

Abstract
Emerging drug resistance in clinical isolates of Staphylococcus aureus might be implicated to the overexpression of NorA efflux pump which is capable of extruding numerous structurally diverse compounds. However, NorA efflux pump is considered as a potential drug target for the development of efflux pump inhibitors. In the present study, NorA model was constructed based on the crystal structure of glycerol-3-phosphate transporter (PDBID: 1PW4). Molecular dynamics (MD) simulation was performed using NAMD2.7 for NorA which is embedded in the hydrated lipid bilayer. Structural design of NorA unveils amino (N)- and carboxyl (C)-terminal domains which are connected by long cytoplasmic loop. N and C domains are composed of six transmembrane α-helices (TM) which exhibits pseudo-twofold symmetry and possess voluminous substrate binding cavity between TM helices. Molecular docking of reserpine, totarol, ferruginol, salvin, thioxanthene, phenothiazine, omeprazole, verapamil, nalidixic acid, ciprofloxacin, levofloxacin, and acridine to NorA found that all the molecules were bound at the large hydrophobic cleft and indicated significant interactions with the key residues. In addition, structure-based virtual screening was employed which indicates that 14 potent novel lead molecules such as CID58685302, CID58685367, CID5799283, CID5578487, CID60028372, ZINC12196383, ZINC72140751, ZINC72137843, ZINC39227983, ZINC43742707, ZINC12196375, ZINC66166948, ZINC39228014, and ZINC14616160 have highest binding affinity for NorA. These lead molecules displayed considerable pharmacological properties as evidenced by Lipinski rule of five and prophecy of toxicity risk assessment. Thus, the present study will be helpful in designing and synthesis of a novel class of NorA efflux pump inhibitors that restore the susceptibilities of drug compounds.
Keywords: Staphylococcus aureus, NorA efflux pump, molecular dynamics, virtual screening, docking
Introduction
Staphylococcus aureus is one of the major emerging multidrug-resistant pathogenic bacteria whose infections have been increasing alarmingly causing 19,000 deaths per year.1 Drug access is overdue by numerous resistant mechanisms such as drug inactivation, target-based mutation, reduced drug access, and efflux pumps. The multidrug efflux systems play an important role in imparting resistance in bacteria and this seems to be a major setback in designing antimicrobial agents. In recent times, the complete genome sequence of S. aureus explored 30 efflux pump genes which belong to the major facilitator super family (MFS).2 MFS transporters reveal that all MFS proteins possess a uniform topology of 12 transmembrane (TM) α-helices which are connected by hydrophilic loops at amino (N) and carboxyl (C) termini in the cytoplasm.3 MFS proteins are dependent on proton motif force that transports the assorted substrates such as ions, sugars, sugar phosphates, drugs, neurotransmitters, nucleosides, amino acids, and peptides across the TM through three distinct transport mechanisms such as uniport, symport, and antiport. However, substrate specificity and transport mechanism of all MFS proteins vary and are less understood. Analysis by using structural, computational, and biochemical techniques reveals that MFS transporters possess a single binding site, alternating access mechanism that involves rocker switch-type movement of the protein.4,5
NorA efflux pump is one of the major overexpressed efflux pumps in the blood stream clinical isolates of S. aueus6,7 and involved in the efflux of multiple diverse drug compounds such as quinolones, fluoroquinolones,8–10 quarternary ammonium compounds and antiseptics,11,12 phenothaizines and thioxanthenes,13 verapamil and omeprazole,14 totarol, reserpine, ferruginol, carnosic acid, and dyes (such as ethidium bromide, rhodamine, acridine, and biocides).15 NorA belongs to MFS transporters which are embedded in the membranes of prokaryotes and eukaryotes.16 Till now, the crystal structure of NorA efflux pump has not been resolved, although the crystal structures of a confined number of MFS proteins, namely GlpT (antiporter),17 LacY (symporter),18 EmrD,19 and OxlT, were resolved.20 Dysfunction of efflux pumps could potentiate the antibacterial activity of the drugs, and this strategy has been attracting many researchers around the globe to develop a novel potent antibacterial compound.21 So far, numerous potent efflux pump inhibitors (EPIs) such as derivatives of piperine,22 boronic species,23 N-caffeyl phenalkymanide,24 2-aryl-5-nitro-1H-indoles,25 aryl benzothiophenes and diarylthiophene,26 6-amino 8-methyl quinolone ester, and some plant-derived compounds27,28 have been reported to target NorA. Conversely, these compounds are unable to enter into the clinical settings due to several pharmacological adverse side effects, especially when administered for longer duration. Besides, the continuous usage of these drugs causes development of drug resistance in pathogenic bacteria. Hence, the present investigation was undertaken to elucidate the structural characteristics and binding mechanism of NorA efflux pump and design potent EPIs for effective clinical management of S. aureus.
Materials and methods
Prophecy of TM helices
NorA protein sequence of S. aureus was retrieved from UniProt database, and TM helices were predicted using various servers such as TMHMM,29 TMpred,30 SOSUI,31 DAS,32 HMMTOP,33 Predict protein,34 and TopPred II35 to confirm origin and end of the helices.
Homology modeling
Three-dimensional (3D) structure of NorA was elucidated on the basis of sequence identity with high score, less e-value, highest resolution, and R-factor of the template structure by performing BLASTP search against Protein Data Bank (PDB). The coordinates for the query structure through pairwise sequence alignment were assigned from template structure by using Clustal X.36 Subsequently, the 3D models were built by using MODELLER 9.14,37 and the least modeler objective (low discrete optimized protein energy score) was chosen. The final model was subjected to molecular dynamics (MD) simulations.
MD simulation
NorA model with the least DOPE score was improved by applying MD simulations using NAMD2.7 software.38 Chemistry at HARvard Macromolecular Mechanics (CHARMM2.7) force field was used for lipids and proteins39 along with the three-site transferable intermolecular potential for water and palmitoyl oleyl phosphatidyl choline for lipids (Figure 1A). Initially, the membrane and the protein complex were minimized and equilibrated with 250,000 runs for 10 picoseconds, and the simulations were performed for 1,000,000 runs for 2 nanoseconds. Integrated motion time step of 2 fs was computed using multiple time step algorithms.40 Short range forces were computed for every two time steps, and long range forces were calculated for every four time steps. The pair list of the nonbonded interactions was computed with a pair list distance of 14.0 Å. Short-range interactions, within 12 Å, were defined as van der Waals and electrostatic interactions. Long-range electrostatic interactions were taken into account using particle mesh Ewald approach.41–43 Pressure was maintained at 1 atm using the Langevin piston and temperature was controlled at 300 K using Langevin dynamics. Covalent interactions between hydrogen and heavy atoms were constrained using SHAKE/RATTLE algorithm.44
Figure 1.

MD simulation of NorA efflux pump.
Notes: (A) NorA is embedded into the lipid bilayer with water molecules. (B) Calculated RMSD graphs of molecular dynamics simulations of NorA efflux pump of Staphylococcus aureus using NAMD software. Time (ps) is taken on X-axis and RMSD (Å) on Y-axis.
Abbreviations: RMSD, root-mean-square deviation; ps, picoseconds.
Model assessment
The quality of the model was assessed by calculating the stereochemical properties, compatibility of the atomic model (3D) with its own amino acid residues (1D), bond lengths, bond angles, and side-chain planarity using SAVES server (http://nihserver.mbi.ucla.edu/SAVES/). Ramachandran plot calculations were performed using PROCHECK to check the stereochemical quality of protein structure.45 Environment profile was developed using Verify3D46 and ERRAT.47 The residue packing and atomic interactions were analyzed using WHATIF, and Ramachandran plot was analyzed using WHATCHECK.48 Root-mean-square deviation (RMSD) of the model was calculated by superimposition of the 3D model with template using Swiss-Pdb Viewer.49
Retrieval of ligands
Phytochemicals such as alkaloids (reserpine), terpenoids (ferruginol, totarol, salvin), xanthenes (thioxanthene, phenothiazone), verapamil, omeprazole, fluoroquinolones (levofloxacin, nalidixic acid, and ciprofloxacin), and dyes (acridine) were downloaded from PubChem. Reserpine analogs were retrieved from PubChem and ZINC database.
Virtual screening and docking
Structure-based virtual screening studies were carried out using AutoDock Vina 4.050 with PyRx.51 Initially, all the ligand molecules were uploaded and energy minimized with universal force field using conjugate-gradient algorithm with 200 run iterations. Virtual screening was carried out against NorA efflux pump by using Lamarkian genetic algorithm. Docking parameters were set as follows: the number of individuals in the population was 150, maximum number of energy evaluations was 25,000, maximum number of generations was 27,000, top individual to survive to next generation was 1, gene mutation rate was 0.02, crossover rate was 0.8, Cauchy beta was 1.0, and genetic algorithm window size was 10.0. The grid was set to the binding pocket at X=29.3901, Y=−42.745, Z=−51.82; dimensions (Å) at X=90.000, Y=105.7097, Z=104.2448; and exhaustiveness at 8. The best docked ligand conformations were saved, and the bond angles, bond lengths, and hydrogen bonding interactions were analyzed using PyMOL.52
Toxicity risk assessment and Lipinski rule of five
Lipinski rule of five, that is, molecular weight (<500 Da), H-bond acceptor (<10), H-bond donor (<5), and cLogP (<5) values; toxicity properties such as mutagenic, tumorogenic, irritant, and reproductive effects; and absorption, distribution, metabolize, excretion, and transport properties were assessed using OSIRIS server (http://www.organic-chemistry.org/prog/peo) and Molinspiration (http://www.molinspiration.com/cgi-bin/properties).
Results and discussion
Prophecy of TM helices
Protein sequence of NorA efflux pump (account number: Q6GIU7) consisting of 387 amino acids was retrieved from UniProt, and prophecy of TM helices has shown different helices at different positions, namely Helix-I (6–30), Helix-II (40–62), Helix-III (69–91), Helix-IV (94–119), Helix-V (127–151), Helix-VI (157–178), Helix-VII (201–227), Helix-VIII (237–259), Helix-IX (262–375), Helix-X (288–351), Helix-XI (326–350), and Helix-XII (357–375) (Table 1). However, Helix-IV, Helix-VI, and Helix-XI by TMpred; Helix-VI and Helix-XII by TMHMM; Helix-XII by SOUSI; and TopPred have not been recognized (Table 1).
Table 1.
Prediction of TM helices of NorA efflux pump using different servers
| TM helixv | HMMTOP
|
SOSUI
|
TMHMM
|
TopPred
|
TMpred
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Start | End | Start | End | Start | End | Start | End | Start | End | |
| Helix-I | 6 | 25 | 6 | 28 | 7 | 29 | 10 | 30 | 8 | 25 |
| Helix-II | 40 | 58 | 39 | 61 | 40 | 62 | 40 | 60 | 42 | 58 |
| Helix-III | 69 | 88 | 69 | 91 | 69 | 91 | 69 | 89 | 71 | 87 |
| Helix-IV | 99 | 116 | 97 | 119 | 95 | 117 | 99 | 119 | – | – |
| Helix-V | 129 | 148 | 127 | 149 | 129 | 151 | 131 | 151 | 131 | 148 |
| Helix-VI | 157 | 176 | 157 | 178 | – | – | 157 | 376 | 357 | 177 |
| Helix-VII | 203 | 221 | 202 | 224 | 203 | 225 | 201 | 221 | 211 | 227 |
| Helix-VIII | 240 | 258 | 237 | 259 | 240 | 259 | 239 | 259 | 240 | 257 |
| Helix-IX | 269 | 288 | 262 | 284 | 266 | 375 | 269 | 289 | 283 | 301 |
| Helix-X | 295 | 313 | 288 | 310 | 292 | 314 | 331 | 351 | 327 | 346 |
| Helix-XI | 326 | 350 | 345 | 367 | 327 | 349 | 356 | 177 | 159 | 111 |
| Helix-XII | 357 | 375 | – | – | – | – | – | – | – | – |
Abbreviation: TM, transmembrane.
NorA model construction
Glycerol-3-phosphate transporter (PDBID: 1PW4) belongs to the MFS and was selected based on highest resolution (3.3 Å), R-value (0.296), and R-free (0.325) as template.53 Based on the spatial restraints, the coordinates of template to structurally conserved regions, structurally variable region, and N- and C-termini have been assigned to query the sequence (Figure 2). Subsequently, all the side chains were fixed by rotamers and 100 models were generated, and the lowest DOPE score structure was selected as the best model for NorA. Variations in secondary structural elements such as irregular helices, sheets, and loops were identified by superimposing with template, and a small loop, observed between Ala20 and Val22 of Helix-I, was renovated using MOD loop server (https://modbase.compbio.ucsf.edu/modloop).
Figure 2.

Pairwise alignment of NorA efflux pump of Staphylococcus aureus with glycerol-3-phosphate transporter (PDBID: 1PW4) of Escherichia coli.
To obtain stable conformation, hydrogens were added to the model initially, and the energy was minimized followed by MD simulations for 2 nanoseconds. Later on, trajectory graph was plotted by taking RMSD of Cα trace on X-axis and time (picoseconds) on Y-axis that showed a sharp increase up to 1.5 nanoseconds and achieved equilibrium at around 2 nanoseconds (Figure 2B). The refined model possesses 12 α-helices and one long cytoplasmic connecting loop, and the quality of the model was checked by SAVES server.
Model assessment
This final model was corroborated with Ramachandran plot calculations using PROCHECK which revealed 308 residues (92.8%) were aligned within the most favored region, 21 residues (6.3%) were located within the additionally allowed regions, three residues (0.9%) were plotted within the generously allowed region, and no residues were aligned within the disallowed region (Figure 3A). The environment profile of Verify3D–1D value was found to be mostly above zero (Figure 3B). The nonbonded interactions between various types of atoms were computed with ERRAT program which showed an overall quality factor of 80.0 (Figure 3C). WHATCHECK program calculated quality indicators such as second-generation packing quality, Ramachandran plot appearance, and chi-1/chi-2 rotamer normality and were found to be 2.7, 0.725, and 2.0, respectively; RMS Z-scores such as bond lengths, bond angles, omega angle restraints, side-chain planarity, improper dihedral distribution, and inside/outside distribution were found to be 0.98, 0.803, 0.564, 1.08, 1.15, and 1.23, respectively. Superimposition of NorA and GlpT recorded an RMSD of 0.86 Å for Cα atoms.
Figure 3.

Assessment of NorA efflux pump structure.
Notes: (A) Ramachandran plot calculations of three-dimensional (3D) model computed using PROCHECK. (B) Compatibility of atomic 3D model of its own amino acid residues (1D) using Verify3D server. (C) 3D profiles of constructed 3D model were verified using ERRAT program. *On the error axis, two lines are drawn to indicate the confidence with which it is possible to reject regions that exceed that error value. **Expressed as the percentage of the protein for which the calculated error value falls below the 95% rejection limit. Good high resolution structures generally produce values around 95% or higher. For lower resolutions (2.5 to 3A) the average overall quality factor is around 91%.
Structure of NorA and binding-site analysis
NorA efflux pump is a single polypeptide chain which exhibits 12 TM α-helices topology with two domains, N- and C-terminal domains, arranged as pseudo-twofold symmetry. Both N- and C-terminal domains are connected by a long cytoplasmic loop (Met172–Lys198) between Helix-VI and Helix-VII (Figure 4A). N-terminal domain consists of six TM α-helices: TM Helix-I (Met1–Ileu30), TM Helix-II (Leu40–Lys64), TM Helix-III (Gly66–Ala86), TM Helix-IV (Phe91–Ileu119), TM Helix-V (Gln124–Ile146), and TM Helix-VI (Arg156–Met172) and C-terminal domain consists of six TM α-helices: TM Helix-VII (Lys198–Tyr225), TM Helix-VIII (Asn234–Phe264), TM Helix-IX (Ser267–Leu286), TM Helix-X (Trp293–Ser318), TM Helix-XI (Gly326–Val353), and TM Helix-XII (Gly356–Arg380). Nevertheless, a large hydrophobic binding cleft is accompanied by eight TM helices of Helix-I, Helix-II, Helix-IV, and Helix-V from N terminal and Helix-VII, Helix-VIII, Helix-X, and Helix-XI from C terminal and composed of nonpolar residues such as Val44, Phe47, Gln51, Phe140, Ile244, Gly248, and Phe303 which are evolutionarily conserved among MFS transporters (Figure 4B).
Figure 4.

3D structure of NorA efflux pump.
Notes: (A) Homology model of NorA efflux pump with N- and C-terminal domains connected by one cytoplasmic connecting loop. (B) 12 transmembrane α-helices such as I–VI from N-terminal domain and VII–XII from C-terminal domain, and large substrate binding cavity (gray) is marked with a circle that is composed of Helix-I, Helix-II, Helix-IV, and Helix-V from N terminal and Helix-VII, Helix-VIII, Helix-X, and Helix-XI from C terminal.
Molecular docking of NorA substrates
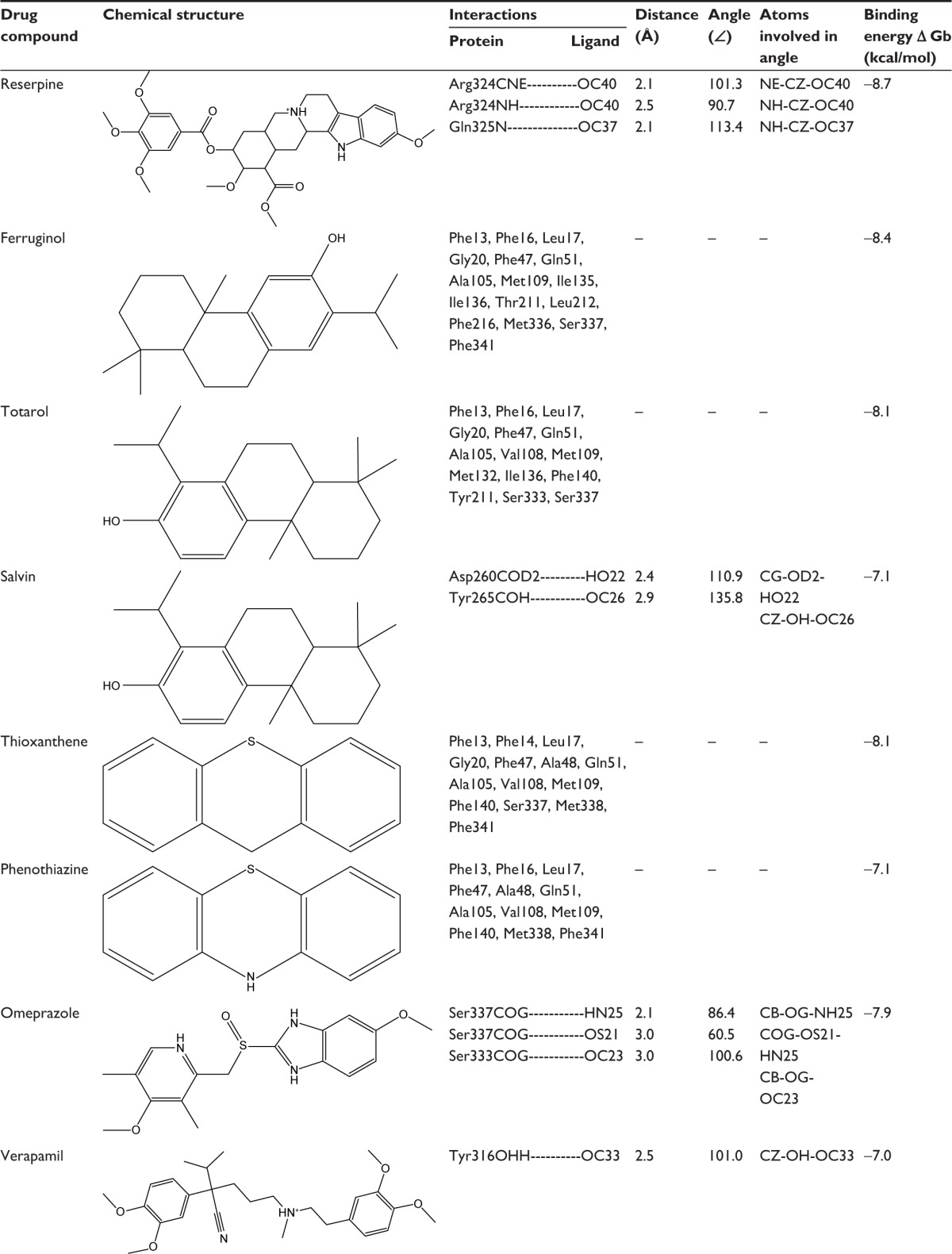
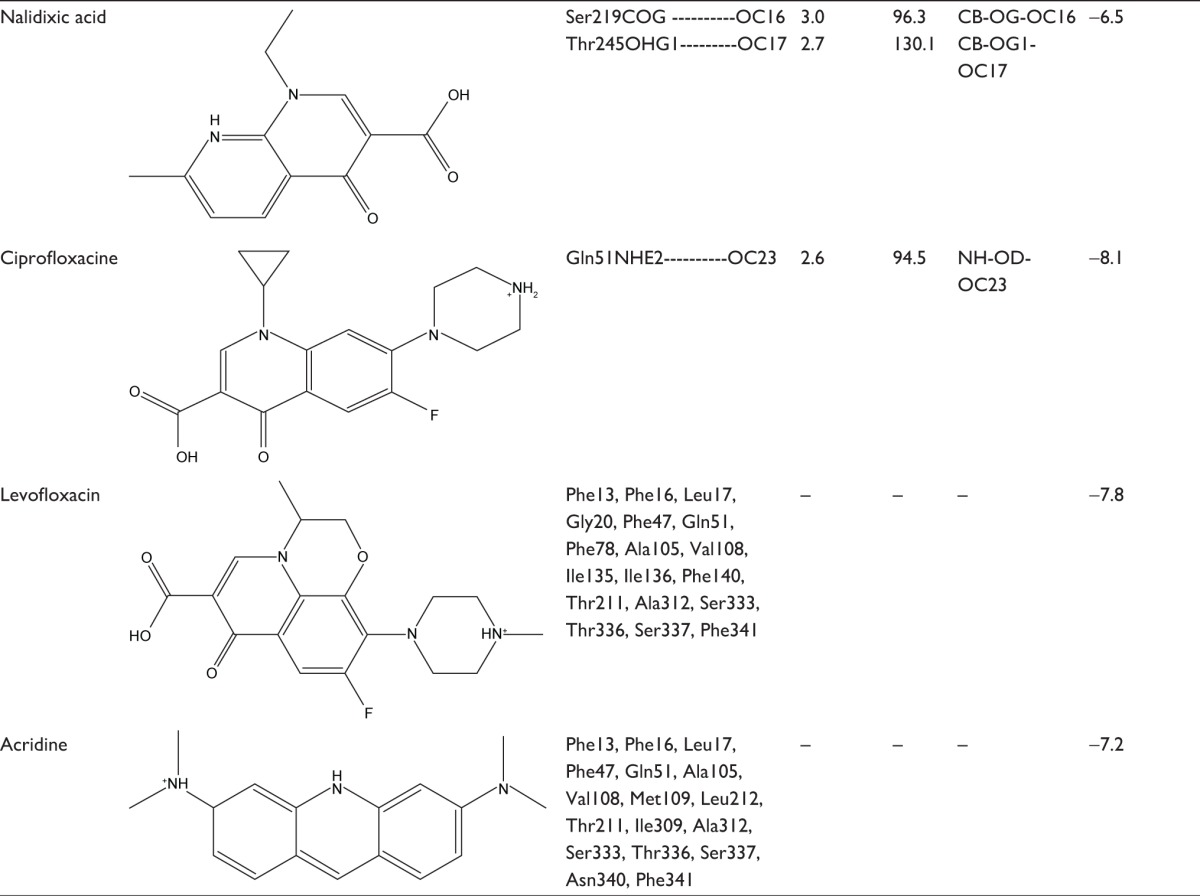
To find out the binding affinity of NorA substrates, molecular docking was performed, and the results are summarized in Table 2. From the docking results, reserpine showed highest binding energy of −8.7 kcal/mol and exhibited three binding interactions by the way of arene–arene interaction with aromatic ring of Phe317, arene–cationic interaction with Lys125, and two hydrogen bonding interactions by accepting the electrons from Arg324 and Phe129. Ferruginol and totarol are meroterpenes which showed binding affinities of −8.4 and −8.1 kcal/mol, respectively. Salvin is triterpene that exerts binding affinity of −7.1 kcal/mol; thioxanthene and phenothiazine have shown binding affinities of −8.1 and −7.1 kcal/mol, respectively, and these compounds were bound at the hydrophobic cleft by dint of hydrophobic interactions with nonpolar residues such as Phe13 and Leu17 of Helix-I, Phe47 and Gln51 of Helix-II, Met109 of Helix-IV, Ile136 and Phe140 of Helix-V, Leu212 and Phe216 of Helix-VII, and Phe341 of Helix-IX. Verapamil and omeprazole are proton pump and L-type calcium channel inhibitors that displayed binding affinities of −7.0 and −7.9 kcal/mol, respectively; verapamil forms one hydrogen bond by accepting the electron from the OH group of Tyr317, and omeprazole exhibits three bonds: two bonds by accepting the electrons from OH groups of Ser333 and one bond with OH group of Ser337. Quinolones such as nalidixic acid, ciprofloxacin, and levofloxacin are synthetic broad spectrum antimicrobial agents; alidixic acid showed lowest binding energy of −6.5 kcal/mol, and ciprofloxacin and levofloxacin showed binding energy of −8.1 and −7.8 kcal/mol, respectively. Nalidixic acid formed two bonds with OH group of Ser219 and Thr245, ciprofloxacin formed one hydrogen bond with amino group of Gln51, and levofloxacin interacted with nonpolar residues of hydrophobic cavity, that is, Phe13, Leu17, Ala105, Val108, Ile136, Phe140, Thr211, and Ala312. Acridine is a dye that shows binding affinity of −7.2 kcal/mol with nonpolar amino acids of the binding pocket (Figure 5).
Table 2.
Binding interactions, bond lengths, bond angles, and binding affinity of NorA substrate with active site residues of NorA efflux pump
|
|
Figure 5.

Binding mode of reserpine, ferruginol, totarol, salvin, phenothiazone, thioxanthene, omeprazole, verapamil, levofloxacin, nalidixic acid, ciprofloxacin, and acridine within the hydrophobic cleft (violet) of NorA efflux pump (gray).
Note: All the ligands are represented in red color.
Virtual screening and docking
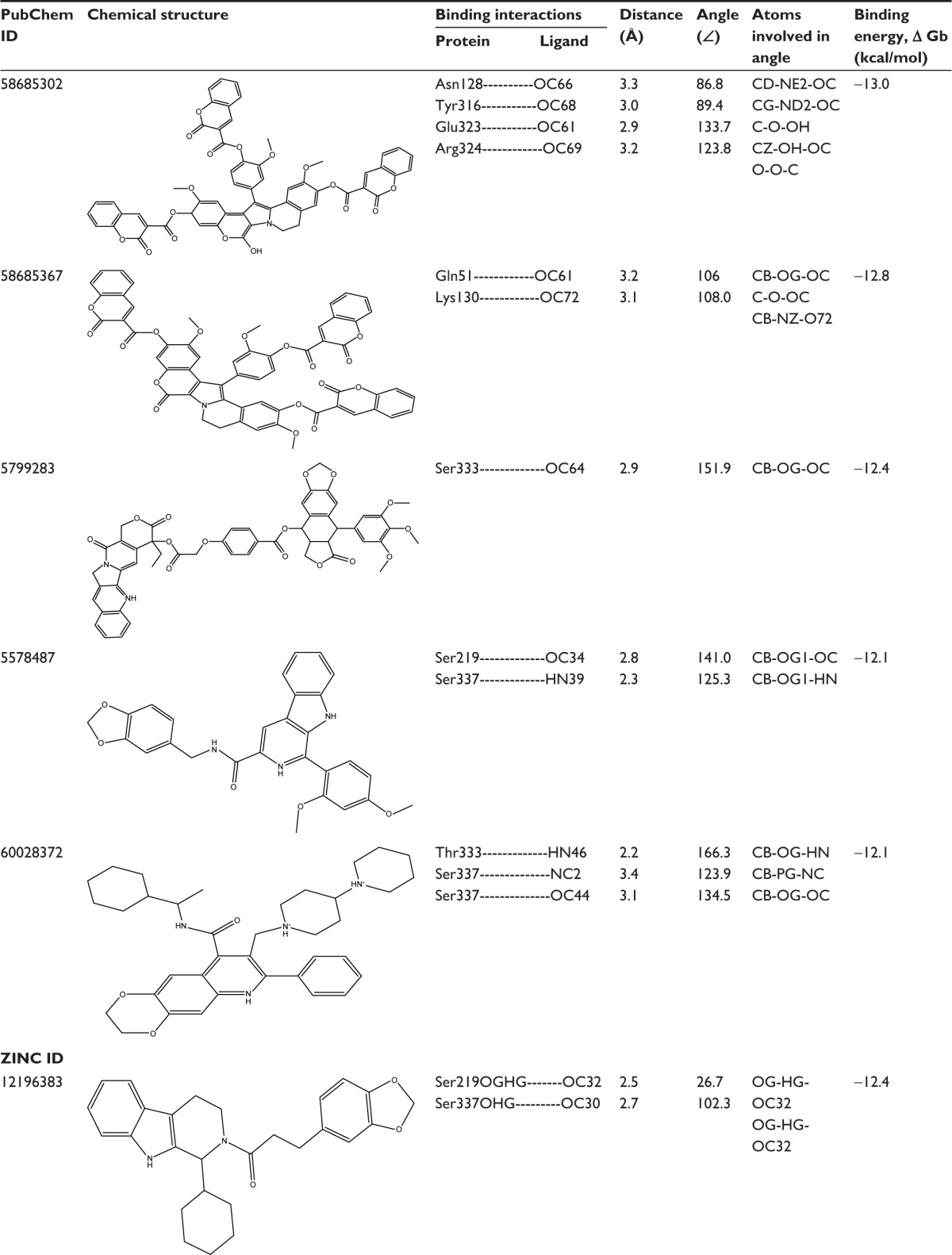
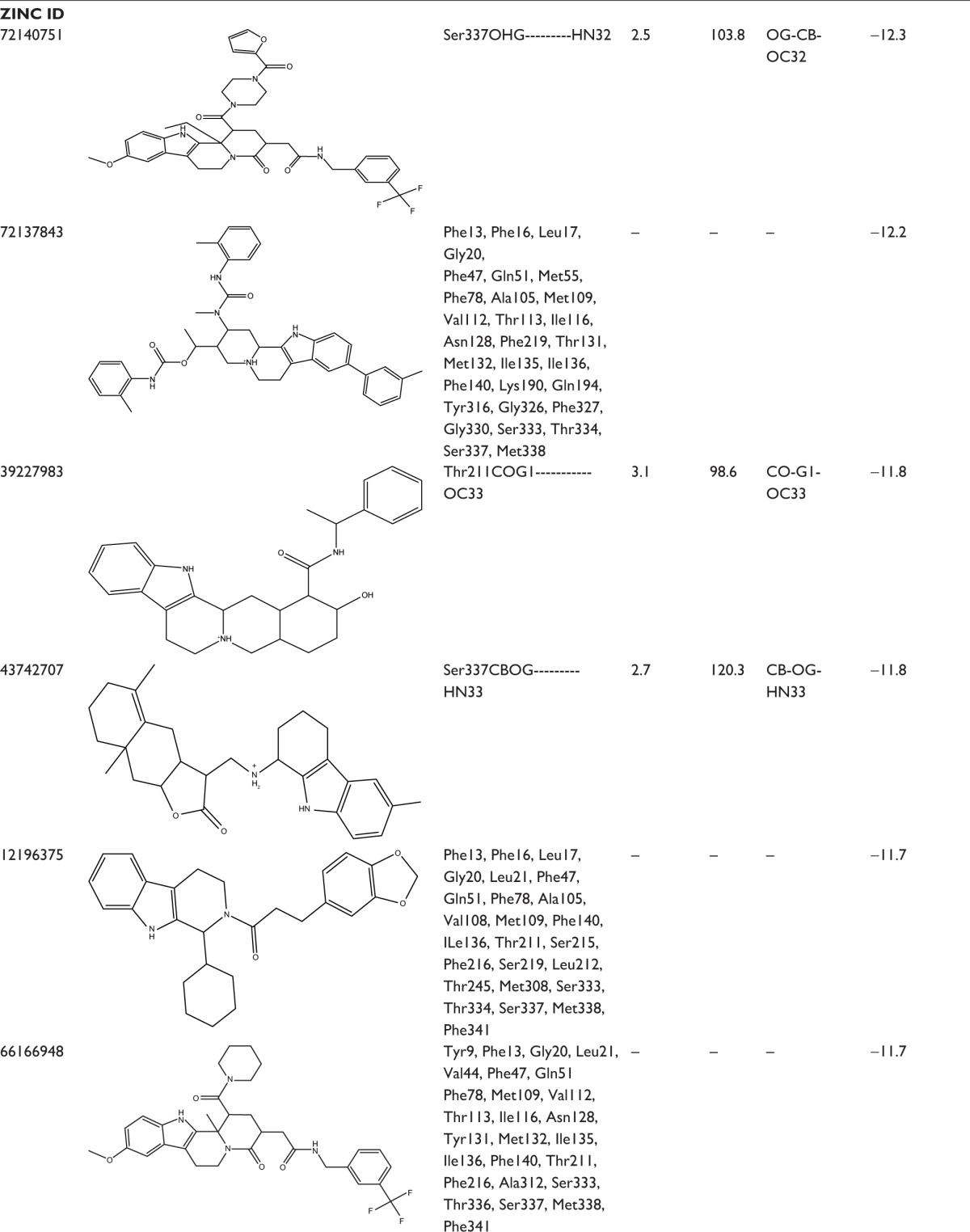
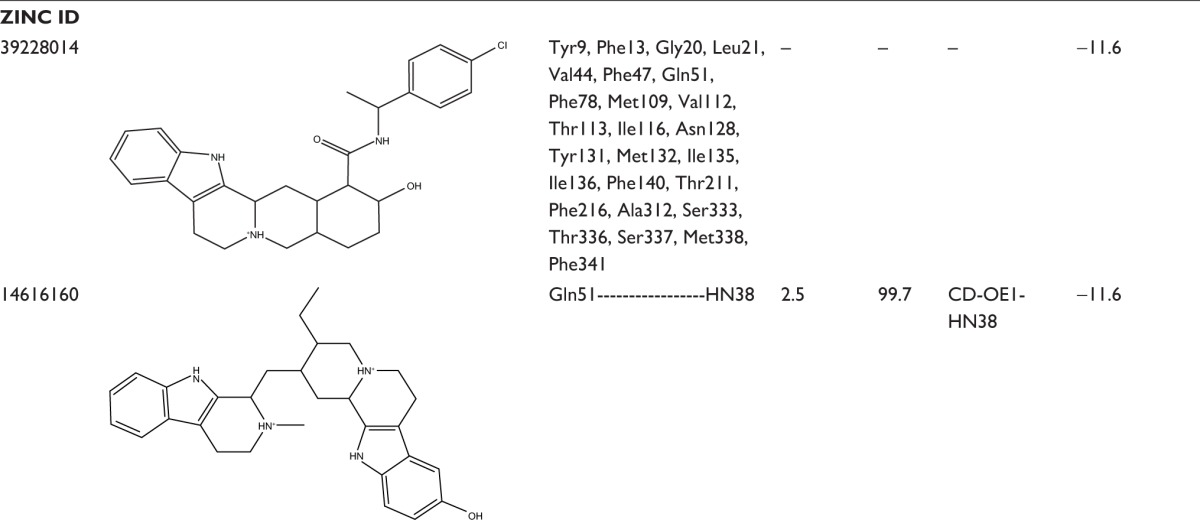
In an effort to discover the selective novel potent NorA EPIs, virtual screening study was performed with the analogs of reserpine that monitored best binding lead molecules from the calculated binding affinities (Table 3). CID58685302 showed highest binding energy of −13.0 kcal/mol and bound at the vestibule of the binding pocket by means of four hydrogen bonding interactions: one with polar amide of Asn128 of Helix-V, one with hydroxyl group of Tyr316 of Helix-X, one with carboxylic group of Glu323, and one with amino group of Arg324 at the connecting loop of X–XI helices (Figure 6A). CID58685367 has shown binding affinity of −12.8 kcal/mol and formed two hydrogen bonds: one with polar amide on Gln51 of Helix-II and one bond with amino group on Lys130 of Helix-V (Figure 6B). CID5799283 showed binding energy of −12.4 kcal/mol and displayed one hydrogen bonding interaction with OH group on Ser333 of Helix-XI (Figure 6C). CID5578487 and CID60028372 have also shown similar binding affinity of −12.1 kcal/mol; CID5578487 showed two hydrogen bonds, that is, CO and NH groups with OH group of Ser219 and Ser337; CID60028372 interacted with three hydrogen bonds, that is, one bond with OH group of Thr333 and two bonds with OH group of Ser337 (Figure 6D and E). ZINC12196383 has shown good binding affinity of −12.4 kcal/mol and conferred two hydrogen bonding interactions formed by accepting electrons from the OH group on Ser219 of Helix-VII and Ser337 of Helix-XI, and its indole and aromatic rings interacted with nonpolar residues of binding cavity such as Phe16, Phe47, Gln51, Ala105, Ile136, and Phe140 (Figure 6F). ZINC72140751 has shown binding energy of −12.3 kcal/mol and displayed one hydrogen bonding interaction with OH group of Ser337 (Figure 6G). ZINC72137843, ZINC12196375, ZINC66166948, and ZINC39228014 have shown binding energy of −12.2, −11.7, −11.7, and −11.6 kcal/mol, respectively, and interacted with nonpolar residues of Phe13, Phe16, Leu17, and Gly20 of Helix-I; Phe47, Gln51, and Met55 of Helix-III; Ala105, Met109, Val112, Thr113, and Ile116 of Helix-IV; Asn128, Phe219, Thr131, Met132, Ile135, Ile136, and Phe140 of Helix-V; Tyr136 of Helix-X; and Gly326, Phe327, Gly330, Ser333, Thr334, Ser337, and Met338 of Helix-XI (Figure 6H, K–M). ZINC39227983, ZINC43742707, and ZINC14616160 compounds have shown best binding affinity of −11.8, −11.8, and −11.6 kcal/mol, respectively; ZINC39227983 exhibited one hydrogen bonding interaction with OH group on Thr211; and ZINC43742707 displayed one hydrogen bonding interaction with OH group of Ser337 (Figure 6I and J). ZINC14616160 displayed one hydrogen bonding interaction with polar residue Gln51 of the binding pocket (Figure 6N).
Table 3.
Binding interactions, bond lengths, bond angles, and binding energies of lead molecules with active pocket residues of NorA efflux pump
|
|
|
Figure 6.

Binding mode of lead compounds.
Notes: Binding mode of lead compounds such as CID58685302 (A), CID58685367 (B), CID5799283 (C), CID5578487 (D), CID60028372 (E), ZINC12196383 (F), ZINC72140751 (G), ZINC72137843 (H), ZINC39227983 (I), ZINC43742707 (J), ZINC12196375 (K), ZINC66166948 (L), ZINC39228014 (M), and ZINC14616160 (N) within the hydrophobic cleft of NorA efflux pump. The lead molecules (slate blue) are represented in the stick model, hydrophobic cleft (forest green) of NorA is represented in the lines and cartoon, and binding interactions are represented in red dotted lines respectively.
Toxicity risk assessment and Lipinski rule of five
Appraisal of pharmacological properties using Lipinski rule of five, such as molecular weight, H-bond donors, H-bond acceptors, cLogP, and toxicity properties reveals that lead molecules are found to be satisfied with these properties and are shown in Table 4. CID58685302, CID58685367, CID58685370, and CID57992283 have shown highest molecular weight. H-bond donors were predicted to be less than five, and H-bond acceptors less than ten. cLogP or partition coefficient plays a major role in accessing the drug in the body and value less than five indicates good absorption and distribution. Most of the compounds are found to be possessing best cLogP values except ZINC72137843, ZINC66166948, CID58685302, CID58685367, CID58685370, and CID57992283. Assessment of toxicity showed that none of the compounds exhibit serious adverse effects, but ZINC66166948 and CID5578487 show very low mutagenic property; CID58685302, CID58685367, and CID58685370 exhibit slight reproductive toxicity. Drug-likeness of the compounds was determined through the fragment of compounds which reveals that all the compounds showed good drug-likeness except ZINC39228014. Drug score was calculated for the above four risk factors which reveal that all the compounds displayed best drug score and were helpful in the development of EPIs for NorA.
Table 4.
Lipinski rule of five and toxicity assessments of the lead compounds were predicted using Molinspiration and OSIRIS server
| S No | PubChem compounds | Mut | Tum | Irr | RE | DL | DS | clogP | MW | Sol | H-acc | H-don | TPSA |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | CID58685302 | − | − | − | + | 1.24 | 0.1 | 7.6 | 1,013 | −12.90 | 10 | 1 | 218.1 |
| 2 | CID58685367 | − | − | − | + | 1.13 | 0.07 | 8.5 | 1,015.0 | −12.2 | 10 | 0 | 216.72 |
| 3 | CID57992283 | − | − | − | − | − | − | 6.7 | 922.88 | − | 12 | 0 | 193.0 |
| 4 | CID5578487 | + | − | − | − | 1.57 | 0.26 | 4.97 | 491.0 | −6.72 | 6 | 2 | 94.7 |
| 5 | CID60028372 | − | − | − | − | − | − | 4.2 | 472.0 | −5.52 | 4 | 3 | 69.33 |
| ZINC compounds | |||||||||||||
| 6 | ZINC12196383 | − | − | − | − | 0.34 | 0.24 | 5.66 | 446.0 | 3 | 1 | 54.6 | |
| 7 | ZINC72140751 | − | − | − | − | 0.67 | 0.23 | 5.99 | 514.0 | −7.59 | 5 | 2 | 128.19 |
| 8 | ZINC72137843 | − | − | − | − | 3.56 | 0.84 | 7.52 | 329.0 | −3.12 | 2 | 4 | 90.90 |
| 9 | ZINC39227983 | − | − | − | − | 4.18 | 0.36 | 3.12 | 469.0 | −5.49 | 2 | 4 | 69.56 |
| 10 | ZINC43742707 | − | − | − | − | 3.02 | 0.21 | 4.79 | 448.0 | −6.04 | 1 | 2 | 58.70 |
| 11 | ZINC12196375 | − | − | − | − | 1.0 | 0.23 | 5.63 | 584.3 | 6.03 | 3 | 1 | 54.56 |
| 12 | ZINC66166948 | + | − | − | − | 5.04 | 0.15 | 6.04 | 612.2 | −5.37 | 4 | 2 | 94.74 |
| 13 | ZINC39228014 | − | − | − | − | 8.8.02 | 0.35 | 3.77 | 521.0 | −5.02 | 2 | 4 | 69.56 |
| 14 | ZINC14616160 | − | − | − | − | 3.42 | 0.24 | 3.2 | 468.0 | −7.05 | 1 | 5 | 60.69 |
Abbreviations: Mut, mutagenic; Tum, tumorigenic; Irr, irritant; RE, reproductive effect; DL, drug-likeness; DS, drug score; clogP, partition coefficient; MW, molecular weight; Sol, solubility; H-acc, hydrogen bond acceptor; H-don, hydrogen bond donor; TPSA, topological polar surface area; S No, serial number.
Conclusion
NorA pump consists of 12 TM helices and a large binding pocket between N- and C-terminal interfaces accommodates numerous drug compounds. Docking simulation studies of NorA substrates reveal that vital residues, namely Phe13, Phe14, Phe16, Leu17, Gly20, Phe47, Ala48, Gln51, Ala105, Gly106, Val108, Met109, Ile135, Ileu136, Phe140, Thr211, Leu212, Phe216, Ala312, and Phe341 present at the binding pocket play a pivotal role in extrusion of numerous compounds. In silico screening affords 14 good binding lead compounds which selectively inhibit the NorA efflux pump by competing with its substrates. Hence, these lead molecules will be useful for designing selective EPIs against NorA efflux pump of S. aureus.
Acknowledgments
The authors are grateful to the coordinator, Bioinformatics Infrastructure Facility, Department of Zoology, Sri Venkateswara University, Tirupati, for providing bioinformatics facilities.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Klevens RM, Morrison MA, Nadle J, et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298(15):1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 2.Hassan KA, Skurray RA, Brown MH. Active export proteins mediating drug resistance in Staphylococci. J Mol Microbiol Biotechnol. 2007;12(3–4):180–196. doi: 10.1159/000099640. [DOI] [PubMed] [Google Scholar]
- 3.Pao SS, Paulsen IT, Saier MH., Jr Major facilitator super family. Microbiol Mol Biol Rev. 1998;62(1):1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 2003;301(5633):616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- 5.Law CJ, Yang Q, Soudant C, Maloney PC, Wang DN. Kinetic evidence is consistent with the rocker switch mechanism of membrane transport by GlpT. Biochemistry. 2007;46(43):12190–12197. doi: 10.1021/bi701383g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Marco CE, Cushin LA, Frempong-Manso E, Seo SM, Jaravaza TA, Kaatz GW. Efflux-related resistance to norfloxacin, dyes, and biocides in bloodstream isolates of Staphylococcus aureus. Antimicrob Agents Chemother. 2007;51(9):3235–3239. doi: 10.1128/AAC.00430-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohammed-Ali MN, Jamalludeen NM. Isolation and characterization of bacteriophage against methicillin resistant Staphylococcus aureus. J Med Microb Diagn. 2015;5:213. [Google Scholar]
- 8.Kaatz GW, Seo SM, Ruble CA. Efflux-mediated fluoroquinolone resistance in Staphylococcus aureus. Antimicrob Agents Chemother. 1993;37(5):1086–1094. doi: 10.1128/aac.37.5.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neyfakh AA, Borsch CM, Kaatz GW. Fluoroquinolone resistance protein NorA of Staphylococcus aureus is a multidrug efflux transporter. Antimicrob Agents Chemother. 2003;37:128–129. doi: 10.1128/aac.37.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.German N, Wei P, Kaatz GW, Kerns RJ. Synthesis and evaluation of fluoroquinolone derivatives as substrate-based inhibitors of bacterial efflux pumps. Eur J Med Chem. 2008;43(11):2453–2463. doi: 10.1016/j.ejmech.2008.01.042. [DOI] [PubMed] [Google Scholar]
- 11.Noguchi N, Tamura M, Narui K, Wakasugi K, Sasatsu M. Frequency and genetic characterization of multidrug-resistant mutants of Staphylococcus aureus after selection with individual antiseptics and fluoroquinolones. Biol Pharm Bull. 2002;25(9):1129–1132. doi: 10.1248/bpb.25.1129. [DOI] [PubMed] [Google Scholar]
- 12.Lewis K, Klibanov AM. Surpassing nature: rational design of sterile-surface materials. Trends Biotechnol. 2005;23(7):343–348. doi: 10.1016/j.tibtech.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Kaatz GW, Moudgal VV, Seo SM, Kristiansen JE. Phenothiazines and thioxanthenes inhibit multidrug efflux pump activity in Staphylococcus aureus. Antimicrob Agents Chemother. 2003;47(2):719–726. doi: 10.1128/AAC.47.2.719-726.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vidaillac C, Guillon J, Arpin C, et al. Synthesis of omeprazole analogues and evaluation of these as potential inhibitors of the multidrug efflux pump NorA of Staphylococcus aureus. Antimicrob Agents Chemother. 2007;51(3):831–838. doi: 10.1128/AAC.01306-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Marco CE, Cushin LA, Frempong-Manso E, Seo SM, Jaravaza TA, Kaatz GW. Efflux-related resistance to norfloxacin, dyes, and biocides in bloodstream isolates of Staphylococcus aureus. Antimicrob Agents Chemother. 2007;51(9):3235–3239. doi: 10.1128/AAC.00430-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saier MH, Beatty JT, Goffeau A, et al. The major facilitator super family. J Mol Microbiol Biotechnol. 1999;1(2):257–279. [PubMed] [Google Scholar]
- 17.Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 2003;301(5633):616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- 18.Guan L, Mirza O, Verner G, Iwata S, Kaback HR. Structural determination of wild-type lactose permease. Proc Natl Acad Sci U S A. 2007;104(39):15294–15298. doi: 10.1073/pnas.0707688104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yin Y, He X, Szewczyk P, Nguyen T, Chang G. Structure of the multidrug transporter EmrD from Escherichia coli. Science. 2006;312(5774):741–744. doi: 10.1126/science.1125629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hirai T, Subramaniam D. Structure and transport mechanism of the bacterial oxalate transporter OxlT. Biophys J. 2004;87(5):3600–3607. doi: 10.1529/biophysj.104.049320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Venter H, Ma S. Efflux pump inhibitors: a novel approach to combat efflux-mediated drug resistance in bacteria. Current Drug Targets. 2016;17(18):702–719. doi: 10.2174/1389450116666151001103948. [DOI] [PubMed] [Google Scholar]
- 22.Nargotra A, Sharma S, Koul JL, et al. Quantitative structure activity relationship (QSAR) of piperine analogs for bacterial NorA efflux pump inhibitors. Eur J Med Chem. 2009;44(10):4128–4135. doi: 10.1016/j.ejmech.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 23.Fontaine F, Hequet A, Voisin-Chiret AS, et al. First identification of boronic species as novel potential inhibitors of the Staphylococcus aureus NorA efflux pump. J Med Chem. 2014;57(6):2536–2548. doi: 10.1021/jm401808n. [DOI] [PubMed] [Google Scholar]
- 24.Michalet S, Cartier G, David B, et al. N-caffeoylphenalkylamide derivatives as bacterial efflux pump inhibitors. Bioorg Med Chem Lett. 2006;17(6):1755–1758. doi: 10.1016/j.bmcl.2006.12.059. [DOI] [PubMed] [Google Scholar]
- 25.Samosoron S, Bremner JB, Ball A, Lewis K. Synthesis of functionalized 2-aryl-5-nitro-1H-indoles and their activity as bacterial NorA efflux pump inhibitors. Bioorg Med Chem. 2005;14(3):857–865. doi: 10.1016/j.bmc.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 26.Fournier D, Chabert J, Marquez B, et al. Synthesis and evaluation of new arylbenzo(b)thiophene and diarylthiophene derivatives as inhibitors of the NorA multidrug transporter of Staphylococcus aureus. Bioorg Med Chem. 2007;15(13):4482–4497. doi: 10.1016/j.bmc.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 27.Pereda-Miranda R, Kaatz GW, Gibbons S. Polyacylated oligosaccharides from medicinal Mexican morning glory species as antibacterials and inhibitors of multidrug resistance in Staphylococcus aureus. J Nat Prod. 2006;69(3):406–409. doi: 10.1021/np050227d. [DOI] [PubMed] [Google Scholar]
- 28.Thai KM, Ngo TD, Phan TV, et al. Virtual screening for novel Staphylococcus aureus NorA efflux pump inhibitors from natural products. Med Chem. 2015;11(2):135–155. doi: 10.2174/1573406410666140902110903. [DOI] [PubMed] [Google Scholar]
- 29.Sonnhammer EL, Von Heijne G, Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol. 1998;6:175–182. [PubMed] [Google Scholar]
- 30.Hofmann K, Stoffel W. TMBASE: a database of membrane spanning protein segments. Biol Chem. 1993;374:166–170. [Google Scholar]
- 31.Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14(4):378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- 32.Cserzo M, Wallin E, Simon I, Von Heijne G, Elofsson A. Prediction of transmembrane alpha-helices in prokaryotic membrane proteins: the dense alignment surface method. Protein Eng. 1997;10(6):673–676. doi: 10.1093/protein/10.6.673. [DOI] [PubMed] [Google Scholar]
- 33.Tusnady GE, Simon I. Principles governing amino acid composition of integral membrane proteins: Application to topology prediction. J Mol Biol. 1998;283(2):489–506. doi: 10.1006/jmbi.1998.2107. [DOI] [PubMed] [Google Scholar]
- 34.Rost B, Sander C. Combining evolutionary information and neural networks to predict protein secondary structure. Protein. 1994;19(1):55–72. doi: 10.1002/prot.340190108. [DOI] [PubMed] [Google Scholar]
- 35.Claros MG, Von Heijne G. TopPred II: an improved software for membrane protein structure predictions. Comput Appl Biosci. 1994;10(6):685–686. doi: 10.1093/bioinformatics/10.6.685. [DOI] [PubMed] [Google Scholar]
- 36.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 38.Kale L, Skeel R, Bhandarkar M, et al. NAMD: greater scalability for parallel molecular dynamics. J Comput Phys. 1999;151:283–312. [Google Scholar]
- 39.Schlick T, Skeel R, Brunger A, Kale L, Board JA, Hermans J. Algorithmic challenges in computational molecular biophysics. J Comput Phys. 1999;151:9–48. [Google Scholar]
- 40.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–934. [Google Scholar]
- 41.Grubmuller H, Heller H, Windemuth A, Schulten K. Generalized Verlet algorithm for efficient molecular dynamics simulations with long-range interactions. Mol Simul. 1991;6:121–142. [Google Scholar]
- 42.Darden TA, Pedersen LG. Molecular modeling: an experimental tool. Environ Health Perspect. 1993;101(5):410–412. doi: 10.1289/ehp.93101410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Essmann U, Berkowitz ML. Dynamical properties of phospholipid bilayers from computer simulation. Biophys J. 1999;76(4):2081–2089. doi: 10.1016/S0006-3495(99)77364-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comp Phys. 1977;23(3):327–341. [Google Scholar]
- 45.Laskoswki RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereo chemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 46.Eisenberg D, Luthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 47.Colovos C, Yeates TO. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993;2(9):1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hooft RW, Sander C, Vriend G. Positioning hydrogen atoms by optimizing hydrogen-bond networks in protein structures. Proteins. 1996;26:363–376. doi: 10.1002/(SICI)1097-0134(199612)26:4<363::AID-PROT1>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 49.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18(15):2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 50.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;1:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolf LK. PyRx. C & EN. 2009;87:31. [Google Scholar]
- 52.DeLano WL. The PyMOL Molecular Graphics System DeLano Scientific. New York, NY: Schrödinger, LLC; version 1.7. [Google Scholar]
- 53.Kalia NP, Mahajan P, Mehra R, et al. Capsaicin, a novel inhibitor of the NorA efflux pump, reduces the intracellular invasion of Staphylococcus aureus. J Antimicrob Chemother. 2012;67(10):2401–2408. doi: 10.1093/jac/dks232. [DOI] [PubMed] [Google Scholar]