

Abstract
The Hemostasis and Thrombosis Research Society (HTRS) Registry was used to monitor the postapproval use of recombinant factor VIIa. The objective of this manuscript is to provide key insights on the demographics of patients with acquired hemophilia in the HTRS Registry. Acquired hemophilia patient registration in HTRS captured age; sex; comorbidities and predisposing conditions; first bleeding location; laboratory parameters; exposure to blood products, factor, and bypassing agents; and initiation of immune suppression/tolerance therapy. Overall, 166 patients with acquired hemophilia were registered in HTRS (83 women, 73 men, median age 70 years); the majority were non-Hispanic whites (61.4%). The most common comorbidities were autoimmune disease (28.4%) and malignancy (14.5%). The most common first site of bleeding was subcutaneous (27.1%); this was more common in whites (29.1%) than blacks (12.5%) and in non-Hispanics (26.4%) than Hispanics (11.8%). Blood product exposure was reported for 33.1% of patients; the most commonly reported product was packed red blood cells (28%). Of the 57 patients with outcome data available for immune tolerance therapy, 26 patients (46%) reported successful treatment, 13 reported unsuccessful treatment (23%), and 18 (32%) were receiving active treatment at the time of registration. The HTRS Registry final analysis provides the only current comprehensive look at acquired hemophilia in the US population, including details on underlying autoimmune diseases and malignancies. Pertinent to recognition and diagnosis of the disease, subcutaneous bleeding as a presenting bleeding symptom was more common in white and non-Hispanic individuals.
Keywords: acquired hemophilia A, comorbidity, immune tolerance, recombinant factor VIIa, registries
Introduction
Acquired hemophilia is a rare disorder marked by the development of autoantibodies to factor VIII (FVIII). Patients present with bleeding and a prolonged activated partial thromboplastin time (aPTT) that does not correct with prolonged incubation mixing with normal plasma (2 h, 37°C).
The first insights into the demographics of acquired hemophilia in the United States were provided in the initial findings by Green and Lechner [1] (215 patients) and showed bimodal distribution around age, with a small peak seen from age 21 to 30 years that correlates with mostly younger patients with postpartum hemorrhage (8.0% of all patients) and a larger peak for older men and women in relatively equal proportions. Nearly half of all patients were aged at least 61 years (61–70 years: 24.5% and 71–80 years: 23.9%). Subsequent findings in the United Kingdom, including a surveillance study through the United Kingdom Haemophilia Centre Doctors’ Organisation (UKHCDO, 172 patients) [2] and a prospective survey of hematologists in south and west Wales (18 patients) [3], have provided more recent data on acquired hemophilia within a national health system with a referral paradigm that assured nearly complete case identification and confirmed that the incidence of acquired hemophilia is roughly 1.3–1.5 per million [4] and that incidence increases with age. Of the 172 patients in the UKHCDO data set, 63% were aged 65 to less than 85 years and an additional 22% were aged at least 85 years. Most recently, the European Acquired Hemophilia Registry (EACH2) provided additional insights on 501 patients with acquired hemophilia from 117 different hemophilia treatment centers in 13 European countries. As seen in other studies, age had a bimodal distribution, with a small peak occurring in younger women with peripartum acquired hemophilia (median age 33.9 years) [5].
Acquired hemophilia is associated with a wide variety of underlying conditions, such as lymphoproliferative or myeloproliferative disorders, solid tumors, autoimmune diseases, drugs (e.g. penicillin, chloramphenicol, phenytoin, INF-α), and graft-versus-host disease after allogeneic bone marrow transplantation. Acquired hemophilia is also known to occur during the postpartum period up to several months after delivery; it was seen in 2% of patients in UKHCDO (acquired hemophilia was pregnancy related in three of four patients aged 21–40 years, and occurred at day 1, week 8, and month 7 postpartum) and in 42 (8.4%) patients in EACH2 who were diagnosed between 21 and 120 days following delivery [2,5,6].
The Hemostasis and Thrombosis Research Society (HTRS) Registry was established in 1999 as a joint effort of the then Hemophilia Research Society (HRS) and Novo Nordisk Inc. The registry's purpose was to serve as a platform for society-based research on bleeding disorders and to monitor the postapproval use of recombinant factor VIIa (rFVIIa, NovoSeven, Novo Nordisk A/S, Bagsvaerd, Denmark). Since rFVIIa was approved in 2006 by the US Food and Drug Administration for the additional indication of treatment of bleeding and prevention of bleeding during surgical procedures in patients with acquired hemophilia, the HTRS Registry has been used for postmarketing surveillance. This article aims to provide key insights on the demographics of acquired hemophilia in the United States based on the HTRS Registry, with specific focus on assessing differences in presentation across sex, race, and ethnicity.
Methods
This was a retrospective review of data from the HTRS Registry, a longitudinal database capturing the demographics and treatment of bleeding episodes and surgery in patients with bleeding disorders. Originally named for the then HRS, the registry was renamed along with the society's name change to HTRS in 2004 and upgraded on a regular basis to include specific questions relating to acquired hemophilia and consolidated demographic data from the HRS and HTRS registries.
Institutional Review Board approval to participate was obtained by all sites, with either patient informed consent obtained prior to patient data entry or Institutional Review Board exemption of consent for retrospective collection of anonymized data. Registration and follow-up data on patients with bleeding disorders were submitted voluntarily by hemophilia treatment centers in the United States and one center in Quebec. Registration forms for patients with acquired hemophilia captured age; sex; comorbidities and predisposing conditions; first bleeding location; laboratory parameters; exposure to blood products, factor, or bypassing agents; and initiation of immune suppression/tolerance. Active prompts for adverse events (i.e. ‘yes’ or ‘no’) on all data entry forms and screens were provided as mandatory affirmation fields to ensure adverse event reporting; mortality and status change forms were completed as needed.
This retrospective analysis examined all patients registered between January 2000 and December 2011 with a reported diagnosis of acquired hemophilia, regardless of whether bleeding episodes or surgical procedures were documented. Statistical analyses were performed on the SAS data set by Quintiles Outcome by Quintiles Inc. (formerly Outcome Sciences) of Cambridge, Massachusetts, USA, the contract research organization that managed the registry. Further descriptive subanalyses by sex, race, and ethnicity were performed by the authors.
Results
Patient disposition and demographics
Between 2000 and 2011, 36 of approximately 100 maximum HTRS sites submitted acquired hemophilia patient cases, with a mean/median (range) of 4.6/3 (1–22) cases per site. A total of 166 patients were registered in the HRS Registry (21 patients, 14 with bleed records; 2000–2003) and HTRS Registry (145 patients, 116 with bleed or surgery records; 2004–2011) databases. The demographics of those entered in the HRS and HTRS registries and those with and without bleeding records were similar; therefore, they are presented as composite data in Table 1.
Table 1.
Summary of patient demographics Hemostasis and Thrombosis Research Society Registry: Demographics
| HRS patients | HTRS patients | |||
| A | B | C | D | |
| Registration only | With bleed records | Registration only | With bleed/surgery records | |
| Number of patients | 7 | 14 | 29 | 116 |
| Age at registration (years) | ||||
| Mean (SD) | 56 (15.82) | 63 (14.60) | 61 (22.11) | 67 (16.81) |
| Median (range) | 59 (36–78) | 64 (22–80) | 68 (13–88) | 73 (13–92) |
| Sex, n (%) | ||||
| Male | 0 (0.00) | 1 (7.14) | 12 (41.38) | 60 (51.72) |
| Female | 7 (100.00) | 3 (21.43) | 17 (58.62) | 56 (48.28) |
| Missing | 0 (0.00) | 10 (71.43) | 0 (0.00) | 0 (0.00) |
| Ethnicity, n (%) | ||||
| White, non-Hispanic | 3 (42.86) | 8 (57.14) | 13 (44.83) | 78 (67.24) |
| White, Hispanic | 1 (14.29) | 3 (21.43) | 4 (13.79) | 7 (7.37) |
| Black, non-Hispanic | 1 (14.29) | 2 (14.29) | 9 (31.03) | 26 (22.41) |
| Black, Hispanic | 0 (0.00) | 0 (0.00) | 1 (3.45) | 1 (0.86) |
| Unknown | 1 (14.29) | 1 (7.14) | 1 (3.45) | 2 (1.72) |
| Other | 1 (14.29) | 0 (0.00) | 1 (3.45) | 2 (1.72) |
| Functional status at registration, n (%) | ||||
| Unrestricted | 4 (57.14) | 3 (21.43) | 14 (48.28) | 30 (25.86) |
| Full school/work, limited recreation | 0 (0.00) | 0 (0.00) | 1 (3.45) | 10 (8.62) |
| Limited school/work/activities | 0 (0.00) | 1 (7.14) | 9 (31.03) | 49 (42.24) |
| Requires assistance, no recreation | 0 (0.00) | 0 (0.00) | 4 (13.79) | 23 (19.83) |
| Unknown | 3 (42.86) | 10 (71.43) | 1 (3.45) | 4 (3.45) |
| Inhibitor titers (BU) | ||||
| Highest human, anti-VIII | ||||
| Mean | 168.24 | 59.90 | 156.20 | 202.29 |
| Median (range) | 61.0 (9.7–665) | 22.0 (3–220) | 64.0 (6.8–960) | 48.0 (1–2969) |
| Lowest human, anti-VIII | ||||
| Mean | 0.83 | 3.54 | 48.04 | 45.17 |
| Median (range) | 0.00 (0–4) | 1.00 (0–18.9) | 0.00 (0–520) | 2.00 (0–878.1) |
| Current human, anti-VIII | ||||
| Mean | ND | ND | 15.90 | 60.30 |
| Median (range) | ND | ND | 0.00 (0–118) | 5.15 (0–878.1) |
| Highest porcine, anti-VIII | ||||
| Mean | 5.50 | 5.71 | ND | 2.80 |
| Median (range) | 5.50 (0–11) | 2.00 (0–20) | ND | 2.25 (0–6.5) |
| Lowest porcine, anti-VIII | ||||
| Mean | 0.00 | 0.50 | ND | 0.73 |
| Median (range) | 0.00 (0–0) | 0.00 (0–5) | ND | 0.30 (0–2.3) |
BU, Bethesda units; HRS, Hemophilia Research Society; HTRS, Hemostasis and Thrombosis Research Society; ND, no data; SD, standard deviation.
Overall, 83 female and 73 male patients were registered with a mean/median age of 65.3/70 years; for 10 patients (6.0%), sex was not recorded. The mean/median ages were similar for male and female patients (Fig. 1a), white and black patients (Fig. 1b), and non-Hispanic/Hispanic patients (Fig. 1c). The majority of registered patients (n = 119, 72%) were aged 61–92 years and were non-Hispanic whites (n = 102, 61.4%). However, nearly 25% (40/166) were black. Most patients (n = 51, 30.7%) had unrestricted functional status at registration, full school or work functional status with limited recreation (n = 11, 6.6%), or limited school, work, or activities (n = 59, 35.5%) as defined by the five-category scale in the Universal Data Collection System (Annual Visit Form Question 34. CDC 59.8C 10/2005) [7]. The median (range) highest human anti-FVIII titer was 50 (1–2969) Bethesda units. The median (range) highest porcine anti-FVIII titer was 2.2 (0–20) Bethesda units.
Fig. 1.
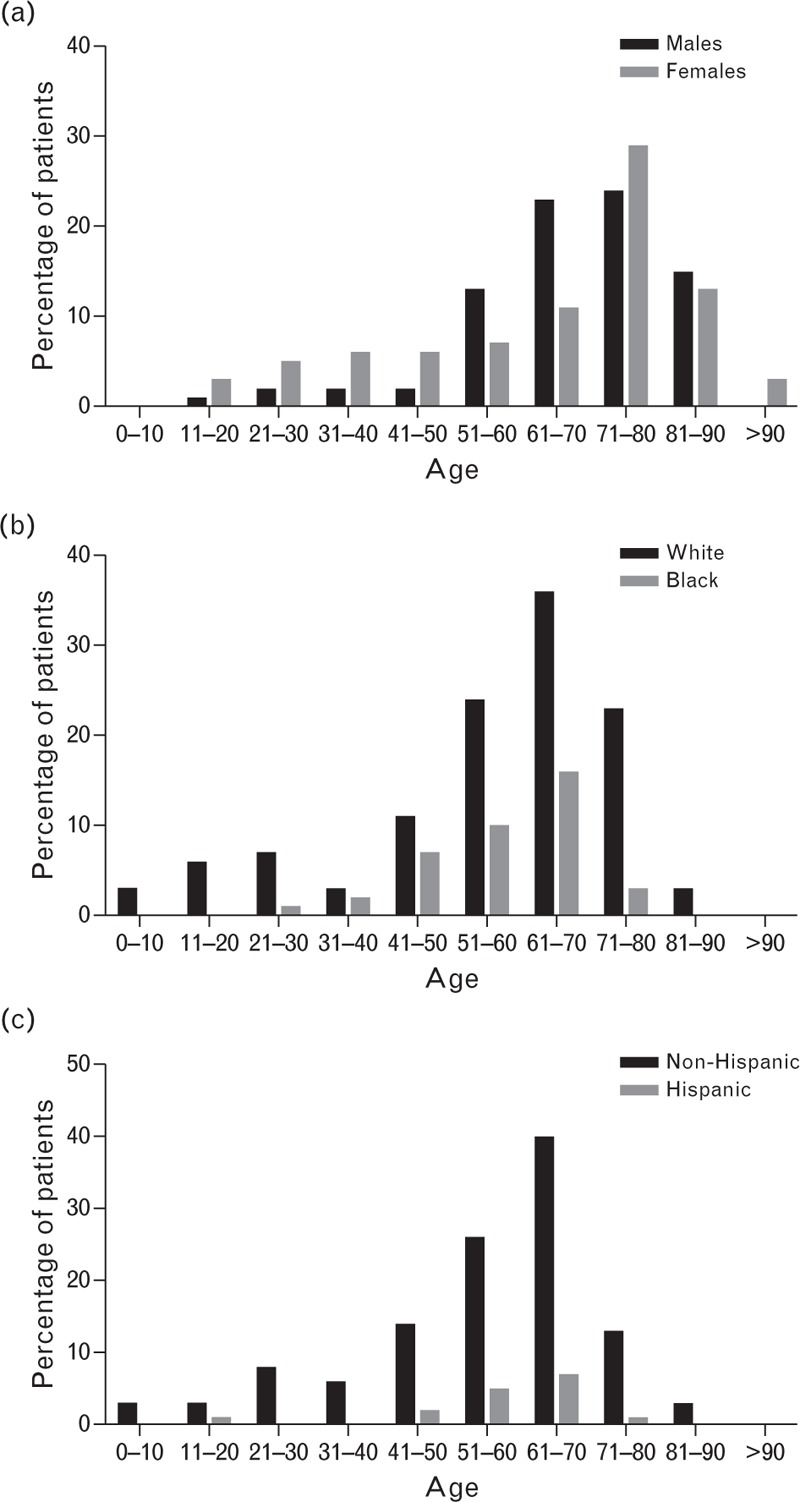
Age distribution of registered patients by sex (a), race (b), and ethnicity (c).
Comorbidities
Comorbidities were recorded for 145 patients with acquired hemophilia in the HTRS Registry. General categories and common subcategories were provided, as were ‘other’ fields for free-text entry. The most frequently recorded illnesses were autoimmune disease (n = 41, 28.4%) and malignancy (n = 21, 14.5%). Other comorbidities included surgery/intervention (11.7%), postpartum hemorrhage (PPH) (3.4%), trauma (1.4%), other (15.9%), and not specified (44.1%) (Tables 2 and 3).
Table 2.
Distribution of comorbidities by sex, race, and ethnicity
| All | Female | Male | White | Black | Non-Hispanic | Hispanic | |
| Number of patients, n | 145 | 73 | 72 | 102 | 37 | 126 | 13 |
| Comorbidities, n (%) | |||||||
| Autoimmune | 41 (28.3%) | 24 (32.9%) | 17 (23.6%) | 28 (27.5%) | 11 (29.7%) | 38 (30.2%) | 1 (7.7%) |
| Excluding diabetes | 27 (18.6%) | 19 (26.0%) | 8 (11.1%) | 20 (19.6%) | 5 (13.5%) | 24 (19.0%) | 1 (7.7%) |
| Diabetes only | 7 (4.8%) | 3 (4.1%) | 4 (5.6%) | 3 (2.9%) | 4 (10.8%) | 7 (5.6%) | 0 (0.0%) |
| Diabetes type 2 only | 7 (4.8%) | 2 (2.7%) | 5 (6.9%) | 5 (4.9%) | 2 (5.4%) | 7 (5.65%) | 0 (0.0%) |
| Malignancy | 21 (14.5%) | 10 (13.7%) | 11 (15.3%) | 18 (17.6%) | 3 (8.1%) | 20 (15.9%) | 1 (7.7%) |
| Postpartum | 5 (3.4%) | 5 (6.8%) | NA | 5 (4.9%) | 0 (0.0%) | 4 (3.2%) | 1 (7.7%) |
| Surgery/procedure/PICC/IV | 17 (11.7%) | 8 (11.0%) | 9 (12.5%) | 14 (13.7%) | 3 (8.1%) | 16 (12.7%) | 1 (7.7%) |
| Trauma | 2 (1.4%) | 2 (2.7%) | 0 (0.0%) | 2 (2.0%) | 0 (0.0%) | 2 (1.6%) | 0 (0.0%) |
| Other | 23 (15.9%) | 12 (16.4%) | 11 (15.3%) | 15 (14.7%) | 8 (21.6%) | 21 (16.7%) | 2 (15.4%) |
| None specified | 64 (44.1%) | 28 (38.4%) | 36 (50.0%) | 42 (41.2%) | 18 (48.6%) | 53 (42.1%) | 7 (53.8%) |
IV, intravenous; NA, not applicable; PICC, peripherally inserted central catheters.
Table 3.
Detailed listing of comorbidities
| Type | Concomitant illness | N | % |
| Autoimmune | Diabetes mellitus | 16 | 11.0 |
| Diabetes mellitus type 2 | 7 | 4.8 | |
| Rheumatoid arthritis | 10 | 6.9 | |
| Systemic lupus erythematosus | 4 | 2.8 | |
| Bullous pemphigoid | 3 | 2.1 | |
| Autoimmune thyroiditis | 2 | 1.4 | |
| Retroperitoneal fibrosis | 1 | 0.7 | |
| Psoriatic arthritis | 1 | 0.7 | |
| Multiple sclerosis | 1 | 0.7 | |
| Lupus anticoagulant | 1 | 0.7 | |
| Erythema nodosum | 1 | 0.7 | |
| Chronic thrombocytopenia | 1 | 0.7 | |
| Celiac disease | 1 | 0.7 | |
| Autoimmune hemolytic anemia | 1 | 0.7 | |
| Any autoimmune | 41 | 28.3 | |
| Any autoimmune (excluding diabetes) | 27 | 18.6 | |
| Malignancy | Breast cancer | 6 | 4.1 |
| Leukemia | 3 | 2.1 | |
| Prostate cancer | 3 | 2.1 | |
| Known metastatic site | 1 | 0.7 | |
| Bladder cancer | 2 | 1.4 | |
| Carcinoma of the intestine, stomach, esophagus, colon, or rectum | 2 | 1.4 | |
| Cervix cancer | 2 | 1.4 | |
| Liver carcinoma | 1 | 0.7 | |
| Multicentric Castleman disease | 1 | 0.7 | |
| Myelofibrosis | 1 | 0.7 | |
| Polycythemia vera | 1 | 0.7 | |
| Squamous cell left hand cancer | 1 | 0.7 | |
| Squamous cell of face | 1 | 0.7 | |
| Total malignancy | 21 | 14.5 | |
| Surgery | Unspecified | 8 | 5.5 |
| Endarterectomy | 2 | 1.4 | |
| Appendectomy | 1 | 0.7 | |
| Cesarean section | 1 | 0.7 | |
| Circumcision | 1 | 0.7 | |
| Hernia repair | 1 | 0.7 | |
| Ileal conduit | 1 | 0.7 | |
| Left hip replacement | 1 | 0.7 | |
| Mediport | 1 | 0.7 | |
| Skin graft from left leg to left elbow | 1 | 0.7 | |
| Tonsillectomy | 1 | 0.7 | |
| Ventral hernia repair | 1 | 0.7 | |
| Total surgery | 12 | 8.3 | |
| Postpartum | PPH | 5 | 3.4 |
| Total postpartum | 5 | 3.4 | |
| Other | Hypertension | 7 | 4.8 |
| Chronic obstructive pulmonary disease | 3 | 2.1 | |
| Hyperlipidemia | 3 | 2.1 | |
| Coronary artery disease | 2 | 1.4 | |
| Cerebrovascular accident | 2 | 1.4 | |
| Hepatitis C | 2 | 1.4 | |
| Herpes zoster | 2 | 1.4 | |
| HIV | 2 | 1.4 | |
| Peripheral vascular disease | 2 | 1.4 | |
| Atrial fibrillation | 1 | 0.7 | |
| Anemia | 1 | 0.7 | |
| Anticardiolipin antibody | 1 | 0.7 | |
| Aortic aneurysm | 1 | 0.7 | |
| Aplastic anemia | 1 | 0.7 | |
| Asthma | 1 | 0.7 | |
| CABG | 1 | 0.7 | |
| Cholecystitis | 1 | 0.7 | |
| Colon perforation | 1 | 0.7 | |
| Chronic renal failure | 1 | 0.7 | |
| Emphysema | 1 | 0.7 | |
| Fell hurting ankle | 1 | 0.7 | |
| Fibroid | 1 | 0.7 | |
| Fibromyalgia | 1 | 0.7 | |
| GERD | 1 | 0.7 | |
| Glaucoma | 1 | 0.7 | |
| Gout | 1 | 0.7 | |
| Hematoma left leg | 1 | 0.7 | |
| Hypertension | 1 | 0.7 | |
| Hyperthyroidism | 1 | 0.7 | |
| Hypothyroidism | 1 | 0.7 | |
| Increased cholesterol | 1 | 0.7 | |
| Left elbow injury | 1 | 0.7 | |
| Monoclonal gammopathy of undetermined significance (not cause of inhibitor) | 1 | 0.7 | |
| On warfarin for history of mesenteric vein thrombosis | 1 | 0.7 | |
| Osteoporosis | 1 | 0.7 | |
| Paroxysmal nocturnal hemoglobinuria | 1 | 0.7 | |
| Pneumonia | 1 | 0.7 | |
| Polymyalgia rheumatica | 1 | 0.7 | |
| Pulmonary nodule not biopsied | 1 | 0.7 | |
| s/p OVA | 1 | 0.7 | |
| Transient ischemic attack | 1 | 0.7 |
CABG, coronary artery bypass grafting; GERD, gastroesophageal reflux disease; PPH, postpartum hemorrhage.
Of 41 patients with reported autoimmune diseases, 16 were reported to have diabetes mellitus (type 2 or unspecified) entered as ‘other’ autoimmune disease. Excluding 14 patients who reported diabetes only, 27 (18.6%) reported other autoimmune disorders, the most common of which were rheumatoid arthritis (n = 10), systemic lupus erythematosus (n = 4), and bullous pemphigoid (n = 3) (Table 3). Autoimmune occurrence was similar across sex and race, but was higher in women (32 vs. 22% in men) and non-Hispanics (29 vs. 8% in Hispanics). This difference held true for autoimmune diseases other than diabetes.
Malignancy occurrence was similar across sex and race, but was higher in non-Hispanics (16 vs. 0% in Hispanics). The most common malignancies reported were breast cancer (six cases), leukemia (three cases, two of which were known by the authors to be chronic lymphocytic leukemia), and prostate cancer (three cases, one of which was reported to be metastatic). Two cases each of bladder, gastrointestinal (including intestine, stomach, esophagus, colon, or rectum), and cervical cancer were reported in the registry. Two cases of myeloproliferative neoplasms (polycythemia vera and myelofibrosis) were reported as ‘other disorders’ but were included in this analysis as malignancies to reflect current understanding of the diseases.
The reported surgery types varied; the dates of surgery and the surgery's relationship to a diagnosis of acquired hemophilia were not uniformly captured in the registry.
Type and locations of bleeds
Frequently recorded bleeding locations included the subcutaneous area (27.1%) and mucosa (21.1%). Other locations of the first bleeding episode for each patient included ‘extremity not otherwise specified (NOS)’ (7.2%), muscle (5.4%), joint (4.2%), retroperitoneal (2.4%), surgery/procedure site (1.8%), postpartum (1.8, 3.6% women), head (1.8%), other (4.2%), and ‘not specified’ (16.3%) (Table 4).
Table 4.
Distribution of first bleed locations by sex, race, and ethnicity
| All | Female | Male | White | Black | Non-Hispanic | Hispanic | |
| Number of patients, n | 166 | 83 | 73 | 117 | 40 | 140 | 17 |
| First bleeding site, n (%) | |||||||
| Subcutaneous | 45 (27.1%) | 25 (30.1%) | 15 (20.5%) | 34 (29.1%) | 5 (12.5%) | 37 (26.4%) | 2 (11.8%) |
| Extremity NOS | 12 (7.2%) | 4 (4.8%) | 8 (11.0%) | 9 (7.7%) | 2 (5.0%) | 9 (6.4%) | 2 (11.8%) |
| Subcutaneous + extremity NOS | 57 (34.3%) | 29 (34.9%) | 23 (31.5%) | 43 (36.8%) | 7 (17.5%) | 46 (32.8%) | 4 (23.6%) |
| Mucosal | 35 (21.1%) | 17 (20.5%) | 17 (23.3%) | 24 (20.5%) | 10 (25.0%) | 29 (20.7%) | 5 (29.4%) |
| Muscle | 9 (5.4%) | 2 (2.4%) | 5 (6.8%) | 8 (6.8%) | 1 (2.5%) | 7 (5.0%) | 2 (11.8%) |
| Joint | 7 (4.2%) | 3 (3.6%) | 4 (5.5%) | 5 (4.3%) | 2 (5.0%) | 7 (5.0%) | 0 (0.0%) |
| Retroperitoneal | 4 (2.4%) | 2 (2.4%) | 1 (1.4%) | 3 (2.6%) | 1 (2.5%) | 4 (2.9%) | 0 (0.0%) |
| Surgery/biopsy/IV-PICC site | 3 (1.8%) | 0 (0.0%) | 3 (4.1%) | 2 (1.7%) | 1 (2.5%) | 3 (2.1%) | 0 (0.0%) |
| Postpartum | 3 (1.8%) | 3 (3.6%) | NA | 3 (2.6%) | 0 (0.0%) | 3 (2.1%) | 0 (0.0%) |
| Head | 3 (1.8%) | 3 (3.6%) | 0 (0.0%) | 3 (2.6%) | 0 (0.0%) | 2 (1.4%) | 1 (5.9%) |
| Other | 7 (4.2%) | 6 (7.2%) | 1 (1.4%) | 3 (2.6%) | 4 (10.0%) | 7 (5.0%) | 0 (0.0%) |
| Not specified | 27 (16.3%) | 27 (32.5%) | 20 (27.4%) | 29 (24.8%) | 18 (45.0%) | 41 (29.3%) | 6 (35.3%) |
IV-PICC, intravenous peripherally inserted central catheters; NA, not applicable; NOS, not otherwise specified.
Subcutaneous bleeding was more common in women (30.1%) than men (20.5%), in whites (29.1%) than blacks (12.5%), and in non-Hispanics (26.4%) than Hispanics (11.8%). Subcutaneous bleeding and ‘extremity NOS’ were not reported in the same patients. If ‘extremity NOS’ is considered an alternate potential classification of subcutaneous bleeding locations, then the aggregate of possible subcutaneous bleeding rises to about one-third of patients and remains more common in white and non-Hispanic individuals (Table 4).
Blood component exposure
Coagulation factor and hemostatic agent exposure were recorded for on-demand and prophylactic therapies for patients with acquired hemophilia in the HRS and HTRS Registry platforms (Table 5). Of the 145 patients for whom information about exposure to blood products was reported, only 48 (33.1%) have actually received blood products. Patient exposure included packed red blood cells (RBCs) (28%), whole blood or fresh frozen plasma (FFP) (14%), platelets (3%), and cryoprecipitate (1%). FVIII exposure was reported for eight (7%) of 128 patients in whom FVIII exposure history was known and for one of 129 patients in whom factor IX exposure history was known (Table 5). Duration of product exposure (in days) was reported for many products but was estimated in at least half of the patients. Although the reported use of packed RBC transfusions was similar across race and ethnicity, the use of whole blood or FFP and the mean/median exposure days was higher in black than white patients and higher in Hispanic than non-Hispanic patients. Whole blood is not readily available in the United States and likely accounts for the use of FFP. Hemostatic products included two different plasma-derived activated prothrombin complex concentrates commercially available in the United States at the time, as well as rFVIIa, aminocaproic acid, desmopressin acetate, antihemophilic factor, and other products.
Table 5.
Distribution of blood product exposure history by race and ethnicity
| All | White | Black | Non-Hispanic | Hispanic | |
| Blood product exposure, n | |||||
| Yes | 48 | 32 | 14 | 41 | 5 |
| No | 62 | 43 | 16 | 54 | 5 |
| Unknown | 35 | 24 | 7 | 29 | 2 |
| Packed RBC, n (%) | 40 (28%) | 28 (28%) | 10 (27%) | 35 (28%) | 3 (25%) |
| Mean (days) | 3.7 | 4.5 | 2.2 | 4.1 | 1.3 |
| Median (days) | 2 | 2 | 1.5 | 2 | 1 |
| IQR (days) | 1–3 | 1–4 | 1–2 | 1–4.5 | 1–1.5 |
| Minimum–maximum (days) | 1–43 | 1–43 | 1–6 | 1–43 | 1–2 |
| Values estimated, n (%) | 22 (60%) | 17 (60%) | 5 (50%) | 19 (50%) | 3 (100%) |
| Whole blood or FFP, n (%) | 21 (14%) | 13 (13%) | 8 (22%) | 17 (14%) | 4 (33%) |
| Mean (days) | 4.2 | 2.7 | 6.8 | 2.1 | 13.5 |
| Median (days) | 2 | 1 | 2 | 1 | 6.5 |
| IQR (days) | 1–3 | 1–3 | 1–3.5 | 1–3 | 2.5–17.5 |
| Minimum–maximum (days) | 0–40 | 1–10 | 0–40 | 0–7 | 1–40 |
| Values estimated, n (%) | 14 (70%) | 8 (60%) | 6 (80%) | 10 (60%) | 4 (100%) |
| Platelets, n (%) | 5 (3%) | 2 (2%) | 2 (5%) | 3 (2%) | 2 (17%) |
| Mean (days) | 21.5 | 2.0 | 80.0 | 2.0 | 41.0 |
| Median (days) | 2.5 | 2 | 80 | 2 | 41 |
| IQR (days) | 1.75–22.25 | 1.5–2.5 | 80–80 | 1.5–2.5 | 21.5–60.5 |
| Minimum–maximum (days) | 1–80 | 1–3 | 80–80 | 1–3 | 2–80 |
| Values estimated, n (%) | 3 (60%) | 2 (70%) | 1 (50%) | 1 (30%) | 2 (100%) |
| Cryoprecipitate, n (%) | 2 (1%) | 1 (1%) | 1 (3%) | 1 (1%) | 1 (8%) |
| Mean (days) | 10.0 | 10.0 | QNS | QNS | 10.0 |
| Median (days) | 10 | 10 | QNS | QNS | 10 |
| Factor 8, n | 8 | 6 | 1 | 6 | 1 |
| Mean | 5.0 | 5.0 | QNS | 5.4 | 3.0 |
| Median | 2 | 2 | QNS | 2 | 3 |
| Factor 9, n | 1 | 1 | 0 | 0 | 1 |
| Mean | 9.0 | 9.0 | NA | NA | 9.0 |
| Median | 9 | 9 | NA | NA | 9 |
FFP, fresh frozen plasma; IQR, interquartile range; NA, not applicable; QNS, quantity not specified; RBC, red blood cells.
Immune tolerance, immunosuppressive therapy, and eradication of the auto-factor VIII antibody inhibitor
The registration case report form (CRF) was designed originally for congenital hemophilia, and it requested history and outcome from immune tolerance therapy (ITT) with FVIII infusions; there were no specific questions about immunosuppression. This series of questions was completed for 65 (39%) of 166 registered patients with acquired hemophilia. It is unlikely that the individual hemophilia center physicians applied immune tolerance regimens in a uniform manner for all the patients in this study analysis. Furthermore, it is difficult to determine if in all cases the physician's use of immunosuppressive therapy was intended to be included as a component of an immune tolerance protocol or simply as an immune system modulator to eradicate the inhibitory antibody. More detailed ITT/immune suppression CRFs were available but were not completed for most registered patients with acquired hemophilia.
Of the 57 evaluable patients with outcome data, 26 patients (46%) reported successful ITT treatment, 13 reported unsuccessful ITT treatment (23%), and 18 (32%) were receiving active treatment at the time of registration.
Discussion
Acquired hemophilia is a very rare disease, occurring in only one in 1 million individuals [1]. Presumably, the 300–350 US patients who develop autoantibodies each year could present acutely to approximately 5700 hospitals [8] and a variety of specialists [9]. Therefore, the primary goal of looking at registry data on rare disorders such as acquired hemophilia is to generate insights that can help improve its diagnosis and treatment in the context of the inability to perform prospective-controlled trials because of the limited number of patients with acquired hemophilia. Toward our goal of generating treatment hypotheses, we sought in this analysis to use the heterogeneous nature of the US population and the HTRS Registry to expand the discussion of demographic contributions to the development and diagnosis of acquired hemophilia.
Comparing acquired hemophilia registries and populations
There are methodological differences in assessing demographics within different acquired hemophilia registries. Within nationalized healthcare systems or within those organized around few hemophilia treatment centers (HTCs), the pattern of patient presentations differs from that seen in the HTRS Registry population. For the UKHCDO study, referral was within a centralized network with a high capture rate under a national health plan. The United Kingdom has 256 hematology departments; 255 of those departments participated in the UKHCDO study and reported on 172 patients over the course of 2 years [2]. The hematology departments also directly oversee coagulation laboratories and hence may have earlier information about patients with abnormal aPTT studies. In the French SACHA Registry, 82 patients were identified over the course of 4 years [10]. In the larger European study (EACH2), 501 patients were recruited through 117 HTCs in 13 countries under informed consent with exceptions; six countries required informed consent for living patients only, and five countries did not require informed consent but did not allow enrollment of patients who were deceased [11,12]. This may have excluded patients who were more severely affected [11].
The HTRS Registry differs from European registries in that the underlying system supporting the care of acquired hemophilia patients is different. Within the United States, not all HTCs care for adult patients. This is due in part to the way hemophilia care is funded, with more HTCs and experts available to facilitate the diagnosis and management of children with congenital bleeding disorders than adults with acquired disorders. Further, multispecialty case-based surveys have identified that physicians who would primarily see a patient with undiagnosed acquired hemophilia (e.g. emergency medicine, hospitalists, critical care, rheumatology, obstetrics/gynecology) are more focused on identifying the underlying site of bleeding than in appreciating that the aPTT values are abnormal, or that they might reflect an underlying bleeding disorder [9]. Given that the US system encourages the presentation of acquired hemophilia in an acute setting, physicians sometimes resist referring patients to hematology [9]; thus, the lack of uniform distribution and availability of specialized coagulation expertise become barriers to diagnosis.
The heterogeneity of the US population allows for an analysis of race and ethnicity as covariates for both the risk of acquired hemophilia and the likelihood of receiving the proper diagnosis. Black patients with acquired hemophilia appear overrepresented (24.1%) compared with the relative percentage of blacks within the US population (13.6%) [13]; the 1.8 times higher ratio of percentage of black patients compared with the total population suggests that blacks may have a higher risk of developing acquired hemophilia. Notably, there has been the suggestion that blacks with hemophilia A have a higher risk of developing FVIII alloantibody inhibitors as well. Through the Universal Data Collection Program, black and Hispanic patients with hemophilia A in the United States have reported twice the FVIII inhibitor rate of white patients in the United States [14]. Hemophilia A inhibitor formation appears to be more frequent in African-American patients, and central nervous system bleeding was seen more frequently as a first bleeding event in this population [15]. The HTRS data are the first to suggest that a similar increased risk might exist for autoantibody FVIII inhibitors. There are some human leukocyte antigen (HLA) haplotypes identified that might predispose to autoantibody formation [16], and perhaps mapping HLA haplotypes may identify racial differences and predispositions. Hispanic patients appear somewhat underrepresented (10.2%) compared with Hispanics in the general US population (16.7%) [17]. However, Hispanic children comprise a much higher percentage of US children (22% of children aged <18 years) [18] but comprise a smaller percentage of the elderly US population (those at risk for acquired hemophilia), which suggests that Hispanic adults are not disproportionally affected by acquired hemophilia.
Predisposing conditions
In the initial study by Green and Lechner [1], acquired hemophilia was commonly associated with malignancy (6.7%) and autoimmune disorders (17.0%). This was also seen in the UKHCDO study (malignancy, 14.7%; autoimmune, 16.7%) and EACH2 (malignancy, 11.8%; autoimmune, 13.4%) [2,5]. In the HTRS Registry, 28.3% of patients with acquired hemophilia reported an autoimmune disorder and 14.5% reported malignancy. Once diabetes (type 2 or unspecified) is excluded as an autoimmune disorder in HTRS, the percentage of patients with an autoimmune disorder drops to 18.6% and is more aligned with previous reports of autoimmune comorbidities in patients with acquired hemophilia. Given the prevalence of diabetes in the elderly population most commonly seen with acquired hemophilia, the coincidence of diabetes and acquired hemophilia is of unclear significance.
In the HTRS Registry, polycythemia vera and myelofibrosis were initially reported as ‘other disorders’ but have been subsequently recategorized as malignancies. Sparse data on patients with cancer are available from the European registries; however, in the United States, there were surprisingly few cases of acute or chronic leukemias. Three cases of leukemia were reported, two of which were known by the authors to be chronic lymphocytic leukemia.
The percentage reporting PPH (3.4%) in the HTRS Registry is similar to that reported in the UKHCDO study (2.0%) [2], but lower than that initially reported by Green and Lechner [1] (7.3%) or that reported in SACHA (7.3%) [10] or EACH2 (8.4%) [5]. The lower incidence of PPH in the HTRS Registry may be because of US obstetricians being hesitant to refer patients to hematologists [9].
There are no longitudinal data from acquired hemophilia that extend long enough to determine if acquired hemophilia could be a harbinger of future autoimmune disease or malignancy. Referral patterns and insurance provider network restrictions within the United States would make long-term follow-up after successful immunosuppression or ITT difficult to accomplish. Anecdotally, there is some evidence that the recurrence of the acquired inhibitor is contemporaneous with the recurrence of malignancy [19]; this suggests that additional study might provide more insight into idiopathic acquired hemophilia.
First bleeding episodes
The HTRS Registry is the first study to examine symptoms by race and ethnicity and to show a racial and ethnic disparity for subcutaneous bleeding episodes, with whites and non-Hispanics being more likely to have subcutaneous bleeding as their first bleed site. With other bleeding disorders, such as immune thrombocytopenic purpura, some have argued that the data show possible racial disparities in the overall incidence of the disease, with blacks having a much lower prevalence than whites [20]. However, this disparity may not exist. Other authors, after reviewing the Veterans Administration population, state that this may be because of the fact that in blacks, the presence of petechiae and ecchymoses may not be recognized [20,21]. A racial disparity among other dermatological disorders, such as skin cancer and atopic dermatitis, is not uncommon [22,23]. In atopic dermatitis, a reliance on erythema scores may mask the severity of disease in black children [22].
Blood product exposure
With data in the HTRS Registry and these analyses, it is assumed that blood product exposure is reported at registration and is therefore historical. However, this may not be totally accurate. Transfusion requirements cannot distinguish between replacement therapy for bleeding occurring before the diagnosis and treatment after diagnosis, reflecting the efficacy of the chosen hemostatic treatment approach. In the HTRS Registry, the collected data on blood product exposure included information on packed RBCs, FFP or whole blood, cryoprecipitate, platelets, and coagulation factors. The higher percentage of patients exposed and the mean/median number of days of FFP exposure observed in blacks and Hispanics may reflect a delay in diagnosis in those patients with abnormal aPTT values, a lack of coagulation experts or laboratory capacity to facilitate diagnostic workup and formulation of a treatment plan, and/or a lack of availability of bypassing agents for acute management outside of HTCs.
Immune tolerance/suppression
The data from the HTRS Registry suggest a very high success rate with immune tolerance induction regimens for acquired hemophilia in the United States. Our data entry form for the acquired hemophilia registry offered ‘immune tolerance’ as a treatment option to eradicate the auto-FVIII neutralizing antibody and did not specify any choices for how immune tolerance was to be approached. Thus, there is a possibility that the data collected may represent a broad array of approaches, including factor VIII (formal immune tolerance induction regimens), corticosteroids, cytotoxic agents, intravenous immunoglobulins, rituximab, and biologic response modifiers. The high rate of reported successful tolerance in this acquired hemophilia registry, the precise regimen(s) employed for best outcome, and the durability of response need to be confirmed in prospective studies. Furthermore, in future studies, the increasing use of rituximab for FVIII autoantibody eradication should be scrutinized closely and longitudinally as this anti-CD20 monoclonal antibody may convey benefits and potential adverse effects for those with acquired hemophilia. For example, rituximab therapy may alter B and T-lymphocyte numbers, resulting in favorable reduction of specific autoantibodies directed against FVIII and/or altering the underlying autoimmune disease process, which was associated with the development of acquired hemophilia.
Limitations
In registries such as HTRS, the periodic entry (batches) of patient cases reflects the reality of rare disorder surveillance; however, this may result in missed patients or missed episodes and may not be truly reflective of the total number of cases of acquired hemophilia. This also leads to difficulty in capturing longitudinal data and especially in the follow-up of patients who become inactive (e.g. spontaneous remissions, successful treatment of underlying comorbid conditions, successful immune tolerance, deceased). Although the HTRS Registry only captures a small percentage of the number of patients diagnosed with acquired hemophilia in the United States, the registry has a high rate of capture in the 36 participating sites. The limited number of sites may reflect regions in which referral networks are more structured and where HTCs exist (e.g. Washington, DC; Philadelphia, Pennsylvania; Chapel Hill, North Carolina). The HTRS Registry has, for acquired hemophilia specifically, missing geographic regions with large populations of elderly patients (e.g. Florida, Southern California) where the racial and ethnic population mix, referral patterns, or availability of adult coagulation experts may be different. The subset of acquired hemophilia-participating sites were predominantly adult HTCs within the HTRS registry, which at one time included up to 100 of the 141 federally designated HTCs in the United States [24]; however, at present, the United States has approximately 5700 hospitals [8] and 10 200 practicing hematologists and oncologists that could potentially see a patient [25]. In an attempt to capture this broader population, the alternative surveillance acquired hemophilia study collected bleed data from 99 patient cases submitted by 92 centers between 2008 and 2011 [26]. Ultimately, the demographics of those captured in HTRS are consistent with those of other acquired hemophilia data sets, suggesting these data are representative of the acquired hemophilia population in the United States.
Conclusions
This analysis of the HTRS Registry represents the largest North American and non-European data set on acquired hemophilia, and provides the only current, comprehensive demographic look at acquired hemophilia in the US population. This article provides the first suggestion that there may be racial and ethnic differences in the risk of autoantibody FVIII inhibitors, similar to the increased risk already identified in blacks who develop FVIII alloantibodies in congenital hemophilia. Such observations highlight the need for a more complete understanding of basic immune response mechanisms and the development of allo- and autoantibodies, and the role of HLA haplotype distributions in different racial and ethnic groups.
As was seen in the UK patients with acquired hemophilia [2,3], the HTRS Registry data indicated that subcutaneous bleeds were the most common bleeding site reported in patients with acquired hemophilia. Interestingly, we see that cutaneous bleeding as a presenting bleeding symptom was most notable in white and non-Hispanic individuals, suggesting that darker skin color may delay the diagnosis of acquired hemophilia. This correlates with a delay in diagnosis of immune thrombocytopenic purpura reported in individuals with dark skin [20,21] and in the diagnosis of other dermatologic disorders in such patients [22,23].
In summary, the hypothesis generation in this and the other acquired hemophilia databases should form the basis of future cooperative and multinational prospective studies that can elucidate specific immunopathophysiologies and factors that influence treatment decisions. This may ultimately lead to standardized treatment approaches and the identification of groups requiring specific targeted approaches. Comparing US and European databases identifies the imperfect referral mechanisms and the increased problems associated with the lack of rare-disease awareness working in the US system of heterogeneously distributed HTCs for adult coagulation disorders.
Acknowledgements
Editorial assistance was provided by Amanda Tricarico, PhD, ETHOS Health Communications, Newtown, Pennsylvania, with financial assistance from Novo Nordisk Inc., Princeton, New Jersey, in compliance with international guidelines on Good Publication Practice. The authors received no remuneration of any kind for the development of this manuscript.
H.A.B.Al-M., A.D.M., and C.M.K. received research funding through their institutions for data entry into the HTRS Registry.
Conflicts of interest
C.M.K. and A.D.M. are consultants to Novo Nordisk. There was no compensation provided for authorship or writing of this manuscript. R.Z.G. and D.L.C. are employees of Novo Nordisk.
References
- 1.Green D, Lechner K. A survey of 215 nonhemophilic patients with inhibitors to Factor VIII. Thromb Haemost 1981; 45:200–203. [PubMed] [Google Scholar]
- 2.Collins PW, Hirsch S, Baglin TP, Dolan G, Hanley J, Makris M, et al. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood 2007; 109:1870–1877. [DOI] [PubMed] [Google Scholar]
- 3.Collins P, Macartney N, Davies R, Lees S, Giddings J, Majer R. A population based, unselected, consecutive cohort of patients with acquired haemophilia A. Br J Haematol 2004; 124:86–90. [DOI] [PubMed] [Google Scholar]
- 4.Collins P, Budde U, Rand JH, Federici AB, Kessler CM. Epidemiology and general guidelines of the management of acquired haemophilia and von Willebrand syndrome. Haemophilia 2008; 14 Suppl 3:49–55. [DOI] [PubMed] [Google Scholar]
- 5.Knoebl P, Marco P, Baudo F, Collins P, Huth-Kuhne A, Nemes L, et al. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost 2012; 10:622–631. [DOI] [PubMed] [Google Scholar]
- 6.Tengborn L, Baudo F, Huth-Kuhne A, Knoebl P, Levesque H, Marco P, et al. Pregnancy-associated acquired haemophilia A: results from the European Acquired Haemophilia (EACH2) registry. BJOG 2012; 119:1529–1537. [DOI] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention. Universal Data Collection (UDC) System. 2013. http://www.cdc.gov/ncbddd/blooddisorders/udc/. [Accessed 12 August 2013] [Google Scholar]
- 8.American Hospital Association Resource Center. Fast Facts on US Hospitals. 2013. http://www.aha.org. [Accessed 9 August 2013] [Google Scholar]
- 9.Reding MT, Cooper DL. Barriers to effective diagnosis and management of a bleeding patient with undiagnosed bleeding disorder across multiple specialties: results of a quantitative case-based survey. J Multidiscip Healthc 2012; 5:277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borg JY, Guillet B, Le Cam-Duchez V, Goudemand J, Levesque H, Group SS. Outcome of acquired haemophilia in France: the prospective SACHA (Surveillance des Auto antiCorps au cours de l’Hemophilie Acquise) registry. Haemophilia 2013; 19:564–570. [DOI] [PubMed] [Google Scholar]
- 11.Collins P, Baudo F, Knoebl P, Levesque H, Nemes L, Pellegrini F, et al. Immunosuppression for acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). Blood 2012; 120:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baudo F, Collins P, Huth-Kuhne A, Levesque H, Marco P, Nemes L, et al. Management of bleeding in acquired hemophilia A: results from the European Acquired Haemophilia (EACH2) Registry. Blood 2012; 120:39–46. [DOI] [PubMed] [Google Scholar]
- 13.Rastogi S, Johnson TD, Hoeffel EM, Drewery MP, US Census Bureau. The Black Population: 2010 Census Brief. US Census Bureau, Department of Commerce. 2010. http://www.census.gov/prod/cen2010/briefs/c2010br-06.pdf. [Accessed 9 August 2013] [Google Scholar]
- 14.Miller CH, Benson J, Ellingsen D, Driggers J, Payne A, Kelly FM, et al. F8 and F9 mutations in US haemophilia patients: correlation with history of inhibitor and race/ethnicity. Haemophilia 2012; 18:375–382. [DOI] [PubMed] [Google Scholar]
- 15.Ragni MV, Ojeifo O, Feng J, Yan J, Hill KA, Sommer SS, et al. Risk factors for inhibitor formation in haemophilia: a prevalent case-control study. Haemophilia 2009; 15:1074–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oldenburg J, Zeitler H, Pavlova A. Genetic markers in acquired haemophilia. Haemophilia 2010; 16 Suppl 3:41–45. [DOI] [PubMed] [Google Scholar]
- 17.Ennis SR, Rios-Vargas M, Albert NG, US Census Bureau. The Hispanic Population: 2010 Census Brief. US Census Bureau. Department of Commerce. 2010. http://www.census.gov/prod/cen2010/briefs/c2010br-04.pdf. [Accessed 9 August 2013] [Google Scholar]
- 18.Foundation KCDCAPotAEC. Child Population by Race. Kids Count Data Center. Population Division, US Census Bureau. 2013. http://datacenter.kidscount.org/data/tables/103-child-population-by-race?loc=1&loct=2#detailed/1/any/false/867,133,38,35,18/66,67,68,69,70,71,12,72/423,424. [Accessed 9 August 2013] [Google Scholar]
- 19.White KT, Aggarwal A, Napolitano M, Kessler CM. Autoantibody inhibitor eradication in acquired hemophilia associated with cancer: a retrospective analysis. Blood 2010; 16:1418. [Google Scholar]
- 20.Terrell DR, Johnson KK, Vesely SK, George JN. Is immune thrombocytopenic purpura less common among black Americans? Blood 2005; 105:1368–1369. [DOI] [PubMed] [Google Scholar]
- 21.Landgren O, Gridley G, Fears TR, Caporaso N. Immune thrombocytopenic purpura does not exhibit a disparity in prevalence between African American and White veterans. Blood 2006; 108:1111–1112. [DOI] [PubMed] [Google Scholar]
- 22.Ben-Gashir MA, Hay RJ. Reliance on erythema scores may mask severe atopic dermatitis in black children compared with their white counterparts. Br J Dermatol 2002; 147:920–925. [DOI] [PubMed] [Google Scholar]
- 23.Taylor SC. Skin of color: biology, structure, function, and implications for dermatologic disease. J Am Acad Dermatol 2002; 46:S41–S62. [DOI] [PubMed] [Google Scholar]
- 24.National Hemophilia Foundation. http://www.hemophilia.org [Accessed 1 April]. Comprehensive Medical Care: Hemophilia Treatment Centers. [Google Scholar]
- 25.Studies AoAMCCfW. 2012 Physician Specialty Data Book. 2012. [Google Scholar]
- 26.Lentz SR, Tandra A, Gut RZ, Cooper DL. A novel supplemental approach to capturing postmarketing safety information on recombinant factor VIIa in acquired hemophilia: the Acquired Hemophilia Surveillance project. J Blood Med 2014; 5:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]