

Abstract
Chemokine release promotes crosstalk between opioid and chemokine receptors that in part leads to reduced efficacy of morphine in the treatment of chronic pain. Based on the possibility that a MOR-CCR5 heteromer is involved in such crosstalk, we have synthesized bivalent ligands (MCC series) that contain mu opioid agonist and CCR5 antagonist pharmacophores linked through homologous spacers (14–24 atoms). When tested on lipopolysaccharide-inflamed mice, a member of the series (MCC22; 3e) with a 22-atom spacer exhibited profound antinociception (i.t. ED50 = 0.0146 pmol/mouse) that was >2000× greater than morphine. Moreover, MCC22 was ~3500× more potent than a mixture of mu agonist and CCR5 antagonist monovalent ligands. These data strongly suggest that MCC22 acts by bridging the protomers of a MOR-CCR5 heteromer having a TM5,6 interface. Molecular simulation studies are consistent with such bridging. This study supports the MOR-CCR5 heteromer as a novel target for treatment of chronic pain.
Introduction
A recent NIH report1, 2 on the role of opioids in the treatment of chronic pain has pointed out that 100-million Americans are living with this condition. Accumulating evidence supports the increased risk for harms associated with long-term opioid therapy due to tolerance that leads to overdosing, dependence, and a variety of other side effects that arise from prolonged use.
As It is presently recognized that tolerance to morphine under chronic inflammatory conditions is in part related to release of chemokines and glutamate that lead to hyperalgesia,3–6 the established functional interactions between opioid receptors and chemokine3 or metabotropic glutamate-5 receptors7 offer an opportunity to uncover new targets to develop treatments of chronic pain without tolerance or other side effects.
Particularly relevant to this problem is a recent report on the extraordinary antinociception produced by a bivalent ligand (MMG22) having both mu opioid agonist and metabotropic glutamate receptor-5 antagonist (mGluR5) pharmacophores.8, 9 As MMG22 targets a MOR-mGluR5 heteromer, it is devoid of tolerance in mice with chronic bone cancer pain. The fact that MMG22 is orders of magnitude more potent than members of its series with shorter spacers or a mixture of monovalent mu agonist and mGluR5 antagonist, suggested bridging of protomers in the heteromer is vastly superior to univalent association.
In view of these results, the present study describes a similar approach for targeting a putative MOR-CCR5 heteromer based on cross-talk of colocalized MOR and CCR5 in neurons and glia in pain processing areas.10–13 Moreover, given the presence of MOR-CCR5 in cultured cells, it appears likely that such heteromers may also exist in vivo.14, 15 Here we report on a series of ligands that contains both mu opioid agonist and chemokine CCR5 antagonist pharmacophores tethered through homologous spacers. A member (MCC22, 3e) of this series having a 22-atom spacer has been found to possess extraordinary potency without tolerance for the treatment of chronic pain.
Design rationale
The pharmacophores for targeting MOR-CCR5 heteromer are derived from the mu opioid agonist oxymorphone 116 and CCR5 antagonist 2 (TAK-220)17 (Figure 1). Oxymorphone has also been employed as precursor for the mu opioid agonist pharmacophore in the synthesis of other bivalent ligands.8, 18, 19 The utility of 2 as a pharmacophore was verified by attaching a short spacer to the nitrogen of its piperidine moiety and determining its ability to antagonize CCL5-stimulated CCR5 expressed in HEK293 cells (Supplementary Figure 1). The derivative 8c of 2 was as effective as 2 in inhibiting CCR5. With that information, members of the MCC series (3a–3f) having different length spacers (14 to 24 atoms) were synthesized (Figure 1). The selection of the spacer length range was based on prior studies that revealed 18–22 atoms are effective in bridging of GPCR protomers for different opioid-containing bivalent ligands.20–24 Additionally, we prepared the monovalent ligands 424 and 5 with attached spacers as controls.
Figure 1.
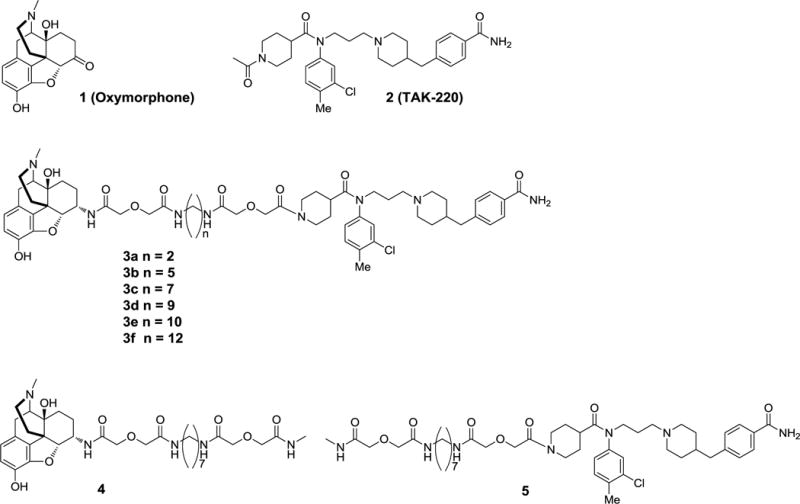
Mu opioid agonist 1 and chemokine CCR5 antagonist 2 pharmacophores have been incorporated into the bivalent ligand series (3a-3f) and monovalent control ligands 4, and 5.
RESULTS
Biological
Antinociception using normal and LPS-prereated mice
As pain is a hallmark of many inflammatory conditions, we have tested the target ligands on lipopolysaccharide (LPS) prereated mice to induce inflammation.8, 25 Antinociception was evaluated using the radiant heat-induced tailflick assay, and morphine was employed as a standard opioid agonist under these conditions. The results are presented in Table 1.
Table 1.
Antinociceptive activity (pmol/mouse) of bivalent and monovalent ligands that contain MOR agonist/CCR5 antagonist pharmacophores.
| Compound | CONTROLS | LPS pretreated (1 mg/kg 24 hours i.p.) | ||||
|---|---|---|---|---|---|---|
| ED50 (CI) | ED50 (CI) | ED50 (CI) | ED50 (CI) | |||
| i.t. | i.c.v. | i.c.v./i.t. ratio | i.t. | i.c.v. | i.c.v./i.t. ratio | |
| Morphine | 27 (21.2–35.6) |
301 (224 – 406) |
11 | 35 c (24.3–50.4) |
281.4 (184.7 – 428.6) |
8.0 |
| 2 | 1000a 34.2 ± 10.8 |
1000 a 26.2 ± 5.9 |
1 | 1000 a 49.0 ± 10.9 |
1000 a 7.6 ± 1.5 |
6.48 |
|
3a (MCC14) |
32.34 b (27.3 – 38.4) |
106.6 c (78.2 – 145.2) |
3.29 | 10.60 b (6.1 – 18.58) |
56.41 c (33.3 – 95.6) |
5.32 |
|
3b (MCC17) |
370.6 c (253.0 – 543.0) |
358.2 c (166.2 – 722) |
3.14 | 10.92 b (6.9 – 17.2) |
187.10 c (92.7 – 377.7) |
6.38 |
|
3c (MCC19) |
166.4 c (54.6 – 506.5) |
250 a 35.9 ± 9.0 |
~1.5 | 11.41 b (7.0 – 18.5) |
692.4 c (432.7 – 1108) |
60.68 |
|
3d (MCC21) |
15.83 b (6.2 – 40.4) |
1000 a 54.5 ± 14.5 |
63.98 | 8.43 (3.0 – 23.8) 24 hours later, there was still 24% MPE |
1000 a 63.1 ± 45.2 |
118.62 |
|
3e (MCC22) |
45.75 b (23.1 – 90.8) |
500a 49.7 ± 14.9 |
~11d | 0.01461 b (0.008 – 0.025) |
500a 41.1 ± 12.9 |
~34,000d |
|
3f (MCC24) |
336.9 c (138.9 – 818.3) |
1000 a 27.6 ± 9.1 |
~2.97 | 122.70 c (80 – 187) |
1000 a 55.7 ± 11.2 |
8.15d |
| 4 | 113.3 c (86.5 – 148.4) |
108.2 c (83.1 – 140.8) |
0.95 | 21.29 c (13.6 – 33.8) |
168.9 c (124.0 – 230.2) |
7.93 |
| 5 | 500a 28.1 ± 11.3 |
500a 43.9 ± 10.9 |
– | 1000a 32.4 ±12.5 |
1000a 51.9 ± 12.3 |
– |
The highest dose measured for antinociception and the corresponding percent maximal possible effect for that dose.
No tolerance was determined, as suggested by the absence of a significant difference in ED50 24 hours after initial testing.
Tolerance was determined by a significant difference in ED50 24 hours after initial testing.
An estimated ratio due to absence of an accurate ED50 value.
When tested on the LPS inflamed mice via i.t. administration, the potencies of homologues 3a–3d with spacers in the range of 14–21 atoms showed minimal potency differences within the LPS pretreated group of mice. However, the homologue (3e) with 22 atoms (MCC22) exhibited a large potency increase. This amounted to ~700-fold for MCC22 compared to its lower homologues 3a-3d. Extending the spacer length to 24 atoms (MCC24) led to a reduced potency that was lower than homologs with shorter spacers (3a–3d). A graphic SAR profile of the MCC series is displayed in Fig. 2.
Figure 2.
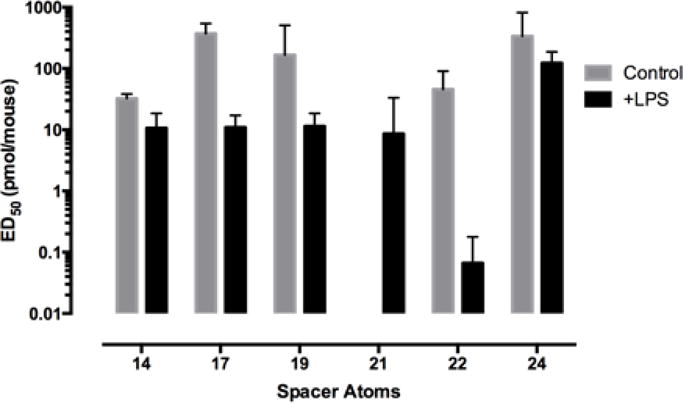
Effect of spacer length in the MCC series (14–24 atoms) on the i.t. ED50 potency in control and LPS-pretreated mice.
Relative to morphine or its monovalent control ligand 4, the potency of MCC22 is ~1500-fold greater. Confirmation for the involvement of a mu opioid receptor in the action of MCC22 was evaluated in inflamed mice pretreated with β-FNA, a selective alkylating agent of the mu opioid receptor.26 The 96.6 ED50 potency ratio (β-FNA-treated/control) supports such involvement.
A comparison of the LPS/tail-flick ED50 of MCC22 with that of the CFA-induced mechanical hyperalgesia mouse model revealed overlapping confidence intervals for ED50 values between the two assays (Table 2). This suggests that the LPS potency enhancement of MCC22 in mice pretreated with LPS is related primarily to inflamation-induced hyperalgesia.
Table 2.
Comparison of antinociception in thermal (LPS) and mechanical (CFA) hyperalgesia mouse models
| Compd. | Thermal vs Mechanical Hypersensitivity | |
|---|---|---|
| (i.t.) | ED50 values (95% CI) pmol/mouse | |
| LPS | CFA | |
| Saline | No effect | No effect |
| Morphine | 35 (24–50) |
15 (10–22) |
| 4 | 21 (14–34) |
27 (18–42) |
| 2 + 4 | No synergy | No synergy |
| 3a | 11 (6–9) |
3 (1–7) |
| 3e | 0.015 (0.009 – 0.025) |
0.019 (0.003 – 0.109) |
| 3f | 123 (80–187) |
58 (40–85) |
The i.c.v. SAR profile in LPS-pretreated mice for 3a–3f differs significantly from the i.t. data, in that there was no potency increase upon lengthening the spacer in the series. This is reflected in the i.c.v./i.t. potency ratio for MCC22 which is >7500, whereas homologues 3a–3d, 3f and monovalent ligands (morphine and 4) have ratios in the 8–60 range.
In contrast to the SAR profile of bivalent series 3a–3f in hyperalgesic mice, the normal control mice exhibited a strikingly different i.t. SAR profile whose distinguishing feature was the absence of enhanced potency for MCC22. Thus, in LPS pretreated mice, MCC22 exhibited ~3000-fold greater potency relative to control mice. Consequently, its icv/i.t. ED50 ratio profile in control mice was substantially lower in LPS pretreated mice. Relatively small differences between ED50 values in normal and inflamed mice were observed for morphine and monovalent agonist 4.
In order to further evaluate the contribution of a 22-atom spacer to the potency of MCC22 in inflamed mice, a mixture of two monovalent ligands consisting of mu opioid agonist 4 with CCR5 antagonist 2 was evaluated in a 1:35 ratio based on their individual ED50 values to permit calculation of the theoretical ED50. The theoretical value was calculated to be 381 pmol/mouse versus the observed ED50 was 52 pmol/mouse, suggesting some potentiation. Based on the observed value, the potency of MCC22 is ~3500× greater relative to the mixture (Table 3). It is noteworthy that bivalents with shorter spacers were ~5-fold more potent, whereas 3f was <1× than the mixture.
Table 3.
Potency of MCC bivalent ligands relative to a mixture of monovalent mu agonist 4 and CCR5 antagonist 2 in LPS-pretreated mice
| Compound | Spacer length (atoms) | Relative Potencya |
|---|---|---|
| 3a | 14 | 4.9 |
| 3b | 17 | 4.8 |
| 3c | 19 | 4.5 |
| 3d | 21 | 6.2 |
| 3e | 22 | 3,559 |
| 3f | 24 | 0.4 |
| 2 + 4 | 1 |
The potencies are relative to the observed ED50 of the mixture (2 + 4) was 52 pmol/mouse (34.6 – 78.2).
Effect of minocycline on MCC22 antinociception in LPS-pretreated mice
It has been reported that LPS induces hyperalgesia via activation of spinal microglia, and it is known that minocycline suppresses such hyperalgesia by inhibiting microglial activation.27 Activated microglia produce proinflammatory chemokines that promote hyperalgesia that lead to the development and maintenance of inflammatory pain. Since chronically activated microglia are known to suppress antinociception produced by opioids,28, 29 we therefore investigated the effect of i.p. minocycline27 on MCC22-induced antinociception in LPS pretreated mice. Significantly, minocycline substantially reduced the antinociception produced by MMC22 (Figure 3). The blockage occurred in a time dependent manner as follows (%MPE): 60 min (100%); 120 min (52.04% ± 17.04); 240min (10.7% ± 5.17). The data suggest that MCC22 reduces LPS-induced hyperalgesia via blockage of the activation of spinal cord microglia.
Figure 3.
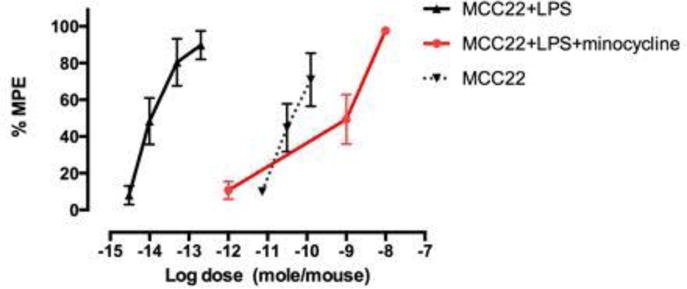
ED50’s of MCC22 on control or LPS-pretreated mice that were injected (45 mg/kg i.p.) with minocycline prior to testing. LPS/MCC22: 0.015 (0.01 – 0.03); naïve/MCC22: 45.75 (23.05 – 90.81); LPS/minocycline/MCC22: 1016 (54-1558)
Modeling and simulation of MCC22 bound to MOR-CCR5 heteromer
For modeling of the MCC bivalents, X-ray crystallographic structures of MOR30 and CCR5 antagonist, Maraviroc, bound to CCR5 were utilized.31 The detailed procedures are presented in the Experimental Section. Docking studies revealed each of the pharmacophores of MCC22 are capable of binding their respective protomers that are interfaced via TM5,6 helices (Figure 4). The binding sites within 10Å surrounding MCC22 are illustrated in Figure 5. Two critical CCR5 residues were found to be essential for binding,32, 33 namely Glu283 on TM7 which make an electrostatic salt-bridge with the central basic nitrogen in the middle of 2, and Ile198 on TM5 which is preserved in a hydrophobic interaction with groups (3-chloro-4-methylphenyl) in 2. The salt bridge interaction of the MOR Asp147 with the charged nitrogen atom on the opioid pharmacophore was also conserved. Neutralization of these two salt bridges led to subsequent dissociation of MCC22 from the heteromer during MD simulation (Figure 6). MCC22 binding to the heteromer reduces overall conformational flexibility of the heteromer as compared to unbound MOR-CCR5 and its protomeric units. Major reduction of flexibility was observed in the extracellular loop (ICL3) region of the TM5:TM6 interface of MOR and in the ECL3 region between TM6 and TM7 of CCR5.
Figure 4.
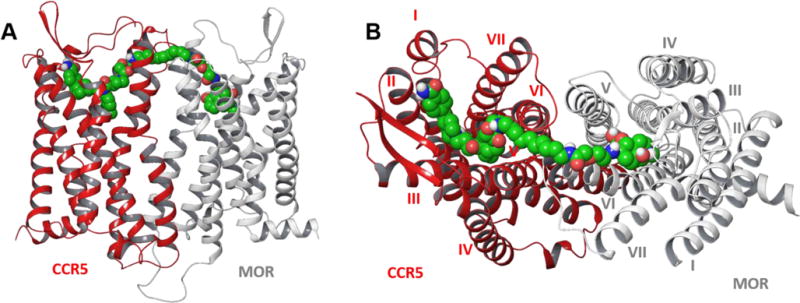
Side (A) and top (B) views of TM5-TM6 interfaced heteromeric model of MOR-CCR5 complex with MCC22.
Figure 5.
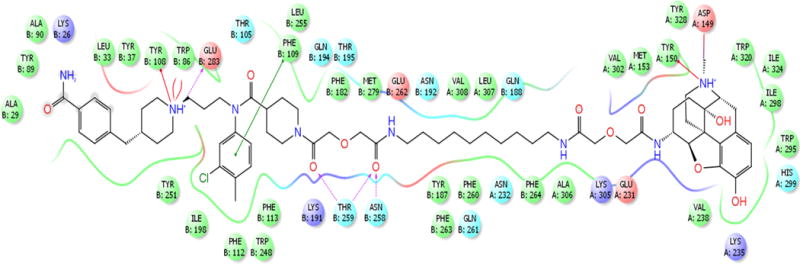
Binding site residues within 5Å of MOR-CCR5 heteromer surrounding MCC22.
Figure 6.
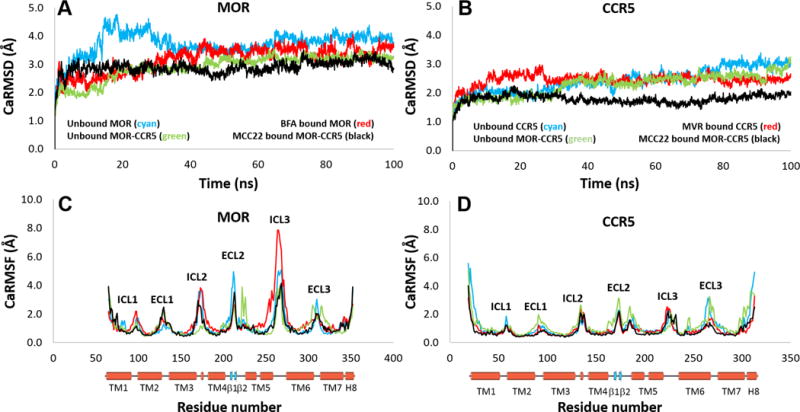
(A) and (B) CαRMSD of MOR and CCR5 showed MCC22 binding reduces the overall conformational flexibility of both MOR and CCR5 in MOR-CCR5 heteromer. (C) and (D) Significant loss of conformational flexibility was observed in the ICL3 region in MOR between TM5 and TM6 as well as in the ECL3 region of CCR5 between TM6 and TM7 due to MCC22 binding.
DISCUSSION AND CONCLUSIONS
The presence of MOR-CCR5 on membranes of human or monkey lymphocytes and Chinese hamster ovary (CHO) cells coexpressing MOR and CCR515 suggest the possibility that such a heteromer may exist in vivo, particularly under inflammatory conditions. In this regard, crosstalk between microglia and neurons via putative MOR-CCR5 may in part be responsible for development of hyperalgesia that may lead to allodynia and chronic pain.34 Since activation of spinal microglia is a key step leading to hyperalgesia via communication with neuronal circuitry, targeting such a heteromer was considered as an approach to developing effective analgesics for treatment of different types of chronic pain. Thus, activation of MOR with concomitant antagonism of CCR5 could represent a promising strategy to develop analgesics with enhanced potency and reduced side effects because it would involve the reduction of hyperalgesia and thereby elevate the pain threshold.
Accordingly, in this study we have synthesized bivalent ligands that contain pharmacophores derived from the mu opioid agonist, oxymorphone 1,16 and CCR5 antagonist 2.17 The design approach involved connecting these pharmacophores through different length spacers (14–24 atoms) to afford the MCC series (3a–3f) of bivalent ligands. The selection of spacer lengths was based on prior studies that suggested effective bridging of heteromeric GPCR protomers in the range of 18–22 atoms,20–24
Members of the MCC series were administered by intrathecal (i.t.) or intracerebroventricular (i.c.v.) routes to both normal and inflamed mice. Lipopolysaccharide (LPS) was injected i.p. to promote inflammation in the mouse model that we employed.35 Ligands 3a–3d with homologous spacers containing 14 – 21 atoms afforded ED50 values for antinociception in the 8–11 pmol range when administered i.t. to LPS pretreated mice. Remarkably, the 22-atom spacer homologue, MCC22 (3e), produced antinociception that was at ~700-fold greater (ED50 = 0.0146 pmol/mouse) than either its lower or higher spacer homologues, and without signs of tolerance (Table 1). Apparently, the 22-atom spacer length is well suited to optimally bridge MOR and CCR5 protomers in the putative heteromer, whereas bivalents with shorter spacers may bind univalently. The longer spacer homologue (MCC24) may bind univalently or in a bridging mode that is less productive, given that its potency is ~10% those of the shorter bivalent ligands 3a–3d. That MCC22 was 3500-times more potent than a mixture of monovalent mu opioid agonist 4 and CCR5 antagonist 2 ligands highlights the critical importance of spacer length (Table 2). It is noteworthy that a similar spacer length-potency relationship has been reported8 for members of the MMG bivalent series that contain mu opioid agonist and metabotropic glutamate receptor5 (mGluR5) antagonist pharmacophores. The 22-atom spacer in MMG22 also is optimal in targeting the MOR-mGluR5 heteromer.8
In view of the ~7500-fold greater potency of i.t. MCC22 in LPS pretreated mice than by the i.c.v route of administration, the putative target (MOR-CCR5 heteromer) of MCC22 appears to be localized in the spinal cord but not the brain. In this regard, the dorsal horn is likely to be the locus of action.
In contrast to the exceptional i.t. potency of MCC22 in LPS pretreated mice, untreated control mice exhibited an i.t. ED50 that was ~3070-fold greater, illustrating the dramatic effect of inflammation in enhancing antinociception of MCC22 (Table 1; Fig 2). The magnitude of this effect may in part be due to the LPS-induced up-regulation of MOR-CCR5 heteromer. Thus, in the LPS-pretreated mouse model, greatly increased spinal expression of cell-surface neuronal or microglial MOR and CCR5 may lead to elevated levels of MOR-CCR5 heteromer36–41 that could also contribute to enhanced potency of MCC22.
Molecular modeling studies are entirely consistent with the bridging of MCC22 with a MOR-CCR5 heteromer that possesses a TM5,6 interface (Figures 4 and 5). This interface was employed in the modeling because it was observed as the more stable dimer in the crystal structure of MOR.30 An alternative TM1-TM2-H8 interfaced dimer also observed in the crystallographic study possesses a 47Å distance between binding sites (Supplementary Figure 2) and is therefore not considered as a putative target, given that the MCC22 length spacer is 26Å. The modeling also revealed that the shorter spacer homologues in the MCC series are incapable of effective bridging of both protomers in the heteromer. Since molecular dynamics simulation studies indicate that MCC22 binding to the heteromer reduces the overall conformational flexibility of MOR-CCR5 heteromer, its exceptional potency may be related to induction of unique conformational changes in binding to the heteromer that are not mimicked by a combination of monovalent ligands. The major reduction of conformational flexibility in MOR-CCR5 that occurs near the TM5-TM6 association interface is consistent with the expected mode of binding of MCC22, and with the finding that it has ~3500-fold greater potency than a mixture of monovalent ligands (2 + 4) in LPS-pretreated mice (Table 3). In this regard, it is likely that the substantially lower potencies of the shorter spacer homologues (3a–3d) are due to univalent association of each of the MCC pharmacophores with their respective protomers in the heteromer. The even greater reduction of potency of MCC24 could be a consequence of either univalent binding mode or to a less productive agonist binding mode when in the bridged state.
It is now established that CNS sensitization arising from a) spinal cord injury, b) chemotherapeutic agents or other medications, c) diabetic neuropathy, and d) a number of diseases that include AIDS, can lead to chronic inflammatory pain associated with hyperalgesia mediated via microglia. In view of the efficacy of MCC22 in blocking both LPS- and CFA-induced hyperalgesia, we conducted studies with minocycline42 because it is well-established that it selectively inhibits the activation of microglia, thereby reducing the production of inflammatory mediators. Significantly, in inflamed mice pretreated i.p with minocycline, the antinociception of i.t. MCC22 was greatly reduced to a potency range observed in the absence of inflammation. Thus, the effectiveness of MCC22 under inflammatory conditions is likely a consequence of the reduction of hyperalgesia due to antagonism of CCR5 and concomitant activation of MOR. That MCC22 is 3500× more potent than a mixture of monovalent mu agonist and CCR5 antagonist supports the necessity of a critical spacer length in achieving this effect by simultaneous binding of the pharmacophores to the protomers in a MOR-CCR5 heteromer. It appears possible that the 22-atom spacer is responsible for the greatly enhanced potency of MCC22 due to a favorable conformational change of the MOR protomer via its TM5,6 interface with the CCR5 protomer maintained in an inactive state by the chemokine antagonist pharmacophore.
The high efficacy of MCC22 in inflammatory and neuropathic pain speaks to its potential utility in the treatment of a variety of conditions associated with chronic pain.43–45 Given the well-established role of CCR5 as a co-receptor in facilitating intracellular transport of HIV into cells, we are presently investigating the efficacy of MCC22 in blocking both chronic pain and the penetration of this virus into the CNS.
EXPERIMENTAL SECTION
Chemistry
Chemical Synthesis
The bivalent series 3a–3f (Fig. 1) in the present study contain pharmacophores derived from the mu opioid agonist 1, and the CCR5 antagonist 2.46 The synthesis of 3a–3f started with the elaboration of intermediates depicted in Scheme 1. With the exception of 8a–8c and 11d–11f, the intermediates 7–12 have been reported previously by us.8, 18, 19, 47 Using literature procedures,46 the amine 8a was prepared and converted into 8c. Calcium mobilization studies in CCR5 expressing HEK cells stimulated by CCL5 revealed that 8c was equally potent at inhibiting calcium release (Supplementary Figure 1). Building block 8b was prepared by reaction of 8a with glycolic anhydride, and used for the preparation of bivalent ligands 3a–3f, and for the preparation of monovalents 5 and 8c as shown in Scheme 1.
Scheme 1.
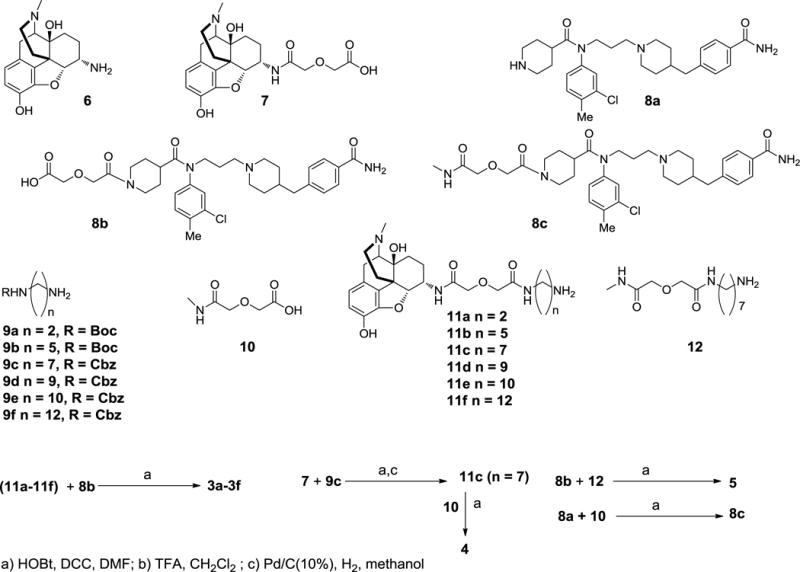
Intermediates employed for the synthesis of bivalent ligands 3a-3f, monovalents 5 and 8c.
General chemistry
Oxymorphone was obtained from Mallinckrodt & Co. All other chemicals and solvents were purchased from Aldrich or Fisher without further purification. 1H and 13C NMR spectroscopy were obtained on 300 MHz on an Oxford Varian VXR 300 MHz NMR Spectrometer. Mass spectroscopy was obtained on Bruker BioTOF II mass spectrometry. Synthesis of CCR5 antagonist 2,46 monovalent 4,24 intermediates 6,48 7,49 9a–9f,49 and 10, 11a–11c49 have been previously reported. Purities of bivalent ligands (3a–3f) and monovalent ligands (4, 5 and 8c) were over 98% based on analysis on HPLC column (Phenomenex Luna SB‐C18 (2) 5u 4.6×250 mm) which was eluted with [H2O/CH3CN; 70:30 with 0.25% TFA; (v/v) at a flow rate of 1 ml/min.]
General procedure for the synthesis of Bivalent ligands 3a–3f
Carboxylic acid 8b (0.97 mmol) was activated with HOBt・H2O (0.74 mmol), DIPEA (168.4 μL, 0.97 mmol), DMAP (9.1 mg, 0.074 mmol) and EDCI (185.4 mg, 0.97 mmol) in DMF (0.5 mL) at 0 °C under N2 atmosphere for 10 min. requisite amines 11a–11f (0.74 mmol) in DMF (2 mL) was added to the above mixture and stirred under N2 atmosphere at room temperature overnight. The reaction mixture was evaporated in vacuum. The residue was purified by column chromatography on silica gel [CH2Cl2/MeOH/NH4OH = 95:4:1 to 92:7.5:0.5 to 89:10:1; (v/v/v)] to give the objective compounds.
N-(3-(4-(4-Carbamoylbenzyl)piperidin-1-yl)propyl)-N-(3-chloro-4-methylphenyl)-1-(14-(((4aS,7S,7aR,12bS)-4a,9-dihydroxy-3-methyl-2,3,4,4a,5,6,7,7a-octahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)amino)-5,10,14-trioxo-3,12-dioxa-6,9-diazatetradecan-1-oyl)piperidine-4-carboxamide, MCC14 (3a)
Yield: ~100%, off-white amorphous solid
1H NMR (400 MHz, CDCl3) δ 7.74 (d, 7.6Hz, 2H), 7.28–7.29 (m, 1H), 7.17–7.19 (m, 3H), 6.96 (d, 8.0Hz, 1H), 6.71 (d, 8.0Hz, 1H), 6.51 (d, 8.0Hz, 1H), 4.53–4.60 (m, 2H), 4.42 (d, 12.8Hz, 1H), 4.18–4.28 (m, 2H), 3.99–4.04 (m, 6H), 3.63 (t, 7.2Hz, 2H), 3.40–3.54 (m, 5H), 3.12 (br d, 18.4Hz, 1H), 2.84 (d, 10.4Hz, 2H), 2.77 (d, 6.4Hz, 2H), 2.59 (d, 6.8Hz, 1H), 2.55 (6.4Hz, 2H), 2.19–2.24 (m, 7H), 2.39 (s, 3H), 2.34 (s, 3H), 1.83 (t, 11.2Hz, 2H), 1.53–1.79 (m, 13H), 1.36–11.42 (m, 1H), 1.21–1.30 (m, 3H), 0.79–1.08 (m, 1H).
13C NMR (100 MHz) δ 173.46, 171.25, 169.35, 169.18, 167.82, 167.24, 145.55, 145.00, 140.76, 138.16, 136.56, 135.23, 131.97, 130.99, 130.80, 129.23, 128.41, 127.36, 126.29, 125.24, 119.23, 117.58, 89.23, 71.50, 71.08, 70.80, 69.62, 69.58, 64.60, 55.88, 53.83, 48.04, 46.38, 45.68, 45.58, 44.85, 43.53, 43.08, 42.92, 41.21, 39.57, 38.87, 38.55, 38.40, 37.64, 33.21, 31.94, 29.04, 28.58, 28.10, 25.21, 22.04, 21.20, 19.76.
MS (ESI): m/z Observed 1069.9 (M+1); 1069.5, Calculated for C56H73ClN8O11 [M+1]+.
N-(3-(4-(4-Carbamoylbenzyl)piperidin-1-yl)propyl)-N-(3-chloro-4-methylphenyl)-1-(17-(((4aS,7S,7aR,12bS)-4a,9-dihydroxy-3-methyl-2,3,4,4a,5,6,7,7a-octahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)amino)-5,13,17-trioxo-3,15-dioxa-6,12-diazaheptadecan-1-oyl)piperidine-4-carboxamide, MCC17 (3b)
Yield: 73%, white solid.
1H NMR (400 MHz, CDCl3) δ 7.73 (d, 6.4Hz, 2H), 7.40–7.50 (m, 1H), 7.27–7.29 (m, 1H), 7.17–7.20 (m, 3H), 6.95–7.00 (m, 2H), 6.71 (d, 8.0Hz, 1H), 6.52 (d, 8.0Hz, 1H), 6.43 (br s, 1H), 5.87 (br s, 1H), 4.50–4.60 (m, 2H), 4.43 (br d, 128 Hz, 1H), 4.23–4.28 (m, 2H), 3.98–4.08 (m, 5H), 3.63 (t, 7.2Hz, 2H), 3.23–3.31 (m, 5H), 3.12 (br d, 18.4Hz, 1H), 2.94–2.96 (m, 1H), 2.84 (d, 10.4Hz, 2H), 2.77 (d, 6.4Hz, 2H), 2.68–2.72 (m, 2H), 2.60 (d, 6.8Hz, 1H), 2.56 (d, 5.6Hz, 2H), 2.17–2.48 (m, 7H), 2.41 (s, 3H), 2.34 (s, 3H), 1.86 (t, 11.2Hz, 2H), 1.53–1.79 (m, 17H), 1.35–1.42 (m, 3H), 1.22–1.30 (m, 4H), 0.94–1.05 (m, 1H).
13C NMR (100 MHz) δ 173.47, 169.96, 169.36, 169.03, 168.16, 167.00, 145.61, 144.93, 140.64, 138.28, 136.49, 135.14, 131.92, 130.87, 130.66, 129.15, 128.28, 127.37, 126.21, 125.09, 119.13, 117.76, 89.31, 71.45, 71.24, 71.06, 69.56, 69.51, 64.50, 55.78, 53.69, 49.84, 47.43, 46.31, 45.64, 44.71, 43.45, 43.02, 42.82, 41.11, 39.84, 39.08, 38.87, 38.13, 37.51, 33.29, 31.90, 296.11, 28.57, 28.18, 28.06, 25.09, 23.49, 21.97, 21.08, 19.90, 19.67.
HRMS (ESI): Observed 1111.5678 (M+1); 1111.5635, Calculated for C59H80ClN8O11 [M+1]+.
N-(3-(4-(4-Carbamoylbenzyl)piperidin-1-yl)propyl)-N-(3-chloro-4-methylphenyl)-1-(19-(((4aS,7S,7aR,12bS)-4a,9-dihydroxy-3-methyl-2,3,4,4a,5,6,7,7a-octahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)amino)-5,15,19-trioxo-3,17-dioxa-6,14-diazanonadecan-1-oyl)piperidine-4-carboxamide, MCC19 (3c)
Yield: 48%, white solid
1H NMR (400 MHz, CDCl3) δ 7.73 (d, 7.2Hz, 2H), 7.17–7.19 (m, 4H), 6.95–6.98 (m, 2H), 6.71 (d, 8Hz, 1H), 6.51 (d, 8Hz, 1H), 5.92 (br s, 1H), 4.56 (br s, 1H), 4.43 (br d, 11.2Hz, 1H), 4.22–4.24 (m, 3H), 3.99–4.07 (m, 4H), 3.82 (br d, 12.8Hz, 1H), 3.55 (br s, 1H), 3.62 (t, 8Hz, 2H), 3.37–3.31 (m, 3H), 3.10 (br d, 18.4Hz, 1H), 2.94 (d, 3H), 2.71–2.87 (m, 4H), 2.54–2.60 (m, 3H), 2.25–2.41 (m, 3H), 2.42 (s, 3H), 2.34 (s, 3H), 1.17–1.90 (m, 27H).
13C NMR (100 MHz) δ 173.80, 169.42, 169.31, 169.19, 168.66, 167.01, 145.67, 145.00, 140.75, 138.23, 136.46, 135.16, 131.98, 130.92, 130.64, 128.22, 128.37, 127.37, 126.29, 125.17, 119.21, 117.90, 89.36, 71.56, 71.39, 71.24, 69.64, 68.99, 64.55, 55.82, 53.72, 50.93, 47.87, 46.35, 45.67, 44.78, 43.91, 43.49, 43.04, 42.85, 40.50, 39.16, 38.94, 38.62, 37.51, 35.98, 35.36, 33.24, 31.84, 28.94, 28.86, 28.30, 28.10, 26.38, 26.20, 25.09, 22.04, 21.11, 19.45.
MS (ESI): m/z Observed 1139.5 [M+H]+; 1139.5 Calculated for C61H84ClN8O11 [M+H]+.
N-(3-(4-(4-Carbamoylbenzyl)piperidin-1-yl)propyl)-N-(3-chloro-4-methylphenyl)-1-(21-(((4aS,7S,7aR,12bS)-4a,9-dihydroxy-3-methyl-2,3,4,4a,5,6,7,7a-octahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)amino)-5,17,21-trioxo-3,19-dioxa-6,16-diazahenicosan-1-oyl)piperidine-4-carboxamide, MCC21 (3d)
Yield: 49%, white foam
1H NMR (400 MHz, CD3OD) δ 7.77 (d, J = 8.4 Hz, 2H), 7.36–7.44 (m, 2H), 7.24 (d, J = 7.6 Hz, 2H), 7.15 (d, J = 6.8 Hz, 1H), 6.64 (d, J = 8.0 Hz, 1H), 6.55 (d, J = 8.0 Hz, 1H), 4.48–4.58 (m, 2H), 4.41 (d, J = 12.8 Hz, 1H), 4.32 (d, J = 14.4 Hz, 1H), 4.26 (d, J = 13.2 Hz, 1H), 3.95–4.12 (m, 6H), 3.60–3.72 (m, 3H), 3.12–3.20 (m, 6H), 2.73–2.92 (m, 4H), 2.53–2.63 (m, 3H), 2.40–2.50 (m, 3H), 2.40 (s, 3H), 2.38 (s, 3H), 2.22–2.31 (m, 3H), 1.89–1.97 (m, 2H), 1.42–1.75 (m, 16H), 1.20–1.40 (m, 12H), 0.99–1.12 (m, 2H).
MS (ESI): m/z 1167.6 [M+H]+.
HRMS (ESI): Observed 1167.6296 [M+H]+; 1167.6256, Calculated for C63H88ClN8O11 [M+H]+.
N-(3-(4-(4-Carbamoylbenzyl)piperidin-1-yl)propyl)-N-(3-chloro-4-methylphenyl)-1-(22-(((4aS,7S,7aR,12bS)-4a,9-dihydroxy-3-methyl-2,3,4,4a,5,6,7,7a-octahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)amino)-5,18,22-trioxo-3,20-dioxa-6,17-diazadocosan-1-oyl)piperidine-4-carboxamide, MCC22 (3e)
Yield: 65%, white foam
1H NMR (400 MHz, CD3OD) δ 7.24 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.4 Hz, 1H), 7.40 (s, 1H), 7.16 (dd, J = 2.0, 8.4 Hz, 1H), 7.16 (dd, J = 2.0, 8.4 Hz, 1H), 6.64 (d, J = 8.4 Hz, 1H), 6.54 (d, J = 8.4 Hz, 1H), 4.50–4.57 (m, 2H), 4.41 (d, J = 13.2 Hz, 1H), 4.33 (d, J = 14.4 Hz, 1H), 4.24 (d, J = 14.4 Hz, 1H), 3.97–4.12 (m, 6H), 3.15–3.72 (m, 3H), 2.75–2.92 (m, 4H), 2.55–2.65 (m, 3H), 2.40–2.50 (m, 3H), 2.45 (d, J = 5.6 Hz, 1H), 2.41 (s, 3H), 2.37 (s, 3H), 2.24–2.32 (m, 3H), 1.91 (t, J = 11.2 Hz, 2H), 1.40–1.75 (m, 17H), 1.20–1.39 (m, 14H), 1.00–1.12 (m, 1H).
MS (ESI): m/z 1181.6 [M+H]+.
HRMS (ESI): Observed 1181.6431; 1181.6412, Calculated for C64H90ClN8O11 [M+H] +.
N-(3-(4-(4-Carbamoylbenzyl)piperidin-1-yl)propyl)-N-(3-chloro-4-methylphenyl)-1-(24-(((4aS,7S,7aR,12bS)-4a,9-dihydroxy-3-methyl-2,3,4,4a,5,6,7,7a-octahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)amino)-5,20,24-trioxo-3,22-dioxa-6,19-diazatetracosan-1-oyl)piperidine-4-carboxamide, MCC24 (3f)
Yield: 87%, pale brown foam
1H NMR (400 MHz, CD3OD) δ 7.77 (d, J = 8.0 Hz, 2H), 7.38–7.44 (m, 2H), 7.24 (d, J = 8.0 Hz, 2H), 7.15 (dd, J = 2.0, 7.6 Hz, 1H), 6.64 (d, J = 8.0 Hz, 1H), 6.53 (d, J = 8.0 Hz, 1H), 4.50–4.60 (m, 2H), 4.41 (d, J = 12.8 Hz, 1H), 4.33 (d, J = 14.8 Hz, 1H), 4.25 (d, J = 14.8 Hz, 1H), 3.98–4.10 (m, 6H), 3.60–3.71 (m, 3H), 3.17–3.28 (m, 4H), 3.12 (t, J = 7.2 Hz, 2H), 2.75–2.90 (m, 4H), 2.56–2.62 (m, 3H), 2.42–2.52 (m, 3H), 2.41 (s, 3H), 2.37 (s, 3H), 2.25–2.34 (m, 3H), 1.91 (t, J = 12.0 Hz, 2H), 1.40–1.75 (m, 18H), 1.20–1.38 (m, 18H), 1.02–1.13 (m, 1H).
MS (ESI): m/z 1209.8 [M+H]+.
HRMS (ESI): Observed 1209.6737; 1209.6725, Calculated for C66H94ClN8O11 [M+H]+.
N-((4aS,7S,7aR,12bS)-4a,9-Dihydroxy-3-methyl-2,3,4,4a,5,6,7,7a-octahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)-2-((3,7,17-trioxo-5-oxa-2,8,16-triazaoctadecan-18-yl)oxy)acetamide (4)
This was synthesized as reported previously.24
N-(3-(4-(4-Carbamoylbenzyl)piperidin-1-yl)propyl)-N-(3-chloro-4-methylphenyl)-1-(3,7,17-trioxo-5,19-dioxa-2,8,16-triazahenicosan-21-oyl)piperidine-4-carboxamide (5)
To the mixture of acid 8b (0.128 g, 0.204 mmol, 1.1 equ) and HBTU (0.141 g, 0.37 mmol, 2.0 equ), a solution of HOBt (0.75 mL, 0.5M in DMF, 0.37 mmol, 2.0 equ) was added at r.t and stirred for 15 min. A clear colorless solution with no precipitate resulted. This mixture was added to a solution of amine 12 (0.055 g, 0.18 mmol, 1.0 equ) which was pre-neutralized with DIPEA (0.105 mL, 0.077 g, 0.59 mmol, 3.20 equ) in 2 mL DMF. The reaction was allowed to stir at r.t for 48 hrs. The solvent was removed in vacuum and further purification was performed over silica gel column chromatography using 97:2.5:0.5 to 95:4:1 to 92:7.5:0.5 to 89:10:1 DCM:MeOH:Ammonium Hydroxide. The final product 5 was isolated as off-white solid (0.06 g, 37% yield).
1H NMR (400 MHz, CDCl3) δ 7.74 (d, 8Hz, 2H), 7.51 (d, 8Hz, 2H), 7.20 (s, 1H), 7.18 (s, 2H), 6.45 (br s, 1H), 4.18–4.30 (m, 2H), 4.04 (s, 2H), 4.02 (s, 2H), 4.00–4.10 (m, 2H), 3.42 (t, 7Hz, 2H), 3.57 (d, 12Hz, 1H), 3.25 (m, 2H), 2.79–2.96 (m, 2H), 2.80 (br d, 16Hz, 3H, methyl on amide nitrogen), 2.57 (d, 6.4Hz, 2H), 2.30–2.50 (4H), 2.41 (s, 3H, methyl on aromatic ring), 1.85–2.00 (m, 2H), 1.40–1.80 (m, 14H), 1.24–1.38 (m, 10H).
13C NMR (100 MHz, CDCl3) δ 173.68, 169.35, 169.25, 169.21, 168.47, 166.88, 144.86, 140.65, 136.63, 135.26, 131.95, 130.99, 129.25, 128.36, 127.37, 126.28, 71.68, 71.09, 71.06, 69.62, 55.70, 53.67, 43.51, 42.78, 41.09, 38.95, 38.75, 37.40, 31.64, 29.22, 29.12, 28.64, 28.53, 28.11, 26.53, 26.43, 25.67, 24.99, 24.95, 19.74.
MS(ESI)-TOF observed 868.4 (M+1), 890.4615 (M+Na+); 867.4 calculated for C45H66ClN7O8.
N-(3-(4-(4-Carbamoylbenzyl)piperidin-1-yl)propyl)-N-(3-chloro-4-methylphenyl)piperidine-4-carboxamide (8a)
The precursor amine tert-butyl 4-((3-(4-(4-carboxybenzyl)piperidin-1-yl)propyl)(3-chloro-4-methylphenyl)carbamoyl)piperidine-1-carboxylate was prepared as yellow oil by elaboration of original procedure of 2.46 To a cold (5 °C) solution of this Boc protected amine (1.130g, 1.847 mmol, 1.0 equ) in 20 mL CH2Cl2 is added trifluroacetic acid (TFA) (2.14 mL, 3.16 g, 27.70 mmol, 15.0 equ) drop wise. The reaction was allowed to warm to room temperature and stirred 18 hrs. when TLC indicated completion of reaction. The light yellow solution is concentrated in vacuum followed by azeotropic distillation from toluene (3 × 50 mL) and trituration with pentane/CH2Cl2 provided TFA salt as amorphous solid. In order to obtain un-protected piperidine, the TFA salt was dissolved in 15 mL water and slowly basified to pH 9~10 with ammonium hydroxide (30%, ~ 15 mL used). The aqueous solution then extracted with CH2Cl2, dried (MgSO4), filtered, concentrated in vacuum to provide crude product. The aqueous layer is brought to pH =7 and rigorously extracted with EtOAc, dried (MgSO4), filtered, concentrated in vacuum to provide more crude product. The combined crude product was further purified on SiO2 column chromatography (pre-treated with 1 % TEA) using EtOAc: MeOH (10:0 to 9:1 to 8:2 to 7:3, all with 1% TEA) as eluent to give 0.76 g (81%) of un-protected piperidine 8a as light yellow oil.
1H NMR (400 MHz, CD3OD) δ 7.78 (d, 8Hz, 2H), 7.41 (m, 2H), 7.24 (d, 8Hz, 2H), 7.15 (d, 8Hz, 1H), 3.66 (t, 7Hz, 2H), 3.31 (t, 1.6Hz, 2H), 3.15 (d, 12Hz, 2H), 2.92 (d, 12Hz, 2H), 2.35–2.61 (m, 7H), 2.41 (s, 3H), 1.96 (t, 12Hz, 2H), 1.61–1.80 (m, 9H), 1.25–1.33 (m, 2H.
13C NMR (100 MHz, CD3OD) δ 180.29, 172.77, 172.72, 146.06, 142.06, 137.82, 136.14, 133.30, 132.58, 130.22, 129.66, 128.69, 127.97, 56.84, 54.62, 44.83, 43.61, 39.76, 38.63, 35.24, 32.50, 28.30, 25.64, 24.28, 22.58, 19.80, 14.75.
MS(ESI)-TOF Observed 511.2465 (M+1); 510.2762, Calculated for C29H39ClN4O2.
2-(2-(4-((3-(4-(4-Carbamoylbenzyl)piperidin-1-yl)propyl)(3-chloro-4-methylphenyl)carbamoyl)piperidin-1-yl)-2-oxoethoxy)acetic acid (8b)
To a mixture of commercially available glycolic anhydride (0.088 g, 0.737 mmol, 1.0 equ) and piperidine 8a (0.380 g, 0.737 mmol, 1.0 equ) at room temperature was added THF (7 mL). The reaction was allowed to stir for 18 hrs. The solvent was removed to provide 8b as a white solid (0.462 g, 100% yield).
MS(ESI)-TOF observed 625.4160 (M-1) (negative mode), 627.3378 (M+1), 649.3180 (M+Na) (positive mode); 626.2871, Calculated for C33H43ClN4O6.
N-(3-(4-(4-Carbamoylbenzyl)piperidin-1-yl)propyl)-N-(3-chloro-4-methylphenyl)-1-(2-(2-(methylamino)-2-oxoethoxy)acetyl)piperidine-4-carboxyamide (8c)
The target compound was obtained by reaction of 8b with commercially available methylamine as described for monovalent 5.
Yield: >90%
1H NMR (400 MHz, CD3OD) δ 7.78(d, 8Hz, 2H), 7.41(m, 2H), 7.23(d, 8Hz, 2H), 7.16(d, 8Hz, 1H), 4.22–4.42(m, 3H), 4.0(s, 2H), 3.66(m, 3H), 3.33–3.34(m, 2H), 2.86(d, 7.2Hz, 2H), 2.76(s, 4H), 2.57(d, 6.4Hz, 2H), 2.42–2.48(m, 2H), 2.40(s, 3H), 2.31(t, 8Hz, 2H), 1.90(t, 12Hz, 2H), 1.58–1.73(m, 9H), 1.22–1.30(m, 2H).
13C NMR (100 MHz, CD3OD) δ 175.97, 172.39, 172.12, 169.24, 146.15, 142.09, 137.85, 136.16, 133.30, 132.56, 130.24, 129.68, 128.67, 127.98, 71.44, 70.09, 56.94, 54.71, 44.65, 43.65, 42.09, 40.68, 38.74, 32.62, 29.74, 29.22, 25.80, 25.74, 19.79.
MS(ESI)-TOF Observed 640.3807 (M+1), 662.3631 (M+Na); 640.3266, Calculated for C34H47ClN5O5 (M+1).
Synthesis of amines (11a-11f)
Synthesis of 11a–11c has been reported previously.49 Using the similar procedures the amines 11d–11f were prepared from their Cbz protected precursors.
N-(9-Aminononyl)-2-(2-(((4aS,7S,7aR,12bS)-4a,9-dihydroxy-3-methyl-2,3,4,4a,5,6,7,7a-octahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)amino)-2-oxoethoxy)acetamide (11d)
Yield: 98% brown oil
1H NMR (400 MHz, CD3OD) δ 6.62 (d, J = 8.4 Hz, 1H), 6.52 (d, J = 8.4 Hz, 1H), 4.48–4.55 (m, 2H), 4.00–4.09 (m, 4H), 3.24 (t, J = 7.2 Hz, 2H), 3.15 (t, J = 18.4 Hz, 1H), 2.80 (d, J = 6.8 Hz, 1H), 2.65 (d, J = 7.6 Hz, 2H), 2.58 (dd, J = 6.8, 18.4 Hz, 1H), 2.40–2.50 (m, 1H), 2.36 (s, 3H), 2.22–2.32 (m, 2H), 1.65–1.76 (m, 1H), 1.41–1.59 (m, 7H), 1.23–1.40 (m, 10H), 1.00–1.12 (m, 1H).
MS (ESI): Observed m/z 559.4 [M+H]+; 559.3, Calculated for C30H46N4O6 [M+H]+.
N-(10-Aminodecyl)-2-(2-(((4aS,7S,7aR,12bS)-4a,9-dihydroxy-3-methyl-2,3,4,4a,5,6,7,7a-octahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)amino)-2-oxoethoxy)acetamide, (11e)
Yield: ~100% pale yellow oil
1H NMR (400 MHz, CD3OD) δ 6.63 (d, J = 8.4 Hz, 1H), 6.52 (d, J = 8.4 Hz, 1H), 4.50–4.55 (m, 2H), 4.07 (s, 2H), 4.04 (s, 2H), 3.24 (t, J = 7.2 Hz, 1H), 3.16 (d, J = 18.8 Hz, 1H), 2.80 (d, J = 6.4 Hz, 1H), 2.64 (d, J = 7.2 Hz, 1H), 2.58 (dd, J = 6.8, 18.8 Hz, 1H), 2.45 (d, J = 5.6 Hz, 1H), 2.36 (s, 3H), 2.22–2.34 (m, 2H), 1.64–1.78 (m, 1H), 1.42–1.60 (m, 7H), 1.22–1.38 (m, 12H), 0.98–1.12 (m, 1H).
MS (ESI): Observed m/z 573.5 [M+H]+; 573.4, Calculated for C31H49N4O6 [M+H]+.
N-(12-Aminododecyl)-2-(2-(((4aS,7S,7aR,12bS)-4a,9-dihydroxy-3-methyl-2,3,4,4a,5,6,7,7a-octahydro-1H-4,12-methanobenzofuro[3,2-e]isoquinolin-7-yl)amino)-2-oxoethoxy)acetamide, (11f)
Yield: 89% white foam
1H NMR (400 MHz, CDCl3) δ 6.99 (br d, J = 8.4 Hz, 1H), 6.68 (d, J = 6.8 Hz, 1H), 6.54 (d, J = 6.8 Hz, 1H), 4.61 (s, 1H), 4.58 (br s, 1H), 3.92–4.10 (m, 4H), 3.28–3.37 (m, 2H), 3.13 (d, J = 18.4 Hz, 1H), 2.68–2.78 (m, 3H), 2.57 (dd, J = 6.4, 18.8 Hz, 1H), 2.38–2.45 (m, 1H), 2.35 (s, 3H), 2.20–2.30 (m, 2H), 1.65–1.80 (m, 1H), 1.20–1.60 (m, 23H), 1.05–1.15 (m, 1H).
MS (ESI): Observed m/z 601.6 [M+H]+; 601.4, Calculated for C33H53N4O6 [M+H]+.
Biology
Intracellular calcium release
Briefly, HEK-293 cells were cultured at 37°C in Dulbelcco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% Penicillin/Streptomycin. When 100 mm2 were transiently transfected with CCR5 receptor cDNA using OptiMEM-1 medium (Invitrogen) and Lipofectamine 2000 (Invitrogen, Carlsbad, CA) reagent according to manufacturer’s protocol (1:2 wt/vol ratio for DNA:Lipofectamine ; 16 μg DNA : 32 μl Lipofectamine). The cells were seeded into 96 well plates (half-area; Corning) at 20,000 cells/well after 24 hours and assayed 48 hours after transfection using the FLIPR calcium kit (Molecular Devices) in a Flexstation-III apparatus (Molecular Devices). Cells were incubated with the calcium dye for 45 minutes at 37°C before the antagonists were added to each well at the concentration listed, and incubated at 37°C for 15 minutes. The control agonist, CCL5 (RANTES), was added at a concentration (100 nM) that elicited an 80% maximal response in untreated cells to each well and the calcium response measured with and without antagonists for 90 seconds. The change in RFU was calculated by subtracting the minimum relative fluorescent unit (RFU) from the maximum RFU response, replicated 6 times and averaged. Each experiment was repeated at least 3 times and the mean ± SEM was plotted for each treatment group. Plates were 80–90% confluent, cells.
Animals for Antinociceptive Studies for Naivie and LPS-Treated mice
Male ICR-CD1 mice (17 – 25g; Harlan, Madison, WI) are housed, 4 to a small box in a temperature- and humidity-controlled environment with unlimited access to food and water. They are maintained on a 12 h light/dark cycle. All experiments are approved by the Institutional Animal Care and Use Committee of the University of Minnesota (Minneapolis, MN).
Antinociceptive Testing
The tail flick assay is used to test for antinociception described by D’Amour and Smith50 and modified by Dewey et. al.51 Mice are held gently in one hand with the tail positioned in the apparatus (Tail Flick Analgesia Meter, Columbus Instruments, Columbus, Ohio) for radiant heat stimulus. The tail-flick response is elicited by applying radiant heat to the dorsal side of the tail. The test latency is measured before drug treatment (control) and again after the drug treatment (test) at the peak time of the compound, a 10 s maximum cut-off time is used to prevent damage to the tail. Antinociception is quantified according to the method of Harris and Pierson52 as the percent maximal possible effect (%MPE) which is calculated as:
A minimum of three groups of eight to ten mice are used for each dose response curve, and each mouse is used only once. ED50 values with 95% confidence intervals (C.I.) are computed with GraphPad Prism 4 by using nonlinear regression methods.
The compounds were tested for acute tolerance by comparing the ED80 – 90 dose measured on day 1 to the same dose administered and retested 24 hours later on the same mouse.
Intrathecal injections
Compounds were dissolved in 5% DMSO and diluted to less than 1% DMSO for injection. These compounds were administered in a volume of 5-μl either i.c.v (Haley and McCormick) or intrathecal (i.t.) as described for mice by Hylden and Wilcox53 in conscious mice. Controls when given either i.c.v. or i.t. with 1% or less DMSO do not show any antinociception. Time course studies, including the times 5, 10, 20, 30 and 60 minutes were used to determine the peak antinociception.
Complete Freund’s adjuvant (CFA)-Mechanical Hyperalgesia54, 55
Before injection of CFA baseline withdrawal thresholds (g) were obtained from a mechanical stimulation using an electronic von Frey anesthesiometer (IITC Life Sciences, Woodland Hills, USA) by applying an accurate force on both left and right hind paw. The left hindpaws were then injected (intraplantar) with a 50% solution of CFA (10 μg, Sigma-Aldrich) in water while the mice are under isoflurane anesthesia. Twenty-four hours later the withdrawal threshold (g) was again measured prior to and after the test compound i.t. at 10, 20, 30 and 60 minutes to determine the peak time and subsequent ED50. % MPE was calculated ((Time-point withdrawal threshold with drug – CFA withdrawal threshold)/(Day 0 withdrawal threshold – CFA withdrawal threshold)) * 100.
Effect of minocycline in mice pretreated by LPS on antinociception of MCC22
Three separate groups of mice (n=8) were pretreated with 1 mg/kg of LPS (i.p.). At 24 hours the baseline antinociception was measured using the tail-flick assay to calculate %MPE. Mice were then injected i.p. with 45 mg/kg of minocycline HCl (Sigma Aldrich) dissolved in distilled water and gently warmed until clear) at three different time points, one, two and four hours, to confirm that minocycline did not cause antinociception by itself. After each group, mice were injected 1 pmol/mouse MCC22 (i.t.) and 5 minutes later the final antinociception was measured. The dose used for MCC22 was hundred times the ED50 dose in LPS pretreated mice. To establish a dose- response curve, a separate group of mice were pretreated with LPS for 24 hours prior to testing. Minocycline (45 mg/kg, i.p) was administered 4 hours before and MCC22 was tested, that resulted in 49.35 ± 14.38 the %MPE. When saline was used instead of MCC22 the %MPE was 6.98 ± 3.55%.
Molecular Modeling
All modeling was performed using the Schrodinger modeling package.56 The approach was adopted from our earlier homology modeling study of GPCR and the design of conjugated bivalent substrate study described in details elsewhere.57, 58 The modeling study of MOR-CCR5 heteromer with its bound MCC22 inhibitor was based on the X-ray crystallographic structures of the beta-FNA-bound MOR (PDB code: 4DKL)30 and Maraviroc (MRV)-bound CCR5 (PDB code: 4MBS)31 fusion protein complexes. Structure preparation involved the removal of the non-native T-4 lysozyme subunit from MOR and the rubredoxin subunit from CCR5. The resultant gaps in the intracellular loop 3 (ICL3) of MOR 264–269 and CCR5 224–227 were homology modeled to generate the contiguous MOR and CCR5 protein model. All missing sidechains and hydrogen atoms were added with standard protein preparation protocols at physiological pH, followed by energy minimization using OPLS-AA 2005 force field59 with Generalized Born implicit solvent model60 to optimize all hydrogen-bonding networks.
Because of the technical challenges of docking an extendable bivalent ligand into a heteromer consisting of two distinctive ligand binding sites, our modeling approach was carried out in two stages. First the conjugated heterobivalent MCC22 ligand was divided into its two monovalent pharmacophore units and a linker unit. Each of the pharmacophore unit was subsequently docked into its corresponding CCR5 and MOR binding sites using Glide at standard SP protocols. The distance between the two bridging atoms involved in the conjugation of the two pharmacophore units was then determined to provide a reasonable estimate in the optimal length for the linker unit. For MCC22, the two docked pharmacophore units were bridged by a 22 atom spacer linker unit. The final complex model with all the amino residues within 5 Å of bivalent ligand was then refined with restraint energy minimization OPLS2005 force field under implicit Generalized Born solvent model to remove all steric clashes.
To determine the potential oligomeric arrangement of the MOR-CCR5 heteromer for MCC22 bivalent ligand binding, super positioning of the CCR5 docked complex onto the previously observed oligomeric arrangements of MOR with either the TM5-TM6 or the TM1-TM2-H8 interface was carried out (Supplementary Figure 2). This approach provided the distal proximity between the two bridging atoms involved in the conjugation of the two docked pharmacophore units and the determination of feasibility for bivalent binding between the two oligomeric arrangements.
Molecular Dynamics Simulation
To gain better insights into the allosteric effect61, 62 of MCC22 binding on the structure of MOR-CCR5 heteromer and the specific interactions involved, six 100 ns molecular dynamics (MD) simulation were carried out for the bound and unbound protomers and MOR-CCR5 heteromer. The study is based on our earlier simulation study of the monomeric MOR receptor in an explicit lipid membrane aqueous system.58 Each of the modeled complex was embedded with a 15Å buffer region from the edge of the complex within a POPC lipid membrane, and explicit TIP3P63 water layer at 0.1M NaCl salt concentration as counter ions inside a rectangular box. The long range electrostatic interactions were evaluated by the Particle-Mesh Ewald method under periodic boundary condition with a dielectric constant of 1. Each MD simulation was carried out using DESMOND64 with default initialization protocol, followed by 100ns unrestraint production simulation run under constant area isothermal isobaric (NPAT) condition at 300K and 1 atm with OPLS-AA 2005 force field. For holo complexes, a force constant of 5kcal/molÅ2 restraint was applied to the heavy atoms of the bound ligand during the initialization to ensure proper equilibrium of the protein-ligand interactions prior to the start of the simulation. The stability of the protein was assessed by evaluating the CαRMSD with respect to the minimized starting structure.
Supplementary Material
Acknowledgments
This research was supported by NIH research grant DA030316. The authors thank Prof. D.A. Simone for his suggestion to use minocycline. We thank the University of Minnesota Supercomputing Institute for providing all the necessary computational resources.
ABBREVIATIONS
- atm
atmospheres
- CFA
complete Freund’s adjuvant
- ED50
dose effective in 50% of test subjects
- equ
equivalent
- HEK-293
human embryonic kidney-293
- icv
intracerebroventricular
- i.p
intraperitoneal
- it
intrathecal
- nM
nanomole
- LPS
lipopolysaccharide
- MD
molecular dynamics
- pmol
picomol
- TL4
toll-like receptor 4
- vol
volume
- wt
weight
Footnotes
CCl5 stimulation of CCR5 receptors expressed in HEK-293 cells; inhibition of Ca2+ by 2 and derivative 2 (8c); Model of TM1-TM2-H8 and TM5-TM6 interfaced MOR-CCR5 heteromer.
Notes “The authors declare no competing financial interest.”
References
- 1.Reuben DB, Alvanzo AAH, Ashikaga T, Bogat GA, Callahan CM, Ruffing V, Steffens DC. National Institutes of Health Pathways to Prevention Workshop: the role of opioids in the treatment of chronic pain. Ann Intern Med. 2015;162:295–300. doi: 10.7326/M14-2775. [DOI] [PubMed] [Google Scholar]
- 2.Chou R, Turner JA, Devine EB, Hansen RN, Sullivan SD, Blazina I, Dana T, Bougatsos C, Deyo RA. The effectiveness and risks of long-term opioid therapy for chronic pain: a systematic review for a National Institutes of Health Pathways to Prevention Workshop. Ann Intern Med. 2015;162:276–286. doi: 10.7326/M14-2559. [DOI] [PubMed] [Google Scholar]
- 3.Parsadaniantz SM, Rivat C, Rostene W, Goazigo AR-L. Opioid and chemokine receptor crosstalk: a promising target for pain therapy. Nat Rev Neurosci. 2015;16:69–78. doi: 10.1038/nrn3858. [DOI] [PubMed] [Google Scholar]
- 4.Hutchinson MR, Shavit Y, Grace PM, Rice KC, Maier SF, Watkins LR. Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol Rev. 2011;63:772–810. doi: 10.1124/pr.110.004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwarz JM, Hutchinson MR, Bilbo SD. Early-life experience decreases drug-induced reinstatement of morphine CPP in adulthood via microglial-specific epigenetic programming of anti-inflammatory IL-10 expression. J Neurosci. 2011;31:17835–17847. doi: 10.1523/JNEUROSCI.3297-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee CW-S, Ho I-K. Pharmacological profiles of oligomerized μ-opioid receptors. Cells. 2013;2:689–714. doi: 10.3390/cells2040689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akgün E, Javed MI, Lunzer MM, Smeester BA, Beitz AJ, Portoghese PS. Ligands that interact with putative MOR-mGluR5 heteromer in mice with inflammatory pain produce potent antinociception. Proc Natl Acad Sci U S A. 2013;110:11595–11599. doi: 10.1073/pnas.1305461110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smeester BA, Lunzer MM, Akgun E, Beitz AJ, Portoghese PS. Targeting putative mu opioid/metabotropic glutamate receptor-5 heteromers produces potent antinociception in a chronic murine bone cancer model. Eur J Pharmacol. 2014;743:48–52. doi: 10.1016/j.ejphar.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee YK, Choi D-Y, Jung Y-Y, Yun YW, Lee BJ, Han SB, Hong JT. Decreased pain responses of C-C chemokine receptor 5 knockout mice to chemical or inflammatory stimuli. Neuropharmacology. 2013;67:57–65. doi: 10.1016/j.neuropharm.2012.10.030. [DOI] [PubMed] [Google Scholar]
- 11.Rogers TJ, Peterson PK. Opioid G protein-coupled receptors: signals at the crossroads of inflammation. Trends Immunol. 2003;24:116–121. doi: 10.1016/s1471-4906(03)00003-6. [DOI] [PubMed] [Google Scholar]
- 12.Mahajan SD, Schwartz SA, Aalinkeel R, Chawda RP, Sykes DE, Nair MPN. Morphine modulates chemokine gene regulation in normal human astrocytes. Clin Immunol (San Diego, CA, U S) 2005;115:323–332. doi: 10.1016/j.clim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Happel C, Steele AD, Finley MJ, Kutzler MA, Rogers TJ. DAMGO-induced expression of chemokines and chemokine receptors: the role of TGF-β1. J Leukocyte Biol. 2008;83:956–963. doi: 10.1189/jlb.1007685. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki S, Chuang LF, Yau P, Doi RH, Chuang RY. Interactions of opioid and chemokine receptors: Oligomerization of mu, kappa, and delta with CCR5 on immune cells. Exp Cell Res. 2002;280:192–200. doi: 10.1006/excr.2002.5638. [DOI] [PubMed] [Google Scholar]
- 15.Chen C, Li J, Bot G, Szabo I, Rogers TJ, Liu-Chen L-Y. Heterodimerization and cross-desensitization between the μ-opioid receptor and the chemokine CCR5 receptor. Eur J Pharmacol. 2004;483:175–186. doi: 10.1016/j.ejphar.2003.10.033. [DOI] [PubMed] [Google Scholar]
- 16.Weiss U. Derivatives of morphine. I. 14-Hydroxydihydromorphinone. J Am Chem Soc. 1955;77:5891–5892. [Google Scholar]
- 17.Takashima K, Miyake H, Kanzaki N, Tagawa Y, Wang X, Sugihara Y, Iizawa Y, Baba M. Highly potent inhibition of human immunodeficiency virus type 1 replication by TAK-220, an orally bioavailable small-molecule CCR5 antagonist. Antimicrob Agents Chemother. 2005;49:3474–3482. doi: 10.1128/AAC.49.8.3474-3482.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le Naour M, Akgün E, Yekkirala A, Lunzer MM, Powers MD, Kalyuzhny AE, Portoghese PS. Bivalent ligands that target μ opioid (MOP) and cannabinoid1 (CB1) receptors are potent analgesics devoid of tolerance. J Med Chem. 2013;56:5505–5513. doi: 10.1021/jm4005219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng Y, Akgün E, Harikumar KG, Hopson J, Powers MD, Lunzer MM, Miller LJ, Portoghese PS. Induced association of μ opioid (MOP) and type 2 cholecystokinin (CCK2) receptors by novel bivalent ligands. J Med Chem. 2009;52:247–258. doi: 10.1021/jm800174p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie Z, Bhushan RG, Daniels DJ, Portoghese PS. Interaction of bivalent ligand KDN21 with heterodimeric delta-kappa opioid receptors in human embryonic kidney 293 cells. Mol Pharmacol. 2005;68:1079–1086. doi: 10.1124/mol.105.012070. [DOI] [PubMed] [Google Scholar]
- 21.Bhushan RG, Sharma SK, Xie Z, Daniels DJ, Portoghese PS. A bivalent ligand (KDN-21) reveals spinal delta and kappa opioid receptors are organized as heterodimers that give rise to delta(1) and kappa(2) phenotypes. Selective targeting of delta-kappa heterodimers. J Med Chem. 2004;47:2969–2972. doi: 10.1021/jm0342358. [DOI] [PubMed] [Google Scholar]
- 22.Daniels DJ, Kulkarni A, Xie Z, Bhushan RG, Portoghese PS. A bivalent ligand (KDAN-18) containing delta-antagonist and kappa-agonist pharmacophores bridges delta2 and kappa1 opioid receptor phenotypes. J Med Chem. 2005;48:1713–1716. doi: 10.1021/jm034234f. [DOI] [PubMed] [Google Scholar]
- 23.Zhang S, Yekkirala A, Tang Y, Portoghese PS. A bivalent ligand (KMN-21) antagonist for mu/kappa heterodimeric opioid receptors. Bioorg Med Chem Lett. 2009;19:6978–6980. doi: 10.1016/j.bmcl.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Natl Acad Sci U S A. 2005;102:19208–19213. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calil IL, Zarpelon AC, Guerrero ATG, Alves-Filho JC, Ferreira SH, Cunha FQ, Cunha TM, Verri WA. Lipopolysaccharide induces inflammatory hyperalgesia triggering a TLR4/MyD88-dependent cytokine cascade in the mice paw. PLoS One. 2014;9:e90013/1–e90013/8. doi: 10.1371/journal.pone.0090013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takemori AE, Larson DL, Portoghese PS. The irreversible narcotic antagonistic and reversible agonistic properties of the fumaramate methyl ester derivative of naltrexone. Eur J Pharmacol. 1981;70:445–451. doi: 10.1016/0014-2999(81)90355-1. [DOI] [PubMed] [Google Scholar]
- 27.Yoon SY, Patel D, Dougherty PM. Minocycline blocks lipopolysaccharide induced hyperalgesia by suppression of microglia but not astrocytes. Neuroscience (Amsterdam, Neth) 2012;221:214–224. doi: 10.1016/j.neuroscience.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watkins LR, Hutchinson MR, Johnston IN, Maier SF. Glia: Novel counter-regulators of opioid analgesia. Trends Neurosci. 2005;28:661–669. doi: 10.1016/j.tins.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 29.Vallejo R, Tilley DM, Vogel L, Benyamin R. The role of glia and the immune system in the development and maintenance of neuropathic pain. Pain Pract. 2010;10:167–184. doi: 10.1111/j.1533-2500.2010.00367.x. [DOI] [PubMed] [Google Scholar]
- 30.Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the μ-opioid receptor bound to a morphinan antagonist. Nature (London, U K) 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, Li T, Ma L, Fenalti G, Li J, Zhang W, Xie X, Yang H, Jiang H, Cherezov V, Liu H, Stevens RC, Zhao Q, Wu B. Structure of the CCR5 chemokine receptor-HIV entry inhibitor Maraviroc complex. Science (Washington DC, U S) 2013;341:1387–1390. doi: 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kondru R, Zhang J, Ji C, Mirzadegan T, Rotstein D, Sankuratri S, Dioszegi M. Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol Pharmacol. 2008;73:789–800. doi: 10.1124/mol.107.042101. [DOI] [PubMed] [Google Scholar]
- 33.Nishikawa M, Takashima K, Nishi T, Furuta RA, Kanzaki N, Yamamoto Y, Fujisawa J-i. Analysis of binding sites for the new small-molecule CCR5 antagonist TAK-220 on human CCR5. Antimicrob Agents Chemother. 2005;49:4708–4715. doi: 10.1128/AAC.49.11.4708-4715.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abbadie C. Chemokines, chemokine receptors and pain. Trends Immunol. 2005;26:529–534. doi: 10.1016/j.it.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 35.Takaki J, Fujimori K, Miura M, Suzuki T, Sekino Y, Sato K. L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: the ‘collusion’ hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. J Neuroinflamm. 2012;9:275–292. doi: 10.1186/1742-2094-9-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mousa SA, Machelska H, Schafer M, Stein C. Immunohistochemical localization of endomorphin-1 and endomorphin-2 in immune cells and spinal cord in a model of inflammatory pain. J Neuroimmunol. 2002;126:5–15. doi: 10.1016/s0165-5728(02)00049-8. [DOI] [PubMed] [Google Scholar]
- 37.Morinville A, Cahill CM, Kieffer B, Collier B, Beaudet A. Mu-opioid receptor knockout prevents changes in delta-opioid receptor trafficking induced by chronic inflammatory pain. Pain. 2004;109:266–273. doi: 10.1016/j.pain.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 38.Ballet S, Conrath M, Fischer J, Kaneko T, Hamon M, Cesselin F. Expression and G-protein coupling of mu-opioid receptors in the spinal cord and dorsal root ganglia of polyarthritic rats. Neuropeptides. 2003;37:211–219. doi: 10.1016/s0143-4179(03)00045-3. [DOI] [PubMed] [Google Scholar]
- 39.Zaringhalam J, Manaheji H, Mghsoodi N, Farokhi B, Mirzaiee V. Spinal μ-opioid receptor expression and hyperalgesia with dexamethasone in chronic adjuvant-induced arthritis in rats. Clin Exp Pharmacol Physiol. 2008;35:1309–1315. doi: 10.1111/j.1440-1681.2008.05009.x. [DOI] [PubMed] [Google Scholar]
- 40.Puehler W, Zoellner C, Brack A, Shaqura MA, Krause H, Schaefer M, Stein C. Rapid upregulation of μ opioid receptor mRNA in dorsal root ganglia in response to peripheral inflammation depends on neuronal conduction. Neuroscience (Oxford U K) 2004;129:473–479. doi: 10.1016/j.neuroscience.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 41.Shaqura MA, Zoellner C, Mousa SA, Stein C, Schaefer M. Characterization of μ opioid receptor binding and G protein coupling in rat hypothalamus, spinal cord, and primary afferent neurons during inflammatory pain. J Pharmacol Exp Ther. 2004;308:712–718. doi: 10.1124/jpet.103.057257. [DOI] [PubMed] [Google Scholar]
- 42.Singh M, Singh P, Vaira D, Amand M, Rahmouni S, Moutschen M. Minocycline attenuates HIV-1 infection and suppresses chronic immune activation in humanized NOD/LtsZ-scidIL-2Rγnull mice. Immunology. 2014;142:562–572. doi: 10.1111/imm.12246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sham YY, Lunzer MM, Powers MD, Javed MI, Cataldo G, Simone DA, Akgün E, Portoghese PS. Modeling and simulation of MCC22, a bivalent ligand that potently inhibits inflammatory and neuropathic pain in mice. Computer Aided Drug Design. Gorden Research Conferance; Mount Snow, Vermont. July 19–24, 2015. [Google Scholar]
- 44.Akgün E, Javed MI, Lunzer MM, Powers MD, Sham YY, Portoghese PS. MCC22 targets putative spinal MOR-CCR5 heteromers in a mouse model of inflammatory pain (MEDI 534). ACS 250th National Meeting; Boston, Massachusetts. August 16–19, 2015. [Google Scholar]
- 45.Portoghese PS, Lunzer MM, Powers MD, Javed MI, Sham YK, Cataldo G, Simone DA, Akgun E. A bivalent ligand (MCC22) potently inhibits inflammatory and neuropathic pain via putative MOR-CCR5 heteromers in mouse spinal cord. 46th meeting of the International Narcotics Research Conference in conjunction with the 77th annual College on Problems of Drug Dependence; Phoenix, Arizona. June 15–19, 2015. [Google Scholar]
- 46.Imamura S, Ichikawa T, Nishikawa Y, Kanzaki N, Takashima K, Niwa S, Iizawa Y, Baba M, Sugihara Y. Discovery of a piperidine-4-carboxamide CCR5 antagonist (TAK-220) with highly potent anti-HIV-1 activity. J Med Chem. 2006;49:2784–2793. doi: 10.1021/jm051034q. [DOI] [PubMed] [Google Scholar]
- 47.Akgün E, Zheng Y, Harikumar KG, Hopson J, Miller LJ, Portoghese PS. Induction of heterodimerization of mu opioid peptide (MOP) and type-2 cholecystokinin (CCK2) receptor by novel bivalent ligands. Drugs Fut. 2008, 33(Suppl A): XXth Int Symp Med Chem; Vienna. Aug31–Sept4, 2008. [Google Scholar]
- 48.Sayre LM, Portoghese PS. Stereospecific synthesis of the 6α- and 6β-amino derivatives of naltrexone and oxymorphone. J Org Chem. 1980;45:3366–3368. [Google Scholar]
- 49.Daniels DJ. Ph D Thesis. University of Minnesota; Minneapolis, MN: 2006. Bivalent ligands as probes for the investigation of opioid receptor dimerization. [Google Scholar]
- 50.D’Amour FE, Smith DL. A method for determining loss of pain senzation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- 51.Dewey WL, Harris LS, Howes JF, Nuite JA. Effect of various neurohumoral modulators on the activity of morphine and the narcotic antagonists in the tail-flick and phenylquinone tests. J Pharmacol Exp Ther. 1970;175:435–442. [PubMed] [Google Scholar]
- 52.Harris LS, Pierson AK. Narcotic antagonists in the benzomorphan series. J Pharmacol Exp Ther. 1964;143:141–148. [PubMed] [Google Scholar]
- 53.Hylden JL, Wilcox GL. Intrathecal morphine in mice: A new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- 54.Wade CL, Krumenacher P, Kitto KF, Peterson CD, Wilcox GL, Fairbanks CA. Effect of chronic pain on fentanyl self-administration in mice. PLoS One. 2013;8:e79239/1–e79239/11. doi: 10.1371/journal.pone.0079239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sorge RE, LaCroix-Fralish ML, Tuttle AH, Sotocinal SG, Austin J-S, Ritchie J, Chanda ML, Graham AC, Topham L, Beggs S, Salter MW, Mogil JS. Spinal cord Toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. J Neurosci. 2011;31:15450–15454. doi: 10.1523/JNEUROSCI.3859-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schrödinger LLC, N. Y., NY. Schrodinger Modeling Suite Package: Maestro, Biolumintate, Glide, Prime, Macromodel, Liaison, Strike, Jaguar. 2013 [Google Scholar]
- 57.Wilson DJ, Shi C, Duckworth BP, Muretta JM, Manjunatha U, Sham YY, Thomas DD, Aldrich CC. A continuous fluorescence displacement assay for BioA: An enzyme involved in biotin biosynthesis. Anal Biochem. 2011;416:27–38. doi: 10.1016/j.ab.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Sham YY, Rajamani R, Gao J, Portoghese PS. Homology modeling and molecular dynamics simulations of the mu opioid receptor in a membrane-aqueous system. ChemBioChem. 2005;6:853–859. doi: 10.1002/cbic.200400207. [DOI] [PubMed] [Google Scholar]
- 59.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 60.Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J Am Chem Soc. 1990;112:6127–6129. [Google Scholar]
- 61.Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, Baell JB, Sexton PM, Christopoulos A, Shaw DE. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature. 2013;503:295–299. doi: 10.1038/nature12595. [DOI] [PubMed] [Google Scholar]
- 62.Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, Thian FS, Kobilka TS, Shaw DE, Mueller L, Prosser RS, Kobilka BK. The dynamic process of β2-adrenergic receptor activation. Cell. 2013;152:532–542. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 64.D. E. Shaw Research N. Y., NY. Desmond Molecular Dynamics System v 3.0. 2011 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.