

Abstract
The recent finding that the human version of a neurodevelopmental enhancer of the Wnt receptor Frizzled 8 (FZD8) gene alters neural progenitor cell cycle timing and brain size is a step forward to understanding human brain evolution. The human brain is distinctive in terms of its cognitive abilities as well as its susceptibility to neurological disease. Identifying which of the millions of genomic changes that occurred during human evolution led to these and other uniquely human traits is extremely challenging. Recent studies have demonstrated that many of the fastest evolving regions of the human genome function as gene regulatory enhancers during embryonic development and that the human‐specific mutations in them might alter expression patterns. However, elucidating molecular and cellular effects of sequence or expression pattern changes is a major obstacle to discovering the genetic bases of the evolution of our species. There is much work to do before human‐specific genetic and genomic changes are linked to complex human traits.
Also watch the Video Abstract.
Keywords: development, evolution, gene regulation, neuroscience
Abbreviation
- HAR
human accelerated region
Introduction
As the human and chimpanzee lineages split, both species have acquired many distinct behaviors, morphological characteristics, and molecular phenotypes 1, 2. Some of the most salient human‐specific traits reside in our brain or involve our unique cognitive abilities 3, 4, 5. Although numerous differences between humans and other primates have been described (2; http://carta.anthropogeny.org/content/about-moca), identifying the DNA alterations responsible for these evolutionary changes is a much more difficult task. This is not because we lack candidates: many human‐specific DNA sequences have been identified. These discoveries began prior to genome sequencing. The first differences between the human and chimp genomes were discovered using chromatin‐stained banding techniques that allowed identification of the fusion of two ancestral ape chromosomes to form human chromosome 2, human‐specific constitutive heterochromatin C bands on chromosomes 1, 9, 16 and Y, and human‐specific pericentric inversions on chromosomes 1 and 18 6. It is now known that many of these structural variants have altered gene expression or downstream phenotypes in humans. For example, the pericentric inversion of chromosome 1 contains copy number increases of developmental genes, and it has been associated with human developmental and neurogenetic diseases 7, 8, 9, 10, 11. After the sequencing of the human genome 12, 13 and many other mammalian genomes, particularly the chimpanzee and the macaque 14, 15, we now have several detailed genome‐wide catalogs of human‐specific genome changes that include chromosome segmental duplications resulting in the appearance of new human genes, differences in splicing, genes that underwent positive selection in humans, and evolutionarily conserved non‐coding sequences with many human‐specific mutations (reviewed in 7, 16, 17, 18). The challenge we now face is how to link specific genetic differences to uniquely human traits.
A number of factors make this a difficult task. First, we have millions of genetic candidates to sort through, and many of these are likely to be dead ends. The neutral theory of molecular evolution, coupled with redundancy in biological networks, suggests that many human‐specific DNA changes had little effect on our biology. On the other hand, most uniquely human traits are complex, and there is no doubt that they are encoded by a combination of mutations in different genomic loci. To make matters worse, hypotheses about causal relationships between human genotypes and phenotypes are difficult to test given the obvious limitations on experimentation and genetic manipulation in humans and non‐human primates. This challenge is partially addressed by engineering human or primate DNA into model organisms, such as mice or fish, and comparing molecular and organismal phenotypes. But the interpretation of human genetic changes in model organisms is challenging, and many hurdles remain 19. Finally, we still have much to learn about the development of all organs, and in particular the complicated architecture of the brain, including how form leads to function. Without this knowledge, it is extremely difficult to predict which mutations have altered our biology during human evolution.
In this context, we carefully analyze a recently published study by Boyd and co‐workers 20, which sheds light on the consequences of human‐specific mutations in a non‐coding region located upstream of the Wnt receptor Frizzled 8 (FZD8). The authors characterize the region as a transcriptional enhancer of FZD8 and then attempt to link its accelerated evolution in humans to changes in neurodevelopment, including spatial and temporal differences in neuronal gene expression leading to a faster cell cycle of neural progenitor cells. The significance of these discoveries is evaluated along side other recent work aiming to functionally characterize uniquely human DNA sequences and elucidate their contributions to the evolution of human traits.
The human brain: What is different about it?
The human brain has all the basic characteristics of a typical mammalian brain such as the six‐layered cortex or neocortex. In addition, our brain also has the typical features of a primate brain: an unusually large neocortex, a big visual cortex and a lateral prefrontal cortex 21. Despite these overall similarities, the human brain has several features that make it unique. The anatomy and development of the human brain diverged in several key ways from those of other primates during evolution. Humans have the largest number of neurons of any primate: approximately 86 billion 22 compared with an estimated 28 and 33 billion neurons in chimpanzee and gorilla brains, respectively 23. On the other hand, our brain is not the largest on earth, being outranked by brains of cetaceans and elephants 24. However, although the elephant brain has about 251 billion neurons, only 5.6 billion (2.2%) are cortical, the majority being concentrated in the cerebellum (97%; 25). In contrast, 20.9% of all neurons in the human brain are cortical, which is more than 10% greater than the cortical proportion in any other mammal 26. Thus, the human cortex is proportionally the largest (84% of the entire brain mass), and it contains the most neurons (85 billion) of any mammal 23, 25, 26, although it is debated whether our neocortex is particularly unique 27, 28.
Beyond numbers, the human brain appears also to be unique in its organization. Non‐invasive brain imaging techniques, such as diffusion‐tensor imaging, made it possible to study long‐range interactions of the cortex and revealed differences in cortical connections in human brains compared with those of chimpanzees and macaques 29. In addition, post‐mortem studies showed that the human brain is also unique in terms of cellular and histological organization of the cortex 30, 31, 32.
To understand the evolution of our species' higher order cognitive abilities, including abstract thinking, long term planning, and an extraordinary ability to produce and elaborate a complex language, we must answer two challenging questions. The first is how to link human cognition to number of neurons, brain size, a highly developed cortex, and particular neuroanatomical differences. The neurobiological bases of our linguistic capacity, for example, are not completely understood, because the main areas controlling language in the brain are also present in chimpanzees 33, 34. The second challenge is to connect DNA changes to uniquely human neurobiology. Although some advances have been made towards understanding the genetics underlying human cognitive traits, such as our spoken language 17, 18, very little is known about the anatomical and molecular mechanisms through which these genetic differences are expressed in the human brain. This is the question addressed by Boyd and colleagues.
An accelerated non‐coding sequence may have altered brain development in human evolution
Boyd et al. designed a study to identify genome sequences involved in the evolution of the unique features of the human cortex. Specifically, they focused on uniquely human gene regulatory enhancers active during neurodevelopment. The authors took advantage of previously identified catalogs of nearly 2,700 human accelerated regions (HARs), which are non‐coding sequences that changed significantly in the human lineage after having been highly conserved across mammalian evolution 35, 36, 37, 38, 39. Conserved non‐coding sequences have been hypothesized to contain much of the regulatory machinery that controls the time, place and mode of expression of genes 40, 41, 42, 43, and human‐specific mutations may alter this function. To provide further evidence of regulatory function and to focus their study on the developing brain, Boyd and co‐workers crossed the list of HARs with publicly available datasets of genome regions displaying epigenetic signatures of enhancer activity in various neurodevelopmental cell types (Supplemental Table S1 in 20). Specifically, the authors used ChIP‐seq data measuring genome sequences bound by (i) the co‐activator p300 (a component of enhancer‐associated protein assemblies) in mouse forebrain tissue at embryonic day 11.5 (E11.5) 44, (ii) the key neurogenesis transcription factors Pax6 and Sox2 in mouse embryonic cortex tissue at E12.5 45 and neural stem cells 46, and (iii) histones with modifications indicative of active enhancers such as H3K4me1 or H3K27ac in neural progenitor cells 47, 48. This analysis allowed them to identify 106 non‐overlapping HARs containing transcriptional enhancer epigenetics marks. From these putative neural enhancers they selected six (HARE1‐6) that were located near genes known or predicted to be involved in corticogenesis, and they performed enhancer assays in transgenic mice. Although three of the selected HAREs displayed enhancer activity in the developing cortex of transgenic mice, the authors selected HARE5, also known as Accelerated Non‐Coding element 516 (ANC516) 35, for further studies because of its highly consistent enhancer activity and location nearby the developmental gene FZD8.
To link the evolution of HARE5 to human neocortical development, Boyd et al. first generated transgenic mice carrying a reporter gene under the control of the human HARE5 enhancer (Hs‐HAR5::LacZ). These mice expressed B‐galactosidase in the lateral forebrain, dorso‐lateral midbrain, spinal cord, and retina (Fig. 1). On the other hand, transgenic mouse embryos carrying the orthologous chimpanzee HARE5 sequence (Pt‐HARE5::lacZ) also displayed B‐gal activity in the same tissues. But at E10.0 and E10.5, Pt‐HARE5 drove weaker and more limited activity in the developing cortex compared with the human version (Fig. 1). There are other genes in the HARE5 region (Fig. 1) that could be the target of this enhancer action: the ankyrin repeat domain 30A (ANKRD30A) (around 1 Mb away), a gene expressed in breast and testis; the gap junction protein, delta 4 (GJD4; located at 350 kb), and CYCLIN Y (CCNY; located at around 400 kb). Then, in order to show which gene is the target of the enhancer activity of HARE5 the authors performed chromosome conformation capture (3‐C) to test for physical association between mouse HARE5 and the core Fzd8 promoter in E12.5 mouse neocortices. They found that mouse HARE5 interacts with the Fzd8 promoter in the neocortex, but it does not interact with this promoter in liver of the same stage embryos, showing that HARE5 is a Fzd8 regulatory region in the developing cortex.
Figure 1.
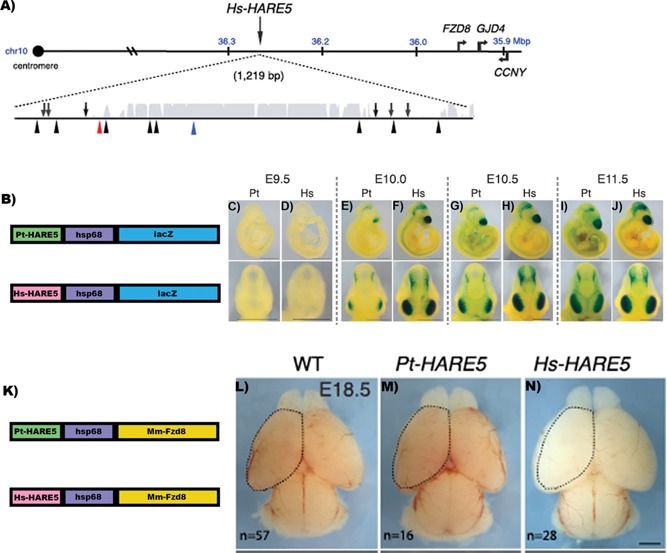
HARE5 gained expression compared with chimpanzee ortholog, driving increases in cortical size in overexpression assays. A: Schematic of Hs‐HARE5 locus on human chromosome 10 (hg19) showing its location relative to genes nearby including FZD8. The 1,219‐bp‐long HARE5 genomic locus with enhancer activity includes the original 619 bp human‐accelerated sequence and flanking sequences. Represented below is a conservation track for the HARE5 locus, showing a region of high‐sequence conservation (gray). Also shown are lineage‐specific mutations for chimpanzee (six; arrows, above line) and human (ten; arrowheads, below line), including one Denisovan (red) and one known human polymorphism (blue). B: Schematic of Pt‐HARE5::lacZ and Hs‐HARE5::lacZ constructs. Representative images of mouse embryos (stable transgenic lines) showing activity (blue LacZ staining) of Pt‐HARE5::LacZ (C, E, G, and I) and Hs‐HARE5::LacZ (D, F, H, and J). K: Schematic of Pt‐HARE5::Fzd8 and Hs‐HARE5::Fzd8 constructs. L to N: Whole‐mount E18.5 mouse brains from the indicated genotypes (n, number of brains examined) showing differences in cortical size. A dotted line was drawn on the WT cortex in (L) to indicate dorsal cortical area and was then superimposed on transgenic cortices in (M) and (N). Modified with permission from 19.
In addition, the authors generated transgenic mice carrying the human or chimp HARE5 sequences driving the expression of the mouse Fzd8 coding sequence. Then they compared several parameters of cortical development, including cortical size, cell cycle state and duration in neural progenitors at E12.5. The results revealed that overexpression of Fzd8 driven by Hs‐HARE5 produced faster progenitor cell cycle in the developing brain and increased neocortical size compared with mice where Fzd8 is controlled by chimpanzee HARE5 or wild‐type mice 20.
Although the findings by Boyd and co‐workers are very suggestive, the further demonstration of the role of HARE5 in human evolution will require additional studies. For example, it would be necessary to use knock‐in strategies, rather than overexpression, to better demonstrate changes in phenotypes. The location of HARE5 near the centromere makes this very challenging. From the data included in the paper, we do not know whether the same number of copies of the Hs‐HARE5‐Fzd8 and Pt‐HARE5‐Fzd8 transgenes were inserted in the genome of compared transgenic mice and if this could influence the differences in phenotype observed. In addition, we lack information about the locus where the transgenes where inserted, and this is another factor to take into account when analyzing transgenic mouse studies. In that sense, locus‐directed insertions of transgenes could produce cleaner results. Another issue is that HARE5 contains 10 human‐specific changes as well as 6 chimpanzee‐specific changes. To determine how the changes in each lineage affected the function of this sequence, it would be ideal to perform functional studies with computationally reconstructed ancestral sequences or sequences from other primates and mammals to definitively show that the human mutations, and not the chimpanzee mutations, altered HARE5 function. Then, it would be helpful to compare brain architecture, as well as counts and ratios of different embryonic cell types, in the context of human versus ancestral HARE5. Another issue is that because of the lack of information about the expression of FZD8 in human and chimpanzee brains, we do not know if this gene is in fact differentially expressed in these two species. Finally, it will be important to generate adult mice carrying human HARE5 and measure brain size, neuronal cell counts, and cognitive and behavioral traits.
Why focus on gene regulatory regions?
Before the genomic era that made it possible to identify conserved non‐coding sequences, most studies searching for the genetic bases of the evolution of human traits focused on protein‐coding regions. These projects identified several brain genes that underwent positive selection in the human genome (for review see 1, 8). In addition, other studies identified new human genes that appeared after the divergence between the human and chimpanzee lineages and linked these genes to the evolution of particular aspects of the human brain 7, 49, 50. So, one might wonder why Boyd and others have looked beyond protein‐coding genes to understand human evolution. First, there are more than twice as many DNA bases in conserved non‐coding regions compared with protein‐coding regions, making them a larger target for evolutionary innovation. Moreover, the high similarity of human and chimpanzee proteins, which has been known for many years 51, 52 and was confirmed with the sequencing of the chimpanzee genome 14, makes it very unlikely that coding changes are the primary source of phenotype differences between the two species. Supporting the potential for the relatively small number of non‐coding mutations in HARs to play a major role in human evolution, studies in other species found that lineage‐specific evolution of regulatory sequences can drive phenotypic changes (reviewed in 53, 54). These studies highlight the importance of studying non‐coding regulatory sequences to unravel the evolution of humans. Boyd and co‐workers pursued this hypothesis by leveraging HAR sequences as raw material upon which to first design studies to test if HARs function as neurodevelopmental enhancers and then attempt to find the possible impact of human mutations on this function.
Many human accelerated non‐coding sequences function as enhancers
Boyd and co‐workers were not the first group to test HARs for gene regulatory function in the developing embryo (Table 1). The first functional study involved HAR1 38, which encodes a long noncoding RNA that is co‐expressed with reelin in Cajal–Retzius neurons in the developing human neocortex from 7 to 19 gestational weeks, a crucial period for cortical neuron specification and migration. Despite extensive sequence changes, the expression pattern of HAR1 in the developing cortex has been highly conserved since the divergence of hominoids and Old World monkeys 38, and no clear phenotypic consequences of HAR1 mutations have been identified to date. The first HAR to be tested as a developmental enhancer was HAR2/HACNS1 55. Using a transgenic mouse enhancer assay, the authors found that the human HAR2/HACNS1 sequence drove strong and reproducible expression in the developing limb bud, pharyngeal arches, ear, and eye at E11.5. In contrast, the chimpanzee and macaque ortholog failed to drive reproducible reporter gene expression in the distal limb bud, while driving expression to the base of the limb 55. Although the authors suggested that this change was produced by adaptive evolution, the pattern of substitutions is more consistent with biased gene conversion, and it has been suggested that the functional change in expression pattern may have been driven by destruction of a repressor binding site 56.
Table 1.
HARs tested in functional assays that showed differences with the chimpanzee ortholog
| HARs | Genome location (hg19) | Closest gene | Experimental approach | Biological function | Domain of expression | Reported functional difference | Postulated phenotypic association | References | |
|---|---|---|---|---|---|---|---|---|---|
| HAR1 | chr20:61,733,447‐61,733,630 | HAR1F, HAR1R | qPCR; in situ hybridization | RNA gene | Cajal‐Retzius neurons | RNA secondary structure | Brain development | 37 | |
| HACNS1/HAR2 | chr2:236773664‐236774209 | AGAP1 (CENTG2), GBX2 | Transgenic mice | Enhancer | Limb, branchial archs | Gain of function | Limb development | 54 | |
| 2xHAR.142 | chr14:34048651‐34048815 | NPAS3 | Transgenic mice | Enhancer | Forebrain, midbrain, spinal cord | Gain of function | Brain development | 57 | |
| HAR202 | chr14:34045570‐34045772 | NPAS3 | Transgenic zebrafish | Enhancer | Central nervous system | Loss of function | Brain development | 56 | |
| ANC516 | chr10:36238421‐36239039 | FZD8 | Transgenic mice | Enhancer | Forebrain, midbrain, spinal cord | Gain of function | Brain development | 19 | |
| 2xHAR.238 | chr2:121832784‐121833140 | GLI2, TFCP2L1 | Transgenic mice | Enhancer | Forebrain, midbrain, hindbrain and spinal cord | Loss of function | Brain development | 58 | |
| 2xHAR.114 | chr20:30425200‐30425393 | MYLK2, FOXS1 | Transgenic mice | Enhancer | Limb and spinal cord | Loss of function | Limb development | 58 | |
| 2xHAR.164 | chr2:133346894‐133347089 | ANKRD30BL, GPR39, LYPD1, NCKAP5 | Transgenic mice | Enhancer | Forebrain, midbrain, hindbrain, spinal cord | Gain of function | Brain development | 58 | |
| 2xHAR.170 | chr5:153640309‐153640363 | GALNT10, SAP30L, HAND1 | Transgenic mice | Enhancer | Isthmus and spinal cord | Loss and gain of function | Brain development | 58 | |
| HAR25 | chr4:182271922‐182272882 | ODZ3 | Transgenic mice | Enhancer | Eye | Loss of function | Eye development | 58 | |
These first functional studies were followed by several investigations that examined bigger sets of predicted HAR enhancers. Analyzing genomic loci with the largest clusters of HARs, Kamm and co‐workers 57 found that the brain transcription factor NPAS3 is associated with 14 HARs, 11 of which act as transcriptional enhancers in a transgenic zebrafish reporter assay. They further showed that human HAR202 lost developmental enhancer capacity compared with the orthologous chimpanzee sequence 57 and human 2xHAR.142 gained developmental enhancer activity in the neocortex compared with the chimpanzee and mouse orthologs 58. Similarly, Capra and collaborators tested 29 HARs with epigenetic signatures of active developmental enhancers in transgenic mouse reporter assays. They found 24 HARs that act as developmental enhancers at E11.5 in specific embrionic tissues, and five of these show expression differences between the human and chimpanzee orthologous sequences 59. Thus, the human‐specific mutations in eight HARs had been shown to alter developmental enhancer function prior to the work of Boyd and colleagues. What was novel about their study was the discovery that mutations that occurred during human evolution can also change molecular and organismal phenotypes downstream of changes in enhancer activity.
Linking human‐specific genetic changes to unique cognitive traits is a long and twisted road
Comparative studies of human and chimpanzee (or ancestral) enhancers are an important step towards linking human genetic changes to unique anatomical and molecular features of our brains. But even if these connections can be ellucidated, the challenge of associating human‐specific brain features with cognative abilities and traits will remain. In the case of HARE5, one specific question is how to link the observed differences in faster progenitor cell cycle in the developing brain and increased neocortical size to cognitive abilities, behavior, or language. One approach to this problem is to look directly for differences in phenotypes in humanized mice in which specific genes or non‐coding sequences have been engineered to match the human, rather than mouse, genome.
An example of this approach is the study of FOXP2, a developmental gene that likely underwent positive selection in the human lineage 60. FOXP2 was dubbed a “language gene” after the discovery that a point mutation in the coding region led to aphasia in an English family 61, 62, 63, although this particular mutation was not one of the two carrying signatures of selection. To explore the functions of the positively selected amino acids, Enard and co‐workers replaced the endogenous mouse gene with a version carrying these two human‐specific changes 64. The resulting humanized mice where phenotipically characterized. Although they are overall healthy, they show some differences in qualitatively different ultrasonic vocalizations, decreased exploratory behavior and decreased dopamine concentrations in the brain, suggesting that the humanized Foxp2 allele affects basal ganglia 64. The major advance of this body of work is that it established a link between human‐specific mutations and several complex organismal phenotypes. What has not yet been done is to show the molecular pathways or developmental processes through which the genetic differences were expressed. This knowledge would advance our understanding of the mechanisms through which human evolution occurred. The study of Boyd and co‐workers is a first step toward filling in this black box between genotype and phenotype for a different human‐specific locus, though the authors have not gone as far as demonstrating effects on downstream phenotypes.
How to link genotype to phenotype?
To fully understand differences between humans and chimpanzees we need to understand how genetic differences impact on phenotype. The studies of Enard et al. and Boyd et al. represent major advances in our understanding of the possible phenotypic effects of human‐specific genetic changes in genes and regulatory regions on neurodevelopmental programs. But we still have a long way to go to fully characterize the functions of these human‐specific genome sequences and how that function was changed during human evolution. Studies designed to link specific genetic differences to uniquely human traits involve many steps. A possible path starts by mapping human‐specific genetic differences and then analyzing the evolutionary forces underlying their appearance. Then it is necessary to assess function (particularly with non‐coding regions) and study anatomical and cellular differences between animals or cells with human versus chimpanzee versions. Finally, the most challenging step is to link molecular differences to human phenotypes.
Our progress on each of these steps is limited in various ways. One major issue is our dependence on model organisms to assess human genotype‐phenotype connections. Working with mice to model human brain evolution allows us to better understand how a particular change in a protein or regulatory region can impact on some aspects of molecular and cellular mechanisms of development leading to morphological differences. Rodents are not as distantly related to primates as commonly believed, as we belong to sister phylogenetic orders that group together in the super‐order Euarchontaglires, which evolved around 85–90 million of years ago 65. Humans and mice share many anatomical similarities that together with other characteristics, such as genetic tools and relatively low cost and easy husbandry, have made mouse the top model organism to study human diseases 66, 67. However, studying human biology in rodents has important limitations. It is clear that mice have differences in their brain anatomy and function that make them an imperfect system to study primate neurobiology. Similarly, they are not ideal for assessing human‐specific morphologies and behaviors. In addition, it is difficult to assess the function of a human regulatory sequence in a mouse, because of the myriad differences between human and mouse in other parts of the genome, including changes in transcription factors that bind to the sequence or changes in other regulatory regions. Hence, the function of a human sequence in the mouse genome cannot truly recapitulate its function in the native context of the human genome.
While it is not easy to imagine a solution for genetic studies at the organismal level there are some technological advances that are advancing this field. Induced pluripotent stem cells (iPSCs) 68 and techniques allowing their in vitro differentiation into various cell lines and tissues (reviewed in 69, 70) make it feasible to do genetics and study downstream cellular and molecular phenotypes in human and chimpanzee cells 71, 72, 3D cultures, and organoids 73, 74, 75, 76, 77. These systems have great potential to allow researchers directly to compare neurodevelopment in the presence and absence of DNA changes that occurred during human evolution. Another challenge is the shear number of HARs and HAR mutations to test for phenotypes. The field has identified a handful enhancers that gained or lost function in humans compared with the chimpanzee sequence. But there are still more than 2,500 HARs that need to be functionally characterized if we want to assess the impact of accelerated evolution in non‐coding regions on the evolution of our species. Massively parallel reporter assays 78, 79, which can be performed in iPSC‐derived cell lines, are one promising high‐throughput approach to screen large numbers of candidate enhancers and enhancer mutations.
Conclusions and outlook
It is clear that no one technique or approach will shed light on the key genetic changes necessary to build a human. We hypothesize that conclusions about the particular developmental pathways and programs that were modified during the evolution of Homo sapiens will only be discovered through the use of a combination of approaches, including transgenic and mutant model organisms, comparative studies on iPSC‐derived cells and organoids from humans and non‐human primates, and human population genetics studies. Mechanistically to understand the evolution of humans will have a tremendous impact on philosophical, religious, and cultural matters. It will also have a fundamental affect on biomedical science, because understanding the molecular and cellular pathways that make us different will provide an unprecedented possibility to also explore whether these modified pathways are responsible for human susceptibility to particular diseases.
References
- 1. Sikela JM. 2006. The jewels of our genome: the search for the genomic changes underlying the evolutionarily unique capacities of the human brain. PLoS Genet 2: e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Varki A, Altheide TK. 2005. Comparing the human and chimpanzee genomes: searching for needles in a haystack. Genome Res 15: 1746–58. [DOI] [PubMed] [Google Scholar]
- 3. Carroll SB. 2003. Genetics and the making of Homo sapiens . Nature 422: 849–57. [DOI] [PubMed] [Google Scholar]
- 4. Herrmann E, Call J, Hernandez‐Lloreda MV, Hare B, et al. 2007. Humans have evolved specialized skills of social cognition: the cultural intelligence hypothesis. Science 317: 1360–6. [DOI] [PubMed] [Google Scholar]
- 5. Leigh SR. 2004. Brain growth, life history, and cognition in primate and human evolution. Am J Primatol 62: 139–64. [DOI] [PubMed] [Google Scholar]
- 6. Yunis JJ, Prakash O. 1982. The origin of man: a chromosomal pictorial legacy. Science 215: 1525–30. [DOI] [PubMed] [Google Scholar]
- 7. Fortna A, Kim Y, MacLaren E, Marshall K, et al. 2004. Lineage‐specific gene duplication and loss in human and great ape evolution. PLoS Biol 2: E207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. O'Bleness M, Searles VB, Varki A, Gagneux P, et al. 2012. Evolution of genetic and genomic features unique to the human lineage. Nat Rev Genet 13: 853–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Davis JM, Searles VB, Anderson N, Keeney J, et al. 2014. DUF1220 dosage is linearly associated with increasing severity of the three primary symptoms of autism. PLoS Genet 10: e1004241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Charrier C, Joshi K, Coutinho‐Budd J, Kim JE, et al. 2012. Inhibition of SRGAP2 function by its human‐specific paralogs induces neoteny during spine maturation. Cell 149: 923–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dennis MY, Nuttle X, Sudmant PH, Antonacci F, et al. 2012. Evolution of human‐specific neural SRGAP2 genes by incomplete segmental duplication. Cell 149: 912–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Consortium HGS. 2004. Finishing the euchromatic sequence of the human genome. Nature 431: 931–45. [DOI] [PubMed] [Google Scholar]
- 13. Lander ES, Linton LM, Birren B, Nusbaum C, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- 14. Consortium CSaA. 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437: 69–87. [DOI] [PubMed] [Google Scholar]
- 15. Gibbs RA, Rogers J, Katze MG, Bumgarner R, et al. 2007. Evolutionary and biomedical insights from the rhesus macaque genome. Science 316: 222–34. [DOI] [PubMed] [Google Scholar]
- 16. Hubisz MJ, Pollard KS. 2014. Exploring the genesis and functions of human accelerated regions sheds light on their role in human evolution. Curr Opin Genet Dev 29: 15–21. [DOI] [PubMed] [Google Scholar]
- 17. Preuss TM. 2012. Human brain evolution: from gene discovery to phenotype discovery. Proc Natl Acad Sci USA 109: 10709–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vallender EJ, Mekel‐Bobrov N, Lahn BT. 2008. Genetic basis of human brain evolution. Trends Neurosci 31: 637–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rossant J. 2015. Mouse and human blastocyst‐derived stem cells: vive les differences. Development 142: 9–12. [DOI] [PubMed] [Google Scholar]
- 20. Boyd JL, Skove SL, Rouanet JP, Pilaz LJ, et al. 2015. Human‐chimpanzee differences in a FZD8 enhancer alter cell‐cycle dynamics in the developing neocortex. Curr Biol 25: 772–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Preuss TM. 2007. Primate Brain Evolution in Phylogenetic Context. Evolution of Nervous Systems. Oxford, UK: Elsevier. [Google Scholar]
- 22. Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, et al. 2009. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled‐up primate brain. J Comp Neurol 513: 532–41. [DOI] [PubMed] [Google Scholar]
- 23. Herculano‐Houzel S, Kaas JH. 2011. Gorilla and orangutan brains conform to the primate cellular scaling rules: implications for human evolution. Brain Behav Evol 77: 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roth G, Dicke U. 2005. Evolution of the brain and intelligence. Trends Cogn Sci 9: 250–7. [DOI] [PubMed] [Google Scholar]
- 25. Herculano‐Houzel S, Avelino‐de‐Souza K, Neves K, Porfirio J, et al. 2014. The elephant brain in numbers. Front Neuroanat 8: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Herculano‐Houzel S. 2012. The remarkable, yet not extraordinary, human brain as a scaled‐up primate brain and its associated cost. Proc Natl Acad Sci USA 109: 10661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barton RA, Venditti C. 2013. Reply to Smaers: getting human frontal lobes in proportion. Proc Natl Acad Sci USA 110: E3683–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Barton RA, Venditti C. 2013. Human frontal lobes are not relatively large. Proc Natl Acad Sci USA 110: 9001–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rilling JK, Glasser MF, Preuss TM, Ma X, et al. 2008. The evolution of the arcuate fasciculus revealed with comparative DTI. Nat Neurosci 11: 426–8. [DOI] [PubMed] [Google Scholar]
- 30. Miller DJ, Duka T, Stimpson CD, Schapiro SJ, et al. 2012. Prolonged myelination in human neocortical evolution. Proc Natl Acad Sci USA 109: 16480–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sherwood CC, Subiaul F, Zawidzki TW. 2008. A natural history of the human mind: tracing evolutionary changes in brain and cognition. J Anat 212: 426–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Preuss TM. 2010. Reinventing Primate Neuroscience for the Twenty‐First Century. Oxford: Oxford University Press. [Google Scholar]
- 33. Cantalupo C, Hopkins WD. 2001. Asymmetric Broca's area in great apes. Nature 414: 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taglialatela JP, Russell JL, Schaeffer JA, Hopkins WD. 2008. Communicative signaling activates 'Broca's' homolog in chimpanzees. Curr Biol 18: 343–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bird CP, Stranger BE, Liu M, Thomas DJ, et al. 2007. Fast‐evolving noncoding sequences in the human genome. Genome Biol 8: R118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bush EC, Lahn BT. 2008. A genome‐wide screen for noncoding elements important in primate evolution. BMC Evol Biol 8: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lindblad‐Toh K, Garber M, Zuk O, Lin MF, et al. 2011. A high‐resolution map of human evolutionary constraint using 29 mammals. Nature 478: 476–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pollard KS, Salama SR, Lambert N, Lambot MA, et al. 2006. An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443: 167–72. [DOI] [PubMed] [Google Scholar]
- 39. Prabhakar S, Noonan JP, Paabo S, Rubin EM. 2006. Accelerated evolution of conserved noncoding sequences in humans. Science 314: 786. [DOI] [PubMed] [Google Scholar]
- 40. Marshall H, Studer M, Popperl H, Aparicio S, et al. 1994. A conserved retinoic acid response element required for early expression of the homeobox gene Hoxb‐1. Nature 370: 567–71. [DOI] [PubMed] [Google Scholar]
- 41. Pennacchio LA, Ahituv N, Moses AM, Prabhakar S, et al. 2006. In vivo enhancer analysis of human conserved non‐coding sequences. Nature 444: 499–502. [DOI] [PubMed] [Google Scholar]
- 42. Prabhakar S, Poulin F, Shoukry M, Afzal V, et al. 2006. Close sequence comparisons are sufficient to identify human cis‐regulatory elements. Genome Res 16: 855–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Visel A, Prabhakar S, Akiyama JA, Shoukry M, et al. 2008. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat Genet 40: 158–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Visel A, Blow MJ, Li Z, Zhang T, et al. 2009. ChIP‐seq accurately predicts tissue‐specific activity of enhancers. Nature 457: 854–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sansom SN, Griffiths DS, Faedo A, Kleinjan DJ, et al. 2009. The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self‐renewal and neurogenesis. PLoS Genet 5: e1000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Engelen E, Akinci U, Bryne JC, Hou J, et al. 2011. Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat Genet 43: 607–11. [DOI] [PubMed] [Google Scholar]
- 47. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA 107: 21931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Meissner A, Mikkelsen TS, Gu H, Wernig M, et al. 2008. Genome‐scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Florio M, Albert M, Taverna E, Namba T, et al. 2015. Human‐specific gene ARHGAP 11B promotes basal progenitor amplification and neocortex expansion. Science 347: 1465–70. [DOI] [PubMed] [Google Scholar]
- 50. Keeney JG, Dumas L, Sikela JM. 2014. The case for DUF1220 domain dosage as a primary contributor to anthropoid brain expansion. Front Hum Neurosci 8: 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. King MC, Wilson AC. 1975. Evolution at two levels in humans and chimpanzees. Science 188: 107–16. [DOI] [PubMed] [Google Scholar]
- 52. Falkiner GH. 1904. Blood Immunity and Blood Relationships. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 53. Carroll SB. 2005. Evolution at two levels: on genes and form. PLoS Biol 3: e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carroll SB. 2008. Evo‐devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell 134: 25–36. [DOI] [PubMed] [Google Scholar]
- 55. Prabhakar S, Visel A, Akiyama JA, Shoukry M, et al. 2008. Human‐specific gain of function in a developmental enhancer. Science 321: 1346–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sumiyama K, Saitou N. 2011. Loss‐of‐function mutation in a repressor module of human‐specifically activated enhancer HAC NS1. Mol Biol Evol 28: 3005–7. [DOI] [PubMed] [Google Scholar]
- 57. Kamm GB, Pisciottano F, Kliger R, Franchini LF. 2013. The developmental brain gene NPAS3 contains the largest number of accelerated regulatory sequences in the human genome. Mol Biol Evol 30: 1088–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kamm GB, Lopez‐Leal R, Lorenzo JR, Franchini LF. 2013. A fast‐evolving human NPAS3 enhancer gained reporter expression in the developing forebrain of transgenic mice. Philos Trans R Soc Lond B Biol Sci 368: 20130019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Capra JA, Erwin GD, McKinsey G, Rubenstein JL, et al. 2013. Many human accelerated regions are developmental enhancers. Philos Trans R Soc Lond B Biol Sci 368: 20130025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Enard W, Przeworski M, Fisher SE, Lai CS, et al. 2002. Molecular evolution of FOXP2, a gene involved in speech and language. Nature 418: 869–72. [DOI] [PubMed] [Google Scholar]
- 61. Fisher SE, Vargha‐Khadem F, Watkins KE, Monaco AP, et al. 1998. Localisation of a gene implicated in a severe speech and language disorder. Nat Genet 18: 168–70. [DOI] [PubMed] [Google Scholar]
- 62. Hurst JA, Baraitser M, Auger E, Graham F, et al. 1990. An extended family with a dominantly inherited speech disorder. Dev Med Child Neurol 32: 352–5. [DOI] [PubMed] [Google Scholar]
- 63. Lai CS, Fisher SE, Hurst JA, Vargha‐Khadem F, et al. 2001. A forkhead‐domain gene is mutated in a severe speech and language disorder. Nature 413: 519–23. [DOI] [PubMed] [Google Scholar]
- 64. Enard W, Gehre S, Hammerschmidt K, Holter SM, et al. 2009. A humanized version of Foxp2 affects cortico‐basal ganglia circuits in mice. Cell 137: 961–71. [DOI] [PubMed] [Google Scholar]
- 65. Bininda‐Emonds OR, Cardillo M, Jones KE, MacPhee RD, et al. 2007. The delayed rise of present‐day mammals. Nature 446: 507–12. [DOI] [PubMed] [Google Scholar]
- 66. Beckers J, Wurst W, de Angelis MH. 2009. Towards better mouse models: enhanced genotypes, systemic phenotyping and envirotype modelling. Nat Rev Genet 10: 371–80. [DOI] [PubMed] [Google Scholar]
- 67. Lieschke GJ, Currie PD. 2007. Animal models of human disease: zebrafish swim into view. Nat Rev Genet 8: 353–67. [DOI] [PubMed] [Google Scholar]
- 68. Yamanaka S. 2012. Induced pluripotent stem cells: past, present, and future. Cell Stem Cell 10: 678–84. [DOI] [PubMed] [Google Scholar]
- 69. Karagiannis P, Yamanaka S. 2014. The fate of cell reprogramming. Nat Methods 11: 1006–8. [DOI] [PubMed] [Google Scholar]
- 70. Yu C, Liu K, Tang S, Ding S. 2014. Chemical approaches to cell reprogramming. Curr Opin Genet Dev 28: 50–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Marchetto MC, Narvaiza I, Denli AM, Benner C, et al. 2013. Differential L1 regulation in pluripotent stem cells of humans and apes. Nature 503: 525–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wunderlich S, Kircher M, Vieth B, Haase A, et al. 2014. Primate iPS cells as tools for evolutionary analyses. Stem Cell Res 12: 622–9. [DOI] [PubMed] [Google Scholar]
- 73. Bae BI, Walsh CA. 2013. Neuroscience. What are mini‐brains? Science 342: 200–1. [DOI] [PubMed] [Google Scholar]
- 74. Bershteyn M, Kriegstein AR. 2013. Cerebral organoids in a dish: progress and prospects. Cell 155: 19–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lancaster MA, Knoblich JA. 2014. Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345: 1247125. [DOI] [PubMed] [Google Scholar]
- 76. Lancaster MA, Renner M, Martin CA, Wenzel D, et al. 2013. Cerebral organoids model human brain development and microcephaly. Nature 501: 373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sasai Y. 2013. Cytosystems dynamics in self‐organization of tissue architecture. Nature 493: 318–26. [DOI] [PubMed] [Google Scholar]
- 78. Melnikov A, Murugan A, Zhang X, Tesileanu T, et al. 2012. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol 30: 271–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, et al. 2012. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol 30: 265–70. [DOI] [PMC free article] [PubMed] [Google Scholar]