

Abstract
Human leucocyte antigen (HLA) compatibility is the main factor determining the occurrence of graft‐vs‐host disease (GVHD) in patients. It has also been shown that minor histocompatibility antigen differences as well as genetic polymorphisms that are not sequenced by standard methodology for HLA typing can play a role. We used mixed lymphocyte cultures (MLCs) as a functional cellular test and investigated gene expression changes driven by HLA incompatibility in an effort to better understand the mechanisms involved in the disease. Gene expression profile of HLA matched and HLA mismatched MLC identified differentially regulated genes and pathways. We found that a great number of genes related to immune function were differentially regulated; these genes were also found to be associated with GVHD and graft rejection. The majority of differentially regulated genes were interferon‐gamma (IFNγ)‐inducible genes and IFNγ neutralisation in MLCs abrogated their induction. The microRNA‐155, a recently identified target for acute GVHD (aGVHD), was also found to be significantly induced in HLA mismatched MLC but not in the matched setting and its induction was not diminished by blocking IFNγ. In this proof‐of‐principle study we show gene expression changes in mismatched MLC that represent alloreactive responses, correlate with markers involved in GVHD and can potentially be useful in the study of the biological processes involved in this disease.
Keywords: graft‐vs‐host, human leucocyte antigen, interferon‐gamma, microRNA‐155, mixed lymphocyte culture
Introduction
Allogeneic haematopoietic stem cell transplantation (allo‐HSCT) can be a successful therapy for many haematological malignancies and dysplasias but its success is limited by graft rejection or the development of graft‐vs‐host disease (GVHD), a major contributor to increased morbidity and mortality of transplanted patients 1. The incidence of GVHD and rejection is significantly lower in patients receiving a graft from a sibling identical for human leucocyte antigens (HLAs), compared with those receiving a graft from an HLA phenotypically identical unrelated donor 2, 3. However, only 35% of patients have an HLA identical sibling donor hence, for the majority of patients an HLA phenotypically identical unrelated donor is the only option.
The classic HLA genetic loci are the best‐characterised determinants for GVHD 4 and donor selection for transplantation is based on matching donor and recipient for HLA by DNA typing for HLA‐class I (HLA‐A, ‐B and ‐C) and II (HLA‐DRB1 and ‐DQB1) loci with an identity of at least 8 of 10 antigens required to proceed with transplantation. However, aGVHD occurs in roughly 40% of patients receiving HLA‐identical grafts 5 due to undetected antigen differences between unrelated individuals which can be recognised by donor T cells following transplantation and lead to the disease. The major histocompatibility complex (MHC) region harbours approximately 200 genes many of which have immune‐related functions and several single nucleotide polymorphisms within the MHC (of the donor or recipient) that are not sequenced by standard methodology for HLA typing confer higher risk for aGVHD 6. In addition, non‐MHC variants are minor histocompatibility antigens that could contribute to GVHD such as CT 60 SNP in CTLA‐4 7, SNP13 in NOD2 8, or cytokines such as interferon‐gamma (IFNγ), interleukin‐6 (IL‐6), IL‐10 and IL‐1Ra 9, 10, 11, 12. Finally, the total number of transplantation antigens encoded within the MHC region remains unknown. Petersdorf et al. have suggested that haplotype matching for unrelated HSCT is beneficial to decrease GVHD‐morbidity 13, but still this approach does not provide functional clues for alloresponses between donor and recipient and does not account for non‐MHC determinants.
Cellular tests such as the mixed lymphocyte culture (MLC) or the frequency analysis of alloreactive helper and cytotoxic T lymphocyte progenitors (HTLp and CTLp, respectively) were established as functional assays representing the cellular response to antigenic determinants and hence the possibility of alloreactions between donor–recipient. These assays rely on measuring cell proliferation or production of cytokines, use radioactive agents and are very laborious and time consuming. In addition, these tests have not been consistently shown to correlate well with the occurrence of aGVHD 14, 15, 16, 17. Therefore, selection procedures in HSCT would greatly benefit from the development of simpler and faster functional assays for example measuring gene expression as a marker of alloreactivity. Gene expression profile in an HLA mismatched setting could be indicative of high‐risk donor–recipient pairs and would allow better donor selection in cases where more than one suitable donor is available for allo‐HSCT; for example haploidentical siblings of a patient.
One study using gene expression profiling suggested that some donors may be ‘stronger alloresponders’ and consequently more likely to elicit GVHD than others 18. By investigating the gene expression profiles of CD4+ and CD8+ cells of 50 allo‐HSCT donors Baron et al. could segregate those whose recipients went on to develop GVHD from those who did not. A limitation of this study, however, was that it looked at the donor cells in isolation and thus did not take into consideration the interactions between the donor and recipient. In addition to genetic background, leading to quantitative or qualitative differences in immune responses, environmental factors and donor/recipient immune system history may contribute to how they will react; hence a functional assay is more favourable than simply sequencing gene variations or viewing donor cells in isolation.
The study presented here was conducted as a proof‐of‐principle that gene expression signatures in MLC of a potential donor–recipient pair could be predictive of incompatibility. To this end we used gene expression microarrays to investigate the genes and pathways differentially regulated in bidirectional MLC from HLA matched vs mismatched pairs. We found that a great number of genes related to immune function were differentially regulated; these genes were also found to be associated with GVHD. The most highly upregulated genes were IFNγ‐inducible genes and IFNγ neutralisation in MLCs abrogated their induction. The microRNA‐155 (miR‐155) was also found to be significantly induced in the mismatched setting but its induction was not diminished by blocking IFNγ. We are proposing that measuring gene expression in MLC could be a simpler and faster method of identifying functional incompatibilities between potential donor–recipient pairs that are not detected by DNA typing techniques, compared with the traditional cellular assays.
Methods
Materials and reagents
Peripheral blood mononuclear cells (PBMCs) were prepared by density gradient centrifugation from healthy volunteer donors. All samples were obtained with written informed consent and the study was approved by the Cyprus National Bioethics Committee. IFNγ neutralising antibody and isotype control were purchased from R&D systems, Abington, UK. Human recombinant IFNγ as well as IFNγ ELISA (enzyme‐linked immunosorbent assay) kits were from PeproTechInc., Rocky Hill, NJ. For real‐time RT‐PCR inventoried TaqMan® Gene expression Assays for IDO1 (Hs00984148_m1), CCL8 (Hs041877715_m1), CXCL9 (Hs001771065_m1), CXCL10 (Hs01124251_g1) and MIR155HG (Hs01374569_m1) and inventoried TaqMan® MicroRNA Assays for miR‐155 (000479) and RNU (001006) were used (Applied Biosystems, Foster City, CA) with SuperScript® III Reverse Transcriptase (Invitrogen Corp., Carlsbad, CA) and TaqMan Universal PCR Mastex Mix (Applied Biosystems, Foster City, CA) and run on an ABI 7500 Real Time PCR System. All other reagents were purchased from Sigma‐Aldrich UK Ltd, Dorset, UK unless otherwise stated.
Mixed lymphocyte cultures
Bidirectional MLCs were set up in roswell park memorial institute (RPMI) media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at a 1:1 ratio and maintained in a humidified incubator at 37°C and 5% CO2. All HLA matched pairs were genotypically matched siblings while all HLA mismatched pairs had at least four out of six mismatches considering HLA‐A, ‐B and ‐DRB1 loci. The HLA types of all individuals included in this study are shown in Table S1, Supporting Information. Typing was performed by combination of standard sequence specific oligonucleotide probes (SSOP) and sequence‐specific primers (SSP) typing methods routinely used for bone marrow transplantation purposes in our european federation for immunogenetics (EFI) accredited laboratory. Unidirectional assays were also performed using three individuals who were either homozygous or heterozygous for the two most frequent haplotypes in the Cypriot population (Table S2). Cells were irradiated with 25 Gy. Cell culture supernatants were harvested at different time points, centrifuged to remove cell debris and stored at −20°C until their use in ELISA assays. Cultures were harvested at different time points for RNA isolation.
Gene expression profiling
The microarray study was performed using the IlluminaHumanHT‐12 v4 Expression BeadChips (Illumina Inc., San Diego, CA). In brief, 1 µg total RNA was reverse transcribed to synthesise first‐ and second‐strand complementary DNA (cDNA), followed by in vitro transcription to synthesise biotin‐labelled complementary RNA (cRNA) using the TotalPrep‐96 RNA amplification kit (Ambion, Austin, TX). Biotin‐labelled cRNA from each sample was then hybridised to the BeadChips at 58°C for 18 h. The hybridised BeadChips were washed, labelled with streptavidin‐Cy3 and then scanned with Illumina BeadScan and images imported into genome studio version 1.5.4 (Illumina Inc.) for data extraction. Analyses were performed using BRB‐Array Tools and MeV 19. Data were log2 transformed and quantile normalised using the lumi package from Bioconductor (http://www.bioconductor.org/) and genes with low variability (less than 20% of expression data values have at least a 1.5‐fold change in either direction from the gene's median value) were filtered out from further analysis. Genes with significantly altered expressions were identified by one‐way analysis of variance (anova) with Benjamini–Hochberg multiple testing correction, with P < 0.01. The entities satisfying the significance analysis were passed on for the fold change analysis. For a given gene its change in expression was calculated by subtracting its average normalised intensity in individuals from the average normalised intensity in MLCs (MLCs were plated at a ratio of 1:1) 20.
Results
Matched and mismatched MLCs have distinct expression profiles
To investigate the differences between matched and mismatched MLC we carried out a whole genome gene expression microarray. We isolated PBMC from a pair of HLA identical siblings (S4 and S5) and a third HLA mismatched individual (S6) which were either cultured alone or in bidirectional matched (S1 = S4 + S5) and mismatched MLCs (S2 = S5 + S6 and S3 = S4 + S6), all in triplicate. After culturing for 72 h RNA was extracted and used for the whole genome expression microarray. Prior to the microarray analysis, it was verified that in the mismatched MLC setting there was increased proliferation as determined by carboxyfluorescein succinimidyl ester (CFSE) dilution assay and upregulation of T‐cell activation markers CD25 and HLA‐DR (data not shown). Unsupervised hierarchical clustering using the set of genes with the most variable mRNA expression (SD > 0.75) showed that the expression profile of mismatched MLC (S2 and S3) was distinct from the matched MLC (S1) defined by two major clusters formed (Figure 1A). Furthermore, the expression profile of the matched MLC (S1) clustered together with the matched individuals (S4 and S5), whereas the mismatched MLC (S2 and S3) was more closely related to the third individual (S6) (Figure 1A). Genes with significantly altered expressions between matched and mismatched MLC were identified by one‐way anova with Benjamini–Hochberg multiple testing correction, with P < 0.01 and the entities satisfying the significance analysis were subsequently used for fold change analysis. Fold change was calculated comparing average normalised intensity of matched or mismatched MLCs to the mean of their respective individuals cultured alone. We identified 193 genes that were differentially regulated between matched and mismatched MLC by at least twofold, of which 142 were upregulated (Tables S3 and 1) and 51 downregulated (Tables 4 and 2). Heatmap of the fold‐changes of regulated genes shows that major differences in gene expression exist in the mismatched MLC as compared with the individuals [S2 − (S5 + S6) and S3 − (S4 + S6)]. Conversely, as shown on the Tables and heatmap no significant differences can be seen in gene expression of the matched MLC compared with the matched individuals cultured alone [S1 − (S4 + S5)]; co‐culturing does not lead to any change in gene expression (Figure 1B).
Figure 1.
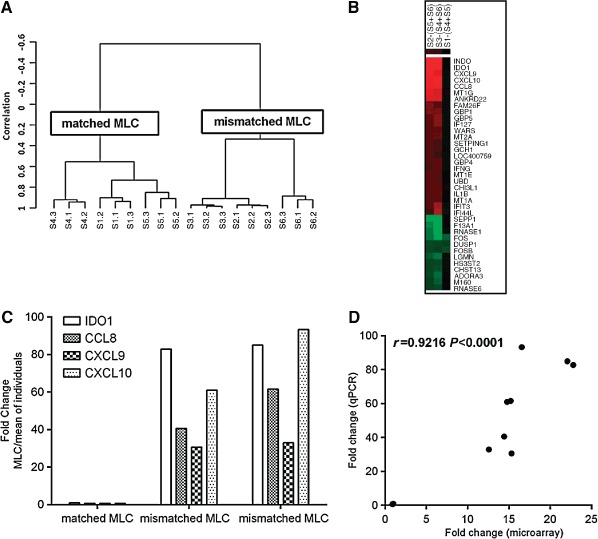
(A) Hierarchical clustering using the set of genes with the most variable mRNA expression (SD > 0.75) defining two major clusters. Clustering was performed using centered correlation and average linkage in BRB‐Array Tools. S1 = S4 + S5 matched mixed lymphocyte culture (MLC), S2 = S5 + S6 and S3 = S4 + S6 mismatched MLC, S4 and S5 matched siblings, S6 mismatched individual. (B). Heatmap of fold‐changes of regulated genes in the mismatched MLC [S2 − (S5 + S6) and S3 − (S4 + S6)] and the matched MLC [S1 − (S4 + S5)]. cluster 3.0 and tree view software were used. Genes with a fold change of ±5 are presented. Red colour indicates upregulated genes, green colour downregulated genes and black colour indicates no change. (C) Upregulation of IDO1, CCL8, CXCL9 and CXCL10 was verified using qPCR shown as fold change in the MLC vs the mean expression of the individuals (MLCs were at a 1:1 ratio). (D) Correlation between fold changes for IDO1, CXCL9, CXCL10 and CCL8 calculated by microarrays and qPCR; Pearson's correlation is shown.
Table 1.
Upregulated genes in matched vs mismatched MLC a
| Gene symbol | S2 − (S5 + S6) mismatched | S3 − (S4 + S6) mismatched | S1 − (S4 + S5) matched | |||
|---|---|---|---|---|---|---|
| FC | Corrected P | FC | Corrected P | FC | Corrected P | |
| INDO | 23.20 | 0.0106 | 21.02 | 0.0100 | 1.14 | 0.9726 |
| IDO1 | 22.78 | 0.0101 | 22.08 | 0.0074 | 1.00 | 1.0003 |
| CXCL9 | 15.31 | 0.0026 | 12.57 | 0.0073 | −1.07 | 1.0076 |
| CXCL10 | 14.78 | 0.0060 | 16.56 | 0.0106 | −1.12 | 1.0131 |
| CCL8 | 14.42 | 0.0107 | 15.21 | 0.0112 | −1.07 | 1.0069 |
| MT1G | 13.33 | 0.0001 | 10.78 | 0.0002 | 1.49 | 0.5269 |
| ANKRD22 | 11.67 | 0.0065 | 11.36 | 0.0070 | −1.01 | 1.0022 |
| FAM26F | 6.66 | 0.0221 | 4.74 | 0.0320 | −1.03 | 1.0123 |
| GBP1 | 6.64 | 0.0120 | 6.42 | 0.0182 | −1.01 | 1.0061 |
| GBP5 | 6.18 | 0.0025 | 7.11 | 0.0064 | −1.09 | 1.0108 |
| IFI27 | 6.00 | 0.0005 | 7.02 | 0.0008 | −1.10 | 1.0076 |
| WARS | 5.98 | 0.0025 | 5.96 | 0.0020 | −1.05 | 1.0177 |
| MT1E | 5.82 | 0.0008 | 4.65 | 0.0001 | 1.13 | 0.9669 |
| MT2A | 5.55 | 0.0006 | 6.27 | 0.0012 | −1.06 | 1.0108 |
| UBD | 5.51 | 0.0044 | 4.67 | 0.0053 | 1.07 | 0.9852 |
| CHI3L1 | 5.43 | 0.0584 | 5.54 | 0.0342 | 1.47 | 0.6315 |
| SERPING1 | 5.00 | 0.0064 | 4.34 | 0.0112 | −1.01 | 1.0047 |
| IFIT3 | 4.89 | 0.0005 | 8.42 | 0.0053 | −1.09 | 0.9901 |
| IL1B | 4.84 | 0.0128 | 6.07 | 0.0048 | 1.27 | 0.9556 |
| GBP4 | 4.72 | 0.0070 | 5.86 | 0.0056 | −1.11 | 0.9540 |
| IFNG | 4.46 | 0.0003 | 5.77 | 0.0016 | −1.09 | 1.0131 |
ANOVA, analysis of variance; FC, fold change; MLC, mixed lymphocyte culture.
Genes identified by microarray to be upregulated more than fivefold in mismatched MLCs with corresponding fold change values in the matched MLC presented for comparison. Genes with significantly altered expressions were identified by one‐way anova with Benjamini–Hochberg multiple testing correction, with P < 0.01 and were passed on for the fold change analysis. For a given gene its change in expression was calculated by subtracting its average normalised intensity in individuals from the normalised intensity in MLCs (P < 0.01).
Table 4.
KEGG pathways overrepresented in matched vs mismatched MLC regulated genesa
| Pathways | Count | % | P‐value | Benjamini |
|---|---|---|---|---|
| Cytokine–cytokine receptor interaction | 17 | 1.4 | 7.6E‐9 | 5.5E‐7 |
| Toll‐like receptor signalling pathway | 8 | 0.7 | 9.2E‐5 | 3.3E‐3 |
| Chemokine signalling pathway | 10 | 0.8 | 1.5E‐4 | 3.6E‐3 |
| Haematopoietic cell lineage | 7 | 0.6 | 2.9E‐4 | 5.3E‐3 |
| Graft‐vs‐host disease | 5 | 0.4 | 7.6E‐4 | 1.1E‐2 |
| Type I diabetes mellitus | 5 | 0.4 | 1.0E‐3 | 1.2E‐2 |
| NOD‐like receptor signalling pathway | 5 | 0.4 | 4.3E‐3 | 4.3E‐2 |
| Antigen processing and presentation | 5 | 0.4 | 1.2E‐2 | 1.0E‐1 |
| JAK‐STAT signalling pathway | 6 | 0.5 | 2.5E‐2 | 1.8E‐1 |
MLC, mixed lymphocyte culture.
KEGG pathways found to be significantly overrepresented within differentially regulated genes between matched and mismatched MLC. Gene set enrichment analysis was performed using the molecular signatures database of DAVID Bioinformatics Resources (National Institute of Allergy and Infectious Diseases, National Institutes of Health). P values are corrected using Benjamini multiple testing correction and a cut‐off of P < 0.05.
Table 2.
Downregulated genes in matched vs mismatched MLC a
| Gene symbol | S2 − (S5 + S6) mismatched | S3 − (S4 + S6) mismatched | S1 − (S4 + S5) matched | |||
|---|---|---|---|---|---|---|
| FC | Corrected P | FC | Corrected P | FC | Corrected P | |
| SEPP1 | −17.20 | 0.0036 | −15.23 | 0.0009 | 1.03 | 1.0020 |
| F13A1 | −8.89 | 0.0003 | −15.95 | 0.00001 | 1.06 | 1.0117 |
| RNASE1 | −7.81 | 0.1433 | −19.80 | 0.0366 | −1.02 | 1.0045 |
| FOS | −7.72 | 0.0101 | −10.77 | 0.0073 | −6.18 | 0.4073 |
| LGMN | −5.96 | 0.0108 | −7.38 | 0.0074 | −1.14 | 0.5846 |
| LGMN | −5.54 | 0.0066 | −6.85 | 0.0030 | −1.22 | 0.4033 |
| DUSP1 | −5.47 | 0.0157 | −4.59 | 0.0327 | −3.99 | 0.5231 |
| FOSB | −5.08 | 0.0551 | −3.79 | 0.0956 | −5.70 | 0.5291 |
| ADORA3 | −4.83 | 0.0047 | −6.53 | 0.0072 | 1.05 | 1.0130 |
| HS3ST2 | −4.61 | 0.0723 | −5.33 | 0.0441 | −1.14 | 0.8104 |
| CHST13 | −4.50 | 0.0029 | −5.66 | 0.0019 | −1.05 | 0.9255 |
ANOVA, analysis of variance; MLC, mixed lymphocyte culture.
Genes found to be downregulated more than fivefold in mismatched MLCs in the microarray study together with corresponding fold change values in the matched MLC presented for comparison. Genes with significantly altered expressions were identified by one‐way anova with Benjamini–Hochberg multiple testing correction, with P < 0.01 and were passed on for the fold change analysis. For a given gene its change in expression was calculated by subtracting its average normalised intensity in individuals from the normalised intensity in MLCs (P < 0.01).
The genes differentially regulated in matched vs mismatched MLC were classified ontologically using PANTHER classification system pathways which also statistically determines over‐ or under‐representation of genes in a particular pathway. Within regulated genes there was significant over‐representation of genes related to immune system response (Table 3). Other significantly represented pathways were angiogenesis and interleukin signalling pathways, transforming growth factor (TGF)‐β signalling pathway, cytoskeletal regulation by Rho GTPase and Toll‐like receptor signalling pathway. Using KEGG pathways these genes were also shown to be significantly associated with GVHD and graft rejection (Table 4). According to the fold change analysis the immunosuppressive enzyme indoleamine 2,3‐dioxygenase (IDO) and chemokines CXCL9, CXCL10 and CCL8 were the most highly upregulated genes in mismatched MLC whereas they were not induced in the matched MLC (Table 1). As validation of the microarray results the expression levels of these four genes were determined by qPCR in the same RNA samples (Figure 1C) and found to be highly correlated to their expression changes as calculated by the microarray (Pearson's correlation r = 0.9216, P < 0.0001) (Figure 1D).
Table 3.
Overrepresented GO biological process pathways in matched vs mismatched MLC a
| Pathways | Homo sapiens genes | Regulated genes twofold matched vs mismatched | Expected | P‐value |
|---|---|---|---|---|
| Immune system process | 2480 | 68 | 21.20 | 2.95E‐17 |
| Response to stimulus | 1767 | 53 | 15.11 | 3.44E‐14 |
| Immune response | 725 | 34 | 6.2 | 1.11E‐13 |
| Response to interferon‐gamma | 113 | 16 | 0.97 | 1.00E‐12 |
| Macrophage activation | 325 | 21 | 2.78 | 1.88E‐10 |
| Cytokine‐mediated signalling pathway | 371 | 19 | 3.17 | 1.13E‐7 |
| Unclassified | 6816 | 25 | 58.28 | 1.33E‐6 |
| Signal transduction | 4019 | 62 | 34.36 | 1.21E‐4 |
| Cell communication | 4224 | 64 | 36.12 | 1.37E‐4 |
| Cellular defence response | 496 | 16 | 4.24 | 1.15E‐3 |
| Cell–cell signalling | 1259 | 26 | 10.76 | 4.83E‐3 |
| Cellular process | 6072 | 77 | 51.92 | 6.42E‐3 |
| Cell surface receptor linked signal transduction | 2049 | 35 | 17.52 | 9.34E‐3 |
| JAK‐STAT cascade | 112 | 7 | 0.96 | 9.94E‐3 |
| JNK cascade | 155 | 8 | 1.33 | 1.12E‐2 |
| Negative regulation of apoptosis | 264 | 10 | 2.26 | 1.80E‐2 |
| MAPKKK cascade | 454 | 13 | 3.88 | 2.79E‐2 |
| Extracellular transport | 138 | 7 | 1.18 | 3.56E‐2 |
| Apoptosis | 978 | 20 | 8.36 | 4.97E‐2 |
GO, gene ontology; MLC, mixed lymphocyte culture; MAPKK, mitogen‐activated protein kinase kinase kinase.
Gene ontology biological process pathways identified to be significantly overrepresented within differentially regulated genes between matched and mismatched MLC. Analysis was performed using the statistical overrepresentation tool of PANTHER classification system. P values are corrected using Bonferroni's multiple testing correction and a cut‐off of P < 0.05.
IDO1, CXCL9, CXCL10 and CCL8 are induced in mismatched MLC
As IDO1, CXCL9, CXCL10 and CCL8 were the most highly upregulated genes in mismatched MLC we chose to further investigate these four genes as they would be more likely to have higher discriminatory value. The temporal regulation of IDO1, CXCL9, CXCL10 and CCL8 in bidirectional mismatched MLC was investigated at 24 h, 48 h, 72 h and day 6 of culture. The mRNA levels of all genes increased with time reaching a peak at 72 h and declining after that (Figure 2A). The induction of these four genes was shown to be a reproducible effect in further HLA mismatched pairs (Figure 2B), while in further pairs of matched MLCs there was no induction of IDO1, CXCL9, CXCL10 or CCL8 showing that these genes are only induced in an HLA mismatched setting (Figure 2B). Across all the 15 different mismatched pairs investigated the induction of the four genes was consistent albeit of different magnitudes probably representing the biological variation of the individuals (Figure 2C).
Figure 2.
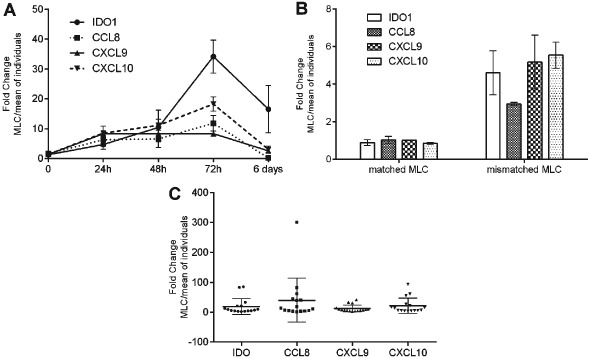
(A) Expression of IDO1, CXCL9, CXCL10 and CCL8 measured by qPCR in bidirectional mismatched mixed lymphocyte cultures (MLCs) at 24 h, 48 h, 72 h and 6 days. (B) Expression of IDO1, CXCL9, CXCL10 and CCL8 in matched and mismatched bidirectional MLCs and (C) in further 16 mismatched MLCs. Gene expression is shown as fold change in the MLC vs the mean expression of the individuals.
To investigate whether the four genes can show directional response we performed selected unidirectional assays using irradiation to complement our findings. We chose three individuals A, B and C based on their HLA haplotypes that were either homozygous or heterozygous for the two most frequent haplotypes in the Cypriot population (Table S2) and performed both bidirectional and unidirectional MLC (AB, BC and AC). Both directions of the unidirectional MLCs were performed by irradiating (ir) the cells from one or the other individual prior to MLC, i.e. AB = bidirectional MLC, irAB = unidirectional MLC were cells from A were irradiated, AirB = unidirectional MLC were cells from B were irradiated. In these assays we found a trend of directional response in the induction of IDO, CCL8, CXCL9 and CXCL10 (Figure S2), although further validation is required.
IFNγ is responsible for IDO1, CXCL9, CXCL10 and CCL8 induction in mismatched MLC
Among the differentially regulated genes in matched and mismatched MLC there was a strong IFNγ signature; indeed IDO1, CXCL9, CXCL10 and CCL8 are IFNγ inducible genes. In addition, IFNγ is known to be an important regulator in T cell alloreaction, extensively studied in the mismatch MLC setting and also as a marker for alloreaction between HLA mismatched donors and recipients. All the significantly regulated genes identified by the microarray in the mismatched MLC were investigated using the INTERFEROME tool, a database of interferon regulated genes 21, revealing that 116 of 193 genes submitted are regulated by IFNγ. Among them were IDO1, CXCL9, CXCL10 and CCL8 as well as other known IFNγ inducible genes such as STAT1 and SOCS1.
We found increased levels of IFNγ in our mismatched MLC peaking at 72 h, which probably accounts for the induction of these genes (Figure 3A). To verify that IFNγ drives the induction of IDO1, CXCL9, CXCL10 and CCL8 in the mismatched MLC we sought to investigate the expression of these genes in the presence of IFNγ neutralising antibody. IFNγ neutralisation significantly decreased the induction of IDO1, CXCL9, CXCL10 and CCL8 in bidirectional mismatched MLCs as compared with isotype matched control (Figure 3B). This finding indicates that the induction of IDO1, CXCL9, CXCL10 and CCL8 in the mismatched MLC setting depends on IFNγ.
Figure 3.
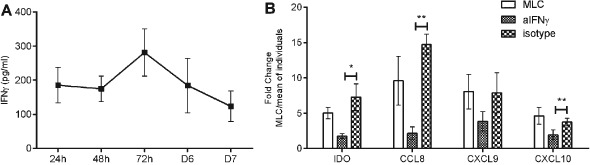
(A) Interferon‐gamma (IFNγ) levels in eight bidirectional mismatched mixed lymphocyte culture (MLC) measured by enzyme‐linked immunosorbent assay (ELISA) at 24 h, 48 h, 27 h, 6 days and 7 days. (B) Fold change expression of IDO, CCL8, CXCL9 and CXCL10 in mismatched MLC in the presence of neutralising antibody to IFNγ or isotype control. Cumulative data of five independent experiments. Gene expression is shown as fold change in the MLC vs the mean expression of the individuals. * P ≤ 0.05, ** P ≤ 0.01.
The miR‐155 is induced in mismatched MLC independently of IFNγ
Among the 77 genes that are not identified by the INTERFEROME database as IFNγ inducible genes the MIR155 Host Gene (MIR155HG) was included. MIR155HG (also known as BIC) consists of three exons and gives rise to miR‐155 which is encoded within exon 3. MIR155HG was identified by our microarray to be induced by 2.55‐fold (corrected P = 0.0009) in the mismatched MLC (S2) (Table S2). MIR155HG induction in the mismatched but not the matched MLC was verified by qPCR (Figure 4A). In the mismatched MLC the expression of MIR155HG increased with time reaching roughly twofold at 72 h and more that fourfold at 6 days of culture (Figure 4B). The expression of miR155 was investigated in further mismatched MLCs at three time points (72 h, day 6 and day 7) and was found to be induced and to be highly correlated to MIR155HG expression (Pearson's correlation r = 0.8864, P = 0.0015, Figure 4C). Even though the INTERFEROME database did not identify MIR155HG as an IFNγ regulated gene there is data showing that MIR155HG and miR‐155 can be induced by IFNγ in vitro 22. To investigate this, mismatched MLCs were repeated in the presence of IFNγ neutralising antibody and the expression of MIR155HG and miR‐155 was measured. Neutralisation of IFNγ did not abrogate the induction of MIR155HG or miR‐155 in mismatched MLC (Figure 4D) and treatment of PBMC with human recombinant IFNγ did not result in the induction of MIR155HG or miR‐155 while inducing IDO1 expression (Figures 4E and S3). These findings suggest that MIR155HG and miR‐155 expression may be induced via alternative pathways in this setting.
Figure 4.
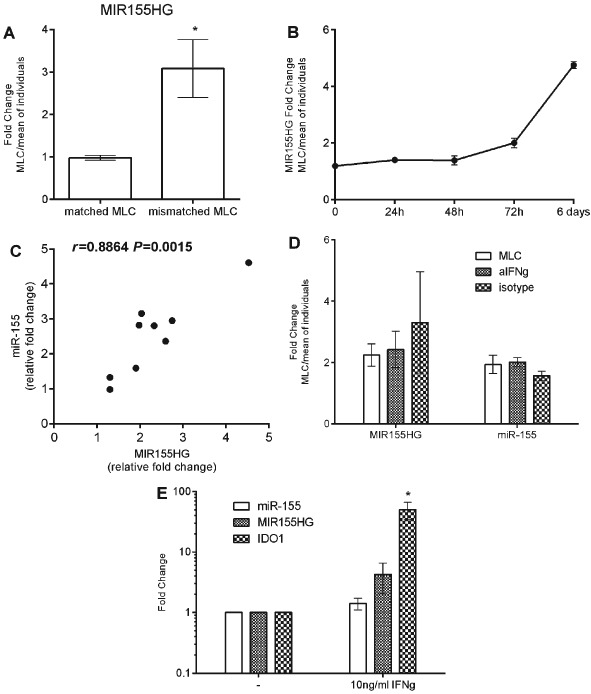
(A) Expression of MIR155HG in matched and mismatched bidirectional mixed lymphocyte cultures (MLCs) at 72 h. (B) Expression of MIR155HG in mismatched MLCs at 24 h, 48 h, 72 h and 6 days. (C) Verification of microRNA‐155 (miR‐155) induction in further mismatched MLCs at three time points (72 h, day 6 and day 7) and correlation to MIR155HG expression, Pearson's correlation is shown. (D) Fold change expression of MIR155HG and miR‐155 in mismatched MLC in the presence of neutralising antibody to interferon‐gamma (IFNγ) or isotype control. Gene expression is shown as fold change in the MLC vs the mean expression of the individuals. (E) Expression of miR‐155, MIR155HG and IDO1 in human peripheral blood mononuclear cell (PBMC) treated with 10 ng/ml of human recombinant IFNγ for 24 h. Fold changes relative to control of untreated PBMC. Cumulative data of three separate experiments; * P ≤ 0.05.
Discussion
Donor selection relies on DNA typing that offers no information about functional responses of the donor and recipient whereas functional tests such as the MLC can complement HLA typing and serve as a more relevant representation of these responses. We used gene expression profiling to study global differences in the HLA mismatched MLC compared with the HLA matched setting and show that gene expression in MLC can reveal mechanisms involved in GVHD and graft rejection; these can potentially serve to better our understanding of the mechanisms involved in the alloresponses during these diseases and reveal therapeutic targets.
Gene expression changes driven by HLA incompatibility were distinctly differentiated from the HLA matched setting and were mostly related to immune function and immune response. The majority of genes regulated in the mismatched setting were identified by the INTERFEROME tool to be IFNγ inducible genes suggesting a very strong influence of INFγ in MLC. Indeed many previous studies have shown the central role of IFNγ in the cytokine network following alloantigen recognition as well as the generation and maintenance of alloresponses associated with the induction of GVHD. In murine models of GVHD IFNγ is elevated early, peaking before clinical symptoms appear 23 and T cells isolated from patients with GVHD have higher levels of IFNγ 24. GVHD results from the recognition of host antigens by donor T cells following allogeneic transplantation and these alloreactive donor T cells are the primary source of IFNγ. Therefore, IFNγ can be used as a surrogate of T‐cell response and many studies have explored the use of IFNγ as a marker for donor selection 25, 26, transplantation‐related complications 27 and GVHD 28. A study by van der Meer et al. attempted to decipher whether levels of IFNγ in MLC, as a marker for deleterious Th1 responses, are sensitive to HLA incompatibility and whether they correlate with GVHD occurrence when the donor–recipient pairs tested proceeded with transplantation. It was shown that high levels of INFγ coincided with severe aGVHD in unrelated bone marrow transplantation, whereas low levels were largely associated with grades 0 and I. In addition, levels of IFNγ in MLC reflected HLA class II differences but not isolated class I differences or mismatches other than HLA (identical siblings), suggesting that measuring IFNγ alone cannot be used for the detection of class I mismatches 26.
By investigating global gene expression changes in mismatched MLC we aimed to identify genes that may constitute good targets to study further as markers for donor–recipient alloresponses. The most highly upregulated genes were IDO1, CXCL9, CXCL10 and CCL8; the regulation of these genes was confirmed by quantitative real‐time PCR and in independent HLA mismatched MLCs. The four selected genes have all been invariably associated with GVHD or allograft rejection (Table S5), supporting that in vitro responses can mirror the in vivo situation. Indeed, gene expression profiling of early changes in hepatic GVHD identified many targets similar to the ones presented here 29. However, this does not exclude the possibility that other genes may show more prominent changes in different MLC, as compared with those studied by microarray in this study, due to the presence of different HLA combinations.
Representing an important immunoregulatory mechanism, IDO catalyses the first and rate‐limiting step of tryptophan catabolism along the kynurenine pathway; decreased tryprophan and increased levels of its metabolites lead to inhibition of T‐cell activation and induce apoptosis 30. IDO is induced by IFNγ in a variety of cells such as epithelial cells and antigen presenting cells and there is extensive literature to support a role for IDO in GVHD. Activation of the kynurenine pathway has been reported in animal models of GVHD as well as in humans following allogeneic stem cell transplantation. Studies in animal models suggest a protective role of IDO as deficiency leads to accelerated GVHD in mice after allogeneic bone marrow transplantation, while inability to induce IDO due to IFNγ deficiency in recipients also leads to exacerbation of disease 31, 32. In contrast to these findings, increased IDO activity and increased levels of metabolites correlate with severity and worse outcome in clinical studies 33, 34. Induction of IDO may represent a counter acting mechanism as expression in host APCs leads to the suppression of experimental GVHD through the induction of regulatory T cells 35.
Chemokines and their receptors play a pivotal role in all phases of GVHD by mediating the organ‐specific homing and activation of effector cells at sites of inflammation. CCL8 was identified as a potential biomarker in murine GVHD using a proteomics approach and was subsequently evaluated in human samples where it was found to be increased during aGVHD and to correlate with disease severity 36. CXCL9 and CXCL10 act through the same receptor CXCR3 and play a central role in the pathogenesis of GVHD via the recruitment of CXCR3+ expressing T cells and natural killer (NK) cells at GVHD target organs. CXCL9 and CXCL10 levels were increased in the liver, intestine and lung of mice with aGVHD 37. CXCL10 and CXCL9 were also markedly increased in the serum of patients with aGVHD and chronic GVHD (cGVHD) 38, with CXCL9 levels suggested to be predictive of cGVHD occurrence 39. Increased CXCL10 expression together with CXCR3+ cells were shown in sections of skin biopsies from GVHD patients 40, supporting an important role in the pathogenesis of skin GVHD in humans.
A number of downregulated genes were also detected in the mismatched MLC which are associated with immune system function, for example, FOS, FOSB, legumain (LGMN), dual specificity phosphatase 1 (DUSP1), CD9 and CD14 among others. It is possible that these genes are involved in immune system regulation by dampening the immune response or mediating tolerance. One example is DUSP1 which was significantly downregulated by approximately fivefold in the HLA mismatched MLC as compared with HLA matched MLC (Table 2). DUSP1 specifically inactivates mitogen‐activated protein kinases (MAPKs) which are critically involved in the immune response and in that way acts as a negative regulator of inflammation 41.
One interesting target to arise from our study is miR‐155. We have shown that MIR155HG and miR‐155 are induced during alloresponses in the HLA mismatched MLC but not in the HLA matched setting. MiR‐155 is currently receiving great interest for its role in GVHD as shown in animal models and a clinical trial is currently recruiting patients to investigate its involvement (NCT01521039, clinicaltrials.gov). MiR‐155 plays a central role in the regulation of T and B cell responses; B and T cells are known to upregulate miR‐155 after B cell receptor (BCR) and T cell receptor (TCR) activation 42, 43. MiR‐155 also regulates T‐cell responses as T cells from miR‐155 knockout mice have an impaired response and lower levels of IL‐2 and IFNγ in response to antigens 44, 45. A role for MiR‐155 in the innate immune response is also supported by the ability of several TLR ligands to strongly induce its levels 22, 46. Such strong evidence of miR‐155 regulation of immune function prompted the investigations into its role in GVHD. It has been shown that miR‐155 is upregulated in T cells from mice developing aGVHD while miR‐155 deficient grafts had markedly reduced lethal aGVHD 47. Furthermore, miR‐155 blockade decreased aGVHD severity and prolonged survival. Upregulation of miR‐155 was also shown in specimens from patients with pathologic evidence of intestinal aGVHD 47, as well as significantly elevated in the serum of GVHD patients where it correlated with disease severity 48.
Although one study reports the induction of miR‐155 by IFNγ in murine bone marrow derived macrophages 22, our findings do not appear to support the same conclusion. MIR155HG was not identified by the INTERFEROME tool as an INFγ inducible gene, IFNγ neutralisation in HLA‐mismatched MLC did not abrogate its induction and finally IFNγ did not induce MIR155HG or miR‐155 expression in human PBMC. IFNγ induction of miR‐155 was shown to require TNFα signalling in the murine macrophages 22, suggesting that miR‐155 may not be a direct target of the canonical JAK/STAT pathway known to transactivate several hundred antiviral genes in response to IFN signalling. MiR‐155 expression was shown to be induced through the JNK MAPK pathway which activates the AP‐1 complex and both human and murine promoter regions of MIR155HG contain binding sites for AP‐1 and NF‐κB family members.
In the absence of significant advances in the treatment of GHVD, early diagnosis and early assessment of treatment response have significant prognostic value for disease outcome and considerable effort has been made to identify such biomarkers. However, predictive markers for the occurrence of GVHD are still lacking and a better understanding of the mechanisms and pathways involved are still needed to provide insight into the pathogenesis of the disease and identify novel targets for therapeutic intervention. We have identified gene expression changes in HLA mismatch MLC which correlate with genes and pathways involved in GVHD. These may be used to study and monitor alloreactive responses in vitro and could also aid in defining the biological processes and molecular cascade involved in GVHD. Finally, they could potentially serve as predictive markers for GVHD but they should firstly be evaluated in human studies to validate their predictive value.
Conflict of interests
The authors have declared no conflicting interests.
Supporting information
Figure S1. Heatmap of regulated genes in mismatched and matched mixed lymphocyte culture (MLC).
Figure S2. Directional response of IDO, CCL8, CXCL9 and CXCL10 in mixed lymphocyte culture (MLC).
Figure S3. Interferon‐gamma (IFNγ) induction of IDO and MIR155HG.
Table S1. Human leucocyte antigen (HLA) types of graft‐vs‐host disease (GVHD) study.
Table S2. Human leucocyte antigen (HLA) types of directional mixed lymphocyte culture (MLC).
Table S3. Upregulated genes in matched vs mismatched mixed lymphocyte culture (MLC).
Table S4. Downregulated genes in matched vs mismatched mixed lymphocyte culture (MLC).
Table S5. Association of IDO1, CXCL9, CXCL10, CLL8 and miR‐155 with graft‐vs‐host disease (GVHD).
Acknowledgments
This work was supported by a grant from the European Regional Development Fund and the Research Promotion Foundation of Cyprus. We thank Dr Costas Koufaris for help with statistical analysis of microarray results and for critical review of the manuscript.
The copyright line for this article was changed on 23 September 2016 after original online publication.
References
- 1. Ferrara JL, Levine JE, Reddy P, Holler E. Graft‐versus‐host disease. Lancet 2009: 373: 1550–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee SJ, Klein J, Haagenson M et al. High‐resolution donor‐recipient HLA matching contributes to the success of unrelated donor marrow transplantation. Blood 2007: 110: 4576–83. [DOI] [PubMed] [Google Scholar]
- 3. Petersdorf EW, Hansen JA, Martin PJ et al. Major‐histocompatibility‐complex class I alleles and antigens in hematopoietic‐cell transplantation. N Engl J Med 2001: 345: 1794–800. [DOI] [PubMed] [Google Scholar]
- 4. Petersdorf EW. Genetics of graft‐versus‐host disease: the major histocompatibility complex. Blood Rev 2013: 27: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jagasia M, Arora M, Flowers ME et al. Risk factors for acute GVHD and survival after hematopoietic cell transplantation. Blood 2012: 119: 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Petersdorf EW, Malkki M, Horowitz MM, Spellman SR, Haagenson MD, Wang T. Mapping MHC haplotype effects in unrelated donor hematopoietic cell transplantation. Blood 2013: 121: 1896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Metaxas Y, Bertz H, Spyridonidis A, Spyroupoulou‐Vlachou M, Porzelius C, Finke J. CT60 single‐nucleotide polymorphism as a surrogate marker for donor lymphocyte infusion outcome after allogeneic cell transplantation for acute leukemia. Bone Marrow Transplant 2012: 47: 411–5. [DOI] [PubMed] [Google Scholar]
- 8. Holler E, Rogler G, Brenmoehl J et al. The role of genetic variants of NOD2/CARD15, a receptor of the innate immune system, in GvHD and complications following related and unrelated donor haematopoietic stem cell transplantation. Int J Immunogenet 2008: 35: 381–4. [DOI] [PubMed] [Google Scholar]
- 9. Bertinetto FE, Dall'Omo AM, Mazzola GA et al. Role of non‐HLA genetic polymorphisms in graft‐versus‐host disease after haematopoietic stem cell transplantation. Int J Immunogenet 2006: 33: 375–84. [DOI] [PubMed] [Google Scholar]
- 10. Cavet J, Dickinson AM, Norden J, Taylor PR, Jackson GH, Middleton PG. Interferon‐gamma and interleukin‐6 gene polymorphisms associate with graft‐versus‐host disease in HLA‐matched sibling bone marrow transplantation. Blood 2001: 98: 1594–600. [DOI] [PubMed] [Google Scholar]
- 11. Middleton PG, Taylor PR, Jackson G, Proctor SJ, Dickinson AM. Cytokine gene polymorphisms associating with severe acute graft‐versus‐host disease in HLA‐identical sibling transplants. Blood 1998: 92: 3943–8. [PubMed] [Google Scholar]
- 12. Rocha V, Franco RF, Porcher R et al. Host defense and inflammatory gene polymorphisms are associated with outcomes after HLA‐identical sibling bone marrow transplantation. Blood 2002: 100: 3908–18. [DOI] [PubMed] [Google Scholar]
- 13. Petersdorf EW, Malkki M, Gooley TA, Martin PJ, Guo Z. MHC haplotype matching for unrelated hematopoietic cell transplantation. PLoS Med 2007: 4: e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. DeGast GC, Mickelson EM, Beatty PG et al. Mixed leukocyte culture reactivity and graft‐versus‐host disease in HLA‐identical marrow transplantation for leukemia. Bone Marrow Transplant 1992: 9: 87–90. [PubMed] [Google Scholar]
- 15. Montagna D, Maccario R, Comoli P et al. Frequency of donor cytotoxic T cell precursors does not correlate with occurrence of acute graft‐versus‐host disease in children transplanted using unrelated donors. J Clin Immunol 1996: 16: 107–14. [DOI] [PubMed] [Google Scholar]
- 16. Segall M, Noreen H, Edwins L, Haake R, Shu XO, Kersey J. Lack of correlation of MLC reactivity with acute graft‐versus‐host disease and mortality in unrelated donor bone marrow transplantation. Hum Immunol 1996: 49: 49–55. [DOI] [PubMed] [Google Scholar]
- 17. Wang XN, Taylor PR, Skinner R et al. T‐cell frequency analysis does not predict the incidence of graft‐versus‐host disease in HLA‐matched sibling bone marrow transplantation. Transplantation 2000: 70: 488–93. [DOI] [PubMed] [Google Scholar]
- 18. Baron C, Somogyi R, Greller LD et al. Prediction of graft‐versus‐host disease in humans by donor gene‐expression profiling. PLoS Med 2007: 4: e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saeed AI, Bhagabati NK, Braisted JC et al. TM4 microarray software suite. Methods Enzymol 2006: 411: 134–93. [DOI] [PubMed] [Google Scholar]
- 20. Buess M, Nuyten DS, Hastie T, Nielsen T, Pesich R, Brown PO. Characterization of heterotypic interaction effects in vitro to deconvolute global gene expression profiles in cancer. Genome Biol 2007: 8: R191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rusinova I, Forster S, Yu S et al. Interferome v2.0: an updated database of annotated interferon‐regulated genes. Nucleic Acids Res 2013: 41: D1040–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA‐155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A 2007: 104: 1604–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Puliaev R, Nguyen P, Finkelman FD, Via CS. Differential requirement for IFN‐gamma in CTL maturation in acute murine graft‐versus‐host disease. J Immunol 2004: 173: 910–9. [DOI] [PubMed] [Google Scholar]
- 24. Yang YG, Wang H, Asavaroengchai W, Dey BR. Role of Interferon‐gamma in GVHD and GVL. Cell Mol Immunol 2005: 2: 323–9. [PubMed] [Google Scholar]
- 25. Danzer SG, Campo AC, Kunze B, Kirchner H, Rink L. Identification of HLA‐DRB1 and HLA‐DQB1 identical individuals by a cytokine‐based mixed lymphocyte culture. Lymphokine Cytokine Res 1994: 13: 303–8. [PubMed] [Google Scholar]
- 26. van der Meer A, Wissink WM, Schattenberg AV, Joosten I. Interferon‐gamma‐based mixed lymphocyte culture as a selection tool for allogeneic bone marrow donors other than identical siblings. Br J Haematol 1999: 105: 340–8. [PubMed] [Google Scholar]
- 27. Tanaka J, Imamura M, Kasai M, Sakurada K. Transplantation‐related complications predicted by cytokine gene expression in the mixed lymphocyte culture in allogeneic bone marrow transplants. Leuk Lymphoma 1995: 19: 27–32. [DOI] [PubMed] [Google Scholar]
- 28. Dickinson AM, Holler E. Polymorphisms of cytokine and innate immunity genes and GVHD. Best Pract Res Clin Haematol 2008: 21: 149–64. [DOI] [PubMed] [Google Scholar]
- 29. Ichiba T, Teshima T, Kuick R et al. Early changes in gene expression profiles of hepatic GVHD uncovered by oligonucleotide microarrays. Blood 2003: 102: 763–71. [DOI] [PubMed] [Google Scholar]
- 30. Fallarino F, Grohmann U, Vacca C et al. T cell apoptosis by kynurenines. Adv Exp Med Biol 2003: 527: 183–90. [DOI] [PubMed] [Google Scholar]
- 31. Jasperson LK, Bucher C, Panoskaltsis‐Mortari A, Mellor AL, Munn DH, Blazar BR. Inducing the tryptophan catabolic pathway, indoleamine 2,3‐dioxygenase (IDO), for suppression of graft‐versus‐host disease (GVHD) lethality. Blood 2009: 114: 5062–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jasperson LK, Bucher C, Panoskaltsis‐Mortari A et al. Indoleamine 2,3‐dioxygenase is a critical regulator of acute graft‐versus‐host disease lethality. Blood 2008: 111: 3257–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Landfried K, Zhu W, Waldhier MC et al. Tryptophan catabolism is associated with acute GVHD after human allogeneic stem cell transplantation and indicates activation of indoleamine 2,3‐dioxygenase. Blood 2011: 118: 6971–4. [DOI] [PubMed] [Google Scholar]
- 34. Xu J, Wei J, Zhu X et al. Increased plasma indoleamine 2,3‐dioxygenase activity and interferon‐gamma levels correlate with the severity of acute graft‐versus‐host disease after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant 2013: 19: 196–201. [DOI] [PubMed] [Google Scholar]
- 35. Tawara I, Shlomchik WD, Jones A et al. A crucial role for host APCs in the induction of donor CD4+ CD25+ regulatory T cell‐mediated suppression of experimental graft‐versus‐host disease. J Immunol 2010: 185: 3866–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hori T, Naishiro Y, Sohma H et al. CCL8 is a potential molecular candidate for the diagnosis of graft‐versus‐host disease. Blood 2008: 111: 4403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hasegawa H, Inoue A, Kohno M et al. Therapeutic effect of CXCR3‐expressing regulatory T cells on liver, lung and intestinal damages in a murine acute GVHD model. Gene Ther 2008: 15: 171–82. [DOI] [PubMed] [Google Scholar]
- 38. Croudace JE, Inman CF, Abbotts BE et al. Chemokine‐mediated tissue recruitment of CXCR3+ CD4+ T cells plays a major role in the pathogenesis of chronic GVHD. Blood 2012: 120: 4246–55. [DOI] [PubMed] [Google Scholar]
- 39. Kitko CL, Levine JE, Storer BE et al. Plasma CXCL9 elevations correlate with chronic GVHD diagnosis. Blood 2014: 123: 786–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Piper KP, Horlock C, Curnow SJ et al. CXCL10‐CXCR3 interactions play an important role in the pathogenesis of acute graft‐versus‐host disease in the skin following allogeneic stem‐cell transplantation. Blood 2007: 110: 3827–32. [DOI] [PubMed] [Google Scholar]
- 41. Abraham SM, Clark AR. Dual‐specificity phosphatase 1: a critical regulator of innate immune responses. Biochem Soc Trans 2006: 34 (Pt 6): 1018–23. [DOI] [PubMed] [Google Scholar]
- 42. Haasch D, Chen YW, Reilly RM et al. T cell activation induces a noncoding RNA transcript sensitive to inhibition by immunosuppressant drugs and encoded by the proto‐oncogene, BIC. Cell Immunol 2002: 217: 78–86. [DOI] [PubMed] [Google Scholar]
- 43. van den Berg A, Kroesen BJ, Kooistra K et al. High expression of B‐cell receptor inducible gene BIC in all subtypes of Hodgkin lymphoma. Genes Chromosomes Cancer 2003: 37: 20–8. [DOI] [PubMed] [Google Scholar]
- 44. Rodriguez A, Vigorito E, Clare S et al. Requirement of bic/microRNA‐155 for normal immune function. Science 2007: 316: 608–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thai TH, Calado DP, Casola S et al. Regulation of the germinal center response by microRNA‐155. Science 2007: 316: 604–8. [DOI] [PubMed] [Google Scholar]
- 46. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF‐kappaB‐dependent induction of microRNA miR‐146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A 2006: 103: 12481–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ranganathan P, Heaphy CE, Costinean S et al. Regulation of acute graft‐versus‐host disease by microRNA‐155. Blood 2012: 119: 4786–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xie LN, Zhou F, Liu XM et al. Serum microRNA155 is increased in patients with acute graft‐versus‐host disease. Clin Transplant 2014: 28: 314–23. [DOI] [PubMed] [Google Scholar]
- 49. Yamamoto M, Ota A, Hori T et al. Early expression of plasma CCL8 closely correlates with survival rate of acute graft‐vs.‐host disease in mice. Exp Hematol 2011: 39: 1101–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Heatmap of regulated genes in mismatched and matched mixed lymphocyte culture (MLC).
Figure S2. Directional response of IDO, CCL8, CXCL9 and CXCL10 in mixed lymphocyte culture (MLC).
Figure S3. Interferon‐gamma (IFNγ) induction of IDO and MIR155HG.
Table S1. Human leucocyte antigen (HLA) types of graft‐vs‐host disease (GVHD) study.
Table S2. Human leucocyte antigen (HLA) types of directional mixed lymphocyte culture (MLC).
Table S3. Upregulated genes in matched vs mismatched mixed lymphocyte culture (MLC).
Table S4. Downregulated genes in matched vs mismatched mixed lymphocyte culture (MLC).
Table S5. Association of IDO1, CXCL9, CXCL10, CLL8 and miR‐155 with graft‐vs‐host disease (GVHD).