs (Figure 6H). Moreover, the frequency of Annexin V positive cells was comparable in Bcl11a WT and KO HSCs, suggesting that decreased cellular output in Bcl11a KO HSCs is not due to increased cell death (Figure 6I).
In summary, deleting Bcl11a in steady-state hematopoiesis did not significantly impact the cell cycle process. However, during regeneration stress such as after transplantation, more Bcl11a-deficient HSCs are recruited into cell cycle but become delayed in G1, most likely due to decreased Cdk6 expression. The prolonged cell cycle transit results in slower cell cycle kinetics compared to WT control cells and ultimately leads to a reduced cellular output.
Bcl11a-deficient HSCs resemble aged HSCs
Physiological aging of the hematopoietic system is associated with changes in HSCs (Geiger et al., 2013; Rossi et al., 2008). These changes include increased HSC numbers but poorer repopulation ability, a decrease in B and T lymphoid development, myeloid lineage skewing and cell cycle alterations. Given the similarity in phenotypes observed between Bcl11a KO HSCs and aged HSCs, we next explored the resemblance in greater detail. We first performed GSEA using two published aging HSC signature gene sets (Flach et al., 2014; D. Sun et al., 2014) and found that Bcl11a KO HSCs significantly enriched for both signatures (Figure 7A). Since the expression of several cell cycle regulators was decreased in Bcl11a KO HSCs (Figure 5E), we next examined the expression of Bcl11a and the cell cycle genes in HSCs isolated from 2-month old young mice and 15–17-month old aged mice. Although the expression of Bcl11a was comparable, there was a significant decrease in Cdk6 expression in the older mice (Figure 7B). In parallel, there were also significant decreases in the expression of Ccna2, cyclin E1 (Ccne1), Ccnb1 and cyclin B2 (Ccnb2) in HSCs from 15–17-month old mice (Figure 7C). Notably, in addition to Cdk6, Ccna2 and Ccnb1 were also significantly reduced in Bcl11a KO HSCs (Figure 5E).
Figure 7. HSCs from Bcl11a-deficient mice resemble the phenotype of aging HSCs.

(A) Gene set enrichment analyses of two aging gene signatures in Bcl11a WT and KO HSCs.
(B) Relative mRNA expression of Bcl11a and Cdk6 in sorted HSCs from 2-month and 15–17-month old C57BL7 mice. n = 4/group.
(C) Relative mRNA expression of cell cycle genes in sorted HSCs from 2-month and 15–17-month old C57BL7 mice. n = 4/group.
(D) Representative FACS profiles of γH2AX staining in HSCs showing mean frequencies from (E).
(E) Frequency of γH2AX in HSCs from Bcl11a WT and KO mice 10 weeks post-p(I:C) and an irradiated control (5 Gy). n = 4 WT, 3 KO, 1 irradiated control.
(F) Relative mRNA expression of Mcm genes in sorted HSCs from 2-month and 15–17-month old C57BL7 mice. n = 4/group.
(G) Gene set enrichment analysis of a Mcm gene signature in Bcl11a WT and KO HSCs.
(H) Relative mRNA expression of Mcm genes in Bcl11a WT and KO HSCs.
(I) Gene set enrichment analyses of upregulated and downregulated genes (≥2-fold, p<0.01) from Bcl11a KO mice in a data set from old (>22 months old) and young (2–3 months old) long-term HSCs acquired from Kowalczyk et al.
(J) Hemoglobin levels (g/dL) in 2-month (2-mo) and 10-month (10-mo) old Bcl11a WT and KO mice. n = 8 WT, 6 KO 2-mo and 15 WT, 8 KO 10 mo.
(K) Frequency of γH2AX in HSCs from 9-month old Bcl11a WT and KO mice and 2-month and 8-month old C57BL6 (B6) mice. n = 3 WT, 3 KO; 3 B6 2-mo, 2 B6 8-mo.
In (A, G, I) n = 3/genotype. Data represent mean ± SD. *p<0.05; **p<0.01; ***p<0.001.
See also Figure S7.
One of the hallmarks of aging is increased occurrence of DNA damage (Geiger et al., 2013; Rossi et al., 2008). Phosphorylation of the histone H2A variant H2AX (γH2AX) is associated with DNA damage and can be used as an early detection marker. Intracellular staining against γH2AX in Bcl11a WT and KO HSCs demonstrated a significantly higher frequency of γH2AX in KO HSCs, indicating increased DNA damage in the absence of Bcl11a (Figures 7D and 7E). As expected, we detected substantial γH2AX signal in HSCs from an irradiated control (5 Gy) and comparable levels of γH2AX between Bcl11afl/fl × Mx1-Cre− (WT) and Bcl11awt/wt × Mx1-Cre+ mice (Figures 7D and 7E; Figure S7A). These findings are consistent with reports of increased DNA damage in aged HSCs (Beerman et al., 2014).
Another mechanism proposed to underlie the cell cycle changes in aged HSCs is replication stress due to decreased expression of mini-chromosome (Mcm) genes (Flach et al., 2014). Six Mcm genes encode the hexameric DNA helicase required for DNA replication (Flach et al., 2014). To determine the age-associated changes in Mcm gene expression, HSCs were purified from 2-month and 15–17-month old mice. Consistent with previous findings (Flach et al., 2014), the expression of all Mcm genes were significantly reduced in older mice (Figure 7F). To examine whether loss of Bcl11a may have a similar effect, we performed GSEA using a Mcm gene set (Flach et al., 2014). The Mcm gene signature was downregulated in Bcl11a KO HSCs (Figure 7G) and gene expression analysis of Mcm genes also showed a general decrease in Bcl11a-deficient HSCs (Figure 7H). These results suggest that Bcl11a-deficiency may induce replication stress, similar to aged HSCs.
To further confirm that Bcl11a-deficient HSCs have an aging phenotype, we assessed whether old WT HSCs associate with a Bcl11a KO gene signature. We compared the gene expression profiles in Bcl11a KO HSCs with published transcriptomic datasets from old (>22 months) and young (2–3 months) HSCs (Kowalczyk et al., 2015; D. Sun et al., 2014). The gene expression change in Bcl11a KO (versus WT) was positively correlated with the change in the aged HSCs (versus young) (Figure S7B). More specifically, the upregulated genes in Bcl11a KO were highly enriched in aged HSCs, whereas the downregulated genes were enriched in young HSCs (Figure 7I), providing further support for the overlapping phenotypes between Bcl11a-deficient and aged HSCs.
Advancing age is often accompanied by the onset of anemia (Geiger et al., 2013; Rossi et al., 2008). Thus, we measured hemoglobin levels in Bcl11a WT and KO mice as an indicator of anemia. As expected, there was no difference in hemoglobin levels in 2-month old Bcl11a WT and KO mice. Hemoglobin levels in 10-month old Bcl11a WT and KO mice were reduced compared to levels in 2-month old mice (Figure 7J). Notably, hemoglobin levels were further decreased in older Bcl11a-deficient mice (P-value = 0.005), whereas the level in Bcl11a WT was comparable to 20-month old C57BL6 mice (Figure 7J; Figure S7C). These findings demonstrate that the anemic condition in older mice is exacerbated by loss of BCL11A.
Given the reported accrual of DNA damage with age (Geiger et al., 2013; Rossi et al., 2008) and our observation that BCL11A loss exacerbates age-associated anemia (Figure 7J; Figure S7C), we hypothesized that phosphorylation of H2AX should be further increased in older Bcl11a KO mice. Indeed, γH2AX was significantly increased from 2-month to 8-month old C57BL6 HSCs and the level of γH2AX was further increased in Bcl11a KO HSCs (Figure 7K).
In summary, our findings strongly suggest that Bcl11a-deficiency results in a phenotypic resemblance to HSCs in the aged hematopoietic system and that typical aging characteristics are more pronounced in aged Bcl11a KO mice.
DISCUSSION
In the hematopoietic system, BCL11A is expressed in definitive but not primitive hematopoiesis (Palis, 2014). In definitive hematopoiesis, BCL11A controls B lymphopoiesis by regulation of apoptosis and cell survival through a p53-dependent pathway (Yu et al., 2012). However, the role of BCL11A in other lineages especially in HSCs remains elusive. Studies have implicated a role of BCL11A in HSC self-renewal and lineage differentiation (Kustikova, 2005; Tsang et al., 2015) that is distinct from its role in B lymphopoiesis. Other studies have suggested that BCL11A has oncogenic potential and can promote leukemia development of both lymphoid and myeloid lineages (Alcalay et al., 2003; Kustikova, 2005; Satterwhite, 2001; Yin et al., 2008). The context-specific role of BCL11A illustrates the importance of a comprehensive analysis of the in vivo function of BCL11A in hematopoiesis as part of ongoing efforts to target BCL11A for HbF induction as well as to develop strategies to generate bona fide definitive HSCs from ESCs or iPSCs.
In this study we have systematically delineated the role of BCL11A in definitive hematopoiesis, particularly in HSCs, through hematopoietic-selective deletion of BCL11A. While our findings indicate that Bcl11a is dispensable for HSCs in hematopoietic homeostasis, apart from B cell defects as previously reported (Liu et al., 2003; Yu et al., 2012), the function of Bcl11a-deficient HSCs is significantly impaired when challenged. We also found that steady-state Bcl11a-deficient HSCs expressed quiescence gene signatures, consistent with recent findings suggesting that steady-state hematopoiesis is mainly sustained by short-term HSCs and other long-lived progenitors (Busch et al., 2015; J. Sun et al., 2014). Moreover, we demonstrate that when challenged, Bcl11a KO HSCs exhibit cell cycle defects resulting in increased cell cycle entry with a concomitant extension of cell cycle transit. By measuring kinetics of cell division in single HSCs, we showed that prolonged cell cycle transit is due to a delay in recruiting cells into active cell cycle and executing the first cell division. The cumulative effect of this delay limits HSC divisions and ultimately leads to a net decrease in cellular output. This likely also explains the increased cell cycle entry in Bcl11a KO HSCs as a compensatory effect. As a result of the cell cycle delay, Bcl11a-deficient HSCs are unable to reconstitute blood lineages and self-renew to the same degree as WT HSCs, and are outcompeted in a transplantation setting. Several cell cycle regulators are decreased in Bcl11a-deficient HSCs, including the G0/G1 regulator CDK6. Notably, CDK6 has been demonstrated to regulate quiescence exit in human HSCs and Cdk6-deficient HSCs are grossly normal until challenged (Laurenti et al., 2015; Scheicher et al., 2015). Thus, our studies suggest that lower Cdk6 expression in Bcl11a-deficient HSCs may be responsible for the delay in cell cycle entry and transit through G1.
Our findings differ from a previously published study, which suggested that cell cycle defects in Bcl11a-deficient HSCs result in hyperproliferation and loss of quiescence (Tsang et al., 2015). It is likely that the discrepancy is due to different experimental protocols. A critical difference is the pan-cellular deletion of Bcl11a through an estrogen-induced (CreERT) system compared to the hematopoietic-selective Mx1-Cre deletion system used in this study. More importantly, administration of estrogen in mice has been shown to increase HSC divisions (Nakada et al., 2014). Thus, it is likely the observed HSC hyperproliferation in the study by Tsang et al is unrelated to Bcl11a gene deletion. In this study, the use of the hematopoietic-selective, inducible Mx1-Cre allele avoids these confounding issues and provides a better and more accurate assessment of the HSC defect in Bcl11a-null mice.
Our findings have significant implications for ongoing efforts to target BCL11A for the major β-hemoglobin disorders. Our results suggest that global targeting of BCL11A in stem/progenitor cells, such as human CD34+ cells, is ill-advised as the absence of BCL11A may adversely affect the function of HSCs. Given BCL11A’s critical requirement in multiple hematopoietic lineages, therapeutic strategies emphasized at disrupting BCL11A selectively in erythroid cells while sparing its expression in non-erythroid lineages need to be considered. Furthermore, our studies illustrate that careful dissection of HSC roles of therapeutic targets for gene therapy or gene editing is required for a complete view of potential in vivo consequences and may help better design the clinical experiments.
HSCs continually replenish blood cells for a lifetime, and the balance between proliferation and quiescence is carefully regulated to ensure blood homeostasis. In the aging hematopoietic system, there is a diminished capacity to adequately maintain homeostasis despite increased numbers of phenotypic HSCs (Morrison et al., 1996; Rossi et al., 2008; Sudo et al., 2000). In our study, loss of Bcl11a in the hematopoietic system exhibited features highly resembling the aged hematopoietic system. The overlap between the phenotypes observed in young Bcl11a-deficient HSCs and aged WT HSCs is striking and extensive. Although we were unable to detect a significant change in Bcl11a mRNA expression in aged WT HSCs, several cell cycle regulators including Cdk6 were decreased in both aged HSCs and Bcl11a-deficient HSCs. Furthermore, aged HSCs have also been shown to exhibit delayed cell cycle kinetics, despite being recruited into active cell cycle to a higher degree compared to young HSCs (Flach et al., 2014). The attenuated cell cycle in Bcl11a KO mice render them unable to sustain hematopoiesis under conditions of stress and regeneration. Although the phenotype is more severe and accelerated in Bcl11a KO mice compared to aged mice, it suggests that common cell cycle pathways may be affected. Furthermore, the increase in HSC numbers in the Bcl11a KO mouse is unable to compensate for the functional defects, similar to the age-associated increase in functionally impaired HSCs. Although the mechanism responsible for this in the aging hematopoietic system remains elusive (Geiger et al., 2013), cell cycle changes and increased symmetrical divisions have been suggested. One of the hallmarks of aging is increased occurrence of γH2AX, which has been proposed to be the earliest marker of DNA damage (Beerman et al., 2014; Rossi et al., 2008) and a sign of replication stress (Flach et al., 2014). The γH2AX level is markedly increased in Bcl11a-deficient HSCs, which also correlated with decreased Mcm gene expression, similar to the phenotype of replication stress observed in the aged HSCs (Flach et al., 2014). Furthermore, phenotypic features typically observed in the aging hematopoietic system, including increased γH2AX and decreased hemoglobin levels, commonly associated with anemia, are exacerbated in aged Bcl11a-deficient mice.
Despite recent progress in defining the characteristic features of aged HSCs, the mechanisms underlying age-associated molecular changes are largely unknown (Rossi et al., 2008). Here, we present a potential connection between Bcl11a and the aging hematopoietic system, mediated through the cell cycle process. The mechanisms by which BCL11A regulates the cell cycle mediator remain to be determined. It is plausible that BCL11A acts indirectly through a mediator that regulates the cell cycle, as we were unable to observe a significant change in Bcl11a expression in aged mice. The gene expression of Bcl11a-deficient and aged HSCs was positively correlated indicating similarity in transcriptomic changes, although comprehensive studies will be needed to determine the extent of the overlap and the specific genes and pathways involved. Nevertheless, in the absence of Bcl11a, HSCs display self-renewal and differentiation defects rendering them unable to sustain long-term hematopoiesis. These findings have revealed a potential role of BCL11A in the aging hematopoietic system and provide mechanistic insights into the regulation of definitive HSCs by BCL11A. Hence, our study not only provides important implications for the design of therapeutic strategies for β-globin disorders, but also benefits current efforts in characterizing molecular mechanisms controlling aging and age-related pathogenesis.
EXPERIMENTAL PROCEDURES
Experimental animals
The Bcl11a floxed (Bcl11afl/fl) mouse strain has previously been described (Ippolito et al., 2014; Sankaran et al., 2009). Bcl11a deletion in Bcl11a × Mx1-Cre mice was achieved by intraperitoneal administration of five doses of poly(I:C) (12.5μg/g body weight) poly(I:C) (InvivoGen). All experiments using adult mice were performed with mouse strains backcrossed on a C57BL6 background (>9 generations) unless stated otherwise. The Institutional Animal Care and Use Committee at Boston Children’s Hospital approved all experiments.
Flow cytometry experiments
For HSC analysis, BM cells were enriched for CD117 using MACS beads (Miltenyi Biotec) prior to staining. In all flow cytometry analyses and purifications, Bcl11a Het and KO mice were always gated positively for eYFP in addition to the indicated immunophenotypes to analyze or isolate Bcl11a-deleted cells. See Table S3 for a complete list of antibodies.
Transplantation experiments
B6.SJL-Ptprca/BoyAiTac (CD45.1; 10–12 weeks old) mice were lethally irradiated (two split doses of 500cGy). Competitive transplantation was performed by the intravenous injection of 200 donor HSCs (LSKCD48−Flt3−CD150+) and 200,000 whole BM cells (CD45.1).
Gene expression experiments
In real-time (RT) PCR experiments the relative expression was quantified using the ΔΔCt method and normalized to Gapdh. See Table S4 for primer sequences. Global gene expression analysis by microarray was performed on the Affymetrix Mouse Gene 2.0 ST platform.
Bioinformatics analyses
Affymetrix CEL files were normalized using RMA (Irizarry et al., 2003). Differentially expressed genes were detected with a threshold of fold change ≥2 and adjusted p-value ≤0.01. Pathway enrichment analyses were performed using GeneGo Metacore from Thomson Reuters (Version 6.24 build 67895, https://portal.genego.com/). GSEA was performed using GSEA software (http://www.broadinstitute.org/gsea) (Subramanian et al., 2005) with default parameters.
Comparative analyses with other published gene expression data derived on different platforms, were performed by comparison of differentially expressed genes (log2 fold-change).
Cell cycle and cell proliferation experiments
Fixation and permeabilization was performed with the BD Cytofix/Cytoperm Kit (BD Biosciences) according to manufacturer’s protocol, followed by incubation with Ki-67 antibody and DAPI before flow cytometry analysis.
Cell proliferation assays were performed by the administration of a single intraperitoneal injection of 5-Bromo-2′-deoxyuridine (BrdU). Fixation and permeabilization and BrdU visualization was performed using the BD BrdU Flow Kit (BD Biosciences) according to manufacturer’s protocol.
Cell division analyses
Evaluation of cell division was performed by incubation with CellTrace Far Red (ThermoFisher Scientific) according to manufacturer’s protocol, followed by cell culturing for 6–7 days before FACS analysis (see Table S5 for growth factor conditions).
Evaluation of cell division kinetics was performed by plating single HSCs in 60-well plates (Nunc). The number of cells in the wells was scored 24 and 48 hours after cell plating with an inverted microscope. Cell counting experiments assessing cellular expansion was performed using CountBright absolute counting beads (ThermoFisher Scientific).
Statistics
Statistical analysis was performed using the unpaired, two-tailed t-test in GraphPad Prism version 6.0h for Mac OS X, (GraphPad Software).
Supplementary Material
Acknowledgments
We thank Dr. H. Tucker (The University of Texas at Austin) for providing the conditional Bcl11a mouse strain, Dr. N. Iscove (Ontario Cancer Institute, University Health Network) for providing the C57BL/6J-Ly5.1-KitW-41/W-41-Gpia/a mice, and Z. Herbert and L. Grimmett from the Molecular Biology Core Facilities at Dana-Farber Cancer Institute for processing microarray samples. We are grateful to Dr. D. Bauer for critical comments. S.L. is supported by a Fellow Award from the Leukemia & Lymphoma Society. J.X. is supported by NIH/NIDDK grants K01DK093543, R03DK101665 and a CPRIT New Investigator award (RR140025). S.H.O. is an investigator of the Howard Hughes Medical Institute (HHMI) and supported by P30DK049216 and R01HL032259.
Footnotes
ACCESSION NUMBER
Microarray data was deposited in the Gene Expression Omnibus under accession number GSE77207.
Supplemental Information includes Figures S1–S7, Tables S1–S5, Supplemental Experimental Procedures and Supplemental References.
AUTHOR CONTRIBUTIONS
Conceptualization, S.L., J.X. and S.H.O.; Methodology, S.L., J.H. and J.X.; Investigation, S.L., J.M., E.S., D.L., C.R., S.M. and S.H.; Writing – Original Draft, S.L., J.X. and S.H.O.; Writing – Review & Editing, S.L., J.X. and S.H.O.; Funding Acquisition, J.X. and S.H.O.; Supervision, S.L., J.X., and S.H.O.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alcalay M, Meani N, Gelmetti V, Fantozzi A, Fagioli M, Orleth A, Riganelli D, Sebastiani C, Cappelli E, Casciari C, Sciurpi MT, Mariano AR, Minardi SP, Luzi L, Muller H, Di Fiore PP, Frosina G, Pelicci PG. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. Journal of Clinical Investigation. 2003;112:1751–1761. doi: 10.1172/JCI200317595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, Shao Z, Canver MC, Smith EC, Pinello L, Sabo PJ, Vierstra J, Voit RA, Yuan GC, Porteus MH, Stamatoyannopoulos JA, Lettre G, Orkin SH. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerman I, Seita J, Inlay MA, Weissman IL, Rossi DJ. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell. 2014;15:37–50. doi: 10.1016/j.stem.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch K, Klapproth K, Barile M, Flossdorf M, Holland-Letz T, Schlenner SM, Reth M, Höfer T, Rodewald H-R. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015 doi: 10.1038/nature14242. [DOI] [PubMed] [Google Scholar]
- Daniel MG, Lemischka IR, Moore K. Converting cell fates: generating hematopoietic stem cells de novovia transcription factor reprogramming. Ann N Y Acad Sci. 2016 doi: 10.1111/nyas.12989. n/a–n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, Alvarez S, Diolaiti ME, Ugarte F, Forsberg EC, Le Beau MM, Stohr BA, Méndez J, Morrison CG, Passegué E. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014;512:198–202. doi: 10.1038/nature13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger H, de Haan G, Florian MC. The ageing haematopoietic stem cell compartment. Nature Publishing Group. 2013;13:376–389. doi: 10.1038/nri3433. [DOI] [PubMed] [Google Scholar]
- Ippolito GC, Dekker JD, Wang Y-H, Lee B-K, Shaffer AL, Lin J, Wall JK, Lee B-S, Staudt LM, Liu Y-J, Iyer VR, Tucker HO. Dendritic cell fate is determined by BCL11A. Proceedings of the National Academy of Sciences. 2014;111:E998–E1006. doi: 10.1073/pnas.1319228111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Jasinski M, Keller P, Fujiwara Y, Orkin SH, Bessler M. GATA1-Cre mediates Piga gene inactivation in the erythroid/megakaryocytic lineage and leads to circulating red cells with a partial deficiency in glycosyl phosphatidylinositol-linked proteins (paroxysmal nocturnal hemoglobinuria type II cells) Blood. 2001;98:2248–2255. doi: 10.1182/blood.v98.7.2248. [DOI] [PubMed] [Google Scholar]
- Kowalczyk MS, Tirosh I, Heckl D, Rao TN, Dixit A, Haas BJ, Schneider RK, Wagers AJ, Ebert BL, Regev A. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015;25:1860–1872. doi: 10.1101/gr.192237.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustikova O. Clonal Dominance of Hematopoietic Stem Cells Triggered by Retroviral Gene Marking. Science. 2005;308:1171–1174. doi: 10.1126/science.1105063. [DOI] [PubMed] [Google Scholar]
- Kühn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- Laurenti E, Frelin C, Xie S, Ferrari R, Dunant CF, Zandi S, Neumann A, Plumb I, Doulatov S, Chen J, April C, Fan J-B, Iscove N, Dick JE. CDK6 Levels Regulate Quiescence Exit in Human Hematopoietic Stem Cells. Cell Stem Cell. 2015:1–41. doi: 10.1016/j.stem.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettre G, Sankaran VG, Bezerra MAC, Araújo AS, Uda M, Sanna S, Cao A, Schlessinger D, Costa FF, Hirschhorn JN, Orkin SH. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proceedings of the National Academy of Sciences. 2008;105:11869–11874. doi: 10.1073/pnas.0804799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Keller JR, Ortiz M, Tessarollo L, Rachel RA, Nakamura T, Jenkins NA, Copeland NG. Bcl11a is essential for normal lymphoid development. Nat Immunol. 2003;4:525–532. doi: 10.1038/ni925. [DOI] [PubMed] [Google Scholar]
- Menzel S, Garner C, Gut I, Matsuda F, Yamaguchi M, Heath S, Foglio M, Zelenika D, Boland A, Rooks H, Best S, Spector TD, Farrall M, Lathrop M, Thein SL. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007;39:1197–1199. doi: 10.1038/ng2108. [DOI] [PubMed] [Google Scholar]
- Moore JD. In the wrong place at the wrong time: does cyclin mislocalization drive oncogenic transformation? Nat Rev Cancer. 2013;13:201–208. doi: 10.1038/nrc3468. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Wandycz AM, Akashi K, Globerson A, Weissman IL. The aging of hematopoietic stem cells. Nature Medicine. 1996;2:1011–1016. doi: 10.1038/nm0996-1011. [DOI] [PubMed] [Google Scholar]
- Nakada D, Oguro H, Levi BP, Ryan N, Kitano A, Saitoh Y, Takeichi M, Wendt GR, Morrison SJ. Oestrogen increases haematopoietic stem-cell self-renewal in females and during pregnancy. Nature. 2014;505:555–558. doi: 10.1038/nature12932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oki T, Nishimura K, Kitaura J, Togami K, Maehara A, Izawa K, Sakaue-Sawano A, Niida A, Miyano S, Aburatani H, Kiyonari H, Miyawaki A, Kitamura T. A novel cell-cycle-indicator, mVenus-p27K−, identifies quiescent cells and visualizes G0–G1 transition. Sci Rep. 2014;4:1–10. doi: 10.1038/srep04012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palis J. Primitive and definitive erythropoiesis in mammals. Front Physiol. 2014;5:3. doi: 10.3389/fphys.2014.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegue E. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J Exp Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietras EM, Warr MR, Passegué E. Cell cycle regulation in hematopoietic stem cells. The Journal of Cell Biology. 2011;195:709–720. doi: 10.1083/jcb.201102131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Jamieson CHM, Weissman IL. Stems Cells and the Pathways to Aging and Cancer. Cell. 2008;132:681–696. doi: 10.1016/j.cell.2008.01.036. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, MIKKOLA HKA, Hirschhorn JN, Cantor AB, Orkin SH. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Xu J, Ragoczy T, Ippolito GC, Walkley CR, Maika SD, Fujiwara Y, Ito M, Groudine M, Bender MA, Tucker PW, Orkin SH. Developmental and species-divergent globin switching are driven by BCL11A. Nature. 2009;460:1093–1097. doi: 10.1038/nature08243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterwhite E. The BCL11 gene family: involvement of BCL11A in lymphoid malignancies. Blood. 2001;98:3413–3420. doi: 10.1182/blood.V98.12.3413. [DOI] [PubMed] [Google Scholar]
- Scheicher R, Hoelbl-Kovacic A, Bellutti F, Tigan AS, Prchal-Murphy M, Heller G, Schneckenleithner C, Salazar-Roa M, Zöchbauer-Müller S, Zuber J, Malumbres M, Kollmann K, Sexl V. CDK6 as a key regulator of hematopoietic and leukemic stem cell activation. Blood. 2015;125:90–101. doi: 10.1182/blood-2014-06-584417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. WIREs Syst Biol Med. 2010;2:640–653. doi: 10.1002/wsbm.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudo K, Ema H, Morita Y, Nakauchi H. Age-associated characteristics of murine hematopoietic stem cells. J Exp Med. 2000;192:1273–1280. doi: 10.1084/jem.192.9.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, Wang H, Le T, Faull KF, Chen R, Gu H, Bock C, Meissner A, Göttgens B, Darlington GJ, Li W, Goodell MA. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell. 2014;14:673–688. doi: 10.1016/j.stem.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, Klein A, Hofmann O, Camargo FD. Clonal dynamics of native haematopoiesis. Nature. 2014;514:322–327. doi: 10.1038/nature13824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang JCH, Yu Y, Burke S, Buettner F, Wang C, Kolodziejczyk AA, Teichmann SA, Lu L, Liu P. Single-cell transcriptomic reconstruction reveals cell cycle and multi-lineage differentiation defects in Bcl11a-deficient hematopoietic stem cells. Genome Biol. 2015;16:178. doi: 10.1186/s13059-015-0739-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, Usala G, Busonero F, Maschio A, Albai G, Piras MG, Sestu N, Lai S, Dei M, Mulas A, Crisponi L, Naitza S, Asunis I, Deiana M, Nagaraja R, Perseu L, Satta S, Cipollina MD, Sollaino C, Moi P, Hirschhorn JN, Orkin SH, Abecasis GR, Schlessinger D, Cao A. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proceedings of the National Academy of Sciences. 2008;105:1620–1625. doi: 10.1073/pnas.0711566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venezia TA, Merchant AA, Ramos CA, Whitehouse NL, Young AS, Shaw CA, Goodell MA. Molecular Signatures of Proliferation and Quiescence in Hematopoietic Stem Cells. Plos Biol. 2004;2:e301–12. doi: 10.1371/journal.pbio.0020301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Peng C, Sankaran VG, Shao Z, Esrick EB, Chong BG, Ippolito GC, Fujiwara Y, Ebert BL, Tucker PW, Orkin SH. Correction of Sickle Cell Disease in Adult Mice by Interference with Fetal Hemoglobin Silencing. Science. 2011;334:993–996. doi: 10.1126/science.1211053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Sankaran VG, Ni M, Menne TF, Puram RV, Kim W, Orkin SH. Transcriptional silencing of -globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev. 2010;24:783–798. doi: 10.1101/gad.1897310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin B, Delwel R, Valk PJ, Wallace MR, Loh ML, Shannon KM, Largaespada DA. A retroviral mutagenesis screen reveals strong cooperation between Bcl11a overexpression and loss of the Nf1 tumor suppressor gene. Blood. 2008;113:1075–1085. doi: 10.1182/blood-2008-03-144436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wang J, Khaled W, Burke S, Li P, Chen X, Yang W, Jenkins NA, Copeland NG, Zhang S, Liu P. Bcl11a is essential for lymphoid development and negatively regulates p53. Journal of Experimental Medicine. 2012;209:2467–2483. doi: 10.1084/jem.20121846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, Li X, Wang W, Zhu P, Zhou J, He W, Ding M, Xiong F, Zheng X, Li Z, Ni Y, Mu X, Wen L, Cheng T, Lan Y, Yuan W, Tang F, Liu B. Tracing haematopoietic stem cell formation at single-cell resolution. Nature. 2016;533:487–492. doi: 10.1038/nature17997. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.