

Abstract
Background and Purpose
Functional measures of human ether‐à‐go‐go‐related gene (hERG; Kv11.1) channel inhibition have been prioritized as an in vitro screening tool for candidate molecules. However, it is unclear how these results can be translated to humans. Here, we explore how data on drug binding and functional inhibition in vitro relate to QT prolongation in vivo. Using cisapride, sotalol and moxifloxacin as paradigm compounds, we assessed the relationship between drug concentrations, binding, functional measures and in vivo effects in preclinical species and humans.
Experimental Approach
Pharmacokinetic–pharmacodynamic modelling was used to characterize the drug effects in hERG functional patch clamp, hERG radio‐labelled dofetilide displacement experiments and QT interval in conscious dogs. Data were analysed in parallel to identify potential correlations between pharmacological activity in vitro and in vivo.
Key Results
An Emax model could not be used due to large variability in the functional patch clamp assay. Dofetilide displacement revealed that binding curves are unrelated to the in vivo potency estimates for QTc prolongation in dogs and humans. Mean in vitro potency estimates ranged from 99.9 nM for cisapride to 1030 μM for moxifloxacin.
Conclusions and Implications
The lack of standardized protocols for in vitro assays leads to significant differences in experimental conditions, making the assessment of in vitro–in vivo correlations unreliable. Identification of an accurate safety window during the screening of candidate molecules requires a quantitative framework that disentangles system‐ from drug‐specific properties under physiological conditions, enabling translation of the results to humans. Similar considerations will be relevant for the comprehensive in vitro pro‐arrhythmia assay initiative.
Abbreviations
- hERG (Kv11.1)
human ether‐à‐go‐go‐related gene
- NPDE
normalized prediction distribution error
- PKPD
pharmacokinetic–pharmacodynamic
- TdP
Torsade de Pointes
Tables of Links
| TARGETS |
|---|
| Cav1.2 |
| Kv11.1 (hERG) |
| Nav1.8 |
| LIGANDS | |
|---|---|
| Astemizole | Penicillin |
| Cisapride | Sotalol |
| Dofetilide http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2768 | Verapamil |
| MK‐499 |
These tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
During the last two decades, a number of drugs have had to undergo labelling revision or market withdrawal due to post‐marketing reports of sudden cardiac death linked to Torsade de Pointes (TdP) (Cavero et al., 2000; Haverkamp et al., 2000; Redfern et al., 2003; Shah and Hondeghem, 2005; Thomsen et al., 2006). Despite major efforts to screen compounds for their pro‐arrhythmic activity in vitro, most compounds still progress into clinical development with an unclear risk of QT interval prolongation in humans (Chen et al., 2006; Ducroq et al., 2007; Gintant, 2008).
From a drug discovery perspective, pharmaceutical research and development has relied on in vitro human ether‐à‐go‐go‐related gene (hERG also known as Kv11.1 channel) assays as a primary screening filter before in vivo experimental protocols are used to evaluate QT/QTc interval prolongation in preclinical species. Multiple approaches have been developed to evaluate drug effects on hERG current in vitro. Gintant et al. (2006) proposed to divide them into two different classes based on whether experimental measures will reflect a direct or indirect effect on the hERG current. Indirect approaches include binding assays, assays measuring ionic flux changes and assays detecting changes in membrane potential. An advantage of binding assays that measure displacement of potent, radiolabelled hERG ligands is that they provide a convenient screening method to detect drug‐hERG channel interactions. These systems use intact cells or cell membranes from heterologous expression systems transfected with the hERG channel and potent, radiolabelled hERG ligands such as dofetilide (Diaz et al., 2004), methanesulfonanilide MK‐499 (Wang et al., 2003) and astemizole (Chiu et al., 2004). Results obtained with the aforementioned indirect assays are considered to be less sensitive than those reported using direct measures of the hERG current but have the advantage of greater throughput, as compared with functional measures.
More recently, newer techniques have been developed to assess hERG currents in a more direct way (Dubin et al., 2005). However, there is no evidence yet of how the results from these assays correlate with the drug‐induced QT‐interval prolongation in humans, even when one considers compounds that show no affinity for other ion channels. In fact, regulatory agencies, academic researchers and pharmaceutical companies appear to have recognized the drawbacks of the existing approach for the evaluation of pro‐arrhythmic properties based on a predominant focus on the hERG channel. As a result, at the conference of the Cardiac Safety Research Consortium (CSRC)–Health and Environmental Sciences Institute (HESI)–Food and Drug Administration (FDA) held in July 2013, a revision of ICH S7B and possible elimination of ICH‐E14 were proposed. The proposal is aimed at shifting the focus from evaluating QT prolongation to evaluating the pro‐arrhythmic activity of a compound using a comprehensive in vitro pro‐arrhythmia assay (CiPA) (Cavero and Holzgrefe, 2015; Sager et al., 2014; Fermini et al., 2016). The approach seems, however, to overlook the importance of a stricter quantitative framework for the translation of in vitro findings and in particular of the potential differences between in vitro and in vivo concentration–effect [pharmacokinetic–pharmacodynamic (PKPD)] relationships.
Here, we evaluated whether a systematic correlation can be found between hERG binding and functional inhibition data in vitro. Subsequently, we attempted to assess how binding and functional inhibition data correlate with the underlying concentration–effect relationship in vivo, both in nonclinical species (dogs) and healthy human subjects (Chain, Dubois et al., 2013). Reference compounds with known clinical QT prolonging effects were used for the purposes of this evaluation, namely cisapride, sotalol and moxifloxacin. Evidence of such a correlation might support the use of hERG binding data in conjunction with PKPD relationships as a screening tool in early drug discovery. The concept might then be expanded to other ion channels. The ultimate goal of this investigation is therefore to assess the feasibility and translational value of binding information as the basis for establishing in vitro–in vivo correlations for drugs with varying affinity for the hERG channel.
Limitations of the assessment of hERG channel blockade
Even though different functional assays are available for screening, the use of in vitro hERG inhibition is based on the assumption that any strong signal, that is, hERG channel inhibition, will be predictive of potential QT prolongation in vivo. Yet, none of the available preclinical in vitro and in vivo methods appear to fully predict the torsadogenic potential in humans (Hoffmann and Warner, 2006). Among other things, there has been limited attention to whether experimental conditions are representative of the physiological milieu in humans (e.g. low K+ concentration, proteins and low rate stimulation imitating bradycardia). Most importantly, the experiments are performed without taking into account the most likely range of drug exposure at the therapeutic dose levels. To better understand the implications of differences in experimental conditions, a brief overview of the hERG assays and their relevance for the evaluation of pro‐arrhythmic effects is provided in the Supporting Information.
Based on the aforementioned hERG channel properties, the use of IC50 values characterizing the potential of a compound to block the hERG current provides a convenient way to compare compounds. However, it should be recognized that potency estimates represent an oversimplification of complex time‐, voltage‐ and state‐dependent processes. Part of this complexity relates to the fact that some compounds will bind in the open phase of the hERG channel (open state blockers), whereas others will bind when the ion channel is closed again, but the channel needs to be activated first. Also, the association rate for the ion channel has an influence (Yu et al., 2015). Thus, the configuration of the voltage clamp waveform (i.e. the time at a certain voltage and the voltage steps) may affect the potency of the drug as well as the time course of inhibition and recovery, reflecting interactions with different states of the channel.
One of the main implications of such differences hERG binding and inhibition is the high incidence of false positive and false negative results in QT prolongation, as previously illustrated in the publications by Chiang et al. (2010), Laverty et al. (2011) and Mirams et al. (2014); all of which provide figures associated with the sensitivity and specificity of experimental protocols used for drug screening. With regard to false positive results, it should be highlighted that despite a considerable debate supporting the views that hERG inhibition in non‐clinical assays are highly predictive of drug effects on the QT interval, exceptions exist, which raise questions about the generalizability of such correlations (e.g. verapamil, which has a high potency for the hERG ion channel but does not prolong the QT interval in vivo) (Wallis, 2010). It has been suggested that false positive results are due to the actions of a compound on currents other than the rapid delayed rectifier current (IKr) such as L‐type Ca2+ channel current or Na+ channel current (Antzelevitch et al., 2004). On the other hand, false negative results in hERG assay data are linked to the fact that various sequentially activated ion channels and transporters may affect the action potential duration. Dumotier et al. (2008) suggest that false negatives in hERG inhibition data arise from (i) effects of other ionic currents; (ii) additional effects such as hERG trafficking inhibition; (iii) drug accumulation in the ventricular myocardium; and (iv) drug metabolite effects on hERG current even if parent drug has no effect.
Another important limitation is the questionable accuracy of the so‐called quantitative parameters. Usually, IC50 values are used to compare and rank compounds. Given the complexities associated with hERG channel inhibition, conclusions drawn from such comparisons may not be accurate. This has potentially important implications for the selection of novel molecules. The bias caused by intrinsic mechanistic differences is often further compounded by other sources of variability in experimental protocols. For instance, temperature has been shown to affect IC50 values for some compounds (Kirsch et al., 2004; Yao et al., 2005). Differences in experimental procedures and techniques contribute to the range of IC50 values reported for any given compound in the literature. These discrepancies indicate that a relative bias remains for different protocol settings even when positive control standards are used to monitor assay sensitivity (Su et al., 2006).
In spite of the fact that different chemotypes can bind with high affinity to the hERG channel and changes in the functional group or structure of a given chemotype can alter the hERG binding profile significantly (Sanguinetti and Tristani‐Firouzi, 2006; Polak et al., 2009; Eichenbaum et al., 2012), here we explore the notion of target occupancy as a screening parameter with potentially direct clinical meaning. Irrespective of whether a systematic relationship between binding and effect can be identified across compounds, we believe that assessment of the correlation between binding and the degree of hERG channel blockade will shed further light on the relevance of parameter estimates such as IC50 arising from functional assays in vitro.
Methods
In vitro displacement and functional assays
[3H]‐dofetilide‐isolated membrane displacement
Dofetilide, moxifloxacin, sotalol and cisapride were purchased from Sigma Aldrich (Zwijndrecht, The Netherlands). [3H] Dofetilide (specific activity 70.0 Ci·mmol−1) was purchased from Perkin Elmer (Groningen, The Netherlands). BSA (fraction V) was purchased from Sigma (St. Louis, MO, USA). G418 (geneticin) was obtained from Stratagene (Cedar Creek, USA). All the other chemicals were of analytical grade and obtained from standard commercial sources. HEK293 cells stably expressing the hERG K+ channel (hERG/HEK293) were kindly provided by Dr. Eckhard Ficker (University of Cleveland, USA).
Cell culture
hERG/HEK293 cells were cultured in a humidified atmosphere at 37°C and 7% CO2 in DMEM, containing 10% FCS, 50 IU·mL−1 penicillin, 50 μg·mL−1 streptomycin and 1.25 μg·mL−1 G418. Initially, cells were sub cultured twice a week (1:8). Then, the cells were sub cultured 1:10 and transferred to large 15 cm diameter plates for membrane preparation.
Membrane preparation
hERG/HEK293 cells were grown to 80–90% confluence and detached from the plates by scraping them into 5 mL of PBS. Then, the detached cells were collected and centrifuged at 250 g for 10 min. The cell pellets were pooled and resuspended in 50 mM ice‐cold Tris–HCl buffer containing 2 mM MgCl2, pH 7.4. An UltraTurrax (Heidolph Instruments, Schwabach, Germany) was used to homogenize the cell suspension. Membranes and the cytosolic fraction were separated by centrifugation at 100 000 g in an Optima LE‐80 K ultracentrifuge (Beckman Coulter, Fullerton, CA, USA) at 4°C for 20 min. The pellets were resuspended using similar procedures in ice‐cold incubation buffer (10 mM HEPES, 130 mM NaCl, 60 mM KCl, 0.8 mM MgCl2, 1 mM EGTA, 10 mM glucose, 0.1% BSA, pH 7.4) using the UltraTurrax. Aliquots (125 or 250 μL) were stored at −80°C. The protein concentration of the membranes was measured using the bicinchoninic acid method (Smith et al., 1985).
Equilibrium radio ligand binding assays
The [3H]‐dofetilide equilibrium binding assays for the hERG K+ channel were performed as described previously (Chadwick et al., 1993; Finlayson et al., 2001; Chiu et al., 2004; Diaz et al., 2004) with minor modifications. In short, membrane aliquots containing 20 μg protein were incubated in a total volume of 100 μL incubation buffer at 25°C for 60 min. Radioligand displacement experiments were conducted using a range of concentrations of the competing ligand in the presence of 5 nM [3H]‐dofetilide. At this concentration, total radioligand binding did not exceed 10% of the initial radioligand added to prevent the ligand depletion. Non‐specific binding was determined in the presence of 10 μM astemizole and represented approximately 15% of the total binding. [3H]‐dofetilide did not bind specifically to membranes prepared from empty HEK293 cells lacking the hERG K+ channel (data not shown). Total binding was determined in the presence of incubation buffer and was set at 100% in all experiments, whereas non‐specific binding was set at 0%. Given the scope of the analysis, data were normalized as percentage (%) dissociation in order to ensure direct comparison across experiments and normalize for eventual differences in assay handling. Incubations were terminated by dilution with ice‐cold wash buffer. Separation of bound from free radioligand was performed by rapid filtration through a 96‐well GF/B filter plate using a Perkin Elmer Filtermate‐harvester (Perkin Elmer). Filters were subsequently washed 12 times with ice‐cold wash buffer. The filter‐bound radioactivity was determined by scintillation spectrometry using the P‐E 1450 Microbeta Wallac Trilux scintillation counter (Perkin Elmer) after addition of 25 μL microscint and 2 h extraction. The protocol was based on triplicates (n = 3), which is considered standard practice for these experiments.
Whole cell patch‐clamp
hERG patch clamp data from reference compounds cisapride, sotalol and moxifloxacin respectively were retrieved from the TI‐Pharma data repository. The hERG assays were all performed using kidney (HEK293) cell lines stably transfected with a (plasmid) expression vector with the cytomegalovirus promoter and a neomycin‐resistance marker (pcDNA3) expressing hERG (University of Wisconsin, USA). These cells are fully characterized (Zhou et al., 1998) and are the most widely used cells in functional isolated whole cell patch‐clamp hERG assays. The extracellular solution consists of (mM): 150 NaCl; 1.8 CaCl2; 1 MgCl2; 5 glucose; 10 HEPES; at pH 7.4. KCl and assay voltage varied in each study (see Table 1 for details). Most protocols were run at 15 s intervals. The amplitude of the tail current following the voltage step back to baseline was measured relative to the holding potential. The signals were corrected for the averaged rundown observed during approximately 10 min exposure to vehicle solution. All values were given as % of control values for each concentration tested. All experiments were performed at room temperature.
Table 1.
Patch clamp assay data available for modelling
| Compound | Data set ID | # of cells tested | Cell line | Temperature | [K+] (mM) | Voltage protocol steps | Concentration range tested (nM) | IC50 observed** |
|---|---|---|---|---|---|---|---|---|
| Cisapride | 225* | 11 | HEK293 | RT | 4 |
−80 mV, +20 mV 5 s, −50 mV 5 s, −80 mV |
0.1, 0.3, 1, 3, 10, 30, 100 | 6.8 |
| 248 | 9 | HEK293 | RT | 10 |
−80 mV, +30 mV 1 s, −80 mV |
19.5, 39.1, 156, 313, 625, 1250, 2500, 5000, 10000, 20000 | 938 | |
| 249 | 4 | HEK293 | RT | 5.3 |
−80 mV, +30 mV 1 s, −80 mV |
3, 10, 30, 100 | 18 | |
| 254 | 7 | HEK293 | RT | 4 |
−80 mV, −60 mV 0.5 s, +60 mV 2 s, −40 mV 6 s |
10, 100, 1000 | 65 | |
| 256 | 3 | HEK293 | RT | 4 |
−80 mV, +20 mV 4 s, −50 mV 3 s, −80 mV 8 s |
1, 10, 100 | 19 | |
| Moxifloxacin | 240* | 17 | HEK293 | RT | 5.4 |
0 mV, −80 mV 20 ms, +40 mV 80 ms |
1000, 10 000, 100 000,300 000, 1 000 000 | 353 600 |
| 252* | 5 | HEK293 | RT | 5.3 |
−80 mV, +30 mV 1 s, −80 mV |
10 000, 30 000, 100 000, 300 000 | 141 000 | |
| 254* | 6 | HEK293 | RT | 4 |
−80 mV, −60 mV 0.5 s, +40 mV 2 s, −40 mv 6 s |
30 000, 100 000, 300 000 | 122 000 | |
| Sotalol | 217* | 4 | HEK293 | RT | 4 |
−80 mV, +20 mV 0.5 s, −50 mV 5 s, −80 mV |
10 000, 30 000, 100 000, 300 000 | 163 000 |
| 222* | 4 | HEK293 | RT | 4 |
−80 mV, +20 mV 0.5 s, −50 mV 5 s, −80 mV |
10 000, 30 000, 100 000, 300 000 | 117 000 |
Data included in the current analysis are marked with an asterisk (*).
For the sake of completeness, IC50 values (nM) derived from the original experimental protocols are also presented along with the experimental protocol details. These estimates may differ from the values obtained by nonlinear mixed effects modelling, which was used to analyse the data in the current investigation.
In vivo PKPD studies in conscious dogs
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). All experiments were approved by the institutional Ethics Committee and conducted according to the ethical standards and Good Laboratory Practice procedures. Telemetric recordings of the ECG were performed in conscious dogs (Ollerstam et al., 2006; Prior et al., 2009). In brief, dogs were implanted with a telemetric transmitter under general anaesthesia. The transmitter was placed in the peritoneal cavity, and electrodes were placed in lead II configuration. A four‐way Latin square crossover design was used for the experiments, during which animals administered vehicle, a sub‐therapeutic, a therapeutic and a supra‐therapeutic oral dose of each compound followed by a washout period between each treatment. The data were recorded and analysed using the Notocord data acquisition system (HEMsoftware, Notocord Inc., Croissy‐sur‐Seine, France). The Dataquest Open ART™ software (St. Paul, MN, USA) was then used to set up and calibrate the telemetry systems.
The QT interval duration was measured for every wave complex. The placement of the ECG callipers was checked and manually corrected if deemed necessary. Blood samples for pharmacokinetics were collected at different times after dosing to ensure accurate characterization of the absorption, distribution and elimination phases. ECG was monitored continuously over the period of 24 h and averaged every 30 sec (cisapride), 1 min (moxifloxacin) or 5 min (d,l‐sotalol). Further details can be found in Chain and Dubois et al. (Chain, Dubois et al., 2013).
Clinical trials in healthy subjects. Data from three Phase I studies were included in this analysis. The studies were conducted in full conformance with the principles of the Declaration of Helsinki and with the local laws and regulations concerning clinical trials. The protocol and the informed consent documents for each study have been formally approved by the relevant research Ethics Committee. For cisapride, a randomized, placebo‐controlled, dose‐escalating design was used in which subjects received up to five doses. Due to safety issues, the study was not continued after the fourth dose. d,l‐Sotalol data was extracted from a double‐blind, randomized, placebo‐controlled, three‐way crossover study in which each subject received one active treatment and two placebo doses. Data for moxifloxacin was available from the positive control arm of a two‐way crossover, single‐blind, randomized, placebo‐controlled trial. Additional information regarding the experimental procedures, including electrode placement and blood sampling for pharmacokinetics, has been previously published and can be found elsewhere (Chain, Dubois et al., 2013).
Data analysis
[3H]‐dofetilide‐isolated membrane displacement
An Imax (maximum inhibitory effect) model was used to describe the displacement of [3H]‐dofetilide (equation (1)):
| (1) |
where Displacement represents the degree of [3H]‐dofetilide displacement. I 0 is the baseline dofetilide binding, Imax the maximum displacement and IC50 the concentration at which 50% displacement is observed. C is concentration of compound tested.
In contrast to functional measures, drug binding was expected to provide information about target occupancy or blockade and as such reflect the differences in the affinity of each ligand for the hERG channel. Parameters of interest are expressed as inhibitory concentrations but reflect the degree of [3H]‐dofetilide displacement: IC20, IC50, IC70 and IC80 represent the concentrations associated with 20, 50, 70 and 80% displacement. The concentrations of competing ligands were analysed using the nonlinear regression function in R. Nonlinear mixed effects modelling was not deemed necessary due to the number of samples and limited variability in the results obtained with the proposed protocol design. It should also be highlighted that no statistical hypothesis testing was performed to compare differences between compounds.
Whole cell patch‐clamp
A sigmoid Imax model was chosen to describe the inhibitory drug effects on hERG channel (equation (2)):
| (2) |
where Inhibition is the effect. I 0 is the baseline inhibition, Imax the maximum inhibition and IC50 the concentration at which 50% inhibition is observed. C is concentration of compound tested, and γ describes the shape or steepness of the curve.
The analysis was performed using nonlinear mixed effects modelling in NONMEM v.7.1.2 (ICON, MD, USA) running on a Windows PC. Model diagnostics were based on graphical and statistical criteria, including goodness‐of‐fit plots, visual predictive checks and normalized prediction distribution errors (NPDE). All three compounds were analysed concurrently, yielding common parameter estimates for I0, and Imax. Baseline and maximum inhibition were deemed to be comparable across the compounds and as such reflect the experimental conditions (i.e. system‐specific properties). By contrast, drug potency varied for each drug. Additive error terms were estimated separately where appropriate.
In vivo and clinical PKPD studies
Estimates of the effects of cisapride, sotalol and moxifloxacin on QT interval prolongation in dogs and healthy subjects, expressed in terms of the concentrations corresponding to a probability of QT interval prolongation ≥10 ms, were used as a clinical reference for establishing potential correlations between in vitro and in vivo experiments. Full details of the experimental protocols and approval by the ethics committees can be found elsewhere (Chain, Dubois et al., 2013). The PKPD analysis was performed using a Bayesian hierarchical model in WinBUGS version 1.4.2 (Lunn et al., 2002). The model comprises three components, including an individual correction factor for RR interval (heart rate), an oscillatory component describing the circadian variation and a truncated Emax model, as shown by equation (3):
| (3) |
where QTc 0 [ms] is the intercept of the QT‐RR relationship (for each individual), RR [s] is the interval between successive R waves, α is the individual heart rate correction factor, A [ms] is the amplitude of circadian rhythm, t is the clock time, Φ is the phase, slope [ms/concentration unit] is the linear pharmacodynamic relationship and C is the observed or, when not available, predicted concentration of the drug at the time of QT measurements.
One of the advantages of this approach is the possibility of characterizing drug effect in a quantitative manner and expressing it in terms of the probability relative to a clinically relevant threshold, irrespective of the baseline QTc values. For the purposes of our analysis, a threshold of ≥10 ms increase in QT was used to assess the relationship between total plasma concentration and the probability of QT prolongation. This reference threshold was selected due to its clinical relevance with regard to the increased risk of TdP. Moreover, there is some evidence that in humans and dogs, QT prolongation does not seem to correlate with baseline QT after correction for differences in heart rate. It should also be noted that plasma protein binding was assumed not to be restrictive for these compounds. Unbound drug concentrations would have yielded similar results, but different apparent parameter values would have been derived after correction for protein binding.
In vitro–in vivo correlation (IVIVC)
The assessment of a potential correlation between binding, functional assay and the probability of QT interval prolongation in dogs and healthy subjects was based on graphical summaries, linear and log‐linear regression techniques. To ensure normalization of the results across different compounds and experimental protocols, the intercept of the log‐linear regression obtained from the IC20, IC50 and IC70 values of the [3H]‐dofetilide‐isolated membrane binding assays was calculated and used as parameters of interest for the in vitro experiments. In principle, the intercept of this regression corresponds to the concentrations associated with the initial, detectable onset of displacement of [3H]‐dofetilide (Oinh). It was compared with the IC50 values obtained from hERG patch clamp assays and concentrations corresponding to 50% probability of QTc prolongation ≥10 ms (i.e. CP50) in dogs and humans. All calculations were performed in R 2.12.1.
Each of the analysis described above was performed in an unblinded manner.
Results
[3H]‐dofetilide‐isolated membrane binding
All three compounds produced a concentration‐dependent displacement of the specific [3H]‐dofetilide binding. Even though experimental data were sampled in triplicates, variability was relatively low. The maximum SD was 12.3%, with only three measurement points over 6%. The displacement curves were best described by a maximum inhibitory effect model. An overview of the concentration versus displacement curves for all three compounds is presented in Figure 1 along with the functional assay results and concentration versus probability of QT prolongation ≥10 ms. The model‐predicted parameter estimates are listed in Table 2. Cisapride had the highest affinity for the hERG K+ channel, displacing [3H]‐dofetilide with an IC50 value of 99.9 nM, whereas moxifloxacin exhibited the lowest affinity of 1030 μM. Sotalol showed displacement with an IC50 value of 56.4 μM.
Figure 1.
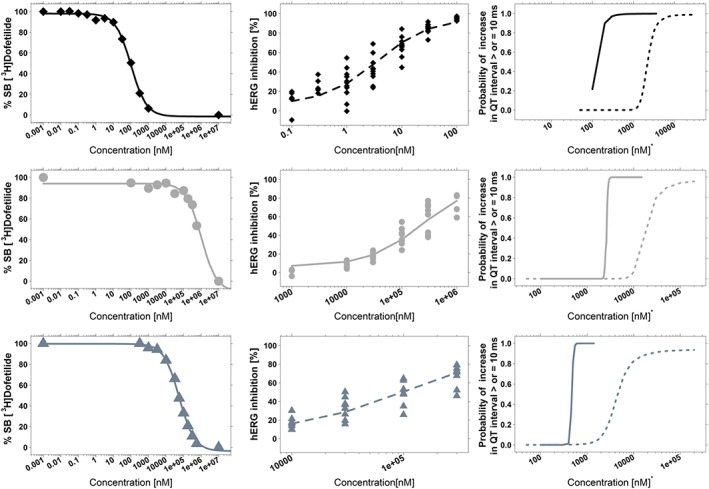
Overview of the concentration–effect relationships derived from in vitro (binding vs. functional hERG assay) and in vivo data (QT prolongation) in dogs and humans for cisapride (top), moxifloxacin (middle) and sotalol (bottom). Left panels show the dofetilide displacement assay; mid panels display the functional hERG patch clamp assay, whereas the curves depicting the concentration versus probability of reaching ≥10 ms QT prolongation in dogs (dotted line) and humans (solid line) are shown on the right panels (Chain, Dubois et al., 2013). In all plots, the lines represent the model‐based population predictions and the symbols the experimental observations. For group size details see Tables 2, 3 and 4.
Table 2.
Mean parameter estimates (90% confidence intervals) and derived PKPD indices (IC20, IC70 and IC80)
| Cisapride | Moxifloxacin | Sotalol | |
|---|---|---|---|
| I0 (%) | 98.1 (96.5–99.7) | 94.0 (90.9–97.2) | 99.7 (97.9–101.6) |
| Imax (%) | 99.5 (96.0–103.0) | 104 (94.0–114.5) | 103 (100.3–106.3) |
| IC50 (μM) | 0.1 (0.086–0.12) | 1030 (752–1466) | 56.4 (50.4–63.2) |
| IC20 (μM) | 0.02 | 160.68 | 13.32 |
| IC70 (μM) | 0.22 | 1649.88 | 117.18 |
| IC80 (μM) | 0.36 | 2543.77 | 190.81 |
Data (n = 3 for each experimental point) from the equilibrium [3H]‐dofetilide binding displacement assay was analysed using an Imax model. I0 represents the percentage [3H]‐dofetilide binding in the absence of a competing molecule. IC20, IC70 and IC80 are the inhibitory concentrations associated with 20, 70 and 80% binding, respectively.
hERG functional assay – whole cell patch clamp
Data from multiple whole cell patch‐clamp experiments, including cisapride, sotalol and moxifloxacin, were pooled and explored for consistency and homogeneity before data fitting. In contrast to the experimental results obtained from the [3H]‐dofetilide‐isolated membrane binding, functional hERG inhibition was considerably variable. Of note was the variability in the experimental data from cisapride, which had apparent IC50 values varying >1000‐fold across protocols. The inhibition curves for all three compounds were best described by a sigmoid maximum inhibitory effect model (Figure 1). Goodness‐of‐fit plots showed that model predictions (population predictions and individual predictions) were able to describe the observed hERG inhibition. Conditional weighted residuals showed no trend for population predictions and only minor trends for individual concentration values. NPDEs were normally distributed with a 0 mean and 1 variance, without a trend (Figures S1 and S2).
Model‐predicted IC50 values ranged from 3.57 nM for cisapride to 103 μM for sotalol and 227 μM for moxifloxacin. I0 and Imax were found to be around physiologically plausible values, that is, 6.4% and 95.7% respectively. High inter‐individual variability (60%) was found for IC50 estimates, with a residual additive error of 23.3% for sotalol and moxifloxacin and 57.7% for cisapride (Table 3). The shape parameter γ was close to 1. Despite the satisfactory model diagnostics, predicted individual inhibition curves could not be derived due to the poor precision of the parameters describing inter‐individual variability.
Table 3.
Population parameter estimates for cisapride, moxifloxacin and sotalol in the hERG patch clamp assay
| Parameter | Cisapride (n = 11) | Moxifloxacin (n = 28) | Sotalol (n = 8) |
|---|---|---|---|
| I0 (%) | 6.43* | 6.43* | 6.43* |
| Imax (%) | 95.7* | 95.7* | 95.7* |
| IC50 (nM) | 3.57 | 227 000 | 103 000 |
| γ | 0.887* | 0.887* | 0.887* |
| Interindividual variability (CV%) | 60 | 60 | 60 |
| Additive error (CV%) | 57.7 | 23.3 | 23.3 |
Experimental data obtained with the different compounds were analysed concomitantly. System‐specific parameters (I0, Imax and Hill coefficient (γ)) were unique to the experimental setting. Therefore, the values with an asterisk (*) have been estimated only once and are applicable to all three drugs. By contrast, IC50 varied for each compound and defined as distinct parameters in the model. A separate additive error term was estimated for cisapride as residual variability in those experiments was significantly higher. CV% indicates the coefficient of variation in percentage.
In vitro–in vivo correlation
In order to establish whether an in vitro–in vivo correlation exists between hERG binding, inhibition and clinically relevant changes in QT interval, the IC20, IC50, IC70 and IC80 values from the in vitro displacement assays (Table 2) and the potency estimates from the functional assay using patch clamp (Table 3) were compared with CP50 estimates previously described by Chain, Dubois et al. (2013) (Table 4). For the sake of clarity, the reader is also advised to assess the overall relationship between drug concentration and probability of QT interval ≥10 ms (Figure 1).
Table 4.
Population PKPD parameter estimates along with 90% credible intervals describing the probability of QT interval prolongation ≥10 ms, as reported by Chain, Dubois et al., 2013
| Primary and derived model parameters | Cisapride | Moxifloxacin | Sotalol | |||
|---|---|---|---|---|---|---|
| Dogs (n = 8) | Healthy subjects (n = 24) | Dogs (n = 8) | Healthy subjects (n = 137) | Dogs (n = 6) | Healthy subjects (n = 30) | |
| Slope [ms·nM−1] | 0.0045 (0.00096–0.0098) | 0.09 (0.087–0.12) | 0.00056 (0.00002–0.0014) | 0.0039 (0.0033–0.0044) | 0.002 (0.0006–0.008) | 0.021 (0.017–0.026) |
| Prob. of ≥10 ms increase at Cmax | 0.75 | 1.0 | 1.0 | 1.0 | 0.9 | 1.0 |
| Cmax (nM) | 2808 | 936 | 112 930 | 10 300 | 22 310 | 5605 |
| CP50 (nM) | 2200 | 140 | 6400 | 2644 | 4600 | 470 |
Data analysis was performed using a Bayesian hierarchical model, which comprises three components, namely: an individual correction factor for RR interval (heart rate), an oscillatory component describing the circadian variation and a truncated Emax model, which is parameterised in terms of a slope.
Figure 2 shows that the slopes describing the displacement curves for cisapride, sotalol and moxifloxacin are of the same order of magnitude. However, no correlation was found between the concentrations at which displacement occurs (i.e. the beginning of linear portion of the curve) and the predicted CP50 values in dogs and humans. For cisapride, this value occurs at lower values than the CP50 in humans, whereas for moxifloxacin and sotalol the onset of displacement occurs at values higher than the CP50 in dogs. Similarly, there was no clear pattern or correlation between IC50 estimates derived from hERG patch clamp and CP50 in dogs and humans. The IC50 of cisapride was around 40‐fold lower than the CP50 in humans, whereas sotalol and moxifloxacin have >80‐fold or >20‐fold difference compared with the CP50 in humans and dogs, respectively. An overview of the parameters used to establish a potential in vitro–in vivo correlation is presented in Table 5.
Figure 2.
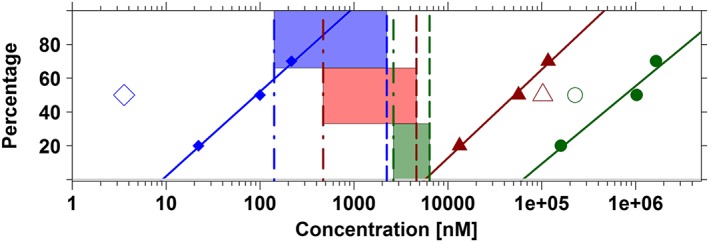
Lack of correlation between in vitro (dofetilide displacement, functional assay) and in vivo (probabilities of QT prolongation in vivo in dogs and humans) data. Ideally, proportional differences should be found between these experiments, reflecting the relative differences in the potency of the different compounds. Data summaries include cisapride (blue diamonds), sotalol (red triangles) and moxifloxacin (dark green dots). Log‐linear regression (solid lines) of the estimates of IC20, IC50 and IC70 obtained from the dofetilide displacement are compared with the IC50 values obtained from hERG patch clamp assays (open symbols) and in vivo (dashed vertical lines) and clinical (dot‐dashed vertical lines) concentrations corresponding to 50% probability of QTc prolongation ≥10 ms (i.e. CP50). The window associated with these concentrations (i.e. CP50) in dogs and humans is depicted using the blue, red and dark green shaded areas for cisapride, sotalol and moxifloxacin, respectively. For instance, for cisapride, IC50 in the binding assay indicates a potency of 99.9 nM. In contrast, potency in the hERG functional assay is much higher, that is, 3.57 nM. These values contrast with CP50 estimate in vivo in dogs and in healthy subjects, which yield estimates of 2233.6 and 141.5 nM, respectively. See Table 5 for further details. Parameters describing the log‐linear regression for cisapride, sotalol and moxifloxacin were respectively y = −49.85 + 49.91*log(x), y = −196.08 + 52.22*log(x) and y = −220.46 + 45.96*log(x).
Table 5.
Summary table showing the comparison of the estimates obtained by the analysis of in vitro (dofetilide displacement, functional assay) and in vivo (probabilities of QT prolongation in vivo in dogs and humans) data
| Cisapride | Moxifloxacin | Sotalol | |
|---|---|---|---|
| CP50D (μM) | 2.23 | 6.39 | 4.62 |
| CP50H (μM) | 0.14 | 2.64 | 0.47 |
| Oinh (μM) | 0.09992 | 62.62 | 5.69 |
| Ratio Oinh/CP50D | 0.004 | 9.8 | 1.2 |
| Ratio Oinh/CP50H | 0.07 | 23.7 | 12.1 |
| IC50 (μM) | 0.00357 | 227 | 103 |
| Ratio IC50/CP50D | 0.0016 | 35.53 | 22.3 |
| Ratio IC50/CP50H | 0.026 | 86.0 | 219.1 |
The onset of inhibition (Oinh) was obtained by log‐linear regression using mean IC20, IC50 and IC70 estimates from the displacement assays. IC50 values are population parameter estimates obtained by nonlinear mixed effects modelling of the patch clamp assays. CP50D and CP50H are the concentrations associated with a 50% probability of reaching ≥10 ms increase in the QT interval in dogs and humans, respectively.
Discussion
Functional inhibition of the hERG channel assays have been used systematically in drug development since the discovery of a link between drug‐induced hERG inhibition and TdP. Typically, IC50 values in μM or nM range are compared with projected plasma concentrations to define a safety margin; that is, the closer these values get the more the results are considered as a liability for QT interval prolongation in humans. As a screening tool, these experiments have evolved to show high sensitivity to changes in hERG function, irrespective of growing evidence about the differences between the degree of inhibition, QT prolongation and TdP (Wallis, 2010; Di Veroli et al., 2014).
This situation is not unique to the screening of candidate molecules and often reflects common practice in experimental safety protocols that are used after candidate selection (Sahota et al., 2015). In addition to the lack of standardization for experimental procedures, a general feature in these protocols is the absence of information regarding the underlying concentration–effect relationships, which can be used as a denominator across experimental conditions and species. Our own group has shown that PKPD relationships can be used to establish in a quantitative manner how changes in drug exposure relate to QT/QTc interval prolongation in preclinical species and in humans. Moreover, we have shown that by using the appropriate parameterization, it is possible to distinguish between drug‐ and system‐specific properties, making it clear how different drugs relate to each other. In fact, a model‐based approach may allow one to disentangle pharmacokinetic from pharmacodynamic differences as well as other intrinsic or extrinsic factors contributing to variability in drug effects (Danhof et al., 2008).
Therefore, the current investigation was aimed at exploring whether binding and functional inhibition could be linked to QTc interval prolongation in preclinical species and in humans using PKPD modelling. Evidence of an in vitro–in vivo correlation for compounds with known QT prolonging effects may provide further insight into the role of differences in receptor density and binding kinetics and consequently facilitate the translation of early findings (Della Pasqua, 2013; France and Della Pasqua, 2015). In addition, evidence of such a correlation might provide the basis for experimental protocol standardization, reducing the rate of false positive and false negative results, for which accurate figures are variable due to differences in experimental protocols and by the discontinuation of compounds (i.e. no clinical data are available for compounds with strong preclinical signals). This problem is illustrated by the case of verapamil, which shows a significant hERG signal, but does not produce QT interval prolongation (Chiang et al., 2010; Laverty et al., 2011; Mirams et al., 2014). If verapamil were developed according to current screening criteria, it might have been discontinued before reaching the clinic.
Clearly, our results contrast with previous publications in which a claim has been made about the predictive performance of the hERG functional assay (Gintant et al., 2006; Wallis, 2010). However, it should be noted that in most investigations data have not been generated or compared in a systematic manner using a model‐based approach. As can be seen from the summary results in Figure 2, it appears that not only the concentration at which effects become evident (i.e. onset of inhibition) varies between compounds but also the relationship between binding and hERG inhibition. Moreover, these inflection points do not correlate with the predicted CP50 values in humans (i.e. Oinh vs. CP50 in Table 5).
Furthermore, it appears that the variability in hERG patch clamp data is large and very sensitive to differences in experimental protocol conditions. Such variability affects the potency estimates and may lead to inaccurate ranking of candidate molecules when comparing functional assay results. Most importantly, potency estimates from whole cell patch clamp do not appear to be strictly predictive of the drug levels associated with QT/QTc interval prolongation in dogs and humans even if one takes into account the potential role of differences in plasma protein binding. Similar conclusions were also drawn by Watson et al. (2011) when comparing modelling results in cynomolgus monkeys with the concentration–response relationships of hERG current derived from in vitro patch clamp experiments.
Given this mismatch between functional measures in vitro and clinical effects, it would be of interest to establish whether binding information, as assessed by [H3]‐dofetilide displacement, bears any correlation with QT interval prolongation in vivo. Surprisingly, this turns out not to be the case, at least for these three compounds. Parameter estimates describing drug affinity for the hERG channel show that displacement occurs at levels, which are much higher than the concentrations associated with QT prolongation ≥10 ms in humans. Furthermore, maximum displacement occurs at very high concentrations, which may represent an important limitation for compounds with poor solubility.
A number of factors may explain these results. First, one should not ignore the fact that hERG ion‐channels are overexpressed in HEK293 cells, which may cause a significant increase in the absolute amount of ligand required to block the available pool of binding sites, yielding apparent estimates, which are unlikely to reflect in vivo conditions. Second, biophase equilibration kinetics and differences in drug‐ion channel interaction may lead to variable signal transduction, yielding different results in functional assays and subsequently discrepancies in action potential duration and QT interval prolongation (Di Veroli et al., 2014; Mirams et al., 2014). Indeed, examples exist in the published literature, which illustrate the implications of delayed equilibration and slow drug‐receptor interactions for the pharmacological effect versus time profile of a compound in vivo and in silico conditions (Yassen et al., 2005; Durdagi et al., 2012; Lee et al., 2016). Another important point to consider explaining the observed discrepancies between in vitro and in vivo experimental data is that hERG blockade is one of a range of factors associated with pro‐arrhythmia. Even though all three compounds are known to bind to hERG channels, it is clearly not the only mechanism underlying drug effects in vivo, as for instance in the case of cisapride, for which other ion channels are known to be involved in the observed QT prolonging effect in dogs and humans (Jonsson et al., 2012). Thus, an evaluation of the potential pro‐arrhythmic effects of drugs based on hERG binding or current block alone may provide an incomplete view of a drug's effects on cardiac repolarization processes.
In summary, it seems that even when considering concentration–effect relationships, there remains a translational gap between drug screening and QT prolongation in the clinic. It appears that screening and ranking procedures based on binding or potency estimates for inhibitory activity on single ion channels may be misleading, even for compounds with known activity on a single ion channel. Our endeavour to discriminate system‐ versus drug‐specific properties in a parametric manner has not yielded the expected results, in that overall measures of target occupancy or inhibition in vitro do not seem to reflect or predict in a quantitative manner the magnitude of drug effects in vivo in dogs or in humans.
Undoubtedly, integrative approaches are needed that account for the multifactorial nature of the pro‐arrhythmic effects in vivo. Among the options available, one should consider the use of a virtual population generator for human cardiomyocyte parameters, as proposed by Polak et al. (2012). The authors propose a computational system including simulations for the evaluation of proarrthymic potential (Tusscher et al., 2004; O'Hara et al., 2011), taking into account the influence of inter‐individual variability in the parameters of interest. In fact, this concept has been recently used to predict the effects of domperidone (Mishra et al., 2014), illustrating how in vitro and more specifically in silico simulations can be used in conjunction with physiologically‐based pharmacokinetic models to predict drug effects in humans. Despite these promising results, some obvious limitations exist, which cannot be overlooked. None of the in silico models currently available account for the contribution of physiological factors such as body temperature, insulin/glucose homeostasis, changes in electrolytes or autonomic tone known to alter QT interval in vivo (Fossa, 2008; van der Linde et al., 2008). These models also ignore the contribution of differences in binding properties and biophase kinetics (Durdagi et al., 2012; Di Veroli et al., 2014; Lee et al., 2016). A comparable situation applies to the extrapolation of QT prolongation from controlled clinical trials to a real‐life setting (Chain et al., 2013). This should be carefully considered by those supporting the CiPA working groups, who are currently responsible for the development and implementation of alternative guidelines for ICH S7B, which will ultimately guide the ranking of compounds in terms of their pro‐arrhythmic risk (Cavero and Holzgrefe, 2015).
We acknowledge a few limitations in our research. First, it is worth reminding the reader that PKPD parameters should be independent from and uncorrelated with the dose and/or experimental protocol design. We have analysed functional hERG assay data available to the TIPharma consortium, which included contributions from five large pharmaceutical companies. However, we cannot exclude the possibility that different protocol settings might have yielded different results. Despite the different protocols (Table 1), it remains unclear whether they represent the optimal experimental conditions for establishing in vitro–in vivo correlations.
Also, others have shown some degree of intra‐lab variability (particularly over time) and inter‐lab variability even when using the same protocols. The ongoing CiPA project is working closely with contributing scientists and vendors to establish standardized protocols for each/all of the ion channel assays under consideration. Similarly, it should be noted that the variability between in vivo pharmacokinetics and pharmacodynamics may contribute to potential inaccuracies in parameter estimates. In this respect, one should be aware that the use of nonlinear mixed effects modelling can take into account the effect of interindividual differences in pharmacokinetics and pharmacodynamics. Moreover, it allows for the discrimination of covariate effects from random variation.
Whereas it is evident that concentration–effect curves and IC50 values do not provide information on binding kinetics, PKPD modelling of these relationships may account for differences in the interaction between drugs and ion channels. Appropriate parameterization and specific sampling requirements would apply to ensure identification of association/dissociation rate constants. Unfortunately, the available experimental data were not suitable for such an approach. We anticipate that a similar mismatch between in vitro binding, functional assay and QT prolongation would have been observed if other PKPD indices were used, for example IC10, or IC20. A first (or even second) order rate constant may be required to describe biophase equilibration processes.
We are also aware of the potential implication of differences in plasma protein binding for the characterization of PKPD relationships. In fact, controversy regarding the correction for protein binding has been highlighted in previous publications (Gintant et al., 2006). Nevertheless, protein binding for the compounds under evaluation is known to be non‐restrictive; that is, the affinity of a compound for the plasma protein does not necessarily alter binding equilibrium relative to the hERG channel or other relevant target channel. We have summarized our results in terms of total concentration as conclusions would not have been different even after correcting for differences in protein binding. It should be noted that the compound with the highest protein binding (i.e. cisapride) also has very high affinity for the hERG channel.
Lastly, we recognize that the results from three reference compounds are not sufficient to allow generalization of the conclusions to a wide class of molecules, as it can be anticipated that changes in a functional group of a given chemotype can alter the hERG binding profile significantly (Polak et al., 2009).
In conclusion, sensitivity and selectivity criteria have driven the development of experimental protocols for the screening of the pro‐arrhythmic potential of candidate molecules in early drug discovery. Whilst our investigation is limited to three reference compounds with known QT‐prolonging effects in humans, the apparent potency obtained from in vitro assays was far higher than the estimates observed in vivo for the same drugs. The progression of molecules may be unintentionally stopped due to the discrepancies or lack of a systematic in vitro–in vivo correlation. Despite the emphasis on the importance of these assays as a screening tool, our findings indicate that results from in vitro protocols are qualitative at best and cannot be used to define the probability of a clinically relevant increase in the QTc interval.
Author contributions
V.F.S.D. performed the data analysis and wrote the manuscript. E.C. contributed to the analysis of hERG functional assay data. M.D. contributed to the research proposal and revision of the manuscript. O.D.P contributed to the research proposal, data analysis and revision of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Goodness‐of‐fit plots. Upper panels show the individual (left) and population (right) predictions versus observed inhibition in the whole cell patch clamp assay. Lower panels show the conditional weighted residuals versus population predicted inhibition values (left) and drug concentration (right). Symbols indicate the different compounds, namely cisapride, sotalol and moxifloxacin.
Figure S2 NPDE summaries for the pharmacokinetic–pharmacodynamic model describing hERG inhibition in the whole cell patch clamp assay. Upper panels show the QQ‐plot of the distribution of the NPDEs for a theoretical N (0, 1) distribution (left) and the histogram of the distribution of the NPDE together with the density of the standard normal distribution (right). Lower panels show the NPDEs versus concentrations (left) and NPDEs versus individual predictions (right).
Supporting info item
Supporting info item
Acknowledgements
We would like to acknowledge Mr Z. Yu (Division of Medicinal Chemistry, LACDR, University of Leiden, The Netherlands) for his valuable contribution to this research and for performing the [3H]‐dofetilide‐isolated membrane binding assays. We also thank Prof A. P. IJzerman (Division of Medicinal Chemistry, LACDR, University of Leiden, The Netherlands) for the valuable discussions and support to the development of a suitable experimental protocol for the evaluation of hERG binding.
Members of the TI Pharma Cardiovascular Safety Project involved in the preparation of this manuscript: AstraZeneca: Sandra Visser, Eli Lilly & Co: Derek Leishman, GlaxoSmithKline: Jackie Bloomer, Nick McMahon, Phil Milliken, Johnson & Johnson: David Gallagher, An Vermeulen, Pfizer: Piet‐Heijn van der Graaf.
Dubois, V. F. S. , Casarotto, E. , Danhof, M. , and Della Pasqua, O. (2016) Pharmacokinetic–pharmacodynamic modelling of drug‐induced QTc interval prolongation in man: prediction from in vitro human ether‐à‐go‐go‐related gene binding and functional inhibition assays and conscious dog studies. British Journal of Pharmacology, 173: 2819–2832. doi: 10.1111/bph.13558.
References
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM et al. (2004). Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation 110: 904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavero I, Holzgrefe H (2015). CiPA: Ongoing testing, future qualification procedures, and pending issues. J Pharmacol Toxicol Methods 76: 27–37. [DOI] [PubMed] [Google Scholar]
- Cavero I, Mestre M, Guillon JM, Crumb W (2000). Drugs that prolong QT interval as an unwanted effect: assessing their likelihood of inducing hazardous cardiac dysrhythmias. Expert Opin Pharmacother 1: 947–973. [DOI] [PubMed] [Google Scholar]
- Chadwick CC, Ezrin AM, O'Connor B, Volberg WA, Smith DI, Wedge KJ et al. (1993). Identification of a specific radioligand for the cardiac rapidly activating delayed rectifier K+ channel. Circ Res 72: 707–714. [DOI] [PubMed] [Google Scholar]
- Chain ASY, Dubois VFS, Danhof M, Sturkenboom MCJ, Della Pasqua O, TI Pharma PKPD Platform Cardiovascular Safety Project Team (2013). Identifying the translational gap in the evaluation of drug‐induced QTc‐interval prolongation. Br J Clin Pharmacol 76: 708–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chain AS, Dieleman JP, van Noord C, Hofman A, Stricker BH, Danhof M et al. (2013). Not‐in‐trial simulation I: Bridging cardiovascular risk from clinical trials to real‐life conditions. Br J Clin Pharmacol 76: 964–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Cordes JS, Bradley JA, Sun Z, Zhou J (2006). Use of arterially perfused rabbit ventricular wedge in predicting arrhythmogenic potentials of drugs. J Pharmacol Toxicol Methods 54: 261–272. [DOI] [PubMed] [Google Scholar]
- Chiang A, Mallinckrodt C, Dmitrienko A, Leishman D (2010). Utility of positive controls in assessing assay sensitivity in ICH S7B and ICH E14 guidance for evaluation of QT/QTc interval prolongation. J Pharmacol Toxicol Methods 62: 143–147. [DOI] [PubMed] [Google Scholar]
- Chiu PJS, Marcoe KF, Bounds SE, Lin C, Feng J, Lin A et al. (2004). Validation of a [3H] astemizole binding assay in HEK293 cells expressing HERG K+ channels. J Pharmacol Sci 95: 311–319. [DOI] [PubMed] [Google Scholar]
- Danhof M, de Lange EC, Della Pasqua OE, Ploeger BA, Voskuyl RA (2008). Mechanism‐based pharmacokinetic‐pharmacodynamic (PK‐PD) modeling in translational drug research. Trends Pharmacol Sci 29: 186–191. [DOI] [PubMed] [Google Scholar]
- Della Pasqua OE (2013). Translational pharmacology: from animal to man and back. Drug Discov Today Technol 10: e315–e317. [DOI] [PubMed] [Google Scholar]
- Di Veroli GY, Davies MR, Zhang H, Abi‐Gerges N, Boyett MR (2014). hERG inhibitors with similar potency but different binding kinetics do not pose the same proarrhythmic risk: implications for drug safety assessment. J Cardiovasc Electrophysiol 25: 197–207. [DOI] [PubMed] [Google Scholar]
- Diaz GJ, Daniell K, Leitza ST, Martin RL, Su Z, McDermott JS et al. (2004). The [3H]dofetilide binding assay is a predictive screening tool for hERG blockade and proarrhythmia: Comparison of intact cell and membrane preparations and effects of altering [K+]o . J Pharmacol Toxicol Methods 50: 187–199. [DOI] [PubMed] [Google Scholar]
- Dubin AE, Nasser N, Rohrbacher J, Hermans ANN, Marrannes R, Grantham C et al. (2005). Identifying modulators of hERG channel activity using the PatchXpress® planar patch clamp. J Biomol Screen 10: 168–181. [DOI] [PubMed] [Google Scholar]
- Ducroq J, Printemps R, Guilbot S, Gardette J, Salvetat C, Le Grand M (2007). Action potential experiments complete hERG assay and QT‐interval measurements in cardiac preclinical studies. J Pharmacol Toxicol Methods56: 159–70. [DOI] [PubMed]
- Dumotier BM, Deurinck M, Yang Y, Traebert M, Suter W (2008). Relevance of in vitro SCREENIT results for drug‐induced QT interval prolongation in vivo: a database review and analysis. Pharmacol Ther 119: 152–159. [DOI] [PubMed] [Google Scholar]
- Durdagi S, Deshpande S, Duff HJ, Noskov SY (2012). Modeling of open, closed, and open‐inactivated states of the hERG1 channel: structural mechanisms of the state‐dependent drug binding. J Chem Inf Model 52: 2760–2774. [DOI] [PubMed] [Google Scholar]
- Eichenbaum G, Pugsley MK, Gallacher DJ, Towart R, McIntyre G, Shukla U et al. (2012). Role of mixed ion channel effects in the cardiovascular safety assessment of the novel anti‐MRSA fluoroquinolone JNJ‐Q2. Br J Pharmacol 166: 1694–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fermini B, Hancox JC, Abi‐Gerges N, Bridgland‐Taylor M, Chaudhary KW, Colatsky T et al. (2016). A new perspective in the field of cardiac safety testing through the comprehensive in vitro proarrhythmia assay paradigm. J Biomol Screen 21: 1–11. [DOI] [PubMed] [Google Scholar]
- Finlayson K, Pennington AJ, Kelly JS (2001). [3H]dofetilide binding in SHSY5Y and HEK293 cells expressing a HERG‐like K+ channel? Eur J Pharmacol 412: 203–212. [DOI] [PubMed] [Google Scholar]
- Fossa AA (2008). The impact of varying autonomic states on the dynamic beat‐to‐beat QT – RR and QT – TQ interval relationships. Br J Pharmacol 154: 1508–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- France NP, Della Pasqua O (2015). The role of concentration − effect relationships in the assessment of QT c interval prolongation. Br J Clin Pharmacol 79: 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gintant GA (2008). Preclinical Torsades‐de‐Pointes screens: advantages and limitations of surrogate and direct approaches in evaluating proarrhythmic risk. Pharmacol Ther 119: 199–209. [DOI] [PubMed] [Google Scholar]
- Gintant GA, Su Z, Martin RL, Cox BF (2006). Utility of hERG assays as surrogate markers of delayed cardiac repolarization and QT safety. Toxicol Pathol 34: 81–90. [DOI] [PubMed] [Google Scholar]
- Haverkamp W, Breithardt G, Camm AJ, Janse MJ, Rosen MR, Antzelevitch C et al. (2000). The potential for QT prolongation and proarrhythmia by non‐antiarrhythmic drugs: clinical and regulatory implications. report on a policy conference of the european society of cardiology. Eur Heart J 21: 1216–1231. [DOI] [PubMed] [Google Scholar]
- Hoffmann P, Warner B (2006). Are hERG channel inhibition and QT interval prolongation all there is in drug‐induced torsadogenesis? a review of emerging trends. J Pharmacol Toxicol Methods 53: 87–105. [DOI] [PubMed] [Google Scholar]
- Jonsson MK, Vos MA, Mirams GR, Duker G, Sartipy P, de Boer TP et al. (2012). van Veen TA application of human stem cell‐derived cardiomyocytes in safety pharmacology requires caution beyond hERG. J Mol Cell Cardiol 52: 998–1008. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch GE, Trepakova ES, Brimecombe JC, Sidach SS, Erickson HD, Kochan MC et al. (2004). Variability in the measurement of hERG potassium channel inhibition: effects of temperature and stimulus pattern. J Pharmacol Toxicol Methods 50: 93–101. [DOI] [PubMed] [Google Scholar]
- Laverty H, Benson C, Cartwright E, Cross M, Garland C, Hammond T et al. (2011). How can we improve our understanding of cardiovascular safety liabilities to develop safer medicines? Br J Pharmacol 163: 675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Mann SA, Windley MJ, Imtiaz MS, Vandenberg JI, Hill AP (2016). In silico assessment of kinetics and state dependent binding properties of drugs causing acquired LQTS. Prog Biophys Mol Biol 120: 89–99. [DOI] [PubMed] [Google Scholar]
- Lunn DJ, Best N, Thomas A, Wakefield J, Spiegelhalter D (2002). Bayesian analysis of population PK/PD models: general concepts and software. J Pharmacokinet Pharmacodyn 29: 271–307. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirams GR, Davies MR, Brough SJ, Bridgland‐taylor MH, Cui Y, Gavaghan DJ et al (2014). Prediction of thorough QT study results using action potential simulations based on ion channel screens. J. Pharmacol. Toxicol. Methods 70: 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra H, Polak S, Jamei M, Rostami‐Hodjegan A (2014). Interaction between domperidone and ketoconazole: Toward prediction of consequent QTc prolongation using purely in vitro information. CPT Pharmacometrics Syst Pharmacol 3: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara T, Virág L, Varró A, Rudy Y (2011). Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol 7: e1002061. doi:10.1371/journal.pcbi.1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollerstam A, Persson AH, Visser SA, Fredriksson JM, Forsberg T , Nilsson LB et al. ( 2007) A novel approach to data processing of the QT interval response in the conscious telemetered beagle dog. J Pharmacol Toxicol Methods 55: 35–48. [DOI] [PubMed] [Google Scholar]
- Ollerstam A, Visser SA, Persson AH, Eklund G, Nilsson LB, Forsberg T et al. (2006) Pharmacokinetic‐pharmacodynamic modeling of drug‐induced effect on the QT interval in conscious telemetered dogs. J Pharmacol Toxicol Methods 53: 174–83. [DOI] [PubMed] [Google Scholar]
- Polak S, Wisniowska B, Brandys J (2009). Collation, assessment and analysis of literature in vitro data on hERG receptor blocking potency for subsequent modelling of drugs' cardiotoxic properties. J Appl Toxicol 29: 183–206. [DOI] [PubMed] [Google Scholar]
- Polak S, Fijorek K, Glinka A, Wisniowska B, Mendyk A (2012). Virtual population generator for human cardiomyocytes parameters: in silico drug cardiotoxicity assessment. Toxicol Mech Methods 22: 31–40. [DOI] [PubMed] [Google Scholar]
- Prior H, McMahon N, Schofield J, Valentin J (2009) Non‐invasive telemetric electrocardiogram assessment in conscious beagle dogs. J Pharmacol Toxicol Method 60: 167–73. [DOI] [PubMed] [Google Scholar]
- Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S et al. (2003). Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res 58: 32–45. [DOI] [PubMed] [Google Scholar]
- Sager PT, Gintant G, Turner JR, Pettit S, Stockbridge N (2014). Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the cardiac safety research consortium. Am Heart J 167: 292–300. [DOI] [PubMed] [Google Scholar]
- Sahota T, Sanderson I, Danhof M, Della Pasqua O (2015). Model‐based prediction of the acute and long‐term safety profile of naproxen in rats. Br J Pharmacol 172: 3861–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Tristani‐Firouzi M (2006). hERG potassium channels and cardiac arrhythmia. Nature 440: 463–469. [DOI] [PubMed] [Google Scholar]
- Shah RR, Hondeghem LM (2005). Refining detection of drug‐induced proarrhythmia: QT interval and TRIaD. Heart Rhythm 2: 758–772. [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD et al. (1985). Measurement of protein using bicinchoninic acid. Anal Biochem 150: 76–85. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to Pharmacology in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Chen J, Martin RL, McDermott JS, Cox BF, Gopalakrishnan M et al. (2006). Block of hERG channel by ziprasidone: biophysical properties and molecular determinants. Biochem Pharmacol 71: 278–286. [DOI] [PubMed] [Google Scholar]
- Thomsen MB, Matz J, Volders PGA, Vos MA (2006). Assessing the proarrhythmic potential of drugs: current status of models and surrogate parameters of torsades de pointes arrhythmias. Pharmacol Ther 112: 150–170. [DOI] [PubMed] [Google Scholar]
- Tusscher KHWJ, Noble D, Noble PJ, Panfilov AV (2004). A model for human ventricular tissue. Am J Physiol Heart Circ Physiol 286: H1573–H1589. [DOI] [PubMed] [Google Scholar]
- van der Linde HJ, van Deuren B, Teisman A, Towart R, Gallacher DJ (2008). The effect of changes in core body temperature on the QT interval in beagle dogs: a previously ignored phenomenon, with a method for correction. Br J Pharmacol 154: 1474–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis RM (2010). Integrated risk assessment and predictive value to humans of non‐clinical repolarization assays. Br J Pharmacol 159: 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Della Penna K, Wang H, Karczewski J, Connolly TM, Koblan KS et al. (2003). Functional and pharmacological properties of canine ERG potassium channels. Am J Physiol Heart Circ Physiol 284: H256–H267. [DOI] [PubMed] [Google Scholar]
- Watson KJ, Gorczyca WP, Umland J, Zhang Y, Chen X, Sun SZ et al. (2011). Pharmacokinetic–pharmacodynamic modelling of the effect of Moxifloxacin on QTc prolongation in telemetered cynomolgus monkeys. J Pharmacol Toxicol Methods 63: 304–313. [DOI] [PubMed] [Google Scholar]
- Yao J‐A, Du X, Lu D, Baker RL, Daharsh E, Atterson P (2005). Estimation of potency of HERG channel blockers: impact of voltage protocol and temperature. J Pharmacol Toxicol Methods 52: 146–153. [DOI] [PubMed] [Google Scholar]
- Yassen A, Olofsen E, Dahan A, Danhof M (2005). Pharmacokinetic‐pharmacodynamic modeling of the antinociceptive effect of buprenorphine and fentanyl in rats: role of receptor equilibration kinetics. J Pharmacol Exp Ther 13: 1136–1149. [DOI] [PubMed] [Google Scholar]
- Yu Z, IJzerman AP, Heitman LH (2015). Kv11.1 (hERG)‐induced cardiotoxicity: a molecular insight from a binding kinetics study of prototypical Kv11.1 (hERG) inhibitors. Br J Pharmacol 172: 940–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA et al. (1998). Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J 74: 230–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Goodness‐of‐fit plots. Upper panels show the individual (left) and population (right) predictions versus observed inhibition in the whole cell patch clamp assay. Lower panels show the conditional weighted residuals versus population predicted inhibition values (left) and drug concentration (right). Symbols indicate the different compounds, namely cisapride, sotalol and moxifloxacin.
Figure S2 NPDE summaries for the pharmacokinetic–pharmacodynamic model describing hERG inhibition in the whole cell patch clamp assay. Upper panels show the QQ‐plot of the distribution of the NPDEs for a theoretical N (0, 1) distribution (left) and the histogram of the distribution of the NPDE together with the density of the standard normal distribution (right). Lower panels show the NPDEs versus concentrations (left) and NPDEs versus individual predictions (right).
Supporting info item
Supporting info item