

Abstract
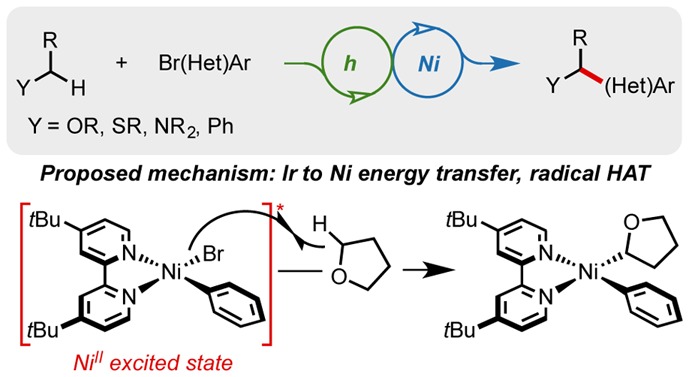
An iridium photocatalyst and visible light facilitate a room temperature, nickel-catalyzed coupling of (hetero)aryl bromides with activated α-heterosubstituted or benzylic C(sp3)–H bonds. Mechanistic investigations on this unprecedented transformation have uncovered the possibility of an unexpected mechanism hypothesized to involve a Ni–Br homolysis event from an excited-state nickel complex. The resultant bromine radical is thought to abstract weak C(sp3)–H bonds to generate reactive alkyl radicals that can be engaged in Ni-catalyzed arylation. Evidence suggests that the iridium photocatalyst facilitates nickel excitation and bromine radical generation via triplet–triplet energy transfer.
Recent efforts in photocatalysis have demonstrated the synthetic utility of employing visible light to facilitate organic reactions. Light-harvesting complexes have been used to activate organic molecules and to evolve challenging intermediates through either electron or energy transfer.1 Perhaps more importantly, photocatalysis has been creatively deployed in the development of organic reactions that operate through mechanistically novel pathways. These mechanistic changes may lower kinetic barriers to target products, providing milder conditions and more selective reactivity than could be achieved via thermal activation. The ability to harness visible light through photocatalysis is particularly attractive for the development of new modes of C–H functionalization, because traditional approaches to catalytic C–H functionalization often require precious metals in relatively high loading, directing groups, stoichiometric additives or oxidants, and/or high temperatures.2 Furthermore, C–H functionalizaton with inexpensive Ni catalysts currently require chelation control, extremely high temperatures (often greater than 140 °C), and/or peroxide reagents that can be hazardous for large scale applications.3
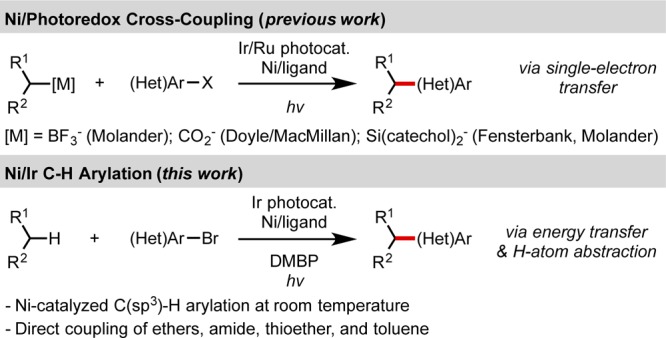
Photoredox dual catalysis has demonstrated the propensity of Ni catalytic systems to facilitate the coupling of alkyl radicals with aryl and alkenyl halides (Scheme 1). Although various alkyl radical precursors, including alkyltrifluoroborates,4 carboxylates,5 and silicates,6 have been developed, the mechanistic considerations are the same.7 Thus, the radical precursor is activated by single electron oxidation to afford an alkyl radical, and the nickel catalyst is subsequently reduced by the photocatalyst to turn over the cycle. Application of this single-electron transfer-based mechanism enables these cross-couplings to proceed under mild conditions with minimal side products compared to traditional cross-coupling reactions. However, two activated partners are required for effective reactivity. In an effort to develop more redox-, atom-, and step-economic processes, we sought to utilize C(sp3)–H bonds as more practical and efficient radical precursors, thereby eliminating the waste generated in the process of oxidative fragmentation and in the synthesis of the requisite redox active fragment.
Scheme 1. Visible Light-Mediated C(sp3)–C(sp2) Cross-Coupling with Ni and Ir/Ru.
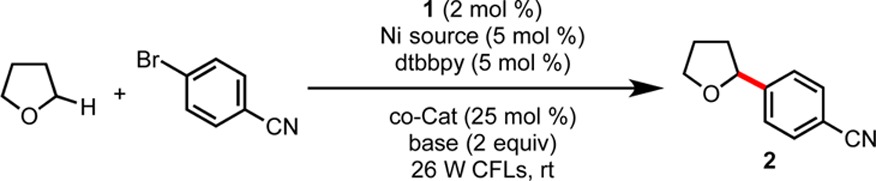
Initially, we hypothesized that incorporation of a diaryl ketone catalyst could generate an alkyl radical from an activated C–H bond through a well-precedented H-atom-transfer (HAT) process from the excited state of the diaryl ketone.8 We anticipated that this radical could be engaged in cross-coupling with an aryl bromide via nickel catalysis. Studies commenced with the reaction of bromobenzonitrile with THF as substrate and solvent (Table 1). Employing benzophenone (1 equiv) with standard Ir/Ni loadings, 2 was detected by GC-MS, although conversion was incomplete after 24 h. Addition of Brønsted bases in an effort to neutralize the HBr formally generated as a byproduct of the reaction improved conversion significantly. In the presence of base, catalytic loadings of benzophenone were tolerated. Further optimization identified 4,4′-dimethoxybenzophenone (DMBP, 4, 25 mol %) as being superior to benzophenone as a co-catalyst. Replacing air-sensitive Ni(COD)2 with bench stable Ni(NO3)2·6H2O provided a modest improvement in yield. Control experiments demonstrated that Ni, Ir, and light were necessary for C–H arylation. However, we unexpectedly observed product formation in the absence of 4, which we had assumed was responsible for the observed hydrogen-atom abstraction. Although this observation has important mechanistic implications (vide infra), the addition of 4 as a substoichiometric additive was nonetheless effective in improving reaction yields.
Table 1. Select Optimization Results and Control Studies.
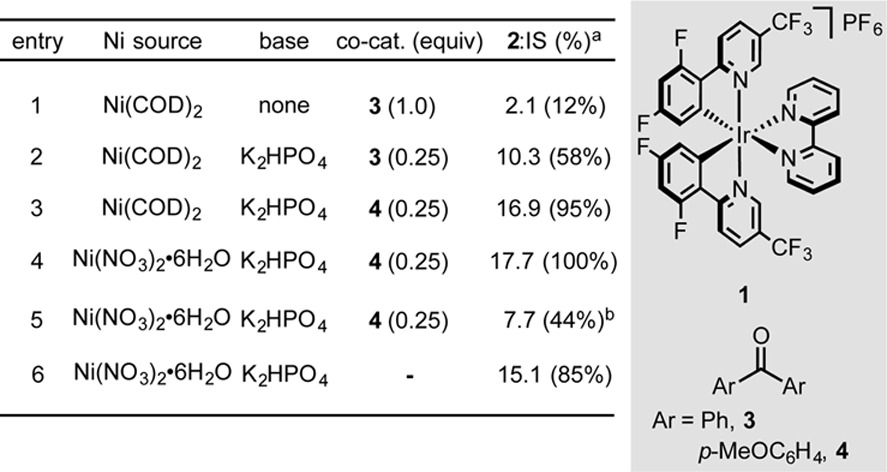
Reactions performed on 0.05 mmol scale. Determined by HPLC analysis relative to 4,4-di-tert-butylbiphenyl as an internal standard.
No ligand.
Next, we elected to explore the types of C(sp3)–H bonds capable of undergoing photochemical Ni/Ir dual catalyzed arylation (Table 2). Importantly, a variety of common solvents were found to participate in the reaction. Most effective were ethereal solvents, including THF (89%, 72 h), DME (6, 91%, 72 h), and Et2O (7, 56%, 72 h). Other ethers, such as 1,4-dioxane and MTBE, afforded cross-coupled products but required extended reaction times with low yields. We attribute this diminished reactivity to higher C–H bond dissociation energies (BDEs) in these substrates, owing to inductive effects in the case of dioxane9 and the reduced stability of the primary alkoxymethyl radical for MTBE. Surprisingly, no cross-coupled product was observed when the reaction was conducted in tetrahydropyran. Some non-ethereal substrates were also found to afford cross-coupled products.N-Methylpyrrolidinone underwent α-arylation with good yield and regioselectivity for the endocyclic secondary position. Tetrahydrothiophene provided an appreciable yield of the desired product 9. Furthermore, toluene effectively coupled at the benzylic position to afford the desired diarylmethane 11 (87%, 72 h).
Table 2. Scope of C(sp3)–H Partners in Ni/Ir Cross-Coupling.
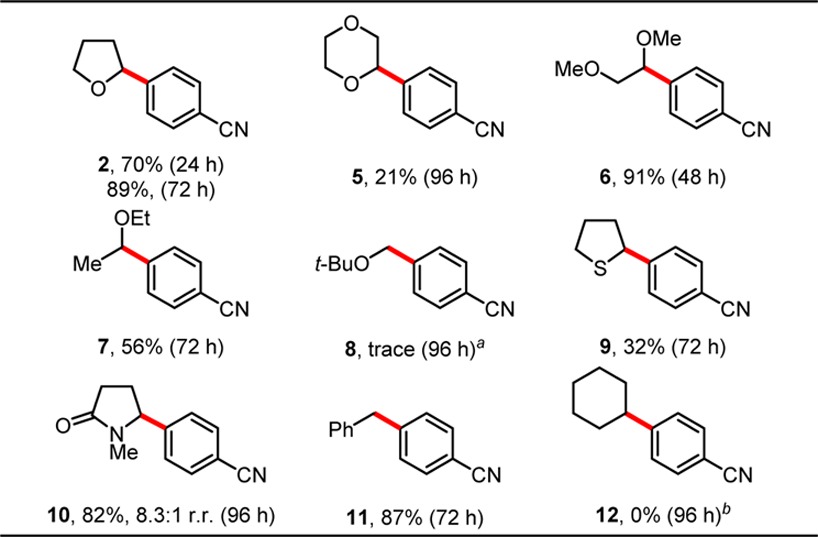
NMR yield relative to DMBP.
No product detected by GCMS analysis. All reactions were performed on 0.35 mmol scale, r.r. = ratio of regioisomers.
Notably, cyclohexane was found to be unreactive, and substrates bearing allylic C–H bonds, such as cyclohexene and 3,4-dihydro-2H-pyran, produced complex product mixtures. Despite the relatively limited scope, the unprecedented, direct coupling of saturated heterocycles with aryl halides provides a complementary approach to C(sp3)–C(sp2) products that are difficult to access through photoredox cross-coupling, because methods to synthesize the corresponding C(sp3)-organometallic partner are not well established.
Further interrogation of the reaction scope demonstrated that the conditions were tolerant of a variety of aryl- and heteroaryl bromides (Table 3). Electron-deficient bromides afforded products in moderate to good yield. Electron-rich bromides also coupled, providing 18 and 19. Furthermore, a series of pyridines (22, 23, and 24) coupled well. Notably, a number of bromides that contained homolytically weak C–H bonds (aldehydes 15 and 21 and ethers 18 and 19) were nonproblematic. Attempts to extend the reaction to alkenyl bromides or aryl iodides, chlorides, and triflates were unsuccessful, returning only unreacted starting material. The ability to couple 1-bromo-4-chlorobenzene selectively at the C–Br bond (17) provides opportunities for further functionalization of reaction products. Reactions were demonstrated to occur successfully in the presence and absence of DMBP at benchtop scale (0.35 mmol). Although the additive is sometimes difficult to separate from the products chromatographically, reactions were often faster, higher yielding, and more reproducible than corresponding reactions performed without the additive.
Table 3. Scope of Aryl and Heteroaryl Bromides in Ni/Ir Cross-Coupling with THF.
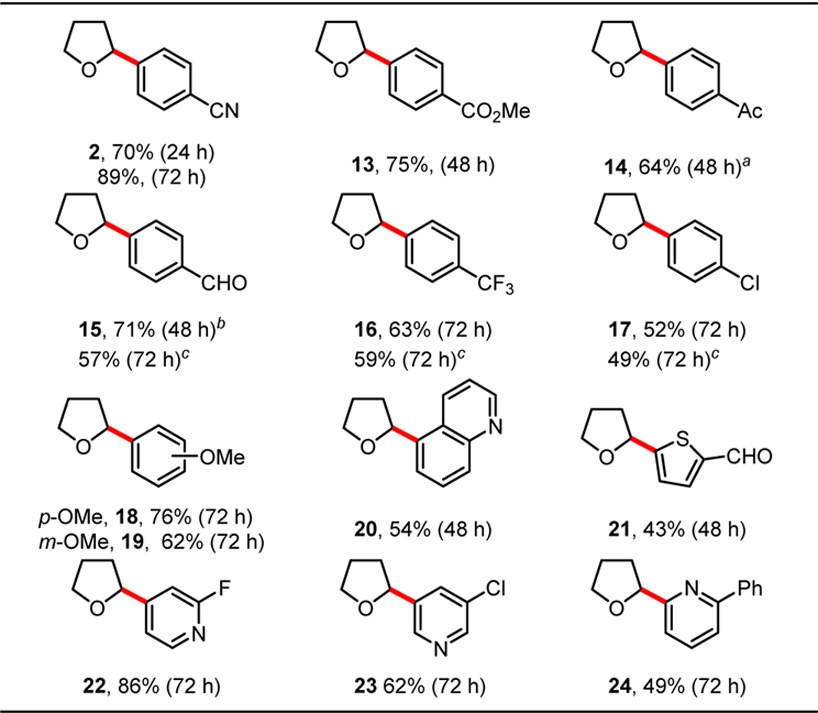
Product was isolated with trace DMBP (14:DMBP = 11.5:1).
Product was inseparable from DMBP.
Reaction was performed in the absence of DMBP. All reactions were conducted at 0.35 mmol scale.
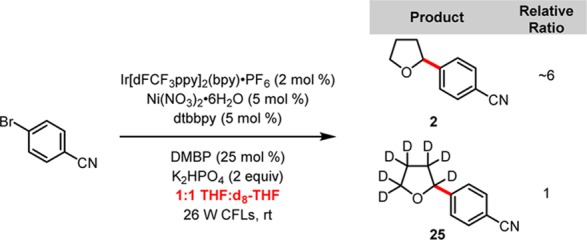
Based upon the unexpected results of our control experiments, we probed the reaction mechanism to achieve a better understanding of the nature of this C–H functionalization process. First, the optimized reaction was performed in a 1:1 mixture of THF and d8-THF (Scheme 2) to observe the effect of heavier hydrogen isotopes on the reaction rate. The ratio of 2 to 25 was 6:1 by 1H NMR spectroscopy. Kinetic isotope effects of this magnitude are typically consistent with a near-thermoneutral radical C–H abstraction (Scheme 2).10
Scheme 2. THF/d8-THF Kinetic Isotope Effect Experiment.
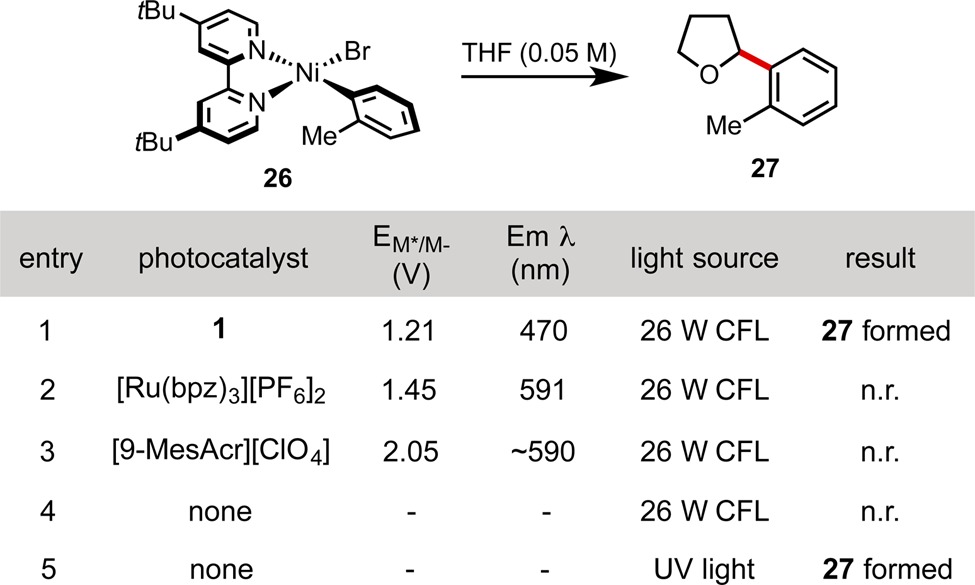
We next undertook a series of experiments aimed at studying the reactivity of stable, isolable Ni(II) oxidative addition complex 26. We first confirmed that treatment of one equivalent of this complex with 1 equiv of photocatalyst 1 in THF under visible light irradiation leads to formation of the C–H cross-coupling product (Table 4, entry 1). Importantly, exposure of the Ni complex to visible light in the absence of photocatalyst leads to no product formation (Table 4, entry 4). We first suspected that the photocatalyst engages the Ni(II) complex in single electron transfer (SET), generating a Ni(III) complex that could catalyze C–H functionalization by homolysis of the bromine–nickel bond to generate a bromine radical. Nocera has observed an analogous Ni–Cl homolysis from an excited-state Ni(III) complex.11 To probe this hypothesis, we treated 26 with more strongly oxidizing photocatalysts Ru(bpz)3·2PF6 (excited state ERed = +1.45 V vs SCE)1a and (9-MesAcr)ClO4 (excited state ERed = +2.06 V vs SCE).12 To our surprise, no C–H cross-coupled product was observed when these catalysts were employed with visible light irradiation. These experiments suggest that a mechanism involving oxidation of Ni(II) to Ni(III) may not be operative in these reactions.
Table 4. Reactions of Ni(II) Oxidative Addition Complexes with THF.
In light of these experiments, we began to suspect that a mechanism involving triplet–triplet energy transfer from the excited-state photocatalyst to the Ni(II) complex might be operative. Importantly, this mechanism would be consistent with our stoichiometric experiments, because, under this manifold, reactivity should be correlated with triplet state energy (i.e., emission wavelength) rather than with excited-state oxidation potential. Indeed, all of the photocatalysts tested exhibit significantly lower triplet-state energies than 1, as evidenced by their longer emission wavelengths. Although energy transfer has been employed in some visible light photocatalytic processes,1a,13 there is limited precedent of this mechanism operating cooperatively with transition metal catalysis.14
The absorption spectrum of 26 exhibits broad, low-intensity features between 400 and 600 nm, which we attribute to Ni d → d* transitions.15 The failure of visible light irradiation to promote the C–H cross-coupling reaction in the absence of photocatalyst suggests that the operative triplet state is not accessible from these excited states. A more intense band appears in the UV region at ∼300 nm. We suspected that irradiation of this absorption band might permit access to the triplet state from which C–H functionalization occurs. Supporting this hypothesis, UV-B irradiation (290–315 nm) of 26 resulted in observation of the arylated tetrahydrofuran product 27 (Table 4, entry 5). A unified mechanistic picture consistent with these data involves a catalytically active triplet excited state that is accessible by two pathways: (1) direct sensitization via triplet–triplet energy transfer in the presence of visible light and photocatalyst 1 (Scheme 3A) or (2) excitation into a higher energy singlet excited state via UV irradiation, which undergoes intersystem crossing and relaxes nonradiatively to the same active excited state. Importantly, in this circumstance, the operative Ni excited state would be inaccessible in the presence of photocatalysts with insufficiently energetic triplet states or by irradiation with insufficiently energetic wavelengths of light (i.e., visible light).
Scheme 3. (A) Plausible Catalytic Cycle, (B) Pathway A: Intermolecular C–H Abstraction, and (C) Pathway B: Concerted Ni–Br Homolysis/C–H Abstraction.
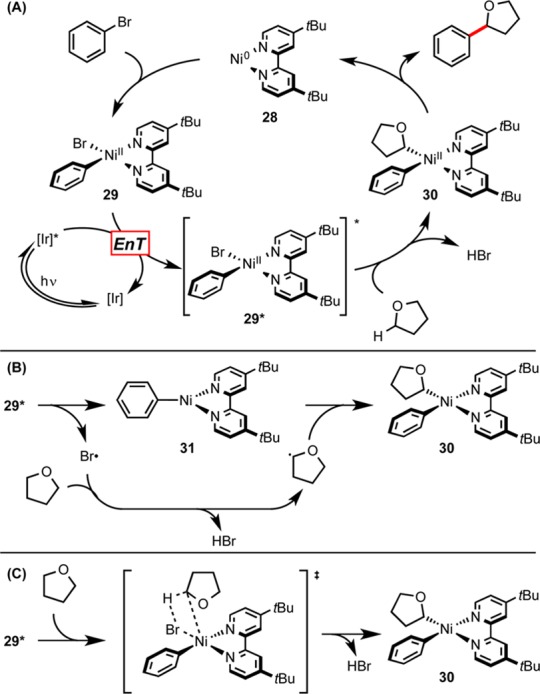
EnT = triplet–triplet energy transfer.
Although we are at this point unsure of the exact nature of the C–H functionalization event, the observed KIE suggests that this complex leads to the formation of a bromine radical through Ni–Br bond homolysis. This radical would then engage the substrate in HAT to form a stabilized carbon-centered radical that adds to the nickel catalyst. The product can be subsequently generated by reductive elimination. We suspect this may occur by one of two different pathways: the first involving the discrete formation of a Ni(I) complex and a bromine radical (Scheme 3B), and the other involving a concerted four-centered transition structure (Scheme 3C). This proposal is consistent with a variety of our experimental observations. Specifically, the weakness of the H–Br bond provides only a mild thermodynamic driving force for abstraction of an H atom from THF, consistent with the measured KIE and our limited ability to engage stronger C–H bonds in this reaction.
A mechanism proceeding through a Ni(III) intermediate would find more direct precedent in Nocera’s report, but the failure of strongly oxidizing photocatalysts to promote the desired reactivity and the ability of the chemistry to proceed in the absence of any oxidant under UV-irradiation lead us to disfavor the involvement of Ni(III) as the major reaction pathway. We are presently pursuing theoretical and spectroscopic support for the proposed pathway. In any event, it appears certain that a previously unknown or underutilized mechanism is at work in these transformations. We anticipate that this pathway could prove impactful for the development of mild, nondirected C–H functionalization reactions. Notably, previous photoredox/nickel dual catalysis C–H functionalization reactions have relied on proximity to a redox active heteroatom to induce activation of the C–H bond.5,16 Utilizing the protocol described, we are able to leverage a HAT manifold to activate C–H bonds that are unreactive in the SET regime.17 Further understanding of the operative mechanism should provide opportunities to extend the reaction to less reactive C–H bonds and/or new halide or pseudohalide partners.
In conclusion, mild conditions for an unprecedented, Ni-catalyzed C(sp3)–H arylation are achieved through the use of an Ir photocatalyst, substoichiometric benzophenone-derivative additive, and visible light. A variety of aryl- and heteroaryl halides are tolerated, including those containing weak C(sp3)–H bonds. Background reactivity in the absence of the presumed co-catalyst led to further mechanistic studies. These experiments suggest a mechanism involving an energy-transfer pathway in which an excited-state nickel complex initiates C–H functionalization through homolysis of a Ni–halide bond. This represents a new approach to C(sp3)–H functionalization by utilizing visible light to access a mechanistically distinct reaction pathway. Further studies focused on mechanistic elucidation are currently underway.
Acknowledgments
This research was generously supported by the NIGMS (R01-GM-113878) and the NSF (CHE-1362841). J.C.T. was supported by a Bristol-Myers Squibb Graduate Fellowship. We thank Professors Joseph Subotnik and Jessica Anna (University of Pennsylvania) for enlightening discussions. We thank Aldrich for iridium salts used in the preparation of the photoredox catalysts. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for collection of HRMS data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b04789.
Experimental details and data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Narayanam J. M. R.; Stephenson C. R. J. Chem. Soc. Rev. 2011, 40, 102. 10.1039/B913880N. [DOI] [PubMed] [Google Scholar]; b Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yoon T. P.; Ischay M. A.; Du J. Nat. Chem. 2010, 2, 527. 10.1038/nchem.687. [DOI] [PubMed] [Google Scholar]; d Reckenthaler M.; Griesbeck A. G. Adv. Synth. Catal. 2013, 355, 2727. 10.1002/adsc.201300751. [DOI] [Google Scholar]
- a Ritleng V.; Sirlin C.; Pfeffer M. Chem. Rev. 2002, 102, 1731. 10.1021/cr0104330. [DOI] [PubMed] [Google Scholar]; b Cho S. H.; Kim J. Y.; Kwak J.; Chang S. Chem. Soc. Rev. 2011, 40, 5068. 10.1039/c1cs15082k. [DOI] [PubMed] [Google Scholar]; c Chen X.; Engle K. E.; Wang D.–H.; Yu J.-Q. Angew. Chem., Int. Ed. 2009, 48, 5094. 10.1002/anie.200806273. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Daugulis O.; Do J.-Q.; Shabashov D. Acc. Chem. Res. 2009, 42, 1074. 10.1021/ar9000058. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Mkhalid I. A. I.; Barnard J. H.; Marder T. B.; Murphy J. M.; Hartwig J. F. Chem. Rev. 2010, 110, 890. 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- a Muto K.; Yamaguchi J.; Itami K. J. Am. Chem. Soc. 2012, 134, 169. 10.1021/ja210249h. [DOI] [PubMed] [Google Scholar]; b Amaike K.; Muto K.; Yamaguchi J.; Itami K. J. Am. Chem. Soc. 2012, 134, 13573. 10.1021/ja306062c. [DOI] [PubMed] [Google Scholar]; c Shiota H.; Ano Y.; Aihara Y.; Fukumoto Y.; Chatani N. J. Am. Chem. Soc. 2011, 133, 14952. 10.1021/ja206850s. [DOI] [PubMed] [Google Scholar]; d Aihara Y.; Chatani N. J. Am. Chem. Soc. 2013, 135, 5308. 10.1021/ja401344e. [DOI] [PubMed] [Google Scholar]; e Aihara Y.; Chatani N. J. Am. Chem. Soc. 2014, 136, 898. 10.1021/ja411715v. [DOI] [PubMed] [Google Scholar]; f Liu D.; Liu C.; Li H.; Lei A. Angew. Chem., Int. Ed. 2013, 52, 4453. 10.1002/anie.201300459. [DOI] [PubMed] [Google Scholar]; g Liu D.; Li Y.; Qi X.; Liu C.; Lan Y.; Lei A. Org. Lett. 2015, 17, 998. 10.1021/acs.orglett.5b00104. [DOI] [PubMed] [Google Scholar]
- a Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Primer D. N.; Karakaya I.; Tellis J. C.; Molander G. A. J. Am. Chem. Soc. 2015, 137, 2195. 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Karakaya I.; Primer D. N.; Molander G. A. Org. Lett. 2015, 17, 3294. 10.1021/acs.orglett.5b01463. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ryu D.; Primer D. N.; Tellis J. C.; Molander G. A. Chem. - Eur. J. 2016, 22, 120. 10.1002/chem.201504079. [DOI] [PubMed] [Google Scholar]; e Amani J.; Sodagar E.; Molander G. A. Org. Lett. 2016, 18, 732. 10.1021/acs.orglett.5b03705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Z.; Ahneman D. T.; Chu L.; Terret J. A.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345, 437. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Corcé V.; Chamoreau L.-M.; Derat E.; Goddard J.-P.; Ollivier C.; Fensterbank L. Angew. Chem., Int. Ed. 2015, 54, 11414. 10.1002/anie.201504963. [DOI] [PubMed] [Google Scholar]; b Jouffroy M.; Primer D. N.; Molander G. A. J. Am. Chem. Soc. 2016, 138, 475. 10.1021/jacs.5b10963. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Patel N. R.; Kelly C. B.; Jouffroy M. Org. Lett. 2016, 18, 764. 10.1021/acs.orglett.6b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez O.; Tellis J. C.; Primer D. N.; Molander G. A.; Kozlowski M. C. J. Am. Chem. Soc. 2015, 137, 4896. 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Walling C.; Gibian M. J. J. Am. Chem. Soc. 1965, 87, 3361. 10.1021/ja01093a014. [DOI] [Google Scholar]; b Leight W. J.; Lathioor E. C.; St. Pierre M. J. J. Am. Chem. Soc. 1996, 118, 12339. 10.1021/ja961973v. [DOI] [Google Scholar]; c Xia J.-B.; Zhu C.; Chen C. J. Am. Chem. Soc. 2013, 135, 17494. 10.1021/ja410815u. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Kamijo S.; Hoshikawa T.; Inoue M. Org. Lett. 2011, 13, 5928. 10.1021/ol202659e. [DOI] [PubMed] [Google Scholar]; e Zhang Y.; Teuscher K. B.; Ji H. Chem. Sci. 2016, 7, 2111. 10.1039/C5SC03640B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallen R. T.; Gleicher G. J.; Mahiou B.; Clapp G. E. J. Phys. Org. Chem. 1989, 2, 367. 10.1002/poc.610020502. [DOI] [Google Scholar]
- a Denés F.; Pichowicz M.; Povie G.; Renaud P. Chem. Rev. 2014, 114, 2587. 10.1021/cr400441m. [DOI] [PubMed] [Google Scholar]; b Simmons E. M.; Hartwig J. F. Angew. Chem., Int. Ed. 2012, 51, 3066. 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- Hwang S. J.; Anderson B. L.; Powers D. C.; Maher A. G.; Hadt R. G.; Nocera D. G. Organometallics 2015, 34, 4766. 10.1021/acs.organomet.5b00568. [DOI] [Google Scholar]
- Ohkubo K.; Mizushime K.; Iwata R.; Souma K.; Suzuki S.; Fukuzumi S. Chem. Commun. 2010, 46, 601. 10.1039/B920606J. [DOI] [PubMed] [Google Scholar]
- Lu Z.; Yoon T. P. Angew. Chem., Int. Ed. 2012, 51, 10329–10332. 10.1002/anie.201204835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo W.-J.; Tsukamoto T.; Kobayashi S. Org. Lett. 2015, 17, 3640. 10.1021/acs.orglett.5b01645. [DOI] [PubMed] [Google Scholar]
- Klein A.; Kaiser A.; Wielandt W.; Belaj F.; Wendel E.; Bertagnolli H.; Zális S. Inorg. Chem. 2008, 47, 11324. 10.1021/ic8007365. [DOI] [PubMed] [Google Scholar]
- Joe C. L.; Doyle A. G. Angew. Chem., Int. Ed. 2016, 55, 4040. 10.1002/anie.201511438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- During the preparation of this manuscript, MacMillan et al. reported a C–H functionalization method for the arylation of similar substrates:Shaw M. H.; Shurtledd V. W.; Terrett J. A.; Cuthbertson J. D.; MacMillan D. W. C. Science 2016, 352, 1304. 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.