

Abstract
Carboxylesterases (CEs) are ubiquitous enzymes responsible for the detoxification of ester‐containing xenobiotics. This hydrolysis reaction results in the formation of the corresponding carboxylic acid and alcohol. Due to their highly plastic active site, CEs can hydrolyze structurally very distinct and complex molecules. Because ester groups significantly increase the water solubility of compounds, they are frequently used in the pharmaceutical industry to make relatively insoluble compounds more bioavailable. By default, this results in CEs playing a major role in the distribution and metabolism of these esterified drugs. However, this can be exploited to selectively improve compound hydrolysis, and using specific in vivo targeting techniques can be employed to generate enhanced drug activity. Here, we seek to detail the human CEs involved in esterified molecule hydrolysis, compare and contrast these with CEs present in small mammals and describe novel methods to improve drug therapy by specific delivery of CEs to cells in vivo. Finally, we will discuss the development of such approaches for their potential application towards malignant disease.
Abbreviations
- CE
carboxylesterase
- CPT‐11
irinotecan (Camptosar), 7‐ethyl‐10‐[4‐(1‐piperidino)‐1‐piperidino]carbonyloxycamptothecin
- ER
endoplasmic reticulum
- hBr3 (CES3)
human brain CE
- hCE1 (CES1)
human liver CE
- hiCE (CES2)
human intestinal CE
- NSC
neural stem cell
- SN‐38
7‐ethyl‐10‐hydroxycamptothecin
Tables of Links
| TARGETS |
|---|
| Enzymes |
| Carboxylesterase 1 |
| LIGANDS |
|---|
| Irinotecan |
| SN‐38 |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
The detoxification of ester‐containing xenobiotics is carried out by carboxylesterases (CEs) (Cashman et al., 1996). These enzymes have been identified in essentially all organisms studied to date, although it is not clear if there are endogenous substrates for these proteins. As a consequence, it is thought that CEs act in a protective manner. Circumstantial evidence that this is the case is provided by the fact that these enzymes tend to be expressed in tissues that might be regularly exposed to xenobiotics, such as liver, kidney and the epithelial linings of the lung and gut (Inkerman et al., 1975; Munger et al., 1991; Brzezinski et al., 1994; Crow et al., 2007; Hatfield et al., 2011). The presence of the ester moiety within natural products is widespread (Figure 1), and it is presumed that this functionality acts to increase the water solubility of these compounds. This is due to the ease with which the oxygen atoms within this chemotype can hydrogen bond with water molecules. This property has been exploited by medicinal chemists in the pharmaceutical industry, where agents that have been identified in high throughput screening approaches can be modified by esterification to yield more soluble, bioactive compounds. As a consequence, many clinically used drugs contain the ester chemotype (Figure 2).
Figure 1.
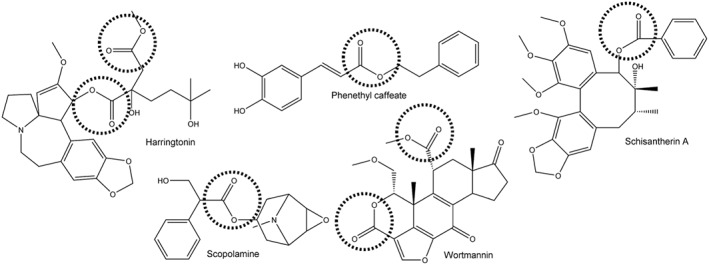
Structures of a wide range of compounds isolated from natural products that contain ester moieties. The ester function is indicated by the dashed circle.
Figure 2.
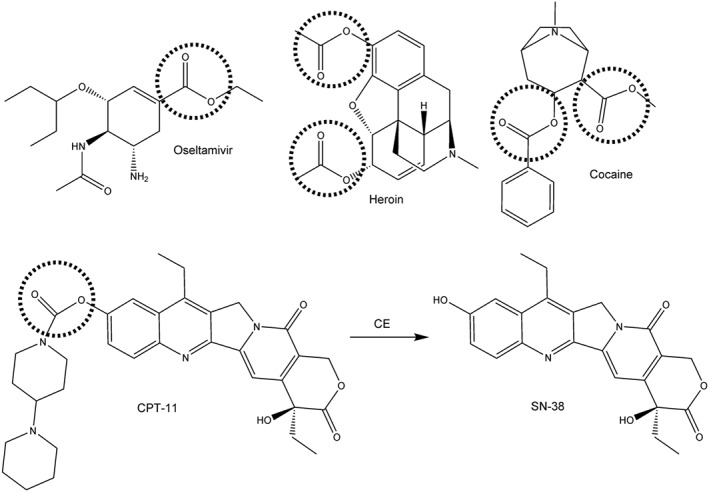
The chemical structures of clinically used and abused drugs that are hydrolyzed by CEs. The ester groups that are cleaved by CEs are indicated by the dashed circles. For CPT‐11, the product of the hydrolysis reaction (SN‐38) is also shown.
The problem with such an approach is that, de facto, this results in the derived compounds being substrates for CEs. If the metabolites of the hydrolysis reaction are inactive, then this results in a detoxification of the drug. Consequently, it is unlikely that these molecules would be active in tissues with high levels of CE. Alternatively, if the carboxylic acid or alcohol that results from the enzymic reaction is more active than the parent molecule, then the latter can be considered a prodrug. In this instance, higher levels of the active drug would be present within cells that have increased levels of the activating CE. By exploiting this property, our group and colleagues have developed specific approaches to selectively deliver drug‐activating enzymes to tumour cells in vivo that, when combined with prodrugs, result in enhanced antitumour activity.
Human CEs
In humans, five potential CE gene coding sequences have been identified in genome sequencing studies. However, to date, only three (hCE1 [CES1]; hiCE (CES2); and hBr3 [CES3]) have been evaluated for their biological activity (Brzezinski et al., 1997; Humerickhouse et al., 2000; Khanna et al., 2000; Sanghani et al., 2004; Quinney et al., 2005; Hatfield et al., 2010; Hatfield et al., 2011). Indeed of these, only hCE1 and hiCE have been extensively analysed with respect to drug activation. While both proteins are ~60 kDa in size and require processing within the endoplasmic reticulum (ER) for functional activity, they tend to be expressed in different locations and have markedly different substrate specificities (Table 1) (Potter et al., 1998b, 1998c). For example, hCE1 is primarily expressed in the liver and tends to hydrolyze small more compact molecules, such as oseltamivir (Figure 2; Wadkins et al., 2001; Shi et al., 2006; Hatfield et al., 2011). In contrast, hiCE is present within the gut and kidney and demonstrates much more variable expression in the liver (Hatfield et al., 2011). This enzyme is capable of hydrolyzing much larger molecules including CPT‐11, cocaine and heroin [Figure 2 (Humerickhouse et al., 2000; Khanna et al., 2000; Sanghani et al., 2004; Hatfield et al., 2010; Hatfield et al., 2011)]. CPT‐11 is an anticancer prodrug that is converted to SN‐38, a potent inhibitor of topoisomerase I, by CEs (Tanizawa et al., 1994). Because hiCE is expressed at high levels in the gut (principally in the duodenum), the delayed diarrhoea that is observed following CPT‐11 treatment (the dose‐limiting toxicity) is likely to reflect the hydrolysis of the drug following secretion into the bile (Morton et al., 2005). In this instance, tissue specific activation of the drug results in toxicity.
Table 1.
Properties of mammalian carboxylesterases
| Property | Enzyme | |||
|---|---|---|---|---|
| hCE1 | hiCE | rCE | hCE1m6 | |
| Gene name | CES1 | CES2 | RLCE | CES1a |
| Protein size | 567aa, 60 kDa | 559aa, 60 kDa | 565aa, 60 kDa | 568aa, 60 kDa |
| Location of expression | Liver, lung epithelia, monocyctes | Intestinal epithelia, liver, kidney | Liver, other? | NAb |
| Cellular location | Microsomes | Microsomes | Microsomes | Microsomes |
| Secretedc | No | No | No | No |
| Stabilityd | Excellent | Poor | Excellent | Excellent |
| Substrate specificity | Small, planar molecules | Large bulky molecules | Large bulky molecules | Large bulky molecules |
| Examples of substrates | NPE, oseltamivir, heroin | NPE, CPT‐11, heroin, cocaine | NPE, CPT‐11 | NPE, CPT‐11 |
| Relative efficiency of CPT‐11 hydrolysis (hCE1 = 1) | 1 | 91 | 650 | 71 |
NPE,nitrophenyl esters.
hCE1m6 was generated by mutagenesis of the hCE1 (CES1) coding sequence. Formally, therefore, it does not have a specific gene name.
Because hCE1m6 represents a mutant form of hCE1, it is not endogenously expressed in vivo.
While all of the enzymes are intracellular, because they are all processed within the ER, they can be engineered to be secreted by removing the C‐terminal amino acid sequence that acts as a signal for localization in the ER.
hCE1, rCE and hCE1m6 are stable for many months, even at room temperature. hiCE rapidly loses activity under the same storage conditions
Using this observation as a platform, we hypothesized that selective delivery of CEs to target cells might yield improved drug activity. For example, if hiCE, or other activating enzymes, could be localized to tumour cells in vivo, then potentially, following CPT‐11 administration, higher levels of the active metabolite (SN‐38) would occur within the tumour milieu. This should result in enhanced antitumour activity, with a minimal increase in toxicity, because the systemic levels of SN‐38 would not change. This then forms the basis of the enzyme/prodrug therapy approach described herein (Figure 3).
Figure 3.
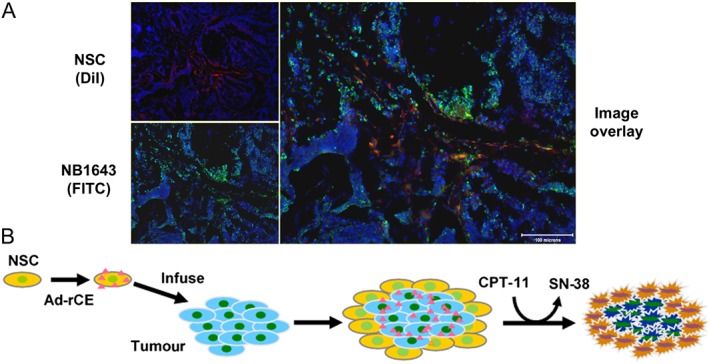
Use of NSCs to target tumour cells in vivo. (A) Co‐localization of NSCs (labelled with DiI – red, top left) to NB1643 neuroblastoma cells (labelled with FITC – green, bottom left) in the liver of a mouse. In this experiment, mice bearing disseminated NB1643 tumours were injected i.v. with DiI‐labelled NSCs, and 48 h later, liver samples were evaluated by immunofluorescence. The image on the right represents an overlay of the left hand panels, indicating that both NSC and tumour cells are present within the same location. (B) A diagram indicating the use of NSC to sensitize cells to CPT‐11. The pink triangles represent secreted rCE. Ad‐rCE, adenovirus expressing rCE.
However, for this methodology to be effective, there are several key requirements. Firstly, the activation of the drug should be minimal in the absence of the activating enzyme. For CPT‐11, while this agent is in widespread use for the treatment of colon cancer, typically, less than 5% of the dose administered to patients is converted to the active metabolite (Mathijssen et al., 2001). This argues that even a relatively modest increase in hydrolysis (say up to 10%) might significantly increase efficacy. Secondly, a specific delivery system that only targets tumour cells must be employed, because any increase in systemic SN‐38 levels is likely to be highly toxic. We have employed the use of neural stem cells (NSCs) because they demonstrate tropism to tumour cells in vivo and can remain localized to these lesions for up to 10 days (Aboody et al., 2000; Brown et al., 2003; Aboody et al., 2006b). Thirdly, these cells must generate sufficient SN‐38, via CPT‐11 hydrolysis, to generate a therapeutic response. To achieve this goal, we have employed viral‐mediated delivery of CE cDNAs to NSCs to yield very high levels of enzyme (Wierdl et al., 2001). Finally, we have developed a suitable animal model in which to assess the efficacy of such an approach. In the following sections, we describe the development and properties of each component of this technology.
Use of CPT‐11
CPT‐11 was developed by Yakult Daiichi as a means of formulating camptothecin for clinical use. The latter molecule was originally employed in clinical trials in cancer patients in the 1970s, but these were stopped due to significant adverse toxicity (Creaven et al., 1972; Muggia et al., 1972). However, by inclusion of the 4‐piperidinopiperidine moiety at position 10 of the molecule (Figure 2), a less toxic prodrug was obtained that required hydrolysis for activation. Indeed, the difference in the cytotoxicity between CPT‐11 and SN‐38 is up to 1000‐fold, with the latter demonstrating IC50 values in the low nanomolar range (Tanizawa et al., 1994). Interestingly, when CPT‐11 is administered to rodents greater than 50% of the drug is converted to SN‐38 within an hour of dosing (Bissery et al., 1996; Morton et al., 2000; Morton et al., 2005). In contrast, less than 5% of the drug is hydrolyzed in man after 24 h (Mathijssen et al., 2001). This argues that the rodent enzymes are considerably more efficient at drug hydrolysis and potentially could be exploited for selective activation in humans. Because CPT‐11 was approved for use in the clinic in the 1990s and demonstrated excellent antitumour activity in a spectrum of adult and paediatric patients (Conti et al., 1996; Furman et al., 1999), we used this as the candidate drug with which to develop the enzyme/prodrug approach.
Choice of CE
Initial efforts to isolate enzymes that were efficient at CPT‐11 hydrolysis were problematic, although a rabbit liver CE (rCE) was ultimately identified and cloned (Potter et al., 1998a) (Table 1). This enzyme readily converted CPT‐11 to SN‐38, conferred enhanced cytotoxicity to the prodrug when expressed in cells and xenografts and was demonstrated to be exceptionally stable. This allowed the crystal structure of the protein to be solved and provided significant insight into substrate hydrolysis (Bencharit et al., 2002; Redinbo and Potter, 2005). Using rCE as a template, the human homologue was isolated (hCE1). However, this enzyme was ~600‐fold less efficient at CPT‐11 activation (Wierdl et al., 2008). Subsequent analysis and comparison of the X‐ray structure of hCE1 with rCE identified several key loops of amino acids that regulated access of the drug to the catalytic residues, and mutagenesis allowed the formation of a mutant hCE1 protein (hCE1m6; Table 1) that was efficient at drug hydrolysis (Wierdl et al., 2008). Finally, by analogy with other ER processed enzymes, we demonstrated that removal of the KDEL sequence at the C‐terminus of both rCE and hCE1m6 allowed for secretion of the active proteins (Potter et al., 1998c; Wierdl et al., 2001; Wierdl et al., 2008). These would presumably result in bystander effects when combined with CPT‐11, that is, where extracellular activation of the drug would result in the active metabolite (SN‐38) diffusing into neighbouring tumour cells, thereby providing an enhanced cytotoxicity. By a combination of different structural and molecular approaches therefore, we have developed an active, secreted human CE that is highly efficient at CPT‐11 activation. That being said, all initial long‐term therapeutic studies in animals were performed using rCE because this enzyme is the most efficient at hydrolyzing CPT‐11 in vitro.
Delivery of CEs
To effect delivery of CEs to tumour cells in vivo, we have employed the use of neural stem cells (NSC) (Aboody et al., 2000; Aboody et al., 2006b). These cells can migrate and localize to areas of abnormal pathology (wounds, stroke lesions, tumour cells, etc.) through an, as yet, unknown mechanism. In naïve animals that demonstrate no such deficits, NSC undergo apoptosis and are cleared from the animal within 72–96 h (Aboody et al., 2006a). In contrast, in mice bearing human tumours, NSC encapsulate the lesion and remain in close proximity for up to 10 days. Use of these cells therefore can yield a therapeutic window in which their clearance from unaffected tissues and organs would ensure that adverse systemic toxicity is minimized following prodrug administration.
To generate high levels of either rCE or hCE1m6 in NSC, we employed adenoviral transduction. This was chosen because it results in transient, high level gene expression without integration into the genome. Using this methodology, large amounts of CE can be generated within cells, and this results in a significant increase in the cytotoxicity of CPT‐11 (Wierdl et al., 2001). Furthermore, media harvested from these cells are also able to activate the drug because the enzyme has been engineered to be secreted. Finally, adenoviral vectors have been used in clinical trials with minimal problems, and there is considerable expertise in working with these agents in the patient population.
Suitable animal models
As indicated above, the activation of CPT‐11 in mice and humans differs markedly. Therefore, developing an approach that modulates that activity of the drug must be interpreted with extreme caution. In an effort to minimize aberrant drug hydrolysis in animals, we identified and developed a plasma esterase‐deficient mouse strain (Morton et al., 2000). This mouse (named Es1e) has considerably reduced levels of a circulating CE that can activate CPT‐11, and PK results indicate that these animals represent a model that much more accurately reproduces the levels of CPT‐11 hydrolysis seen in humans. To allow for xenotransplanation, the Es1e mice were crossed with a Scid (severe combined immune deficient) strain to yield animals (Es1e/scid) that were plasma esterase‐deficient and would permit growth of human tumour cells (Morton et al., 2005).
Finally, because we believe that this drug activation approach would be unlikely to be effective towards large solid tumours, but much more efficacious against small metastatic lesions, we used disseminated disease models for paediatric neuroblastoma (Thompson et al., 2001). Patients diagnosed with the latter frequently demonstrate a complete response to chemotherapy, but subsequently relapse 2–4 years later (Park et al., 2008). This argues that residual tumour cells that escape the initial treatment, reside in these individuals and it is at this stage that the enzyme/prodrug approach would be employed. Therefore, a series of animal models were developed with i.v. injection of low numbers (1 × 105–1 × 106) of human neuroblastoma cells into Es1e/scid mice (Aboody et al., 2006a; Danks et al., 2007). This allows for a long latency with regard to tumour development and mimics what is observed in patients who are apparently free of disease. The efficacy of the enzyme/prodrug approach using CE/CPT‐11 was evaluated in these animals.
CE/CPT‐11 prevents disseminated neuroblastoma
Having developed all of the individual components necessary for assessing selective drug activation, therapeutic studies were initiated. In these experiments, mice were injected with varying numbers of tumour cells, and the latter allowed to grow for 14 days. At this time point, NSCs expressing rCE were infused into the animals. CPT‐11 administration was started 4 days later to provide time for maximal CE expression and for free NSCs to clear the animals (see the diagram in Figure 4). The drug was given daily for 5 days, repeated the following week and, after a week for recovery, this complete process was repeated. As indicated in Figure 4, administration of NSC expressing rCE resulted in a significant increase in animal survival, and this occurred in drug dose‐dependent fashion (Aboody et al., 2006a; Danks et al., 2007). This argues that this was truly a pharmacological effect based upon selective drug activation, and not related to any intrinsic property of the NSCs. Additional studies confirmed that the circulating levels of SN‐38 were the same in animals receiving the drug alone and those given the drug + NSC, demonstrating that local activation of CPT‐11 was responsible for the antitumour activity (Danks et al., 2007). Indeed, when using 15 mg kg−1 CPT‐11, 90% of the animals survived in the NCS/CE group and were essentially cured of the disease. As exemplified by the significantly extended time frame of these experiments (note the scale on the abscissa axis), these mice live for more than 1 year following tumour cell infusions, representing over 50% of their lifespan. This significant increase in survival is rarely observed in patients and, although bone marrow transplants, the use of therapeutic antibodies and extremely toxic chemotherapy can extend patient survival, such procedures nearly always fail. Cleary, therefore, adaptation of this enzyme/prodrug approach to a patient population would be highly desirable.
Figure 4.
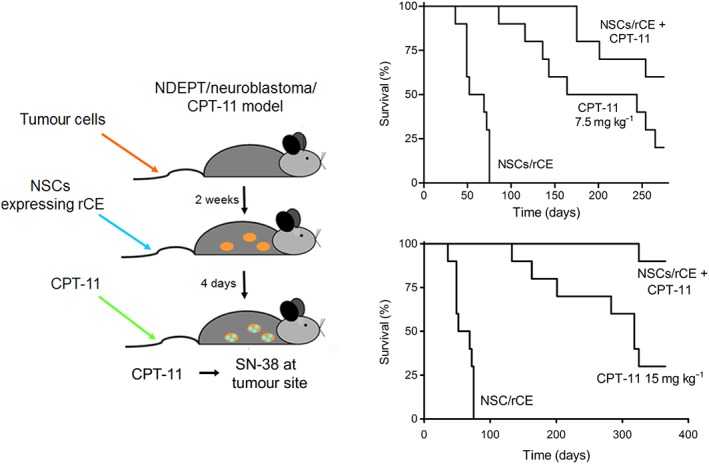
A diagram indicating the experimental design and Kaplan–Meier curves indicating the efficacy of NSC CE/CPT‐11 in a mouse model of disseminated neuroblastoma. Two different doses of CPT‐11 were used in these studies: 7.5 mg kg−1 (upper curves) and 15 mg kg−1 (lower curves).
As an alternative, this enzyme/prodrug approach may allow dose reduction of CPT‐11, thereby minimizing the systemic toxicity (principally delayed diarrhoea), without compromising antitumour activity. It is likely that the doses of drug that would be used in clinical studies would saturate the exogenous NSC‐delivered CE present within the tumour milieu, and potentially therefore, the same efficacy may be achieved using considerably lower doses of the prodrug. While this has not been explored in detail in animal models, careful optimization of the number of NSCs and the schedule for administration, as well as the dose of CPT‐11, may well yield regimens that minimize the non‐specific toxicity of this agent.
Implementation of NSC CE/CPT‐11 enzyme/prodrug therapy in humans
Clearly, the implementation of this approach in humans is problematic. It is exceedingly unlikely that authorities and institutions would allow such studies to be undertaken in paediatric patients initially, and hence, all of the necessary safety and toxicity trials would need to be performed in adults. Unfortunately, it is not obvious which adult disease would be the best suited for such an approach. However, initial studies have begun in collaboration with the City of Hope Medical Center, and virally modified NSCs have been infused into individuals diagnosed with malignant brain tumours. Patients were pre‐screened for antibodies against NSC and if they tested positive, they were not eligible for this trial. Additional experiments have confirmed that NSC‐mediated drug activation can be observed in this setting, and future trials will continue to assess the safety of this approach. To date, no adverse events or effects have been seen that would contra‐indicate the use of this approach in either adults or children.
Summary
The use of enzyme/prodrug approaches has long been recognized as a potential method for improving drug therapy, but to date, both chemical and biological issues have significantly limited the efficacy of such methodology. With CE/CPT‐11, we believe that the vast majority of the areas of concern have been significantly reduced by careful optimization of each component of the system. This includes the design of a human CE that can efficiently activate the drug, as well as being capable of being secreted to provide a bystander effect; the use of tumour‐tropic NSC that are amenable to viral transduction; the use of adenovirus that yield very high levels of CE proteins; and effective and predictive animal models that more accurately reflect esterified drug metabolism in humans. Ultimately of course, the success of this technology will be determined using very specific clinical trials that select the appropriate patient population in which such an approach would be effective. While this technique will not be generally applicable to all individuals diagnosed with cancer, for the patients who have minimal residual disease that contribute to relapse, such as paediatric neuroblastoma patients, this approach may be highly beneficial.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
Work in the Potter lab has been supported in part by NIH grants CA76202, CA79763, CA98468, CA108775, CA113446, AT007531, NS58089, a Cancer Center Core grant CA21765, and by the American Lebanese Syrian Associated Charities (ALSAC) and St. Jude Children's Research Hospital (SJCRH).
Wierdl, M. , Tsurkan, L. , Hatfield, M. J. , and Potter, P. M. (2016) Tumour‐selective targeting of drug metabolizing enzymes to treat metastatic cancer. British Journal of Pharmacology, 173: 2811–2818. doi: 10.1111/bph.13553.
References
- Aboody KS, Brown A, Rainov NG, Bower KA, Liu S, Yang W et al. (2000). Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Natl Acad Sci 97: 12846–12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboody KS, Bush RA, Garcia E, Metz M, Najbauer J, Justus KA et al. (2006a). Development of a tumor‐selective approach to treat metastatic cancer. PLoS One 1: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboody KS, Najbauer J, Schmidt NO, Yang W, Wu JK, Zhuge Y et al. (2006b). Targeting of melanoma brain metastases using engineered neural stem/progenitor cells. Neuro Oncol 8: 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA et al. (2015). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bencharit S, Morton CL, Howard‐Williams EL, Danks MK, Potter PM, Redinbo MR (2002). Structural insights into CPT‐11 activation by mammalian carboxylesterases. Nat Struct Biol 9: 337–342. [DOI] [PubMed] [Google Scholar]
- Bissery MC, Vrignaud P, Lavelle F, Chabot GG (1996). Preclinical antitumor activity and pharmacokinetics of irinotecan (CPT‐11) in tumor‐bearing mice. Ann N Y Acad Sci 803: 173–180. [DOI] [PubMed] [Google Scholar]
- Brown AB, Yang W, Schmidt NO, Carroll R, Leishear KK, Rainov NG et al. (2003). Intravascular delivery of neural stem cell lines to target intracranial and extracranial tumors of neural and non‐neural origin. Hum Gene Ther 14: 1777–1785. [DOI] [PubMed] [Google Scholar]
- Brzezinski MR, Abraham TL, Stone CL, Dean RA, Bosron WF (1994). Purification and characterization of a human liver cocaine carboxylesterase that catalyzes the production of benzoylecgonine and the formation of cocaethylene from alcohol and cocaine. Biochem Pharmacol 48: 1747–1755. [DOI] [PubMed] [Google Scholar]
- Brzezinski MR, Spink BJ, Dean RA, Berkman CE, Cashman JR, Bosron WF (1997). Human liver carboxylesterase hCE‐1: binding specificity for cocaine, heroin, and their metabolites and analogs. Drug Metab Dispos 25: 1089–1096. [PubMed] [Google Scholar]
- Cashman J, Perroti B, Berkman C, Lin J (1996). Pharmacokinetics and molecular detoxification. Environ Health Perspect 104: 23–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti JA, Kemeny NE, Saltz LB, Huang Y, Tong WP, Chou TC et al. (1996). Irinotecan is an active agent in untreated patients with metastatic colorectal cancer. J Clin Oncol 14: 709–715. [DOI] [PubMed] [Google Scholar]
- Creaven PJ, Allen LM, Muggia FM (1972). Plasma camptothecin (NSC‐100880) levels during a 5‐day course of treatment: relation to dose and toxicity. Cancer Chemother Rep 1 56: 573–578. [PubMed] [Google Scholar]
- Crow JA, Borazjani A, Potter PM, Ross MK (2007). Hydrolysis of pyrethroids by human and rat tissues: examination of intestinal, liver and serum carboxylesterases. Toxicol Appl Pharmacol 221: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danks MK, Yoon KJ, Bush RA, Remack JS, Wierdl M, Tsurkan L et al. (2007). Tumor‐targeted enzyme/prodrug therapy mediates long‐term disease‐free survival of mice bearing disseminated neuroblastoma. Cancer Res 67: 22–25. [DOI] [PubMed] [Google Scholar]
- Furman WL, Stewart CF, Poquette CA, Pratt CB, Santana VM, Zamboni WC et al. (1999). Direct translation of a protracted irinotecan schedule from a xenograft model to a Phase I trial in children. J Clin Oncol 17: 1815–1824. [DOI] [PubMed] [Google Scholar]
- Hatfield MJ, Tsurkan L, Garrett M, Shaver T, Edwards CC, Hyatt JL et al. (2011). Organ‐specific carboxylesterase profiling identifies the small intestine and kidney as major contributors of activation of the anticancer prodrug CPT‐11. Biochem Pharmacol 81: 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfield MJ, Tsurkan L, Hyatt JL, Yu X, Edwards CC, Hicks LD et al. (2010). Biochemical and molecular analysis of carboxylesterase‐mediated hydrolysis of cocaine and heroin. Br J Pharmacol 160: 1916–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humerickhouse R, Lohrbach K, Li L, Bosron W, Dolan M (2000). Characterization of CPT‐11 hydrolysis by human liver carboxylesterase isoforms hCE‐1 and hCE‐2. Cancer Res 60: 1189–1192. [PubMed] [Google Scholar]
- Inkerman PA, Scott K, Runnegar MT, Hamilton SE, Bennett EA, Zerner B (1975). Carboxylesterases (EC 3.1.1). Purification and titration of chicken, sheep, and horse liver carboxylesterases. Can J Biochem 53: 536–546. [DOI] [PubMed] [Google Scholar]
- Khanna R, Morton CL, Danks MK, Potter PM (2000). Proficient metabolism of CPT‐11 by a human intestinal carboxylesterase. Cancer Res 60: 4725–4728. [PubMed] [Google Scholar]
- Mathijssen RHJ, van Alphen RJ, Verweij J, Loos WJ, Nooter K, Stoter G et al. (2001). Clinical pharmacokinetics and metabolism of irinotecan (CPT‐11). Clin Cancer Res 7: 2182–2194. [PubMed] [Google Scholar]
- Morton CL, Iacono L, Hyatt JL, Taylor KR, Cheshire PJ, Houghton PJ et al. (2005). Metabolism and antitumor activity of CPT‐11 in plasma esterase‐deficient mice. Cancer Chemother Pharmacol 56: 629–636. [DOI] [PubMed] [Google Scholar]
- Morton CL, Wierdl M, Oliver L, Ma M, Danks MK, Stewart CF et al. (2000). Activation of CPT‐11 in mice: Identification and analysis of a highly effective plasma esterase. Cancer Res 60: 4206–4210. [PubMed] [Google Scholar]
- Muggia FM, Creaven PJ, Hansen HH, Cohen MH, Selawry OS (1972). Phase I clinical trial of weekly and daily treatment with camptothecin (NSC‐100880): correlation with preclinical studies. Cancer Chemother Rep 56: 515–521. [PubMed] [Google Scholar]
- Munger JS, Shi GP, Mark EA, Chin DT, Gerard C, Chapman HA (1991). A serine esterase released by human alveolar macrophages is closely related to liver microsomal carboxylesterases. J Biol Chem 266: 18832–18838. [PubMed] [Google Scholar]
- Park JR, Eggert A, Caron H (2008). Neuroblastoma: biology, prognosis, and treatment. Pediatr Clin North Am 55: 97–120. [DOI] [PubMed] [Google Scholar]
- Potter PM, Pawlik CA, Morton CL, Naeve CW, Danks MK (1998a). Isolation and partial characterization of a cDNA encoding a rabbit liver carboxylesterase that activates the prodrug Irinotecan (CPT‐11). Cancer Res 52: 2646–2651. [PubMed] [Google Scholar]
- Potter PM, Wolverton JS, Morton CL, Whipple DO, Danks MK (1998b). In situ subcellular localization of epitope tagged human and rabbit carboxylesterases. Cytometry 32: 223–232. [PubMed] [Google Scholar]
- Potter PM, Wolverton JS, Morton CL, Wierdl M, Danks MK (1998c). Cellular localization domains of a rabbit and a human carboxylesterase: influence on irinotecan (CPT‐11) metabolism by the rabbit enzyme. Cancer Res 58: 3627–3632. [PubMed] [Google Scholar]
- Quinney SK, Sanghani SP, Davis WI, Hurley TD, Sun Z, Murry DJ et al. (2005). Hydrolysis of capecitabine to 5′‐deoxy‐5‐fluorocytidine by human carboxylesterases and inhibition by loperamide. J Pharmacol Exp Ther 313: 1011–1016. [DOI] [PubMed] [Google Scholar]
- Redinbo MR, Potter PM (2005). Mammalian Carboxylesterases: from drug targets to protein therapeutics. Drug Discov Today 10: 313–325. [DOI] [PubMed] [Google Scholar]
- Sanghani SP, Quinney SK, Fredenburg TB, Davis WI, Murry DJ, Bosron WF (2004). Hydrolysis of irinotecan and its oxidative metabolites, 7‐ethyl‐10‐[4‐N‐(5‐aminopentanoic acid)‐1‐piperidino] carbonyloxycamptothecin and 7‐ethyl‐10‐[4‐(1‐piperidino)‐1‐amino]‐carbonyloxycamptothecin, by human carboxylesterases CES1A1, CES2, and a newly expressed carboxylesterase isoenzyme, CES3. Drug Metab Dispos 32: 505–511. [DOI] [PubMed] [Google Scholar]
- Shi D, Yang J, Yang D, LeCluyse EL, Black C, You L et al. (2006). Anti‐influenza prodrug oseltamivir is activated by carboxylesterase human carboxylesterase 1, and the activation is inhibited by antiplatelet agent clopidogrel. J Pharmacol Exp Ther 319: 1477–1484. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: 1054–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanizawa A, Fujimori A, Fujimori Y, Pommier Y (1994). Comparison of topoisomerase I inhibition, DNA damage, and cytotoxicity of camptothecin derivatives presently in clinical trials. J Natl Cancer Inst 86: 836–842. [DOI] [PubMed] [Google Scholar]
- Thompson J, Guichard SM, Cheshire PJ, Richmond LB, Poquette CA, Ragsdale ST et al. (2001). Development, characterization and therapy of a disseminated model of childhood neuroblastoma in SCID mice. Cancer Chemother Pharmacol 47: 211–221. [DOI] [PubMed] [Google Scholar]
- Wadkins RM, Morton CL, Weeks JK, Oliver L, Wierdl M, Danks MK et al. (2001). Structural constraints affect the metabolism of 7‐ethyl‐10‐[4‐(1‐piperidino)‐1‐piperidino]carbonyloxycamptothecin (CPT‐11) by carboxylesterases. Mol Pharmacol 60: 355–362. [DOI] [PubMed] [Google Scholar]
- Wierdl M, Morton CL, Weeks JK, Danks MK, Harris LC, Potter PM (2001). Sensitization of human tumor cells to CPT‐11 via adenoviral‐mediated delivery of a rabbit liver carboxylesterase. Cancer Res 61: 5078–5082. [PubMed] [Google Scholar]
- Wierdl M, Tsurkan L, Hyatt JL, Edwards CC, Hatfield MJ, Morton CL et al. (2008). An improved human carboxylesterase for enzyme/prodrug therapy with CPT‐11. Cancer Gene Ther 15: 183–192. [DOI] [PubMed] [Google Scholar]