

Graphical abstract

Keywords: NMR, Spectroscopy, Coffee, Authenticity
Highlights
-
•
A new screening method for identifying the species in ground roast coffees using low-field NMR spectroscopy.
-
•
A calibration-free statistical approach to detecting adulteration of arabica with robusta coffee.
-
•
A study of time-varying spectral changes in the 16-OMC component of lipophilic extracts following sample preparation.
Abstract
This work reports a new screening protocol for addressing issues of coffee authenticity using low-field (60 MHz) bench-top 1H NMR spectroscopy. Using a simple chloroform-based extraction, useful spectra were obtained from the lipophilic fraction of ground roast coffees. It was found that 16-O-methylcafestol (16-OMC, a recognized marker compound for robusta beans) gives rise to an isolated peak in the 60 MHz spectrum, which can be used as an indicator of the presence of robusta beans in the sample. A total of 81 extracts from authenticated coffees and mixtures were analysed, from which the detection limit of robusta in arabica was estimated to be between 10% and 20% w/w. Using the established protocol, a surveillance exercise was conducted of 27 retail samples of ground roast coffees which were labelled as “100% arabica”. None were found to contain undeclared robusta content above the estimated detection limit.
1. Introduction
Coffee beans are one of the most widely traded commodities in the world, and as such, are vulnerable to fraud within the supply chain (Toci, Farah, Pezza, & Pezza, 2016). The two main species grown are Coffea arabica L. (around 70% of the market) and Coffea canephora Pierre ex A. Froehner (variety robusta) (Belitz, Grosch, & Schieberle, 2009). Arabica beans are the most expensive, and are prized for their smooth, rounded flavour, whilst the more disease-resistant robusta plants produce beans that yield a rougher brewed drink, and thus command a lower price. There is potential, therefore, for unscrupulous traders to make economic gain by partially or wholly substituting arabica with robusta beans, deceiving other parties in the supply chain and, ultimately, the consumer. Objective methods are needed for the reliable identification of both species, and for the estimation of their contents in coffee products. Whole beans may be distinguished by inspection (International Coffee Organization, 2016, Mendonca et al., 2009), but chemical analysis is required to confirm the identity of ground roast products, for example to detect the adulteration of arabica by amounts of robusta.
Coffee contains a complex mixture of hundreds of different organic compounds, present in concentrations ranging from trace quantities up to tens of percent by weight. Major components are carbohydrates, amino acids and lipids. Potentially more characteristic of the individual species, however, are minor components such as the diterpenes of the kaurane family, whose presence in different coffee products is relatively well-documented (Kurzrock and Speer, 2001, Scharnhop and Winterhalter, 2009). These include cafestol, found in both bean types, and kahweol, found in arabica beans and in some, but not all, robusta beans. A further diterpene, 16-O-methylcafestol (16-OMC), is found exclusively in robusta beans, and has thus been proposed as a reliable marker for distinguishing between the two bean types (Speer & Mischnick, 1989). The stability of 16-OMC with respect to the roasting process means that it can also be used to detect the presence of robusta in processed coffee products (Speer & Koelling-Speer, 2006). An official method exists for the determination of 16-OMC in roasted coffee by HPLC, but it requires a time-consuming sample preparation (“DIN 10779, 2011”). Alternative methods that are rapid and low-cost would increase the uptake of authenticity testing and be of benefit to the sector.
High-field 1H NMR spectroscopy has been previously reported for the analysis of coffee. The majority of studies have examined aqueous extracts of coffee, in a variety of applications including ascertaining species and geographical origin (Cagliani et al., 2013, Charlton et al., 2002, Consonni et al., 2012, Schievano et al., 2014, Wei et al., 2011, Wei et al., 2012). In contrast, a recent study (Monakhova et al., 2015) focused on the analysis of lipophilic extracts from coffee beans and products, and their potential for addressing issues of authenticity in arabica and robusta coffees. It was shown that many minor components, including kahweol and 16-OMC, produce clearly identifiable peaks in 400 MHz spectra, and further, that integration of the 16-OMC peaks can be used to estimate the amount of robusta in coffee blends with an approximate detection limit of 1–3% w/w. The authors concluded that high-field NMR spectroscopy has potential as a screening tool for identifying coffee species, for example in advance of applying the more time-consuming official method.
The present paper explores whether a recent development in NMR technology, low-field (“benchtop”) spectroscopy, can similarly be used to address issues of coffee authentication. Compared with high-field instruments (Blümich, 2016, Blümich et al., 2009), benchtop spectrometers are smaller and more robust. Capital and maintenance costs are lower, as these instruments utilise permanent rather than superconducting magnets and thus do not need any cryogens. Modern benchtop spectrometers are also high-resolution instruments, capable of capturing as many data points per frequency interval as their high-field counterparts. However, their lower magnetic field strengths (typically 40–100 MHz) mean that resonances appear broader and more overlapped (Gerdova et al., 2015, Jakes et al., 2015). Although the chemical shifts of protons on the ppm scale are invariant to field strength, frequency separations (in Hz) are not. For instance, a chemical shift difference of 0.1 ppm translates into a separation of 60 Hz in a 600 MHz spectrum, but of only 6 Hz at an operating frequency of 60 MHz. Furthermore, second order effects on multiplet intensities are more important at lower fields, since chemical shift differences (in Hz) are reduced relative to J-couplings (typical J = 4–12 Hz: the J-coupling is invariant to field strength). Thus, when displayed on a conventional chemical shift scale, spectra that contain many resonances exhibit substantially different profiles at low- and high-field strengths. Consequently, it is not obvious that an analysis developed using high-field spectra will translate readily to low-field measurements.
As in the work by Monakhova et al. (2015), the present paper focuses on analysis of the lipophilic fraction extracted from samples of ground roast coffee beans. The aim has been to determine whether low-field NMR spectroscopy can offer the specificity and sensitivity needed to distinguish between arabica and robusta samples, and further, to quantitatively characterize mixtures of the two. Spectra obtained at both low (60 MHz) and high (600 MHz) field strengths are compared and contrasted, and the previously unreported low-field spectrum annotated. A protocol is described for detecting the presence of ground robusta beans in a sample, through a distinct spectral signature arising from the marker compound 16-OMC. Finally, results are reported from application of the low-field method to a collection of retail samples of ground roast coffees, all of which carried the labelling claim “100% arabica”.
2. Materials and methods
2.1. Samples
17 samples of roast coffee beans were obtained from a range of UK retailers and from the British Coffee Association, as detailed in Table1(a). The authenticity of these intact bean samples was confirmed by inspection. Combinations of these samples were used to produce an assortment of 54 mixtures, as detailed in Table1(b). In addition, 27 samples of ground roast coffees, all of which displayed the labelling claim “100% arabica” on their packaging (and two of which were also labelled decaffeinated), were purchased from UK retailers (Table1(c)). 16-OMC and deuterated chloroform were purchased from Sigma Aldrich (Gillingham, UK).
Table 1.
Description of coffee samples.
| (a) Whole bean samples | ||||
| Number of samples | Number of extracts | Comments | ||
| Arabica | 7 | 14 | Purchased from UK retailers. Includes one decaffeinated sample. Two extracts prepared per sample | |
| Geographic origins of the beans, as stated on labels: Kenya, Peru (×2), Indonesia, Africa & South America (blend), Africa & Central & South America (blend), 1 × origin not stated | ||||
| 4 | 4 | Supplied by the British Coffee Association | ||
| Geographic origins of the beans: Colombia, Honduras, Nicaragua, Brazil | ||||
| Robusta | 3 | 6 | Purchased from UK retailers. Two extracts prepared per sample | |
| Geographic origins of the beans, as stated on labels: India, Tanzania, 1 × origin not stated | ||||
| 3 | 3 | Supplied by the British Coffee Association | ||
| Geographic origins of the beans: Vietnam, India, Uganda | ||||
| (b) Mixtures prepared from whole bean samples | ||||
| % w/w arabica | % w/w robusta | Number of samples | Number of extracts | Comments |
| 90, 80, 70,… 10 | 10, 20, 30,… 90 | 18 | 18 | Two mixture series, each prepared from a randomly selected pair of whole bean samples |
| 90, 80, 60 | 10, 20, 40 | 36 | 36 | Twelve partial mixture series, each prepared from a different pairwise combination of whole bean samples |
| (c) Surveillance samples (retail-purchased ground roast coffees) | ||||
| Number of samples | Number of extracts | Comments | ||
| Labelled “100% arabica” | 27 | 32 | Purchased from UK retailers. Includes two decaffeinated samples. Two extracts prepared from five of the samples | |
| Geographic origins of the beans, as stated on the labels: Indonesia, Central & South America (blend), Africa & Asia & South America (blend), Africa & Brazil & Central America (blend), Guatamala (×3), Latin America, Brazil, Indonesia & Africa & Latin America (blends, ×2), Sumatra, Java, Columbia (×2), Kenya, Java & Sumatra (×2), Costa Rica, “multiple countries of origin” (×3), origin not stated (×5) | ||||
2.2. Sample preparation
Whole bean coffee samples were ground with a pestle and mortar, to produce a particulate sample visually comparable to purchased ground roast coffees. Mixture samples were prepared from these grounds, as detailed in Table1(b). The surveillance samples were all purchased as ground roast coffees, so no further grinding step was needed. Gradation tests (0.1, 0.3, 0.5 and 1 mm sieves) determined that ground sample particle sizes were typically distributed across the range 0.3–1 mm, for both the purchased and in-house ground coffees.
To extract the lipophilic fraction, 1 g of ground sample was mixed with 3.0 ml deuterated chloroform and agitated in a shaker bath for 5 min. The extract was then filtered through cotton wool directly into an NMR tube. For a subset of samples only (some of the whole bean and surveillance samples, as detailed in Table 1), the extraction procedure was carried out in duplicate.
To prepare a sample of 16-OMC for spectral analysis, 2 mg were diluted in 500 μl deuterated chloroform.
2.3. Spectral acquisition
2.3.1. Low-field 1H NMR spectroscopy
60 MHz 1H NMR spectra were acquired on a Pulsar low-field spectrometer (Oxford Instruments, Tubney Woods, Abingdon, Oxford, UK) running SpinFlow software (v1, Oxford Instruments). The sample temperature was 37 °C, and the 90° pulse length was 7.2 μs as determined by the machine’s internal calibration cycle. For each sample, 256 free induction decays (FIDs) were collected using a filter width of 5000 Hz and recycle delay of 2 s, resulting in an acquisition time of approximately 40 min per extract. These parameters represent an acceptable compromise between speed and spectral quality. FIDs were zero-filled to give spectra of 65,536 points. The linewidth was maintained between 0.5 and 0.9 Hz by daily checking of the chloroform FWHM and shimming as and when necessary.
In all cases, the FIDs were Fourier-transformed, co-added and phase-corrected using SpinFlow and MNova (Mestrelab Research, Santiago de Compostela, Spain) software packages to present a single frequency-domain spectrum from each extract. Where spectra were examined qualitatively, apodization (1 Hz exponential) was additionally applied to the FIDs. The chemical shift scale in all spectra was referenced to the residual chloroform peak at 7.26 ppm.
2.3.2. High-field 1H NMR Spectroscopy
600 MHz 1H NMR spectra were collected from selected extracts using a Bruker Avance III HD spectrometer running TopSpin 3.2 software and equipped with a 5 mm TCI cryoprobe. The probe temperature was regulated at 27 °C. For each spectrum 64 scans were collected using 30° pulses with a spectral width of 20.5 ppm, acquisition time of 2.67 s and recycle delay of 3 s. FIDs were zero-filled and transformed using exponential line broadening (0.3 Hz) to give spectra of 65,536 points. The spectra were referenced to the residual chloroform peak at 7.26 ppm.
2.4. Data analysis
All data visualization and processing of the frequency-domain spectra was carried out using Matlab (The Mathworks, Cambridge, UK) installed along with the “Statistics and Machine Learning” and “Signal Processing” toolboxes, making use of a range of inbuilt functions.
To visualize or quantify individual peaks or groups of peaks, the relevant region was locally baseline corrected using a second order polynomial fit. Spectra and any calculated peak integrals were then normalized through division by the integrated glyceride region (3.9–4.6 ppm) to compensate for unavoidable variation in sample concentration (see Parker et al., 2014), which in the present case arises from variable extraction efficiency at the sample preparation stage, as well as in the original lipid content of different coffees. Simple linear regression with no intercept term was used to model normalized 16-OMC peak integrated areas as a function of sample robusta concentration.
A method based upon the well-known signal processing method of matched filtering (Turin, 1960) was employed as a means of detecting the presence of 16-OMC in the mixture and surveillance samples. It exploits the fact that the spectral region around the 16-OMC peak position can be expected to contain only Gaussian noise unless there is some robusta coffee present in the sample. Matched filtering involves cross-correlating a “signal” with a “template” with the aim of detecting the template within the signal. Briefly, in the present work, the template is a pure Lorentzian peak at the expected location of the 16-OMC resonance. The distribution of a test statistic (the maximum normalized cross-correlation of the signal and template) was determined under conditions of the null hypothesis (no 16-OMC peak present in the signal) using Gaussian noise to simulate the signal (100,000 resamples). To test for the presence of 16-OMC in an extract, the same statistic was calculated from a signal comprising a baseline-corrected section (3.05–3.30 ppm) of the low-field spectrum, and used in conjunction with the established distributional parameters to decide whether to accept or reject the null hypothesis at the desired probability level. A graphical description of the method is given in Supplementary material.
3. Results and discussion
A 60 MHz spectrum of an extract prepared from one of the whole bean arabica samples is shown in Fig.1(a), along with the 600 MHz spectrum collected from the same extract. The spectra have been independently scaled and offset to facilitate comparison. A key point to note is that although the resonances in the 60 MHz spectrum are broader and more overlapped than in the 600 MHz spectrum, the spectra nevertheless contain analogous information. Lipids are present in roast coffee beans in concentrations up to 14% w/w and comprise mainly of triglycerides (but also di- and mono-glycerides as well as free fatty acids). Thus, at both field strengths, the spectral profile is dominated by resonances attributable to the triglyceride component of the lipophilic extract. Detailed annotation of these peaks has been given previously (see Parker et al. (2014) for low-field annotations; Guillen and Ruiz (2001) for high-field annotations).
Fig. 1.
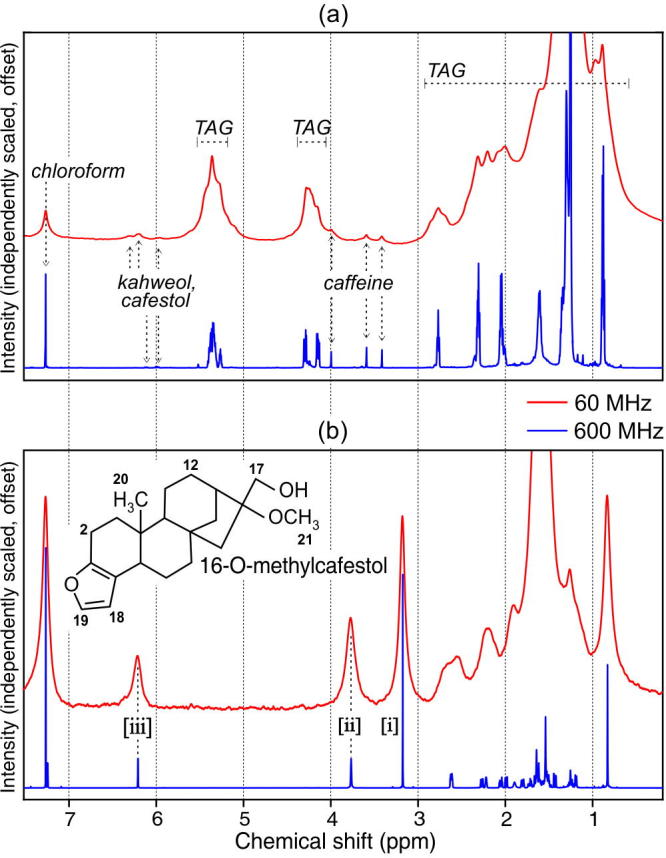
(a) 60 MHz and 600 MHz spectra of a lipophilic extract prepared from arabica coffee beans. Major spectral features include the solvent reference peak at 7.26 ppm, and several groups of features attributed mainly to triglycerides (TAGs). Subplot (b) shows 60 MHz and 600 MHz spectra of 16-OMC in chloroform. The isolated peaks labelled [i]–[iii] are potential marker peaks for the compound, discussed further in the text. The chemical structure of 16-OMC is also shown: numbers indicate protons involved in the main spectral features of interest. The chemical shift scale is common to (a) and (b).
Where there is no overlap from the triglyceride signals, small resonances arising from more minor constituents of the extract can be discerned. Detailed annotation of these features in high-field (400 MHz) spectra of lipophilic extracts from coffee was conducted by Monakhova et al. (2015), which assists in annotating the features as they appear in the low-field spectrum: the peaks at 3.42, 3.59 and 3.99 ppm arise from caffeine, and the somewhat more overlapped features at 5.95, 6.19, 6.30 ppm from the main diterpenes found in arabica coffees, kahweol and cafestol.
Because the extract was prepared from arabica beans, absent from the spectra are any signals arising from another diterpene, 16-OMC, the recognized marker compound found in robusta beans only. However, in Monakhova et al. (2015), isolated resonances from this compound were seen in 400 MHz spectra of robusta extracts. It was decided, therefore, to investigate whether any marker signals from 16-OMC could likewise be identified in spectra collected at the much lower field strength of 60 MHz.
A spectrum of the pure compound in chloroform was collected at 60 MHz, and for comparison purposes, at 600 MHz also (Fig.1(b)). The majority of the 16-OMC resonances are found in the 0.8–2.6 ppm range, which is also where the most prominent triglyceride resonances occur. However, the peaks centered at 3.16, 3.75 and 6.20 ppm (respectively labelled [i]–[iii] in Fig.1(b)) are comparatively more isolated. These peaks have been previously assigned in 300 MHz (Scharnhop & Winterhalter, 2009) and 600 MHz (Schievano et al., 2014) spectra.
Peak [iii] at 6.20 ppm is a doublet arising from the H18 proton. The splitting is seen in the 600 MHz spectrum, but cannot be resolved at 60 MHz. Other diterpenes found in coffees exhibit resonances at similar chemical shifts that also arise from the H18 proton: a doublet at 6.21 ppm in cafestol and dehydrocafestol, and singlets at 6.30 ppm in kahweol and 16-O-methylkahweol and at 6.31 ppm in dehydrokahweol. At high-field strengths, these resonances are resolved into discrete signals, but at 60 MHz one must expect considerable overlap, as indeed is seen in Fig.1(a). This limits the usefulness of peak [iii] as a marker for the presence of 16-OMC in a coffee extract.
Peak [ii] at 3.75 ppm is attributed to the two, non-equivalent H17 protons. Note that the resonances appear here as a singlet, but under certain sample conditions (such as a different concentration) a doublet may be obtained even in low-field spectra (data not shown). This is believed to be a second order effect (Schievano et al., 2014). More importantly, the 16-OMC found in robusta coffee beans is present mostly in esterified rather than free form: in this case, the non-equivalent H17 protons give rise to two doublets at 4.28 and 4.45 ppm instead of at 3.75 ppm (Kolling-Speer, Strohschneider, & Speer, 1999). Since in the low-field spectrum, these signals would be overlapped by the much more intense triglyceride peaks, they have no potential for use as markers for robusta.
Peak [i] at 3.16 ppm is the strongest of the isolated signals seen in the spectrum of pure 16-OMC. It is a singlet arising from the H21 protons in the methyl functional group that distinguishes 16-OMC from cafestol. Crucially, in the 60 MHz spectrum of the arabica extract, no other resonances are seen in this region, making this peak a candidate for further exploration as a marker signal for the presence of robusta coffee using low-field NMR spectroscopy.
Fig. 2 shows 60 MHz spectra from the arabica extract and two further extracts prepared from robusta and from decaffeinated arabica beans, in the region between 3.0 and 6.5 ppm, using a greatly expanded and offset y-scale for clarity. Note the caffeine resonances in the spectra of both the arabica and robusta extracts but not in that from the decaffeinated beans, and the kahweol peaks in the arabica extracts but not in that from robusta.
Fig. 2.
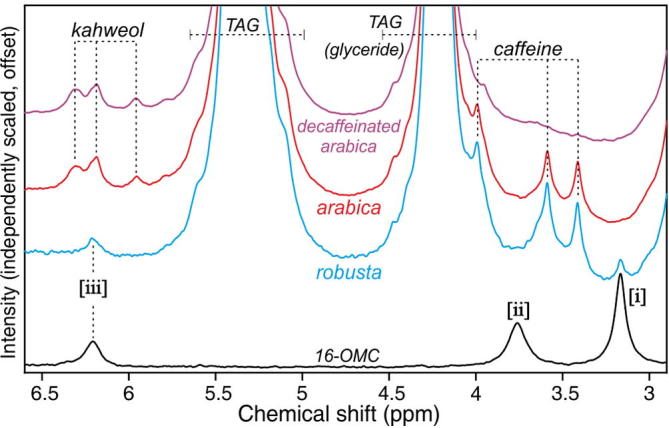
Expansions of the 3–6.5 ppm region in 60 MHz spectra of extracts prepared from robusta, arabica and decaffeinated arabica beans. Also shown for comparison is the spectrum of 16-OMC in chloroform.
The plot also includes the 16-OMC spectrum: the three peaks [i]–[iii] occur in this region of the chemical shift scale. It is clear that of the coffee bean extracts, only that from robusta shows evidence of the presence of 16-OMC. In particular, peak [i] at 3.16 ppm, an isolated feature in the 16-OMC spectrum, is clearly visible in that from the robusta extract. Note the absence of peak [ii] in the robusta spectrum, a consequence of the 16-OMC being esterified in coffee, as discussed above. Peak [iii] is also apparently isolated in the robusta spectrum, but is coincident with the kahweol features seen here in the arabica extracts (but known to also sometimes be present in small amounts in robusta coffees); moreover, cafestol resonances are known from high-field assignments to occur at around this chemical shift (D’Amelio et al., 2013, Scharnhop and Winterhalter, 2009, Schievano et al., 2014).
In the context of detecting the presence of robusta in a coffee extract, peak [i] at 3.16 ppm is evidently the best potential marker signal. To explore this idea further, it was examined more closely in sets of spectra obtained from two mixture series as detailed in Table1(b), along with the authentic arabica and robusta samples used to prepare the mixtures in each case. Expansions of the 3.16 ppm region in 60 MHz spectra, following baseline correction and normalization, are shown for one of the mixture series in Fig.3(a). The progression from 0% robusta (in which the peak is absent) to 100% is clear to see.
Fig. 3.
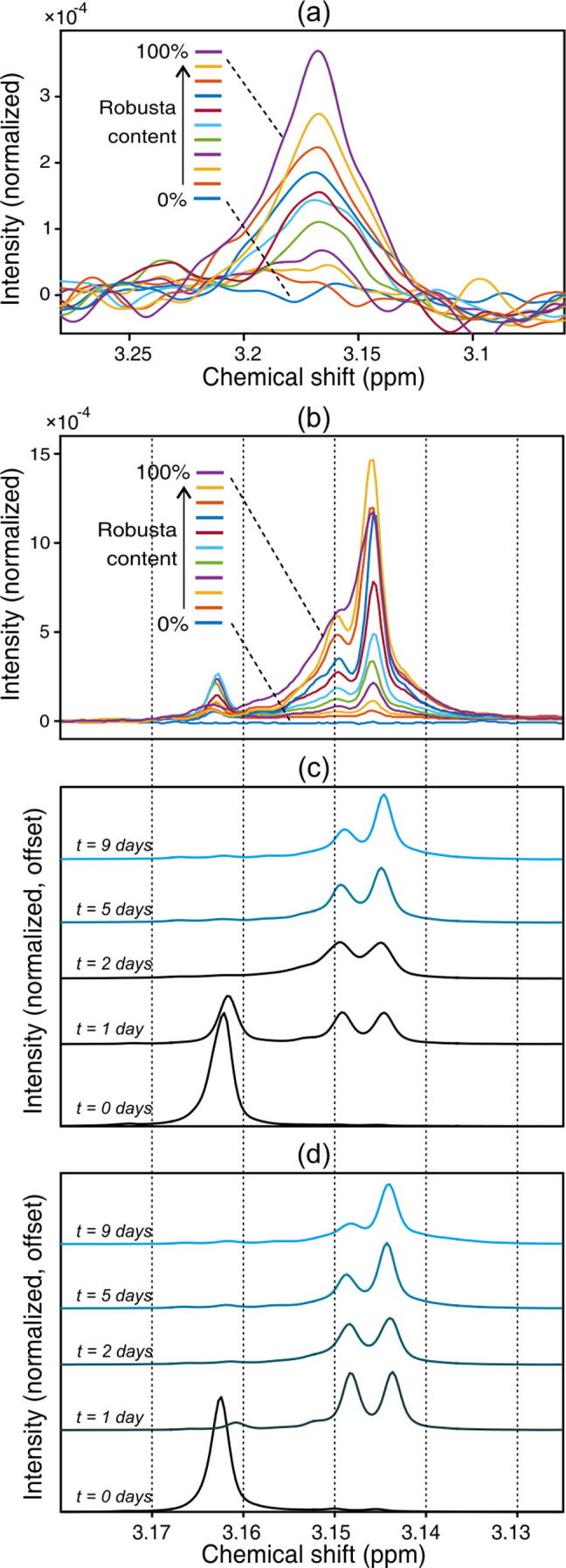
Spectra of extracts from samples containing 0, 10, 20,…, 100% w/w robusta in arabica, acquired by (a) 60 MHz, and (b) 600 MHz NMR, shown for regions around the resonance at 3.16 ppm. Subplots (c) and (d) show the changes observed in this region in 600 MHz spectra as a function of extract age, for two different preparations from robusta beans. The chemical shift scale is common to (b), (c), and (d).
According to Scharnhop and Winterhalter (2009) in the spectrum of pure 16-OMC this peak appears as a singlet originating from the three H21 protons. However, Monakhova et al. (2015) suggested that in coffee extracts, this feature is sometimes observed shifted to a slightly lower ppm value. They proposed that this was due to the chemical breakdown of 16-OMC in the coffee sample, arising for example from exposure to light. This is broadly consistent with what is seen in the 60 MHz spectra in the present work: the shape of the feature suggests it may comprise multiple overlapped resonances, although signal-to-noise and field strength limitations prevent these from being fully resolved. A much clearer picture of this effect is gained, however, from the 600 MHz spectra collected from the same mixture series (Fig.3(b)). In addition to the peak at 3.162 ppm, there are at least two further significant resonances at 3.147 and 3.150 ppm, and potentially some minor features also (note that for clarity, the chemical shift range shown in Fig.3(b) is narrower than that in Fig.3(a)).
A further investigation was conducted, using high-field spectroscopy only, to examine the behaviour of these different features as a function of sample ageing. One extract was prepared from each of two robusta coffees (the authentic beans used to prepare the two mixture series). 600 MHz spectra were obtained of each extract immediately after preparation (t = 0 days) and after t = 1, 2, 5, and 9 days. The extracts were left in ambient light and at room temperature between measurements.
The region of interest is shown in Fig.3(c) and (d). It can be seen that there are three main resonances involved in the 3.16 ppm region. For freshly prepared samples (t = 0), the only signal present is a single peak at 3.162 ppm. One day after preparation (t = 1), two additional peaks of approximately equal intensity are seen at slightly lower chemical shifts (3.150 and 3.147 ppm) and the original peak at 3.162 ppm is substantially smaller for both samples. Two days after preparation (t = 2), the 3.162 ppm peak has disappeared entirely. As the sample ages further, a progressive difference in the intensities of the peaks at 3.150 and 3.147 ppm is observed, with the former becoming relatively smaller and the latter increasingly larger, although significantly, the integrated area of the complete region remains constant over time. It is proposed that the peak at 3.150 ppm arises from an intermediate and at 3.147 ppm from a final breakdown product of 16-OMC.
Note also that the mixture spectra shown in Fig.3(b) are qualitatively most similar to the spectra obtained nine days after preparation (Fig.3(c) and (d), t = 9). Since spectral recording of this mixture series by high-field NMR took place 12–14 days after sample preparation, this finding adds weight to the hypothesis that 16-OMC as present in lipophilic coffee extracts exhibits consistent breakdown over time. Furthermore, spectra from the second mixture series, which was prepared 3–4 days in advance of recording by high-field NMR, exhibited variable proportions of the three peaks, consistent with the 16-OMC in these extracts being at an intermediate stage of breakdown (data not shown).
Additional changes in the high-field spectra (data not shown) as the extracts aged were observed as follows: disappearance of the 16-OMC H18 and H19 peaks at 6.21 and 7.24 ppm, of the H2 peak at 2.62 ppm, and of the H20 peak at 0.83 ppm; appearance of peaks at 1.11, 4.43, 4.62, 5.51, 9.49 and 10.02 ppm; and the diminishing and slight shifting of the H12 peak at 1.67 ppm to a lower chemical shift. The disappearance of the H18 and H19 signals and the appearance of signals in the aldehyde region (9.49 and 10.02 ppm) suggest the changes involve the opening of the furan ring. There is evidence of such a phenomenon in chemical reactions involved in the production of tricalysiolide from cafestol (Bigi, Liu, Zou, Houk, & White, 2012) as well as in human digestion of cafestol and kahweol (acidic conditions) (De Lucia et al., 2009). Given that the spectrum of pure 16-OMC in chloroform exhibits long term stability (data not shown), it is proposed that the instability of the compound as observed in robusta extracts arises from the opening of the furan ring under the acidic conditions present in coffee.
This study indicates that to properly represent the 16-OMC content of coffee extracts, both the intact 16-OMC signal and those of its breakdown products need to be taken into account. With this is in mind, a region covering the 3.16 ppm resonance as well as the breakdown product peaks at 3.150 and 3.147 ppm was integrated in the baseline corrected and normalized spectra from both mixture series collected at both field strengths. The peak areas are plotted versus the concentration of robusta in Fig.4(a) and (b). The calibrations serve to illustrate the potential for mixture quantitation using 60 MHz NMR spectroscopy. For both series, the root-mean-square error (RMSE) in predicting the compositional values was 7% w/w. This compares well with 4% w/w obtained by 600 MHz spectroscopy, considering the relative cost and complexity of the two techniques.
Fig. 4.

Integrated area of the 3.16 ppm region plotted versus robusta content for the two 0, 10, …, 100% w/w mixture series indicated by ■ and  (a) by 60 MHz, and (b) by 600 MHz NMR.
(a) by 60 MHz, and (b) by 600 MHz NMR.
The different regression line gradients arise from the difference in the 16-OMC contents of the robusta beans used to prepare each series. This can vary considerably between coffees (Speer & Koelling-Speer, 2006). The area of the 16-OMC peak is thus a proxy for, rather than a direct measure of, the robusta content. In the context of adulteration detection, however, quantitation is not essential and may be unnecessarily demanding. A more intuitive approach is merely to look for evidence of a 16-OMC peak. If it is found, then the presence of robusta beans in the sample is indicated, since the compound is entirely absent from arabica coffees.
In terms of data analysis, this suggests the use of a signal detection approach, as described in Section 2 and in Supplementary material. Fig.5(a) shows the values of the matched filtering test statistic obtained for the authentic (whole bean) coffee samples. Also marked on the plot are the 0.5 and 99.5 percentiles for the statistic’s distribution established by simulation. It is seen that all of the values for the authentic arabica coffees occur between these percentiles, consistent with the expected type I error rate and validating the assumptions of the statistical method. Furthermore, all of the robusta samples lie far above the 99.5% percentile meaning that for these samples the null hypothesis (no 16-OMc peak present) can be confidently rejected; in each case, a peak consistent with a 16-OMC signal has been detected in the data. This shows conclusively that low-field NMR spectroscopy is capable of distinguishing reliably between pure arabica and pure robusta ground roast coffees.
Fig. 5.

Values for the test statistic (maximum value of the normalized cross-correlation between signal and template, within a pre-defined tolerance window around zero lag; see Supplementary material) for (a) the whole bean extract, (b) the extracts from mixtures, and (c) the surveillance sample extracts. In subplots (a) and (c), data points from replicate extracts are joined by dotted lines. In subplot (b), dotted lines distinguish mixture series (and partial series) prepared from different pairwise combinations of arabica and robusta beans.
Fig.5(b) shows the values of the test statistic obtained from the mixture samples, which includes both the mixture series and the partial mixture series (prepared with 10, 20 and 40% w/w robusta contents). All samples with a robusta content of ⩾20% w/w fall above the 99.5% percentile, indicating correctly that a 16-OMC peak and thus some robusta has been detected in the extract. Of the mixtures containing 10% w/w robusta, five are correctly identified as containing some robusta coffee. For the remaining seven extracts, the 16-OMC signal was not large enough to be detected, indicating the limits of sensitivity have been reached. Potentially this could be improved upon by changing aspects of the experimental protocol to give better spectral signal-to-noise, such as the co-addition of more FIDs, or a longer extraction step to give a more concentrated sample. However, the penalty would be an increased turnaround time per sample, which is undesirable in the context of a high-throughput screening scenario. In summary, the outcomes for the mixture samples suggest that samples containing at least 20% w/w robusta are very likely to be detected by the present protocol, and that a substantial proportion of samples containing only 10% w/w robusta will also be identified.
Finally, the outcomes from the surveillance samples (retail purchased “100% arabica” ground coffees) are shown in Fig.5(c). The values for all samples fall between the 0.5 and 99.5 percentiles, with the exception of two extracts which fall just above the 99.5% boundary (although their replicates do not, which suggests that random spectral noise is the likely cause of this outcome). It is concluded that, with an estimated detection limit of 10–20% w/w robusta in arabica, no evidence of fraudulent substitution has been found.
4. Conclusion
This work explored the use of low-field NMR spectroscopy in the analysis of lipophilic extracts prepared from ground roast coffees. Hitherto unreported 60 MHz spectra from various arabica and robusta samples have been annotated, by drawing on analogous information in high-field (600 MHz) spectra collected from the same samples.
The low-field approach is shown to be sensitive and selective enough to monitor a key marker compound, 16-OMC, found only in robusta coffees. This compound gives rise to an isolated peak at 3.16 ppm in the 60 MHz spectrum, which can be used as a direct indicator of the presence of 16-OMC (and thus a proxy for robusta) in a sample. A further study of 16-OMC as it manifests in coffee extracts was carried out using 600 MHz spectroscopy, to examine changes over time that occur following sample preparation. This phenomenon has been previously hinted at in the literature but not fully described.
A signal processing technique, matched filtering, was employed in a protocol for detecting the 3.16 ppm marker peak in 60 MHz spectra of coffee extracts. Aiming for peak detection rather than quantitation allows for a calibration-free adulteration test, with the obvious advantage that a database of reference samples is not required. By analysing an assortment of laboratory-prepared coffee mixtures, the effective limit of detection was estimated to be 10–20% w/w robusta in arabica, recognizing that stating a precise detection limit is not meaningful, since the concentration of 16-OMC in different robusta beans can vary considerably. Further development work is ongoing to improve the sensitivity of the method via a number of routes, and early results are encouraging. For example, a main factor determining the detection limit is the spectral signal-to-noise, which can readily be increased though the co-addition of more scans, albeit at the cost of a longer turnaround time per sample.
In common with many authentication issues of a similar nature, setting the detection threshold amounts to striking a balance between type I and type II errors, or in other words, between the proportion of authentic samples that are erroneously flagged as adulterated, and the level of adulteration that passes undetected. In the present work, a boundary was chosen to give a low type I error rate for authentic arabica extracts, whilst correctly detecting 16-OMC in all test mixtures that contained ⩾20% w/w, and around half of those that contained 10% w/w robusta. Under these conditions, a survey of 27 UK retail purchased “100% arabica” ground coffees revealed no evidence of fraud.
In conclusion, this work has shown that low-field NMR spectroscopy has the potential for addressing the issue of species authenticity in ground roast coffees. The experimental methodology described here could readily be adapted for other compounds with isolated marker peaks, the most obvious being caffeine. A patent based upon this approach has been applied for.
Author contributions
The manuscript was written through contributions of all authors, with primary input as follows: experimental work: EW, YG, IJC, GLG; data analysis: MD, EKK; initiation and planning: MD, ADW, DW, EKK; writing up: MD, ADW, IJC, EKK.
Conflict of interest
There are no conflicts of interest associated with this work.
Acknowledgments
The authors acknowledge financial support from Innovate UK (project number 101250) and the Institute of Food Research BBSRC Core Strategic Grant fund (project number BBS/E/F/00042674). The authors thank the British Coffee Association for the supply of authenticated coffee samples.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.foodchem.2016.08.028.
Contributor Information
Marianne Defernez, Email: marianne.defernez@ifr.ac.uk.
Ella Wren, Email: ella.wren@ifr.ac.uk.
Andrew D. Watson, Email: andrew.watson@ifr.ac.uk.
Yvonne Gunning, Email: yvonne.gunning@ifr.ac.uk.
Ian J. Colquhoun, Email: ian.colquhoun@ifr.ac.uk.
Gwénaëlle Le Gall, Email: gwenaelle.legall@ifr.ac.uk.
David Williamson, Email: David.Williamson@oxinst.com.
E. Kate Kemsley, Email: kate.kemsley@ifr.ac.uk.
Appendix A. Supplementary data
Supplementary Fig. 1.

References
- Belitz H.D., Grosch W., Schieberle P. 4 ed. Springer; Berlin, Heidelberg: 2009. Food chemistry. [Google Scholar]
- Bigi M.A., Liu P., Zou L., Houk K.N., White M.C. Cafestol to tricalysiolide B and oxidized analogues: Biosynthetic and derivatization studies using non-heme iron catalyst Fe(PDP) Synlett. 2012;(19):2768–2772. doi: 10.1055/s-0032-1317708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blümich B. Introduction to compact NMR: A review of methods. Trends in Analytical Chemistry. 2016:1–10. [Google Scholar]
- Blümich B., Casanova F., Appelt S. NMR at low magnetic fields. Chemical Physics Letters. 2009;477:231–240. [Google Scholar]
- Cagliani L.R., Pellegrino G., Giugno G., Consonni R. Quantification of Coffea arabica and Coffea canephora var. robusta in roasted and ground coffee blends. Talanta. 2013;106:169–173. doi: 10.1016/j.talanta.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Charlton A.J., Farrington W.H.H., Brereton P. Application of H-1 NMR and multivariate statistics for screening complex mixtures: Quality control and authenticity of instant coffee. Journal of Agricultural and Food Chemistry. 2002;50(11):3098–3103. doi: 10.1021/jf011539z. [DOI] [PubMed] [Google Scholar]
- Consonni R., Cagliani L.R., Cogliati C. NMR based geographical characterization of roasted coffee. Talanta. 2012;88:420–426. doi: 10.1016/j.talanta.2011.11.010. [DOI] [PubMed] [Google Scholar]
- D’Amelio N., De Angelis E., Navarini L., Schievano E., Mammi S. Green coffee oil analysis by high-resolution nuclear magnetic resonance spectroscopy. Talanta. 2013;110:118–127. doi: 10.1016/j.talanta.2013.02.024. [DOI] [PubMed] [Google Scholar]
- De Lucia M., Panzella L., Melck D., Giudicianni I., Motta A., Napolitano A., d’Ischia M. Differential reactivity of purified bioactive coffee furans, cafestol and kahweol, with acidic nitrite: Product characterization and factors controlling nitrosation versus ring-opening pathways. Chemical Research in Toxicology. 2009;22(12):1922–1928. doi: 10.1021/tx900224x. [DOI] [PubMed] [Google Scholar]
- DIN 10779 . German Institute for Standardization (DIN); 2011. Analysis of coffee and coffee products – Determination of 16-O-methyl cafestol content of roasted coffee – HPLC-method. [Google Scholar]
- Gerdova A., Defernez M., Jakes W., Limer E., McCallum C., Nott K.…Kemsley E.K. 60 MHz 1H NMR spectroscopy of triglyceride mixtures. In: Capozzi F., Laghi L., Belton P.S., editors. Magnetic resonance in food science: Defining food by magnetic resonance. Royal Society of Chemistry; London: 2015. pp. 17–30. [Google Scholar]
- Guillen M.D., Ruiz A. High resolution H-1 nuclear magnetic resonance in the study of edible oils and fats. Trends in Food Science & Technology. 2001;12(9):328–338. [Google Scholar]
- International Coffee Organization (2016). International Coffee Organization Annual Review 2014–2015.
- Jakes W., Gerdova A., Defernez M., Watson A.D., McCallum C., Limer E.…Kemsley E.K. Authentication of beef versus horse meat using 60 MHz H-1 NMR spectroscopy. Food Chemistry. 2015;175:1–9. doi: 10.1016/j.foodchem.2014.11.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolling-Speer I., Strohschneider S., Speer K. Determination of free diterpenes in green and roasted coffees. Journal of High Resolution Chromatography. 1999;22(1):43–46. [Google Scholar]
- Kurzrock T., Speer K. Diterpenes and diterpene esters in coffee. Food Reviews International. 2001;17(4):433–450. [Google Scholar]
- Mendonca J.C.F., Franca A.S., Oliveira L.S. Physical characterization of non-defective and defective Arabica and Robusta coffees before and after roasting. Journal of Food Engineering. 2009;92(4):474–479. [Google Scholar]
- Monakhova Y.B., Ruge W., Kuballa T., Ilse M., Winkelmann O., Diehl B.…Lachenmeier D.W. Rapid approach to identify the presence of Arabica and Robusta species in coffee using H-1 NMR spectroscopy. Food Chemistry. 2015;182:178–184. doi: 10.1016/j.foodchem.2015.02.132. [DOI] [PubMed] [Google Scholar]
- Parker T., Limer E., Watson A.D., Defernez M., Williamson D., Kemsley E.K. 60 MHz H NMR spectroscopy for the analysis of edible oils. Trends in Analytical Chemistry. 2014;57(100):147–158. doi: 10.1016/j.trac.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharnhop H., Winterhalter P. Isolation of coffee diterpenes by means of high-speed countercurrent chromatography. Journal of Food Composition and Analysis. 2009;22(3):233–237. [Google Scholar]
- Schievano E., Finotello C., De Angelis E., Mammi S., Navarini L. Rapid authentication of coffee blends and quantification of 16-O-methylcafestol in roasted coffee beans by nuclear magnetic resonance. Journal of Agricultural and Food Chemistry. 2014;62(51):12309–12314. doi: 10.1021/jf505013d. [DOI] [PubMed] [Google Scholar]
- Speer K., Koelling-Speer I. The lipid fraction of the coffee bean. Brazilian Journal of Plant Physiology. 2006;18(1):201–216. [Google Scholar]
- Speer K., Mischnick P. 16-O-methylcafestol – A new diterpene in coffee – Discovery and identification. Zeitschrift Fur Lebensmittel-Untersuchung Und-Forschung. 1989;189(3):219–222. [Google Scholar]
- Toci A.T., Farah A., Pezza H.R., Pezza L. Coffee adulteration: More than two decades of research. Critical Reviews in Analytical Chemistry. 2016;46(2):83–92. doi: 10.1080/10408347.2014.966185. [DOI] [PubMed] [Google Scholar]
- Wei F.F., Furihata K., Hu F.Y., Miyakawa T., Tanokura M. Two-dimensional H-1-C-13 nuclear magnetic resonance (NMR)-based comprehensive analysis of roasted coffee bean extract. Journal of Agricultural and Food Chemistry. 2011;59(17):9065–9073. doi: 10.1021/jf201716w. [DOI] [PubMed] [Google Scholar]
- Wei F., Furihata K., Koda M., Hu F., Miyakawa T., Tanokura M. Roasting process of coffee beans as studied by nuclear magnetic resonance: Time course of changes in composition. Journal of Agricultural and Food Chemistry. 2012;60(4):1005–1012. doi: 10.1021/jf205315r. [DOI] [PubMed] [Google Scholar]