

Summary
Mitochondrial ribosomes translate membrane integral core subunits of the oxidative phosphorylation system encoded by mtDNA. These translation products associate with nuclear-encoded, imported proteins to form enzyme complexes that produce ATP. Here, we show that human mitochondrial ribosomes display translational plasticity to cope with the supply of imported nuclear-encoded subunits. Ribosomes expressing mitochondrial-encoded COX1 mRNA selectively engage with cytochrome c oxidase assembly factors in the inner membrane. Assembly defects of the cytochrome c oxidase arrest mitochondrial translation in a ribosome nascent chain complex with a partially membrane-inserted COX1 translation product. This complex represents a primed state of the translation product that can be retrieved for assembly. These findings establish a mammalian translational plasticity pathway in mitochondria that enables adaptation of mitochondrial protein synthesis to the influx of nuclear-encoded subunits.
Keywords: mitochondrial translation, cytochrome c oxidase, assembly, MITRAC, mitochondrial ribosome, COX1, C12ORF62, translation regulation, OXPHOS, translational plasticity
Graphical Abstract
Highlights
-
•
Mitochondrial ribosomes display translational plasticity
-
•
COX1 translation in mitochondria is stalled in the absence of nuclear-encoded COX4
-
•
A ribosome nascent chain complex of COX1 is a primed state for complex IV assembly
-
•
MITRAC regulates translation via COX1 ribosome nascent chain complexes interaction
Mitochondrial translation displays plasticity, allowing the adaptation to the availability of a nuclear-encoded complex subunit.
Introduction
Mitochondria play a vital role in cellular energy production and intermediate metabolism. Consequently, defects in mitochondrial function cause severe disorders in human and initiate cellular stress response and signaling pathways that alter nuclear gene expression (Chandel, 2015, Raimundo, 2014, Shadel and Horvath, 2015). Among such mitochondrial malfunctions, oxidative phosphorylation (OXPHOS) disorders define a group of defects of the respiratory chain and the F1Fo ATP-synthase causing severe and frequently fatal neuromuscular or cardiac disorders (Antozzi and Zeviani, 1997, DiMauro and Schon, 2003, Smeitink et al., 2006). The 13 core subunits of the OXPHOS complexes I, III, IV, and V, all of which are integral membrane proteins, are encoded by the mtDNA. Therefore, one quarter of the mitochondrial proteome is dedicated to the expression and inheritance of mtDNA (Meisinger et al., 2008). Besides the core subunits of the oxidative phosphorylation system, the human mitochondrial genome encodes 2 rRNAs and 22 tRNAs, required for their translation (Hällberg and Larsson, 2014). Accordingly, mitochondrial ribosomes are devoted to the synthesis of membrane proteins for which they associate to the inner membrane. The membrane inserted mitochondrial translation products associate with nuclear-encoded subunits in a defined order, eventually forming stoichiometric, functional enzyme complexes (Fernández-Vizarra et al., 2009, Richter-Dennerlein et al., 2015, Soto et al., 2012). Obviously, this assembly process requires that the products of two independent genetic systems have to be carefully balanced against each other to avoid an accumulation of idle subunits in the inner membrane. In case of cytosolic proteins, the synthesis of components of multiprotein complexes has been shown to be balanced through translational or transcriptional control mechanisms, so that subunits are synthesized in proportion to their stoichiometry (Li et al., 2014). However, in contrast to nuclear gene expression processes, we lack insight into how gene expression and especially mitochondrial translation is regulated in metazoa.
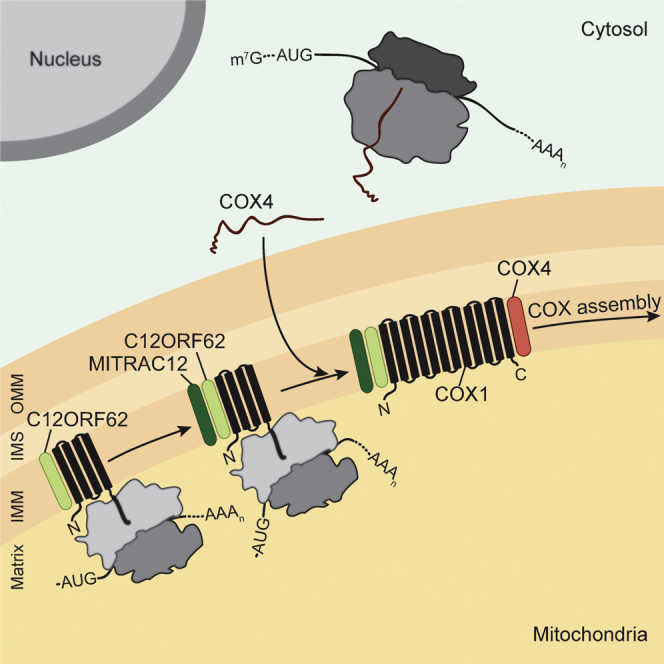
With regard to the assembly of respiratory chain complexes, the cytochrome c oxidase (complex IV) has been best analyzed. A plethora of chaperone-like factors, termed assembly factors, participate in formation and stabilization of transient intermediates in the biogenesis process. The assembly process commences with the synthesis of the mitochondrial-encoded COX1 that serves as a headstone on which the complex is built. Newly synthesized and membrane integrated COX1 engages with the assembly factors C12ORF62 (hCOX14) and MITRAC12 (hCOA3), forming assembly intermediates termed MITRAC (Clemente et al., 2013, Dennerlein et al., 2015, Mick et al., 2012, Ostergaard et al., 2015, Weraarpachai et al., 2012). Loss of C12ORF62 or MITRAC12 leads to complex IV-deficiency and concomitantly neuromuscular disorders (Ostergaard et al., 2015, Weraarpachai et al., 2012). The lack of C12ORF62 or MITRAC12 has been found to affect COX1 synthesis, suggesting that these proteins may link complex IV assembly to translational performance of mitochondrial ribosomes in an undefined manner (Clemente et al., 2013, Mick et al., 2012, Szklarczyk et al., 2012, Weraarpachai et al., 2012). This finding lead to the central open question as to how mitochondrial translation can be adjusted to provide sufficient amounts of mitochondrial-encoded proteins to match the demands of OXPHOS biogenesis.
The available information on the early steps of cytochrome c oxidase biogenesis provides us with a unique opportunity to assess mitochondrial translation in the context of assembly between mitochondrial- and nuclear-encoded subunits. Here, we demonstrate that a ribosome nascent chain complex selectively translating COX1 mRNA associates with early assembly factors of the cytochrome c oxidase. Translation of COX1 progresses through defined membrane-integrated translation intermediates that engage with assembly factors in the inner mitochondrial membrane cotranslationally. Most importantly, our analyses reveal translational plasticity in mitochondria. A block in early steps of cytochrome c oxidase assembly stalls translation. The concomitantly accumulated ribosome COX1 nascent chain complex represents an assembly primed state of the translation product. We propose that this mechanism of translational plasticity enables a spatially precise control of translation that eventually allows adaptation to the influx of imported subunits according to cellular demands.
Results
Mitochondrial Ribosomes Associate with the MITRAC Assembly Intermediate
MITRAC represents a group of early assembly intermediates of COX1 (Richter-Dennerlein et al., 2015). C12ORF62 and MITRAC12 are genuine constituents of the MITRAC complexes, required for cytochrome c oxidase assembly and translation of COX1. When mitochondrial translation products are labeled with [35S]methionine prior to immunoisolation of C12ORF62 or MITRAC12, both proteins are found in association with newly synthesized full-length COX1 (Figure 1A). However, in contrast to C12ORF62, MITRAC12 coisolated COX2, which is recruited to COX1 after the first nuclear-encoded subunits have engaged with COX1 (Mick et al., 2011, Soto et al., 2012). This finding suggested that C12ORF62 and MITRAC12 are bound to COX1 at different stages of the COX1 biogenesis process and that C12ORF62 potentially acts earlier. To assess the function of C12ORF62 in cytochrome c oxidase assembly and translation, we determined its interaction network. We generated a stable HEK293T cell line utilizing a genomically integrated C12ORF62FLAG-encoding cassette enabling expression of the tagged protein at physiological level (Figure 1C). C12ORF62FLAG-containing complexes were purified after stable isotope labeling with amino acids in cell culture (SILAC) and copurified proteins identified by quantitative mass spectrometry. As expected, we recovered the known constituents of MITRAC and early assembling structural subunits of complex IV. Unexpectedly, the large and small subunits of the mitochondrial ribosome copurified with C12ORF62 and were detected with a subunit coverage of 91% (Amunts et al., 2015, Greber et al., 2015) (Figure 1B; Table S1). We confirmed ribosome association to C12ORF62 by western blot analyses (Figure 1C). Interestingly, when cells were treated with the translational inhibitor, thiamphenicol, prior to isolation, the ribosome-C12ORF62 association was lost while MITRAC complex constituents, including the previously synthesized COX1 and the mitochondrial export protein OXA1L, remained associated. This suggested that only translating ribosomes engage with C12ORF62, while the interaction with the OXA1L insertase was maintained in the absence of ribosome association (Figure 1C). Based on this, we assessed whether ribosome association was specific to C12ORF62 or common to COX1-associated assembly factors. In immunoprecipitation analyses, MITRAC12 and also MITRAC15, which is required for complex IV as well as complex I biogenesis (Mick et al., 2012), displayed ribosome-association, but to a lesser extent than C12ORF62. In contrast, the late-acting MITRAC7 displayed COX1- and MITRAC-association, but no ribosome interactions (Figure 1D). We concluded that COX1 assembly factors associate with translating ribosomes in early steps of the assembly process.
Figure 1.

C12ORF62 Interacts with Translating Mitochondrial Ribosomes
(A) [35S]methionine-labeled mitochondrial translation products were subjected to immunoprecipitation with indicated antibodies. Total, 5%; eluates, 100%.
(B) Equal amounts of isolated mitochondria from differentially SILAC-labeled HEK293T wild-type cells or cells expressing C12ORF62FLAG were subjected to FLAG-immunoisolation. Purified protein complexes were natively eluted and analyzed by liquid chromatography-mass spectrometry (LC-MS) (n = 4). Thresholds: p < 0.05; mean ratios >5 or >10 (dashed lines).
(C) Isolated mitochondria from HEK293T cells expressing C12ORF62FLAG were treated with thiamphenicol (TAP) (50 μg/mL) for 24 hr where indicated and subjected to FLAG-immunoprecipitation. Total, 3%; eluates, 100%.
(D) FLAG-immunoprecipitation experiments were performed from isolated mitochondria purified from HEK293T cells expressing FLAG tagged mL62, C12ORF62, MITRAC12, MITRAC15, or MITRAC7. Total, 3%; eluates, 100%.
Mitochondrial Ribosomes Associated with C12ORF62 Translate COX1 mRNA
Because C12ORF62 interacted specifically with translating ribosomes, we hypothesized that the MITRAC-associated ribosomes were selectively dedicated to translation of COX1. To assess this directly, we first analyzed the level of COX1 mRNAs that copurified with early and late MITRAC components using real-time PCR. In agreement with our hypothesis, C12ORF62-associated ribosomes contained significant levels of COX1 mRNA and only negligible amounts of COX2 transcript (Figure 2A). To further support and extend this finding by quantitative data, we assessed RNAs that were bound to C12ORF62-associated ribosomes by NanoString analysis. The recovery of 12S and 16S rRNA supported the ribosome isolation shown by western blot analysis (Figure 1D) as both RNAs specifically copurified with C12ORF62 and MITRAC12. Moreover, COX1 mRNA was mainly isolated with C12ORF62 and to a lesser extent with MITRAC12 (Figure 2B). Other mitochondrial-encoded transcripts were only negligibly recovered with C12ORF62. In contrast, when ribosomes were isolated via mL62 (ICT1), a core component of the mitochondrial ribosome (Richter et al., 2010), all tested mitochondrial transcripts copurified (Figure 2C).
Figure 2.

C12ORF62-Associated Mitochondrial Ribosomes Are Committed to COX1 Synthesis
(A–C) Immunoprecipitation from indicated cell lines was performed as in Figure 1D. Isolated RNA samples of total and eluates were quantified by real-time PCR (A) and NanoString technology (B and C). Levels of copurified RNA were calculated relative to mL62 (A) or total (B and C), respectively (mean ± SEM; n = 3).
In yeast mitochondria, mRNAs possess 5′UTRs that are recognized by specific translational activators, which are essential to facilitate translation (Herrmann et al., 2013). In human mitochondria, 5′UTRs and translational activator proteins are not conserved, indicating that different mechanisms for translational regulation have to be in place (Hällberg and Larsson, 2014, Herrmann et al., 2013, Soto et al., 2012). Based on the observed association between ribosomes and early assembly intermediates in human mitochondria, we tested whether Cox14 and Coa3, early assembly factors of Cox1 (Barrientos et al., 2004, Fontanesi et al., 2011, Mick et al., 2011), similarly associated with ribosomes in yeast. Both Cox14 and Coa3 copurified each other and Cox1. However, our analyses did not reveal evidence for an interaction between these assembly factors and mitochondrial ribosomes (Figure S1). In contrast, mitochondrial ribosome copurified efficiently with Mba1, a ribosome receptor in yeast mitochondria (Ott et al., 2006). In summary, in human mitochondria, early COX1 assembly factors engage with mitochondrial ribosomes selectively translating COX1 mRNA. We conclude that, among the known assembly factors, C12ORF62 represents the major ribosome-interacting constituent of the MITRAC complex.
Figure S1.

Yeast Cox14 and Coa3 Assembly Factors Do Not Associate with Mitochondrial Ribosomes, Related to Figure 1
Mitochondria from wild-type (WT) and Mba1ProtA expressing strains were solubilized in digitonin buffer and protein complexes isolated via immunoisolation (IgG chromatography or Cox14- or Coa3-specific antiserum). Eluates were separated by SDS-PAGE and analyzed by western-blotting. Total 1%, eluate 100%.
Distinct COX1 Translation Intermediates Associate with C12ORF62
COX1 consists of 12 transmembrane spans (TM) exposing both termini into the mitochondrial matrix (Tsukihara et al., 1996). To test whether C12ORF62 required a full-length COX1-protein for association, we [35S]methionine pulse-labeled mitochondrial translation products in a cell line carrying a G to A transition in COX1 (G6930A). This mutation generates a “hungry codon” that leads to expression of a main translation product truncated after leucine 342 within the ninth membrane span (COX11–342) (D’Aurelio et al., 2001, Temperley et al., 2010) and minute amounts of full-length COX1 (Figure 3A). Despite the lack of its C-terminal portion, COX11–342 copurified efficiently with C12ORF62 (Figure 3A). Similarly, the 35S-labeled COX11–342 could be coimmunoprecipitated with MITRAC12 and MITRAC7, indicating that the truncated protein enters into early cytochrome c oxidase assembly intermediates (Figure 3B).
Figure 3.

C12ORF62 Associates with COX1 during Synthesis
(A and B) [35S]methionine-labeled mitochondrial translation products from 143B wild-type (WT) and COX1G6930A mutant (G6930A) cell lines were subjected to immunoisolation using indicated antibodies. Total, 5%; eluates, 100%.
(C and D) HEK293T cells were incubated in [35S]methionine-containing medium for 30 min and treated with puromycin (Pur) (2 μg/mL) where indicated for 10 or 20 min. Cell lysates were subjected to C12ORF62 immunoisolation. Total, 5%; eluates, 100%. COX1 fragments (COX1f1–f3), indicated by asterisks, were quantified in (D) (mean ± SEM; n = 3).
(E) HEK293T cells were labeled and treated as in (C). Puromycin was added for 20 min where indicated prior to immunoisolation using C12ORF62 and MITRAC12 antibodies. Samples were analyzed as in (C).
(F and G) Experiments were performed as in (C). Radiolabeled samples were subjected to immunoisolation using antibodies against MITRAC7 (F) and TIM21 (G), in comparison to C12ORF62.
See also Figure S2.
The observed interactions of C12ORF62 with ribosomes (Figure 1) and with the truncated translation-product of COX1 suggested to us that C12ORF62 might associate with nascent COX1. To test this directly, we treated mitochondria during pulse-labeling with puromycin to release nascent chains from mitochondrial ribosomes. Instead of an array of translation products, COX1 (fully translated during pulse) and three distinct shorter translation products (COX1f1–f3) copurified with C12ORF62 (Figure 3C and quantification in Figure 3D). These three translation products also associated with MITRAC12 (Figure 3E). However, we were unable to recover COX1f1–f3 in immunoprecipitations of the later-acting MITRAC7 or TIM21 (Figures 3F, 3G, S2A, and S2B). In contrast, in yeast mitochondria nascent Cox1 did not copurify with the early assembly factor Coa3 upon puromycin treatment (Figure S2C), which is in agreement with the absence of an interaction between Coa3 and mitochondrial ribosomes. In summary, our findings suggest that the early assembly factors, C12ORF62 and MITRAC12, interact with nascent COX1, whereas components of a late MITRAC only associate with the full-length COX1.
Figure S2.

Late COX Biogenesis Factors Do Not Interact with Nascent Chain Fragments as well as Yeast Coa3, Related to Figure 3
(A and B) Longer exposures of experiments shown in Figures 3F and 3G.
(C) Yeast Coa3 does not interact with Cox1 nascent chain. [35S]methionine labeling of mitochondrial translation products was carried out in organello mitochondria. Mitochondrial translational products were labeled for 25 min. Samples were treated with 0.1 mM puromycin (Pur) for 15 min and mitochondria solubilized in digitonin buffer and subjected to immunoisolation using Coa3-specific antiserum. Samples were analyzed by SDS-PAGE and digital autoradiography or western blot. Total 10%, eluate 100%.
Defining Ribosome Nascent Chain Complexes of COX1
Due to its hydrophobicity, COX1 displays aberrant migration on SDS-PAGE. Hence, we estimated the length of the largest truncated translation product by comparing its migration to COX11–342. COX1f3 migrated slightly faster than COX11–342, indicating that translation was terminated upstream of transmembrane span (TM) 9 (Figures 4A and 4C). This was supported by the observation that upon puromycin treatment of the COX1G6930A cell line, the same truncated COX1 products copurified with C12ORF62 as in the wild-type control (Figure 4B). We concluded that C12ORF62 interacted with N-terminal fragments of COX1 that contain less than nine TM helices.
Figure 4.

Membrane Topology and Interactions of COX1 Fragments
(A and B) Mitochondrial translation products were labeled with [35S]methionine in 143B wild-type (WT) and COX1G6930A mutant cells, treated with puromycin (Pur) (2 μg/mL) where indicated and subjected to C12ORF62 immunoprecipitation. Total, 5%; eluates, 100%. Asterisks and a red arrowhead indicate puromycin-released COX1 fragments COX1f1–f3 and mutant COX11–342, respectively.
(C) Schematic presentation of COX1 topology. The position of premature stop codons for in vitro COX1 synthesis, as in (D), are indicated by arrowheads. Relative position of mutation of COX1G6930A in transmembrane domain nine is indicated by a red arrow. N, amino-terminal; C, C-terminal; IMS, intermembrane space; IMM, inner mitochondrial membrane.
(D) HEK293T cells, 143B wild-type (WT) and the COX1G6930A mutant were treated and radiolabeled as in (A). COX1 peptides of defined lengths (indicated by the number of amino acids) were synthesized and radiolabeled in vitro using rabbit reticulocyte lysates. Puromycin (Pur)-released COX1 fragments are indicated by asterisks and aligned with in vitro synthesized COX1 peptides (asterisk). Mutant COX11–342 protein in the COXG6930A cell line is marked by red arrowheads.
(E) Schematic presentation of discontinuously synthesized fragments in relation to COX1 membrane topology.
To define the lengths of the truncated translation products with a molecular ruler, we synthesized a recoded COX1 gene and expressed defined COX1 fragments (Figure 4C) in reticulocyte lysate and compared their migration to the COX1 fragments that interacted with C12ORF62 after puromycin treatment (Figure 4D). We estimated that COX1f3 represented a fragment of ∼280 amino acid (aa), COX1f2 ended after ∼212 aa, and COX1f1 was even slightly shorter (Figure 4D). Taking the topology of COX1 into consideration and the fact that ∼30–60 aa are covered by the ribosomal exit tunnel (Kramer et al., 2009), C12ORF62 appears to recruit ribosome nascent chain complexes when helices 1–4 or 1–6 are translated and membrane inserted. Accordingly, COX1 translation occurs in a discontinuous manner. Three major translation products, which are partially membrane-inserted, are generated (Figure 4E). Moreover, these nascent COX1 chains associate with C12ORF62 in a cotranslational manner. Our results are in agreement with previous findings by Rooijers et al. (2013) using ribosome profiling to show that mitochondrial ribosomes are not equally distributed on mitochondrial transcripts. These data suggest hotspots for ribosome pausing, which agrees with an accumulation of distinct COX1 fragments when released with puromycin.
C12ORF62 and MITRAC12 Act Consecutively
Our analyses showed that both C12ORF62 and MITRAC12 engage with ribosome-nascent chain complexes translating COX1. However, they apparently associate to a different extent (see above). To assess the functional interplay of the two proteins in COX1 translation, we depleted C12ORF62 by small interfering RNA (siRNA)-mediated knockdown (Figure 5A). C12ORF62-deficient cells displayed a severe growth defect compared to the non-targeting control that could be overcome by expression of a siRNA-resistant version of C12ORF62FLAG (scC12ORF62FLAG) (Figure 5B), demonstrating specificity of used siRNA and functionality of FLAG-tagged C12ORF62. When we pulse-labeled mitochondrial translation products with [35S]methionine, a drastic reduction in the amount of COX1 was apparent (Figure 5C). The phenotype observed here agreed with previous analyses on the loss of C12ORF62 (Mick et al., 2012, Weraarpachai et al., 2012). Because COX1 is a target of the m-AAA protease (Hornig-Do et al., 2012), we tested whether the decreased level of newly synthesized COX1 was a result of an increased turnover by the quality control machinery. To this end, we downregulated the m-AAA protease subunit AFG3L2 together with C12ORF62 (Figure S3A). However, when AFG3L2 was depleted, we did not observe a significant increase in the amount of newly synthesized COX1. Accordingly, the decreased level of newly synthesized COX1 observed upon C12ORF62 knockdown is apparently not a result of increased turnover but rather of reduced translation. Interestingly, compared to cells depleted for MITRAC12 (Figure S3B), the amount of COX1 synthesized in 1 hr in the C12ORF62 knockdown was clearly exacerbated suggesting that COX1 synthesis was more sensitive to a loss of C12ORF62 (Figures 5D and S3C). To further ascertain that the observed reduction in COX1 reflected a reduced synthesis rather than an increased turnover, we followed the fate of newly synthesized COX1 in a chase experiment over time. While COX1 displayed a half-life of 24 hr upon MITRAC12 knockdown (Mick et al., 2012), in C12ORF62-deficient cells, turnover of COX1 was indistinguishable from the non-targeting control (Figures 5E and S3D). Accordingly, the reduction in the amount of COX1 observed during pulse labeling cannot be reconciled with the half-life of the protein but rather reflects a reduction in translation.
Figure 5.

C12ORF62 Acts as the Initial COX1 Maturation Protein
(A and B) Depletion efficiency and specificity of C12ORF62 siRNA-mediated downregulation. HEK293T cells were treated with siRNA oligonucleotides against C12orf62 or with control siRNA for 72 hr followed by western blot analysis (A). Expression of a siC12orf62-1-resistant form of FLAG tagged C12ORF62 restores the growth phenotype upon loss of endogenous C12ORF62. Cells inducibly expressing scC12ORF62FLAG were treated with siC12orf62-1 for 72 hr in the presence of tetracycline (Tet) where indicated. Dashed line represents the starting cell number (mean ± SEM, n = 6) (B).
(C–E) COX1 synthesis and stability in C12ORF62-deficient cells. C12ORF62 was depleted from HEK293T cells for 72 hr followed by radiolabeling for 1 hr (C). Levels of newly synthesized COX1 were quantified, internally standardized to ATP6, and normalized to non-targeting control (dashed line) in C12ORF62- and MITRAC12-deficient cells (mean ± SEM, n = 3) (D). COX1 stability was determined after 24 hr chase (mean ± SEM, n = 3) (E).
(F) FLAG immunoisolation of C12ORF62 and MITRAC12. HEK293T cells were [35S]methionine labeled, treated with puromycin (Pur) where indicated, and subjected to immunoprecipitation. Protein complexes were natively eluted and analyzed by SDS-PAGE (left panel) and BN-PAGE (right panel). COX1 fragments are indicated by asterisks and pre-complexes of MITRAC are labeled with MITRACPre1 and MITRACPre2 (# undefined complex).
(G) COX1 translation intermediates can be retrieved for translation. HEK293T cells were incubated with [35S]methionine and chased after media replacement for indicated time points with cold methionine. All samples were treated with puromycin for 20 min prior to MITRAC12FLAG immunoisolations (left panel). COX1 fragments and full-length COX1 isolated by MITRAC12FLAG were quantified (mean ± SEM, n = 4) (right panels).
See also Figure S3.
Figure S3.

Mitochondrial Translation Is Affected upon Reduction of C12ORF62 and MITRAC12, Related to Figure 5
(A) Downregulation of the m-AAA protease does not restore COX1 levels. AFG3L2 was depleted by siRNA application for 72 hr and COX1 quantified after SDS-PAGE and digital autoradiography (mean ± SEM; n = 3).
(B) Steady-state levels of mitochondrial proteins after siRNA-mediated downregulation of MITRAC12. HEK293T cells were treated with siRNA molecules against Mitrac12 for 72 hr followed by western blot analysis.
(C and D) COX1 synthesis and stability in C12ORF62- and MITRAC12-deficient cells. C12ORF62 and MITRAC12 were depleted from HEK293T cells for 72 hr followed by radiolabeling for 1 hr. The cells were either harvested (C) or after media replacement, chased for indicated time points (D). Samples were analyzed by SDS-PAGE and digital autoradiography.
Our results were in agreement with the idea that C12ORF62 acted upstream of MITRAC12 in COX1 biogenesis. To assess directly whether the translation intermediates of COX1 associated with MITRAC12 or C12ORF62 were present in the same protein complex, we released COX1f1–f3 from ribosomes with puromycin, isolated MITRAC12, or C12ORF62 under native conditions, and analyzed the purified complexes by BN-PAGE (Figure 5F). Both proteins purified the MITRAC intermediate containing full-length COX1 (Dennerlein et al., 2015, Mick et al., 2012). In addition, both proteins purified a faster migrating protein complex (MITRACPre2). However, an even smaller complex (MITRACPre1) was unique to the C12ORF62 purified sample (Figure 5F). We conclude that the COX1 translation intermediates initially associate with C12ORF62, and MITRAC12 is recruited sequentially. To assess whether the COX1 nascent chain intermediates could be retrieved for further COX1 synthesis, we performed [35S]methionine-labeling pulse chase analyses and determined the level of nascent and full-length COX1 isolated with MITRAC12FLAG (Figure 5G). As expected, the COX1f1-3 nascent chains decreased during chase, while full-length COX1 increased in a time-dependent manner. Hence, COX1 nascent chain intermediates can mature to full-length COX1 and thus represent productive translation intermediates.
Association of COX4 to Newly Synthesized COX1 Occurs Posttranslationally
During the assembly process of the cytochrome c oxidase, COX4 (yeast Cox5, see Table S2) is the first nuclear-encoded subunit that associates with newly synthesized COX1. Subsequently, COX5A (yeast Cox6) associates with COX1 and COX4 to form an assembly intermediate prior to addition of the mitochondrial-encoded COX2 (Dennerlein and Rehling, 2015, Fontanesi, 2013, Soto et al., 2012). In the mature cytochrome c oxidase, COX4 bridges TM1 and TM12 of COX1, but also contacts COX1 with its matrix and intermembrane space domains (Figures 6A and S4). At steady state, the majority of COX4 resides in complex IV and only a fraction is present in MITRAC assembly intermediates (Figure 6B). Upon puromycin treatment, import of [35S]COX4 into mitochondria and subsequent purification of MITRAC12 or C12ORF62, we did not recover COX4 in MITRACPre1 or MITRACPre2 (Figure 6C). This finding suggested that COX4 only associated with full-length COX1, which agreed well with the available structural data.
Figure 6.

COX4 Interacts with Premature COX1
(A) Structure of COX1 and COX4 in the mature bovine complex IV. PDB: 3ASN; COX4, red; COX1, green with TM1, blue; TM12, orange (Suga et al., 2011).
(B) Two-dimensional BN/SDS-PAGE analysis of COX4. RCS, respiratory chain supercomplexes; IV, complex IV.
(C) HEK293T cells were treated with puromycin (Pur) for 20 min where indicated. Mitochondria were isolated and [35S]methionine-labeled COX4 was imported for 45 min. Samples were subjected to FLAG immunoprecipitation followed by BN-PAGE (upper panel) and SDS-PAGE (lower panel) analyses.
(D) Isolated mitochondria from HEK293T cells expressing COX4FLAG were subjected to FLAG-immunoprecipitation. Total, 1%; eluates, 100%.
(E) Mitochondrial translation products were radiolabeled in HEK293T cells. Cell lysates were subjected to immunoisolation using indicated antibodies. Mitochondrial translation was terminated with puromycin (Pur) prior to isolation where indicated. #, undefined mitochondrial translation product. Total, 5%; eluates, 100%.
(F) Mitochondrial translation products were labeled with [35S]methionine in 143B wild-type (WT) and COX1G6930A mutant cells. Samples were subjected to immunoisolation and analyzed as in (E). Total, 5%; eluates, 100%.
See also Figure S4.
Figure S4.

Interaction Interface Map of COX4 to COX1 in the Mature Bovine Complex IV, Related to Figure 6
Highlighted are COX4 (red), COX1 TM2 and TM11 (green), COX1 TM1 (blue) and COX1 TM12 (orange). PDB: 3ASN (Suga et al., 2011).
Remarkably, we found that readily detectable amounts of mitochondrial ribosomes copurified with COX4, indicating that ribosomes are engaged with MITRAC complexes at the stage of COX4 assembly to COX1 (Figure 6D).
This finding led us to assess whether the first nuclear-encoded structural subunit associated with COX1 cotranslationally. To this end, we tested the interaction of COX4 with nascent COX1f1–f3 by immunoisolation of COX4 after [35S]methionine-labeling. While COX4 copurified full-length COX1, the nascent translation products COX1f1–f3 were not found (Figure 6E). Surprisingly, when COX4 was isolated from COX1G6930A cells, the fully synthesized shortened version COX11–342 was found in complex with COX4 (Figure 6F). This finding demonstrates that TM10–TM12 were dispensable for the initial binding of COX4 to COX1. Despite ribosome association with COX4-containing MITRAC intermediates, COX4 only binds to COX1 upon completion of its synthesis. We conclude, that association of the first nuclear-encoded cytochrome c oxidase subunit, COX4, to COX1 occurs posttranslationally. However, a full-length COX1 molecule is not required for an interaction with COX4.
Defects in COX1 Assembly Cause Accumulation of a Ribosome Nascent Chain Complex
In contrast to COX4, C12ORF62, and MITRAC12 engage with COX1 cotranslationally. A stabilizing function for full-length COX1 has been attributed to MITRAC12, which remains COX1-associated beyond the stage of COX4 assembly (Clemente et al., 2013, Dennerlein et al., 2015, Mick et al., 2012, Ostergaard et al., 2015). At the same time, loss of C12ORF62 or MITRAC12 affects COX1 translation (Figure 5D) (Mick et al., 2012, Ostergaard et al., 2015, Weraarpachai et al., 2012). These observations led us to hypothesize that mitochondrial translation could be coupled to the formation of COX1-containing biogenesis intermediates. Therefore, we assessed whether ribosome performance was affected when the assembly process was compromised. To this end, we immunoisolated C12ORF62 after pulse-labeling of mitochondrial translation products under conditions of siRNA-mediated knockdown of MITRAC12. MITRAC12 depletion specifically led to accumulation of ribosome nascent chain complex containing the COX1f1–f3 fragments in a C12ORF62-associated state (Figure 7A and quantification in Figure 7B). This finding was in agreement with the idea of a link between the COX1 assembly process and mitochondrial ribosome function.
Figure 7.

Nascent Chain Ribosome Complexes Accumulate on Early Assembly Factors upon Block of Complex IV Assembly
(A and B) MITRAC12 was depleted in HEK293T cells by siRNA treatment for 72 hr prior to [35S]methionine labeling and immunoisolation using antibodies as indicated (A). COX1 fragments are labeled by asterisks. Total, 5%; eluates, 100%. (B) Quantification of COX1 fragments upon MITRAC12 depletion (mean ± SEM; n = 3).
(C) Cell lysates (50 μg for MITRAC12, C12ORF62, SDHA, and TIM44; 25 μg for all other proteins) were analyzed by SDS-PAGE and western blotting. Quantification of COX4, COX1, and C12ORF62 protein levels (mean ± SEM, n = 4) after 72 hr knock-down of COX4.
(D) Quantification of newly synthesized COX1 upon COX5B and COX4 depletion. Level of COX1 synthesis (standardized to ATP6) were calculated and normalized to non-targeting control (mean ± SEM; n = 3).
(E) COX4 was ablated in HEK293T cells prior to radiolabeling of mitochondrial translation products. Samples were subjected to immunoprecipitation using indicated antibodies. Radiolabeled protein bands were quantified along the gel lanes and the differential profile (Δintensity) plotted along the gel section of interest. Bottom graph, increase of COX1f1/f2 fragments relative to the control (mean ± SEM; n = 4).
(F) COX4 and MITRAC12 were depleted in HEK293T cells expressing C12ORF62FLAG by siRNA treatment for 72 hr prior to [35S]methionine labeling and FLAG-immunoisolation. Upper panel: western blot analysis of cell lysates. Lower panel: BN-PAGE analysis of natively eluted immunoisolation samples. As a control, one sample was treated with puromycin prior to isolation omitting siRNA treatment.
See also Figure S5.
To analyze whether mitochondrial ribosomes display translational plasticity to adapt translation in response to the availability of nuclear-encoded subunits, we depleted COX4 by siRNA. Western blot analyses of cell lysates demonstrated efficient COX4 depletion. In addition, the level of COX1 was decreased leading to the loss of functional complex IV (Figures 7C and S5A). Obviously, this steady-state analysis did not discriminate between the synthesis and turnover of COX1 under conditions of COX4 depletion. Therefore, we assessed translation of COX1 by radiolabeling of mitochondrial translation products under conditions of siRNA-mediated downregulation of the early assembling COX4 and as a control the later assembling COX5B (yeast Cox4). While depletion of COX5B had little effect on COX1 translation, a depletion of COX4 noticeably reduced COX1 synthesis (Figure 7D). To assess directly whether the reduced synthesis of COX1 correlated with the formation of the COX1 ribosome nascent chain complex, we immunoisolated C12ORF62 after radiolabeling of translation products in COX4-deficient cells. As expected, ablation of COX4 stalled COX1 translation and led to accumulation of the nascent COX1 fragments in a C12ORF62-bound state (Figure 7E). In contrast, ablation of COX6C (yeast Cox9), a late assembling nuclear-encoded structural subunit, and loss of the assembly factors SCO2 or SURF1, which participate in copper- and heme-insertion respectively, did not lead to an accumulation of nascent COX1 associated with C12ORF62 (Figures S5B–S5D). We concluded that only the first steps in the COX assembly process impact translational plasticity.
Figure S5.

Reduction of COX6C, SCO2, and SURF1 Does Not Lead to an Accumulation of Nascent Chain Ribosome Complexes, Related to Figure 7
(A) Depletion of COX4 leads to cytochrome c oxidase deficiency. Isolated mitochondria from siRNA mediated COX4 ablated HEK293T cells (72 h) were subjected to BN-PAGE and activity staining.
(B) Steady state of mitochondrial proteins after siRNA-mediated downregulation (72 h) of COX5B, COX6C, SCO2 and in Surf1−/− cells.
(C) Mitochondrial protein synthesis in COX6C- and SCO2-depleted cells, and in Surf1−/− HEK293T cells. Mitochondrial translation products were radiolabeled for 1 hr in HEK293T cells treated with siRNAs against Cox6C and Sco2 (72 h) and in Surf1−/− cells. Cells were harvested and samples analyzed by SDS-PAGE and digital autoradiography.
(D) Nascent COX1 does not accumulate in a C12ORF62 complex upon loss of COX6C, SCO2 or SURF1. COX6C and SCO2 were ablated in HEK293T cells by siRNA application for 72h prior to radiolabeling of mitochondrial translation products. Samples were subjected to immunoprecipitation using indicated antibodies and eluates analyzed by SDS-PAGE and autoradiography. Total, 5%; eluates, 100%
To address whether COX4 was required for the formation of MITRACPre complexes, we depleted COX4 and isolated COX1-intermediates via C12ORF62FLAG after [35S]methionine labeling (Figure 7F). Interestingly, COX4 depletion led to accumulation of MITRACPre1 and a concomitant loss of MITRACPre2. Similar results were obtained when MITRAC12 was downregulated. Hence, loss of COX4 or MITRAC12 stalls nascent COX1 in association with C12ORF62 in the MITRACPre1 complex. In summary, we find that upon loss of the nuclear-encoded COX4, translation of COX1 is stalled in a ribosome-nascent chain complex, together with C12ORF62. As a result, the amount of newly synthesized full-length COX1 decreases demonstrating translational plasticity in response to the presence or absence of the nuclear-encoded structural subunit COX4.
Discussion
Protein complexes of the mitochondrial oxidative phosphorylation system are built from subunits of dual genetic origin. A central and unanswered question is whether regulatory mechanisms exist that allow cells to control the supply and to avoid unproductive accumulation of nuclear- and mitochondrial-encoded subunits in mitochondria (Barrientos et al., 2002, Greber and Ban, 2016, Herrmann and Funes, 2005, Richter-Dennerlein et al., 2015). Recent work showed that, in general, the transport of nuclear-encoded proteins into mitochondria is integrated into metabolic and cell-cycle control pathways (Harbauer et al., 2014a, Harbauer et al., 2014b, Opalińska and Meisinger, 2015). With regard to the synthesis of mitochondrial-encoded proteins, micro RNAs have recently been found to stimulate mitochondrial translation during muscle differentiation (Zhang et al., 2014). However, it is currently unknown whether and how translation in mitochondria can be balanced against the influx of nuclear-encoded respiratory chain subunits. Here, we demonstrate that mitochondrial ribosomes are not unrestrained protein production enzymes but rather display translational plasticity to cope with an altered requirement of mitochondrial-encoded subunits. Ribosomes that are specifically dedicated for COX1 translation associate with early MITRAC components, C12ORF62 and MITRAC12. C12ORF62 acts upstream of MITRAC12 and associates with COX1 in a ribosome nascent chain complex. It is interesting to note that the conserved protein insertase OXA1L (Funes et al., 2011) copurifies with C12ORF62. This association is also observed under conditions of thiamphenicol treatment when translation is blocked and ribosomes are lost from the complex. It is tempting to speculate that in the inner membrane, a pool of OXA1L molecules is dedicated to bind a specific ribosome, translating a dedicated mRNA, and linking this translation process to an assembly route. For this to occur, specific accessory factors, in the case of COX1 the C12ORF62, would have to associate with OXA1L to facilitate selectivity in the membrane insertion of a single defined mitochondrial-encoded polypeptide chain.
Our analyses demonstrate that a block in the assembly process of the cytochrome c oxidase by depletion of the first nuclear-encoded subunit that engages with COX1 stalls translation of COX1. The ribosome nascent chain complex maintains COX1 translation intermediates in a partially membrane integrated (≥TM1–TM4) and C12ORF62-bound state. We show that this ribosome-arrested pool of COX1 represents the assembly primed state that can be released to continue for full translation when COX1 is recruited into subsequent assembly steps. Interestingly, C12ORF62 deficiency leads to decreased COX1 synthesis, however, COX1 stability remains unaltered (shown here and in Mick et al., 2012, Weraarpachai et al., 2012), suggesting a direct effect of C12ORF62 on COX1 translation performance by MITRAC-associated ribosomes. In contrast, ablation of MITRAC12 has a milder effect on translation and causes reduced stability of newly synthesized COX1. This finding is in agreement with the idea that two pathways contribute to balancing the amount of COX1. Ribosome translational plasticity allows to regulate the supply of subunits for assembly in response to early steps of the assembly pathway and proteolytic quality control mechanisms act subsequently (Dennerlein et al., 2015, Quirós et al., 2015). The observed interaction between mitochondrial ribosomes and early assembly intermediates in human mitochondria reveals a functional and physical link between formation of stalled COX1 nascent chains attached to the ribosome and the cytochrome c oxidase biogenesis process.
Apparently, mitochondrial translational plasticity is not limited to metazoa. A recent study by Couvillion et al. (2016) demonstrated a synchronization of mitochondrial and cytosolic translation processes in Saccharomyces cerevisiae. Upon nutrient-induced stimulation of nuclear-encoded gene expression, translation of mitochondrial-encoded respiratory chain subunits is enhanced. Interestingly, stimulation of mitochondrial translation relies on protein import into mitochondria. This finding indicates that a flux of molecules from the cytosol to mitochondria triggers the increase in translation. In contrast to human mitochondrial mRNAs, yeast mRNA possess long 5′UTRs, which are bound by mRNA-specific translational activator proteins to promote their translation. In case of yeast, Cox1, one of the translational activators is Mss51. Mss51 activity is regulated in a feedback cycle. Upon accumulation of Cox1 assembly intermediates, Mss51 is recruited to the newly synthesized Cox1 in complex with the assembly factors Coa3 and Cox14. This recruitment and potentially additional assembly factors inactivate Mss51 to stall translation of the COX1 mRNA (for review see Dennerlein and Rehling, 2015, Herrmann et al., 2013, Soto et al., 2012). Here, we find that under our experimental conditions, the assembly factors Coa3 and Cox14 do not bind to mitochondrial ribosomes. This is in contrast to what we observed for MITRAC constituents in human. Accordingly, the mechanistic bases for translational plasticity in yeast and human must be very different because 5′UTRs and translational activators are not conserved. Our finding of a direct physical link of the translation machinery with assembly intermediates in human might provide some insight as to why human mitochondrial mRNAs have lost significant 5′UTRs and RNA-binding translation activators to regulate protein expression.
The metazoan mechanism of translational plasticity identified here represents an elegant way to adapt mitochondrial protein production to the cellular demands. In principle, many different mechanisms have been identified to regulate elongation of nascent polypeptide chains (Rodnina, 2016). Given the limited genetic accessibility of mitochondria and the fact that many basic aspects of mitochondrial translation are still enigmatic, it will be a challenging task to assess how processivity of the mitochondrial ribosome is modulated at the molecular level.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal antibody to human C12ORF62 | This paper | PRAB4845 |
| Rabbit polyclonal antibody to human MITRAC12 | This paper | PRAB3762 |
| Rabbit polyclonal antibody to human MITRAC7 | This paper | PRAB4843 |
| Rabbit polyclonal antibody to human COX1 | This paper | PRAB3035 |
| Rabbit polyclonal antibody to human COX6A | This paper | PRAB3283 |
| Rabbit polyclonal antibody to human uL1m | This paper | PRAB4964 |
| Mouse monoclonal antibody to human bL12m | Abcam | ab58334 |
| Rabbit polyclonal antibody to human uL23m | This paper | PRAB1716 |
| Rabbit polyclonal antibody to human uS15m | Proteintech | 17006-1 |
| Rabbit polyclonal antibody to human mS40 | Proteintech | 16139-1 |
| Mouse monoclonal antibody to human OXA1L | Proteintech | 66128-1 |
| Rabbit polyclonal antibody to human MITRAC15 | This paper | PRAB4814 |
| Mouse monoclonal antibody to FLAG tag | Sigma Aldrich | F3165; RRID: AB_259529 |
| Mouse monoclonal antibody to human COX2 | Abcam | ab110258; RRID: AB_10887758 |
| Rabbit polyclonal antibody to human TIM21 | This paper | PRAB3674 |
| Rabbit polyclonal antibody to human COX4 | This paper | PRAB1522 |
| Mouse monoclonal antibody to human SDHA | Novex | 459200 |
| Rabbit polyclonal antibody to human Rieske | This paper | PRAB1512 |
| Rabbit polyclonal antibody to human ATP5B | This paper | PRAB4826 |
| Rabbit polyclonal antibody to human TIM44 | Proteintech | 13859-1 |
| Rabbit polyclonal antibody to human NDUFB8 | This paper | PRAB3764 |
| Rabbit polyclonal antibody to human TIM23 | This paper | PRAB1527 |
| Rabbit polyclonal antibody to human COX5B | Proteintech | 11418-2 |
| Rabbit polyclonal antibody to human SCO2 | This paper | PRAB4982 |
| Rabbit polyclonal antibody to human COX6C | This paper | PRAB4949 |
| Rabbit polyclonal antibody to human SURF1 | This paper | PRAB1528 |
| Rabbit polyclonal antibody to yeast Mba1 | This paper | PRAB0263 |
| Rabbit polyclonal antibody to yeast Coa3 | This paper | PRAB2046 |
| Rabbit polyclonal antibody to yeast Cox14 | This paper | PRAB1544 |
| Rabbit polyclonal antibody to yeast Cox1 | This paper | PRAB0372 |
| Rabbit polyclonal antibody to yeast Mrp51 | Green-Willms et al., 1998 | N/A |
| Rabbit polyclonal antibody to yeast uL29m | Gruschke et al., 2010 | N/A |
| Rabbit polyclonal antibody to yeast Cox4 | This paper | PRAB0578 |
| Rabbit polyclonal antibody to yeast Tim17 | This paper | PRAB0302 |
| Rabbit polyclonal antibody to yeast Tom70 | This paper | PRAB3530 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Anti-FLAG M2 Affinity Gel | Sigma-Aldrich | A2220 |
| L-lysine:2HCl - 13C6; 15N2 | Cambridge Isotope Lab | 13L-177 |
| L-arginine:HCl - U-13C6; U-15N4 | Cambridge Isotope Lab | 13C-450 |
| Protein-A Sepharose CL-4B | GE Healthcare | 17-0963-03 |
| [35S]methionine | Hartmann Analytic | SCM-01 |
| Puromycin | invivoGen | Ant-pr-1 |
| Emetine dihydrochloride hydrate | Sigma-Aldrich | 219282 |
| Anisomycin | AppliChem | A7650,0025 |
| Hygromycin B | Invitrogen | 10687010 |
| Lipofectamine RNAiMax | Invitrogen | 13778-150 |
| GeneJuice | Novagen | 70967-3 |
| Critical Commercial Assays | ||
| SensiMix Sybr Low-Rox Kit | Bioline | QT62-05 |
| First Strand cDNA Synthesis kit | ThermoFisher Scientific | K1612 |
| Rapid DNA Ligation Kit | ThermoFisher Scientific | K1422 |
| mMESSAGE mMACHINE® SP6 Transcription Kit | Ambion | AM1340 |
| Flexi rabbit reticulocyte lysate system | Promega | L4540 |
| Experimental Models: Cell Lines | ||
| Human osteosarcoma 143B cells | D’Aurelio et al., 2001 | N/A |
| HEK293-Flp-In T-Rex | ThermoFisher Scientific | R78007 |
| HEK293-Flp-In T-Rex-C12ORF62FLAG | This study | N/A |
| HEK293-Flp-In T-Rex-COX4FLAG | This study | N/A |
| HEK293-Flp-In T-Rex-MITRAC7FLAG | Dennerlein et al., 2015 | N/A |
| HEK293-Flp-In T-Rex-MITRAC12FLAG | Mick et al., 2012 | N/A |
| HEK293-Flp-In T-Rex-MITRAC15FLAG | Mick et al., 2012 | N/A |
| HEK293-Flp-In T-Rex-mL62FLAG | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| YPH499 - Mat a, ade2-101 his3-Δ200 leu2-Δ1 ura3-52 trp1-Δ63 lys2-801 | Sikorski and Hieter, 1989 | N/A |
| Mba1ProtA - Mat a, ade2-101 his3-Δ200 leu2-Δ1 ura3-52 trp1-Δ63 lys2-801; mba1::mba1-TEV-ProtA-7HIS-HIS3MX6 | Lorenzi et al., 2016 | N/A |
| Recombinant DNA | ||
| pOG44 Flp-Recombinase Expression Vector | ThermoFisher Scientific | V600520 |
| pcDNA5/FRT/TO | ThermoFisher Scientific | V6520-20 |
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | Addgene | 42230 |
| pEGFP-N1 | Clontech | N/A |
| Sequence-Based Reagents | ||
| See Table S4 for primer sequences | N/A | N/A |
| Software and Algorithms | ||
| MaxQuant - version 1.5.1.0 | Cox and Mann, 2008 | http://www.coxdocs.org/doku.php?id=:maxquant:start |
| ImageQuantTL 7.0 software | GE Healthcare | http://www.gelifesciences.com/webapp/wcs/stores/servlet/catalog/en/GELifeSciences-de/products/AlternativeProductStructure_16016/ |
| ImageJ 1.47v | NIH | https://imagej.nih.gov/ij/notes.html |
| PyMOL | DeLano et al., 2000 | https://www.pymol.org/ |
| CCP4Interface - version 7.0.015 | Winn et al., 2011 | http://www.ccp4.ac.uk/index.php |
Contact for Reagent and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Peter Rehling (Peter.Rehling@medizin.uni-goettingen.de).
Experimental Model and Subject Details
Cell Lines and Culture Conditions
Human embryonic kidney cell lines (HEK293-Flp-In T-Rex; HEK293T), 143B wild-type, and 143B COX1G6930A (D’Aurelio et al., 2001) were cultured in DMEM (Dulbecco’s modified eagle medium) supplemented with 10% (v/v) fetal bovine serum (FBS), 2 mM L-glutamine and 50 μg/ ml uridine at 37°C under 5% CO2 humidified atmosphere.
An inducible HEK293T cell line expressing C-terminal FLAG tagged C12ORF62, mL62 (ICT1) or COX4 were generated as described (Mick et al., 2012). In brief, HEK293T cells were transfected with pOG44 and pcDNA5/FRT/TO containing the respective FLAG construct using GeneJuice as transfection reagent. Selection was started two days after transfection using hygromycin B (100 μg/ ml). After two weeks single clones were isolated.
Surf1−/− HEK293T cell line was generated applying the CRISPR/Cas9 technology as previously described (Ran et al., 2013). Briefly, oligonucleotides (Table S4) containing the guide sequences were annealed and ligated into the pX330 vector. HEK293T cells were transfected with pX330-Surf1 and with the pEGFP-N1 plasmid. After three days single cells expressing GFP were sorted by flow cytometry into 96 well plates. After colony expansion single colonies were screened by immunoblotting.
For SILAC experiments, cells were cultured for five passages in DMEM lacking arginine and lysine and supplemented with 10% (v/v) dialyzed FBS, 600 mg/ l proline, 42 mg/ l arginine hydrochloride (13C615N4-arginine “heavy”) and 146 mg/ l lysine hydrochloride (13C615N2-lysine “heavy”). Cell counts were performed using a Neubauer chamber.
Yeast Strains and Culture Conditions
S. cerevisiae strains derived from YPH499 were used in this study (Sikorski and Hieter, 1989). The Mba1ProtA expressing strain was generated by genomic insertion of a HIS3MX6 marker-containing cassette as described previously (Janke et al., 2004). Expression of the fusion protein was confirmed by western blot analysis.
Yeast cells were grown in liquid media containing 1% yeast extract, 2% peptone and 3% glycerol at 30°C with shaking. Isolation of yeast mitochondria was performed as previously described (Meisinger et al., 2006). For this yeast cells were grown to an OD600 of 2 at 30°C. After isolation of yeast mitochondria, the mitochondrial pellet was suspend in SEM buffer (250 mM saccharose, 1 mM EDTA and 10 mM MOPS) at a concentration of 10 mg protein/ml. Mitochondria were then aliquoted and snap-frozen in liquid nitrogen and store at −80°C.
Method Details
siRNA Transfection
HEK293T cells were transfected with siRNA oligonucleotides (8.25 to 33 nM final) (Eurogentec) using Lipofectamine RNAiMAX (Invitrogen) following the reverse transfection protocol provided by the company. Cells were cultured for 72h prior analyses. Sequences for the sense strands of oligonucleotides are provided in Table S4.
In Vitro Protein Synthesis and Import Analysis
Cloning, RNA and protein synthesis were performed as previously described (Dennerlein et al., 2015). Briefly, cDNA was generated from isolated RNA using First Strand cDNA Synthesis kit (Thermo Scientific). PCR amplicons were digested for 30 min at 37°C using Fast Digest Restriction enzymes (Thermo Scientific) and ligated into appropriate vectors using the Rapid DNA Ligation Kit (Thermo Scientific).
Protein import into human mitochondria isolated from HEK293T cells was performed as described by Lazarou et al. (2009). In brief, to generate radiolabeled precursor proteins, RNA was synthesized from PCR products, which were amplified from cDNA. Proteins were synthesized in the presence of [35S]methionine using rabbit reticulocyte lysates (Promega) following the manufacture instructions. Radiolabelled proteins were incubated with freshly isolated mitochondria in import buffer (250 mM sucrose, 20 mM HEPES/KOH [pH 7.4], 5 mM MgOAc, 15 mM sodium succinate, 15 mM malate, 3 mM ATP and 3 mM NADH) for 45 min, at 37°C, 450 rpm. Mitochondria were pelleted at 10,000 × g, washed in 150 μl SEM buffer (250 mM sucrose, 1 mM EDTA, 10 mM MOPS [pH 7.2]) and solubilized for either immunoprecipitation or BN-PAGE analysis. After immunoprecipitation experiments eluates were separated either by SDS- or BN-PAGE and analyzed by digital autoradiography using Phosphorimager Screens and Storm 820 scanner from GE Healthcare. For quantifications the ImageQuant TL software was used (GE Healthcare). To generate in vitro truncated COX1 peptides, a premature stop codon (UAA) was inserted where indicated. Primers are listed in Table S4. Numbers represent the length of the synthesized peptides. The genetic code was adjusted for expression on cytosolic ribosomes of rabbit.
In Vivo [35S]Methionine Labeling of Mitochondrial Translation Products
The labeling of mitochondrial-encoded peptides was performed in 25 cm2 flasks as previously described (Chomyn, 1996) with some modifications. Cytosolic translation was inhibited with 100 μg/ml emetine or anisomycin. Mitochondrial-encoded proteins were synthesized in the presence of 200 μCi/ml [35S]methionine for the indicated time intervals. Mitochondrial translation was inhibited in pulse experiments using 2 μg/ml puromycin for 10 or 20 min where indicated prior to sample analyses. For chase experiments to measure COX1 stability the media was replaced by standard growth media and cells were further cultured for indicated time points. Radioactive signals were detected using Phosphor Screens and a Storm 820 scanner from (GE Healthcare). Eluted COX1 fragments were quantified in relation to full-length isolated COX1 and are presented relative to the control.
To measure the kinetic reduction of COX1 fragments and increase in full-length COX1 associated with MITRAC12, HEK293T-MITRAC12FLAG cells were pulse-labeled (10 min) with [35S]methionine as described. The chase was performed with standard growth media, supplemented with 200 ng/ml L-methionine. Cells were further cultured for indicated time points followed by puromycin treatment for 20 min. Cells were lysed and applied to immunoisolation. Eluted full-length COX1 and COX1 fragments after chase (5, 10 and 20 min) were quantified in relation to isolated full-length COX1 or COX1 fragments, respectively after 10 min pulse (chase = 0).
Affinity Purification Procedures
Affinity purification experiments were performed as described (Dennerlein et al., 2015) with modifications. Briefly, isolated mitochondria or whole cell extracts were suspended in lysis buffer (50 mM Tris/HCl [pH 7.4]; 150 mM NaCl; 20 mM MgCl2; 10% glycerol; 1 mM PMSF and 1% digitonin) and incubated for 30 min at 4°C prior to centrifugation at 16,000 × g at 4°C for 15 min. Supernatants were incubated either with anti-FLAG M2 Affinity Gel (Sigma) or with specific or control antibodies conjugated to ProteinA-sepharose (GE Healthcare) for 1h. After washing of the resin, proteins were eluted with FLAG peptides or by pH shift (0.1 M Glycin [pH 2.8]).
BN-PAGE
Standard protocols for BN-PAGE were used as described previously (Mick et al., 2012). In brief, native protein complexes were separated on 4%–14% BN-PAGE. Prior to the gel run, mitochondria or cells were solubilized in 1% digitonin-containing buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 10% Glycerol, 20 mM MgCl2, 1 mM PMSF) at a concentration of 1 mg/ml. Insoluble material was removed by centrifugation (16,000 × g, 15 min, at 4°C). The supernatant was mixed with BN-loading buffer (0.5% Coomassie Brilliant Blue G-250, 50 mM 6-aminocaproic acid, 10 mM Bis-Tris/HCl [pH 7]) and subjected to BN-PAGE analysis.
RNA Isolation, Quantitative Real-Time PCR, and NanoString Analyses
RNA was isolated from cell extracts or elution fractions after performing FLAG affinity purification experiments using TRIzolR reagent (Invitrogen), following the manufacturers instructions.
cDNA was synthesized using random hexamer primer with the First strand cDNA Synthesis kit from Thermo Scientific, following manufactures instructions. Reactions for quantitative Real-Time (RT)-PCR analysis were prepared using the SensiMix SYBR Low-Rox One Step kit (Bioline). Samples of each experiment were quantified in triplicate and compared to mL62FLAG eluates (cDNA of mL62 eluates were used in a 1/10 dilution). Primer pairs are listed in Table S4.
NanoString measurements were performed by the Microarray and Deep-Sequencing Core Facility, University Medical Center Göttingen (UMG), following the instructions provided by NanoString Technologies. Data of elution fractions was normalized to the input sample. Sequences of probes are listed in Table S3.
Mass Spectrometric Analyses
Equal amounts of mitochondria isolated from differentially SILAC-labeled HEK293T wild-type cells or cells expressing C12ORF62FLAG were mixed followed by FLAG-immunoisolation (n = 4 including label switch). Purified proteins were separated on a NuPAGE Novex 4%–12% Bis-Tris gradient gel (Thermo Fisher Scientific) and visualized with colloidal Coomassie Brilliant Blue G-250 followed by cutting of gel lanes into 12 equal slices. Gel slices were destained, cysteine residues reduced and alkylated, proteins in-gel digested using trypsin, and peptides prepared for LC-MS analysis as described previously (Schwarz et al., 2015). LC-MS analyses were performed on an Orbitrap Elite instrument (Thermo Fisher Scientific, Bremen, Germany) directly coupled to an UltiMate 3000 RSLCnano system (Thermo Scientific, Dreieich, Germany). Peptides were washed and preconcentrated for 5 min on a C18 μ-precolumn (Acclaim PepMap μ-Precolumn Cartridge; 0.3 mm × 5 mm, particle size 5 μm, Thermo Scientific) at a flow rate of 30 μl/min using 0.1% trifluoroacetic acid. Peptides were separated on a C18 reversed-phase nano LC column (Acclaim PepMap RSLC analytical column; 75 μm × 50 cm; particle size 2 μm; pore size 100 A°; Thermo Scientific) at a temperature of 40°C using a binary solvent system consisting of 4% DMSO in 0.1% formic acid (solvent A) and 50% methanol, 30% acetonitrile, and 4% DMSO in 0.1% formic acid (solvent B). For peptide elution, a 30 min linear gradient of 1%–65% solvent B followed by 65%–95% solvent B in 5 min was employed. The flow rate was 250 nl/min.
The Orbitrap Elite was weekly calibrated using standard compounds and operated with the following settings: acquisition of full MS scans (m/z 370 to 1,700) in the orbitrap at a resolution of 120,000 (at m/z 400) with an automatic gain control (AGC) of 1 × 106 ions and a maximum injection time of 200 ms; collision-induced dissociation (CID) of up to 15 of the most intense precursor ions (charge ≥ +2) in the linear ion trap applying a signal threshold of 2,500, an AGC of 5 × 103, a maximum injection time of 150 ms, a normalized collision energy of 35%, an activation q of 0.25, and an activation time of 10 ms. The dynamic exclusion time for previously fragmented precursor ions was set to 45 s.
Mass Spectrometric Data Analysis
Mass spectrometric raw data were processed with MaxQuant (version 1.5.1.0) (Cox and Mann, 2008) and its integrated search engine Andromeda (Cox et al., 2011). Data were searched using the organism-specific UniProt human protein database including protein isoforms (version 2015_02; 89,796 entries) and a set of common contaminants provided by MaxQuant. The database search was performed with the following parameters: tryptic specificity with a maximum of two missed cleavages; precursor mass tolerances of 20 ppm for the first and 4.5 ppm for the main search; fragment ion mass tolerance of 0.5 Da; oxidation of methionine and acetylation of protein N-termini as variable, carbamidomethylation of cysteine as fixed modification; 13C6/15N2-lysine (Lys8) and 13C6/15N4-arginine (Arg10) as heavy labeled amino acids; false discovery rate of < 1% on peptide and protein level. For protein identification, at least one unique peptide with a minimum of six amino acids was required.
SILAC-based relative protein quantification, performed using MaxQuant default settings including “match between runs,” was based on unique peptides and a minimum ratio count of one. Ratios, reported as C12ORF62/WT values (Extended Data Table S1), were log10-transformed. The mean log10 ratio across all replicates was calculated for each protein and p values using a one-sided Student’s t test were determined. Proteins with a SILAC ratio ≥ 5 and a p value < 0.05 were considered to be specifically associated with C12ORF62 complexes.
In Organello Labeling of Mitochondrial Translation Products and Isolation of Yeast Mitochondrial Proteins
Mitochondrial translation products were labeled for 25 min with 20 μM [35S]methionine at 30°C as previously described (Mick et al., 2010). Samples were treated with 0.1 mM puromycin for 15 min at 30°C. Per reaction, mitochondria were mixed with freshly prepared translation buffer (900 mM Sorbitol, 225 mM KCl, 22.5 mM Kpi [pH 7.4], 30 mM Tris [pH 7.5], 4.5 mg/ml BSA, 6 mM ATP, 0.75 mM GTP, 9 mM 2-Ketoglutarat, 12 mM Creatin Phosphate, 19 mM MgSO4, 7.5 μg/ml Cycloheximide, 0.15 mM amino acid mixture without Methionine) containing 2.5 mg/ml of creatine kinase. Labeling reaction was started by addition of [35S]methionine. Prior to coimmunoprecipitation, mitochondria were washed with SEM buffer (250 mM sucrose, 1 mM EDTA and 10 mM MOPS). To detect radioactive signals, Storage Phosphor Screens (GE Healthcare) were used and images were digitized using a Storm820 scanner (GE Healthcare).
Mba1ProtA isolation and immunoisolation of Coa3 and Cox14 were performed as described previously (Bareth et al., 2013). Briefly, the mitochondrial fraction was solubilized in 1% digitonin buffer containing 150 mM NaCl, 50 mM Tris-HCl [pH 7.5], 10% glycerol, 2 mM phenylmethylsulfonyl fluoride (PMSF) and 20 mM MgCl2 for 30 min on ice. Samples were clarified by centrifugation at 20,000 × g at 4°C for 10 min and added to beads. Binding was carried out for 2 hr with shaking at 4°C. Beads were washed with washing buffer containing 0.3% digitonin. For the Mba1ProtA isolation, bound material was eluted by overnight cleavage at 4°C with 0.4 mg/ml AcTEV (tobacco etch virus [TEV]) protease. For the Coa3 and Cox14 isolation elution was performed with 0.1 M Glycin (pH 2.8). Eluates were subsequently analyzed by SDS-PAGE and western blotting.
Data and Structural Analysis
Autoradiograms were quantified using ImageQuantTL 7.0 software (GE Healthcare). Western blots were quantified with a LAS3000 imaging system and the ImageJ 1.47v software. For the cytochrome c oxidase structure the Protein Data Bank entry 3ASN und PyMOL were used. The interaction between COX4 and COX1, residues with a maximal distance of 5 Å between both proteins were identified using CCP4Interface (version 7.0.015). Selected residues were highlighted within the structure using PyMOL.
Quantification and Statistical Analysis
Statistical parameters including exact value of n (number of independent experimental replications), the definition of center and dispersion and precision measures (mean ± SEM) are reported in the figures and the figure legends. Mass spectrometric data are judged to be statistically significant when p < 0.05 by one-sided Student’s t test.
Author Contributions
Conceptualization, R.R.-D., S.D., and P.R.; Validation, S.D.; Formal Analysis, S.D., A.S., S.O., and B.W.; Investigation, R.R.-D., S.D., I.L., B.B., C.R., and C.W.; Writing – Original Draft, R.R.-D., S.D., and P.R.; Writing – Review & Editing, R.R.-D., S.D., and P.R.; Visualization, R.R.-D. and S.D.; Supervision, S.D. and P.R.
Acknowledgments
We thank M. Rodnina and S. Callegari for critical reading, G. Manfredi and J. Montoya for the G6930A mutant cell line, M. Ott and T.D. Fox for antibodies, B. Knapp for technical assistance, G. Salinas-Riester for support with NanoString analyses, S. Jakobs and A. Dickmanns for support, and A. Barrientos, T.D. Fox, and E.A. Shoubridge for discussion. Supported by the Deutsche Forschungsgemeinschaft (B.W. and P.R.), SFB860, GGNB, EXC 294 BIOSS (B.W.), ERC (ERCAdG 339580) to P.R., MWK FoP 88b (P.R.), and the Max Planck Society (P.R.).
Published: September 29, 2016
Footnotes
Supplemental Information includes five figures and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.09.003.
Supplemental Information
References
- Amunts A., Brown A., Toots J., Scheres S.H.W., Ramakrishnan V. Ribosome. The structure of the human mitochondrial ribosome. Science. 2015;348:95–98. doi: 10.1126/science.aaa1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antozzi C., Zeviani M. Cardiomyopathies in disorders of oxidative metabolism. Cardiovasc. Res. 1997;35:184–199. doi: 10.1016/s0008-6363(97)00141-7. [DOI] [PubMed] [Google Scholar]
- Bareth B., Dennerlein S., Mick D.U., Nikolov M., Urlaub H., Rehling P. The heme a synthase Cox15 associates with cytochrome c oxidase assembly intermediates during Cox1 maturation. Mol. Cell. Biol. 2013;33:4128–4137. doi: 10.1128/MCB.00747-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos A., Barros M.H., Valnot I., Rötig A., Rustin P., Tzagoloff A. Cytochrome oxidase in health and disease. Gene. 2002;286:53–63. doi: 10.1016/s0378-1119(01)00803-4. [DOI] [PubMed] [Google Scholar]
- Barrientos A., Zambrano A., Tzagoloff A. Mss51p and Cox14p jointly regulate mitochondrial Cox1p expression in Saccharomyces cerevisiae. EMBO J. 2004;23:3472–3482. doi: 10.1038/sj.emboj.7600358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel N.S. Evolution of mitochondria as signaling organelles. Cell Metab. 2015;22:204–206. doi: 10.1016/j.cmet.2015.05.013. [DOI] [PubMed] [Google Scholar]
- Chomyn A. In vivo labeling and analysis of human mitochondrial translation products. Methods Enzymol. 1996;264:197–211. doi: 10.1016/s0076-6879(96)64020-8. [DOI] [PubMed] [Google Scholar]
- Clemente P., Peralta S., Cruz-Bermudez A., Echevarría L., Fontanesi F., Barrientos A., Fernandez-Moreno M.A., Garesse R. hCOA3 stabilizes cytochrome c oxidase 1 (COX1) and promotes cytochrome c oxidase assembly in human mitochondria. J. Biol. Chem. 2013;288:8321–8331. doi: 10.1074/jbc.M112.422220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvillion M.T., Soto I.C., Shipkovenska G., Churchman L.S. Synchronized mitochondrial and cytosolic translation programs. Nature. 2016;533:499–503. doi: 10.1038/nature18015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- D’Aurelio M., Pallotti F., Barrientos A., Gajewski C.D., Kwong J.Q., Bruno C., Beal M.F., Manfredi G. In vivo regulation of oxidative phosphorylation in cells harboring a stop-codon mutation in mitochondrial DNA-encoded cytochrome c oxidase subunit I. J. Biol. Chem. 2001;276:46925–46932. doi: 10.1074/jbc.M106429200. [DOI] [PubMed] [Google Scholar]
- DeLano W.L., Ultsch M.H., de Vos A.M., Wells J.A. Convergent solutions to binding at a protein-protein interface. Science. 2000;287:1279–1283. doi: 10.1126/science.287.5456.1279. [DOI] [PubMed] [Google Scholar]
- Dennerlein S., Rehling P. Human mitochondrial COX1 assembly into cytochrome c oxidase at a glance. J. Cell Sci. 2015;128:833–837. doi: 10.1242/jcs.161729. [DOI] [PubMed] [Google Scholar]
- Dennerlein S., Oeljeklaus S., Jans D., Hellwig C., Bareth B., Jakobs S., Deckers M., Warscheid B., Rehling P. MITRAC7 acts as a COX1-specific chaperone and reveals a checkpoint during cytochrome c oxidase assembly. Cell Rep. 2015;12:1644–1655. doi: 10.1016/j.celrep.2015.08.009. [DOI] [PubMed] [Google Scholar]
- DiMauro S., Schon E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- Fernández-Vizarra E., Tiranti V., Zeviani M. Assembly of the oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochim. Biophys. Acta. 2009;1793:200–211. doi: 10.1016/j.bbamcr.2008.05.028. [DOI] [PubMed] [Google Scholar]
- Fontanesi F. Mechanisms of mitochondrial translational regulation. IUBMB Life. 2013;65:397–408. doi: 10.1002/iub.1156. [DOI] [PubMed] [Google Scholar]
- Fontanesi F., Clemente P., Barrientos A. Cox25 teams up with Mss51, Ssc1, and Cox14 to regulate mitochondrial cytochrome c oxidase subunit 1 expression and assembly in Saccharomyces cerevisiae. J. Biol. Chem. 2011;286:555–566. doi: 10.1074/jbc.M110.188805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funes S., Kauff F., van der Sluis E.O., Ott M., Herrmann J.M. Evolution of YidC/Oxa1/Alb3 insertases: three independent gene duplications followed by functional specialization in bacteria, mitochondria and chloroplasts. Biol. Chem. 2011;392:13–19. doi: 10.1515/BC.2011.013. [DOI] [PubMed] [Google Scholar]
- Greber B.J., Ban N. Structure and function of the mitochondrial ribosome. Annu. Rev. Biochem. 2016;85:103–132. doi: 10.1146/annurev-biochem-060815-014343. [DOI] [PubMed] [Google Scholar]
- Greber B.J., Bieri P., Leibundgut M., Leitner A., Aebersold R., Boehringer D., Ban N. Ribosome. The complete structure of the 55S mammalian mitochondrial ribosome. Science. 2015;348:303–308. doi: 10.1126/science.aaa3872. [DOI] [PubMed] [Google Scholar]
- Green-Willms N.S., Fox T.D., Costanzo M.C. Functional interactions between yeast mitochondrial ribosomes and mRNA 5′ untranslated leaders. Mol. Cell. Biol. 1998;18:1826–1834. doi: 10.1128/mcb.18.4.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruschke S., Gröne K., Heublein M., Hölz S., Israel L., Imhof A., Herrmann J.M., Ott M. Proteins at the polypeptide tunnel exit of the yeast mitochondrial ribosome. J. Biol. Chem. 2010;285:19022–19028. doi: 10.1074/jbc.M110.113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hällberg B.M., Larsson N.-G. Making proteins in the powerhouse. Cell Metab. 2014;20:226–240. doi: 10.1016/j.cmet.2014.07.001. [DOI] [PubMed] [Google Scholar]
- Harbauer A.B., Opalińska M., Gerbeth C., Herman J.S., Rao S., Schönfisch B., Guiard B., Schmidt O., Pfanner N., Meisinger C. Mitochondria. Cell cycle-dependent regulation of mitochondrial preprotein translocase. Science. 2014;346:1109–1113. doi: 10.1126/science.1261253. [DOI] [PubMed] [Google Scholar]
- Harbauer A.B., Zahedi R.P., Sickmann A., Pfanner N., Meisinger C. The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab. 2014;19:357–372. doi: 10.1016/j.cmet.2014.01.010. [DOI] [PubMed] [Google Scholar]
- Herrmann J.M., Funes S. Biogenesis of cytochrome oxidase-sophisticated assembly lines in the mitochondrial inner membrane. Gene. 2005;354:43–52. doi: 10.1016/j.gene.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Herrmann J.M., Woellhaf M.W., Bonnefoy N. Control of protein synthesis in yeast mitochondria: the concept of translational activators. Biochim. Biophys. Acta. 2013;1833:286–294. doi: 10.1016/j.bbamcr.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Hornig-Do H.-T., Tatsuta T., Buckermann A., Bust M., Kollberg G., Rötig A., Hellmich M., Nijtmans L., Wiesner R.J. Nonsense mutations in the COX1 subunit impair the stability of respiratory chain complexes rather than their assembly. EMBO J. 2012;31:1293–1307. doi: 10.1038/emboj.2011.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C., Magiera M.M., Rathfelder N., Taxis C., Reber S., Maekawa H., Moreno-Borchart A., Doenges G., Schwob E., Schiebel E., Knop M. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- Kramer G., Boehringer D., Ban N., Bukau B. The ribosome as a platform for co-translational processing, folding and targeting of newly synthesized proteins. Nat. Struct. Mol. Biol. 2009;16:589–597. doi: 10.1038/nsmb.1614. [DOI] [PubMed] [Google Scholar]
- Lazarou M., Smith S.M., Thorburn D.R., Ryan M.T., McKenzie M. Assembly of nuclear DNA-encoded subunits into mitochondrial complex IV, and their preferential integration into supercomplex forms in patient mitochondria. FEBS J. 2009;276:6701–6713. doi: 10.1111/j.1742-4658.2009.07384.x. [DOI] [PubMed] [Google Scholar]
- Li G.-W., Burkhardt D., Gross C., Weissman J.S. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014;157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi I., Oeljeklaus S., Ronsör C., Bareth B., Warscheid B., Rehling P., Dennerlein S. The ribosome-associated Mba1 escorts Cox2 from insertion machinery to maturing assembly intermediates. Mol. Cell. Biol. 2016 doi: 10.1128/MCB.00361-16. Published online August 22, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisinger C., Pfanner N., Truscott K.N. Isolation of yeast mitochondria. Methods Mol. Biol. 2006;313:33–39. doi: 10.1385/1-59259-958-3:033. [DOI] [PubMed] [Google Scholar]
- Meisinger C., Sickmann A., Pfanner N. The mitochondrial proteome: from inventory to function. Cell. 2008;134:22–24. doi: 10.1016/j.cell.2008.06.043. [DOI] [PubMed] [Google Scholar]
- Mick D.U., Vukotic M., Piechura H., Meyer H.E., Warscheid B., Deckers M., Rehling P. Coa3 and Cox14 are essential for negative feedback regulation of COX1 translation in mitochondria. J. Cell Biol. 2010;191:141–154. doi: 10.1083/jcb.201007026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mick D.U., Fox T.D., Rehling P. Inventory control: cytochrome c oxidase assembly regulates mitochondrial translation. Nat. Rev. Mol. Cell Biol. 2011;12:14–20. doi: 10.1038/nrm3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mick D.U., Dennerlein S., Wiese H., Reinhold R., Pacheu-Grau D., Lorenzi I., Sasarman F., Weraarpachai W., Shoubridge E.A., Warscheid B., Rehling P. MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation. Cell. 2012;151:1528–1541. doi: 10.1016/j.cell.2012.11.053. [DOI] [PubMed] [Google Scholar]
- Opalińska M., Meisinger C. Metabolic control via the mitochondrial protein import machinery. Curr. Opin. Cell Biol. 2015;33:42–48. doi: 10.1016/j.ceb.2014.11.001. [DOI] [PubMed] [Google Scholar]
- Ostergaard E., Weraarpachai W., Ravn K., Born A.P., Jønson L., Duno M., Wibrand F., Shoubridge E.A., Vissing J. Mutations in COA3 cause isolated complex IV deficiency associated with neuropathy, exercise intolerance, obesity, and short stature. J. Med. Genet. 2015;52:203–207. doi: 10.1136/jmedgenet-2014-102914. [DOI] [PubMed] [Google Scholar]
- Ott M., Prestele M., Bauerschmitt H., Funes S., Bonnefoy N., Herrmann J.M. Mba1, a membrane-associated ribosome receptor in mitochondria. EMBO J. 2006;25:1603–1610. doi: 10.1038/sj.emboj.7601070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirós P.M., Langer T., López-Otín C. New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 2015;16:345–359. doi: 10.1038/nrm3984. [DOI] [PubMed] [Google Scholar]
- Raimundo N. Mitochondrial pathology: stress signals from the energy factory. Trends Mol. Med. 2014;20:282–292. doi: 10.1016/j.molmed.2014.01.005. [DOI] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter R., Rorbach J., Pajak A., Smith P.M., Wessels H.J., Huynen M.A., Smeitink J.A., Lightowlers R.N., Chrzanowska-Lightowlers Z.M. A functional peptidyl-tRNA hydrolase, ICT1, has been recruited into the human mitochondrial ribosome. EMBO J. 2010;29:1116–1125. doi: 10.1038/emboj.2010.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter-Dennerlein R., Dennerlein S., Rehling P. Integrating mitochondrial translation into the cellular context. Nat. Rev. Mol. Cell Biol. 2015;16:586–592. doi: 10.1038/nrm4051. [DOI] [PubMed] [Google Scholar]
- Rodnina M.V. The ribosome in action: Tuning of translational efficiency and protein folding. Protein Sci. 2016;25:1390–1406. doi: 10.1002/pro.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooijers K., Loayza-Puch F., Nijtmans L.G., Agami R. Ribosome profiling reveals features of normal and disease-associated mitochondrial translation. Nat. Commun. 2013;4:2886. doi: 10.1038/ncomms3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz J.J., Wiese H., Tölle R.C., Zarei M., Dengjel J., Warscheid B., Thedieck K. Functional proteomics identifies acinus L as a direct insulin- and amino acid-dependent mammalian target of rapamycin complex 1 (mTORC1) substrate. Mol. Cell. Proteomics. 2015;14:2042–2055. doi: 10.1074/mcp.M114.045807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadel G.S., Horvath T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163:560–569. doi: 10.1016/j.cell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski R.S., Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeitink J.A., Zeviani M., Turnbull D.M., Jacobs H.T. Mitochondrial medicine: a metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab. 2006;3:9–13. doi: 10.1016/j.cmet.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Soto I.C., Fontanesi F., Liu J., Barrientos A. Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim. Biophys. Acta. 2012;1817:883–897. doi: 10.1016/j.bbabio.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga M., Yano N., Muramoto K., Shinzawa-Itoh K., Maeda T., Yamashita E., Tsukihara T., Yoshikawa S. Distinguishing between Cl- and O2(2-) as the bridging element between Fe3+ and Cu2+ in resting-oxidized cytochrome c oxidase. Acta Crystallogr. D Biol. Crystallogr. 2011;67:742–744. doi: 10.1107/S0907444911022803. [DOI] [PubMed] [Google Scholar]
- Szklarczyk R., Wanschers B.F., Cuypers T.D., Esseling J.J., Riemersma M., van den Brand M.A., Gloerich J., Lasonder E., van den Heuvel L.P., Nijtmans L.G., Huynen M.A. Iterative orthology prediction uncovers new mitochondrial proteins and identifies C12orf62 as the human ortholog of COX14, a protein involved in the assembly of cytochrome c oxidase. Genome Biol. 2012;13:R12. doi: 10.1186/gb-2012-13-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temperley R., Richter R., Dennerlein S., Lightowlers R.N., Chrzanowska-Lightowlers Z.M. Hungry codons promote frameshifting in human mitochondrial ribosomes. Science. 2010;327:301. doi: 10.1126/science.1180674. [DOI] [PubMed] [Google Scholar]
- Tsukihara T., Aoyama H., Yamashita E., Tomizaki T., Yamaguchi H., Shinzawa-Itoh K., Nakashima R., Yaono R., Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- Weraarpachai W., Sasarman F., Nishimura T., Antonicka H., Auré K., Rötig A., Lombès A., Shoubridge E.A. Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am. J. Hum. Genet. 2012;90:142–151. doi: 10.1016/j.ajhg.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Zuo X., Yang B., Li Z., Xue Y., Zhou Y., Huang J., Zhao X., Zhou J., Yan Y. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell. 2014;158:607–619. doi: 10.1016/j.cell.2014.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.