

Abstract
Cell transformation and tumor progression involves a common set of acquired capabilities, including increased proliferation, failure of cell death, self-sufficiency in growth, angiogenesis, and tumor cell invasion and metastasis (1). The stromal environment consists of many cell types, including fibroblasts, macrophages, and endothelial cells, in addition to various extracellular matrix (ECM) proteins that function to support normal tissue maintenance, but have also been implicated in tumor progression (2). Both the chemical and mechanical properties of the ECM have been shown to influence normal and malignant cell behavior. For instance, mesenchymal stem cells differentiate into specific lineages that are dependent on matrix stiffness (3), while tumor cells undergo changes in cell behavior and gene expression in response to matrix stiffness (4). ECM remodeling is implicated in tumor progression and includes changes in both the chemical and mechanical properties of the ECM (5) that can be a result of 1.) increased deposition of stromal ECM, 2.) enhanced contraction of ECM fibrils, and 3.) altered collagen alignment and ECM stiffness. In addition, remodeling of the ECM may alter whether tumor cells employ proteolytic degradation mechanisms during invasion and metastasis. Tumor cells respond to such changes in ECM remodeling through altered intracellular signaling and cell cycle control that lead to enhanced proliferation, loss of normal tissue architecture, and local tumor cell migration and invasion into the surrounding stromal tissue (6). This review will focus on the bi-directional interplay between the mechanical properties of the ECM and changes in integrin-mediated signal transduction events in an effort to elucidate cell behaviors during tumor progression.
Keywords: Mechanotransduction, mechanosensor, integrins, morphogenesis, invasion, tumor progression, matrix remodeling, RhoA, FAK, extracellular matrix, bi-directional signaling
Integrin Signaling in Tumor Progression
Integrins are transmembrane cell surface receptors that provide external links to the ECM and are the major receptors responsible for cell-matrix adhesions (7, 8). Integrins consist of one α subunit and one β subunit that assemble into 24 different heterodimers, each with different binding properties for a variety of ECM proteins (7, 9, 10). The large ectodomain of integrins is responsible for ligand binding, while the cytoplasmic tails of integrin heterodimers form multi-molecular complexes containing adaptor proteins involved in signaling pathways and scaffolding proteins that couple the ECM to the actin cytoskeleton (7, 8, 11, 12).
Extracellular ligand binding to the ectodomain induces integrin clustering and conformational rearrangements of the cytoplasmic tail, resulting in changes in binding interactions with the actin cytoskeleton and signal transduction pathways (13, 14). Corresponding conformational changes in the cytoplasmic tail are transmitted to the ligand-binding regions of the ectodomain, via the transmembrane domain, resulting in enhanced binding affinity and additional integrin clustering (7, 8, 14). For adherent cells, integrin clustering can lead to local remodeling of the actin cytoskeleton to form focal adhesions. Consequently, cell-generated contractile forces transmitted through integrins can remodel the surrounding matrix and increase matrix stiffness (15–18). This bi-directional signaling through outside-in and inside-out mechanisms has important implications in tumor progression. For instance, matrix deposition can occur through a feedback loop involving both outside-in and inside-out signaling. Specifically, intracellular actin-myosin generated force applied to α5β1 integrin results in an increase in fibronectin matrix formation (19, 20). Thus, as the tumor microenvironment undergoes changes in both the chemical and mechanical composition of the ECM integrins play a key role in bidirectional signaling.
Integrins can be considered multisensory receptors that sense two general elements of the ECM; chemical composition (which ligands are present) and mechanical properties (what is the physical context of the ligands that are present). The variety of chemical cues that comprise the ECM of the basement membrane and stroma has been extensively reviewed elsewhere (21, 22). Changes in ECM composition associated with mammary carcinoma alter the expression profile of integrins and downstream signaling to enhance cell invasion and tumor metastasis (23). The expression profile of many integrins is altered in the transition of cells from normal to neoplastic and this is believed to be a necessary step for tumor cell invasion (23, 24). Specifically, adhesion to periostin, osteopontin and tenascin-C alter cell signaling pathways to enhance cancer cell invasion (25–27). Despite changes in integrin levels, integrins remain necessary, as tumorigenic mammary cells treated with a function-blocking antibody for β1 integrin demonstrated a decrease in cell proliferation and an increase in apoptosis (28). Ultimately, modifications in tissue composition alter the local microenvironment, resulting in dramatic changes in integrin engagement and downstream signaling associated with the progression of many types of cancers (29–32).
Increased Deposition of Stromal ECM during Tumor Progression
One of the single biggest risk factors for breast carcinoma is an increase in mammographic tissue density. While the underlying cause of increased mammographic tissue density remains largely unknown, there are several factors associated with changes in breast density, including diet, hormone exposure, genetics, age, body mass index, and parity (33–35). In the normal adult mammary gland, several types of collagen (I, III, and V) and collagen crosslinking proteins are found abundantly and are regulated by reproductive state (36). Dense breast tissue is comprised primarily of type I collagen (37–39) but also involves upregulation of other ECM proteins, such as, type III and V collagens, fibronectin (FN), tenascin-C, and periostin. Increased deposition of ECM results in tumor tissue being stiffer than normal tissue, as determined by Atomic Force Microscopy (AFM) measurements in vivo comparing normal tissue to tumor centers (40). Consequently, the increased deposition of ECM proteins alters both the chemical composition and the mechanical properties of the ECM. An increase in the stiffness of the tumor microenvironment is functionally significant, as it promotes tumor progression through a variety of signaling pathways (41, 42).
During tumor progression the deposition of these additional matrix proteins, a process termed desmoplasia, is associated with poor patient prognosis (43). Thus, these extracellular proteins can be used as predictive markers for carcinoma. For example, Jahkola et al. determined that tenascin-C found at invasive mammary tumor borders is a predictor of both local and distant recurrence (44–46). Additionally, periostin expression has also been associated with tumor size and with poor outcome of ER-positive tumors (47, 48). Consequently, current research is aimed at better understanding the cellular mechanisms underlying the association of altered ECM composition and matrix stiffness with patient prognosis.
Matrix Stiffness and Integrin Signaling
Mechanical parameters of the ECM, such as ligand density, porosity, cross-linking, and ECM orientation, all influence matrix stiffness and the counter-balancing tensional forces that the matrix exerts on cells. However, the mechanisms by which matrix tension regulates integrin-mediated changes in signaling and cytoskeletal reorganization are not known. Normal tissue homeostasis requires reciprocal interactions between the counter-balancing forces produced by the matrix and cell-generated contractile forces. Mechanotransduction is the process by which mechanical forces are converted into biochemical signals and thus the mechanism by which cells adjust to changes in the microenvironment during tumor progression.
Studies have shown that matrix stiffness strengthens integrin-cytoskeletal linkages and integrin clustering (49–53), as well as increases integrin expression, activity, and focal adhesion formation (42, 54, 55). As depicted in Figure 1, mechanosensitive proteins must undergo conformational changes that alter signal transduction events, intracellular localization, or cytoskeletal reorganization in response to changes in the mechanical properties of the matrix. Cells sense external forces via integrin adhesions and respond through actomyosin contractile forces that are equal to that of the surrounding matrix to maintain normal tissue architecture (56–59). However, an imbalance in the reciprocal force interactions between the matrix and the cells can result in pathological conditions, such as fibrosis, atherosclerosis, and cancer (56, 57, 60–63).
Figure 1.
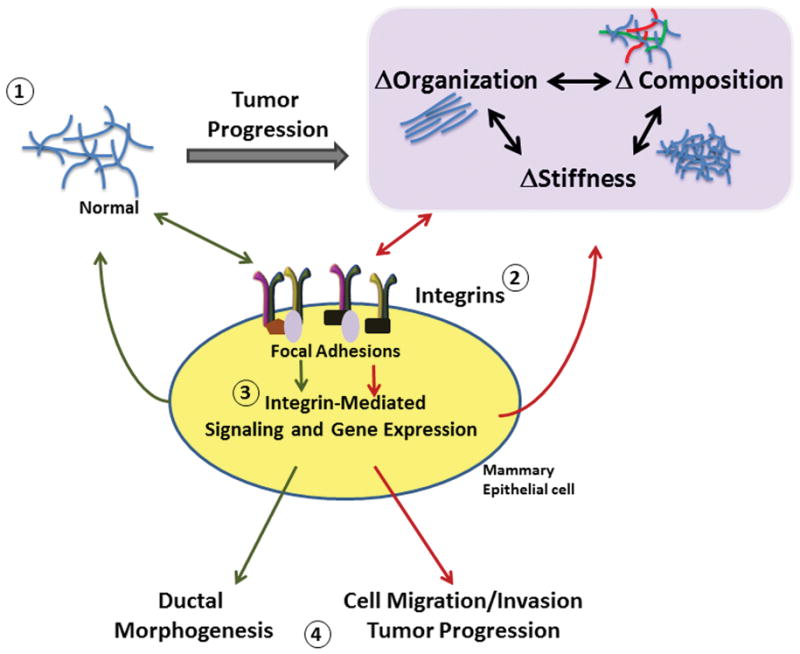
Bi-directional signals from integrins and the ECM during mammary tumor progression. 1. The extracelluar matrix changes in both physical and chemical composition during breast cancer progression. 2. The integrins respond to and signal back to the matrix through altered integrin engagement, increased adhesion, and focal adhesion signaling. 3. Downstream integrin signaling changes in response to cues from the ECM. Integrin signaling pathways feed back to the matrix through enhancing deposition of matrix components and by altering cell contractility to induce matrix organization. 4. Ultimately, mammary epithelial cells respond to the ECM by tuning their phenotype toward ductal morphogenesis under normal matrix conditions or toward enhanced proliferation, migration and invasion in response to abnormal matrix conditions.
Changes in the mechanical properties of the matrix, such as increased stiffness or ECM protein density, have been shown to enhance malignant and non-malignant cell growth and proliferation through integrin-mediated mechanisms (42, 58, 64–67). Tissue morphogenesis is also regulated by the biophysical properties of the ECM in vitro and in vivo through integrin-mediated mechanisms. For instance, human breast cancer cells cultured in compliant matrices exhibit cell phenotypes similar to normal differentiated structures (42, 55, 58, 65, 68). However, when the cells are cultured in a stiffer matrix, their tissue architecture is altered. Provenzano et al. (2008) demonstrate that high mammary collagen density promotes tumor formation in vivo, as quantified by an increased area of hyperplasia (69). However, manipulating β1 integrin function using function-blocking antibodies causes tumorigenic mammary cells to undergo phenotypic reversion, such that they form structures similar to normal mammary acini (70), while genetic ablation of β1 integrin inhibits mammary tumorigenesis in vivo (71).
Matrix stiffness has also been shown to regulate integrin signaling during cell motility. Cell migration and integrin adhesions are influenced by matrix stiffness (52, 66, 72, 73), while mechanical stretching of a collagen matrix or increased matrix stiffness can enhance cancer cell invasion (74, 75). Moreover, Levental et al., (2009) showed that enhanced collagen cross-linking, which increases matrix stiffness, promotes breast tumorigenesis through increased invasion and enhanced integrin signaling. Data suggest that mechanotransduction in breast cancer cells involves β1, β3 and β4 integrins, which are major receptors for cell-matrix interactions involved in migration and invasion in 2D and 3D models (28, 76). Furthermore, transformed mammary epithelial cells treated with either a β1 integrin function-blocking antibody or manipulated to decrease integrin signaling exhibit reduced cell invasion when cultured in cross-linked collagen gels. The outcome of this in vivo is likely to be enhanced metastasis, as changes in mammary collagen density and resulting stiffness are positively correlated with an increased number of lung metastases in vitro and in vivo (69, 77).
Integrin Signaling and Cell Tuning
Increased matrix stiffness activates integrin-mediated signaling events to increase traction forces in cells through actomyosin contractility (42, 73, 78). The amount by which cells pull on the surrounding matrix is influenced by the stiffness of the matrix through a feedback mechanism (32, 62). Solon et al. have demonstrated that fibroblasts alter the amount of actomyosin contractility to match the stiffness of the ECM (79). If the stiffness of the ECM exceeds the abilities of cells to tune their contractility in order to produce equally reciprocal forces, then cell behavior is altered. For instance, when cell contractility is reduced through inhibition of RhoA or myosin, there is a disruption of integrin clustering, focal adhesion formation, and breast cell morphogenesis in response to matrix stiffness (29, 42). However, studies have shown that even when increased matrix tension is reciprocated by cell-generated contractile forces, there is less matrix contraction and remodeling, which correlates with disrupted tissue phenotype (58, 65, 68). These observations suggest cells constantly evaluate the stromal environment and modify, or tune, their internal tension to match that of the external environment (42, 57).
While it is established that integrins play a role in responding to mechanical properties in the ECM, it is not clear how mechanical cues facilitate changes in integrin-based multimolecular complexes to initiate biochemical signaling events and actin cytoskeletal reorganization in order to regulate cell behavior during cancer progression. External force induces tension-dependent conformational changes in many integrin-associated proteins, such in vinculin, p130Cas, FAK, src, and filamin A (50, 54, 80–82), suggesting these intracellular signaling molecules play an important role in the mechanosensing capabilities of integrins. For instance, Friedland et al. demonstrated that mechanical tension on α5β1 integrin increases integrin affinity and FAK phosphorylation, while the stretch-sensitive protein p130Cas undergoes conformational extension in response to cell stretching through activation of Rap1 GTPase (54, 83). In addition to regulating biochemical signaling events, multimolecular complexes have also been shown to couple integrins to the cytoskeleton in response to mechanical cues. For instance, talin has been shown to be required for the reinforcement of integrin-cytoskeletal linkages in response to external force application (84, 85), while zyxin, which directly interacts with p130Cas, has been shown to mobilize from focal adhesions to actin filaments in response to mechanical force (86). These studies support a role for multiple proteins in assembling a mechanosensitive complex to initiate biochemical signaling pathways and to regulate the actin cytoskeleton in response the external force.
Filamin A (FLNa), an actin binding protein that directly couples β1 integrin to the actin cytoskeleton, has been suggested to play a mechanosensitive role in tuning tissue morphogenesis (87). Gehler et al. (2009) demonstrated that FLNa exhibits increased binding interactions with β1 integrin when breast epithelial cells are cultured in a stiffer collagen matrix (68). Interestingly, when β1 integrin-FLNa interactions are reduced, then cells cultured in a compliant matrix (which usually promotes normal tissue morphogenesis) exhibit reduced matrix contraction and disrupted tissue architecture. However, when β1 integrin-FLNa interactions are enhanced above that normally found in a stiff matrix, cells exhibited increased matrix contraction and displayed a reverted phenotype similar to normal morphogenesis (Figure 2).
Figure 2.
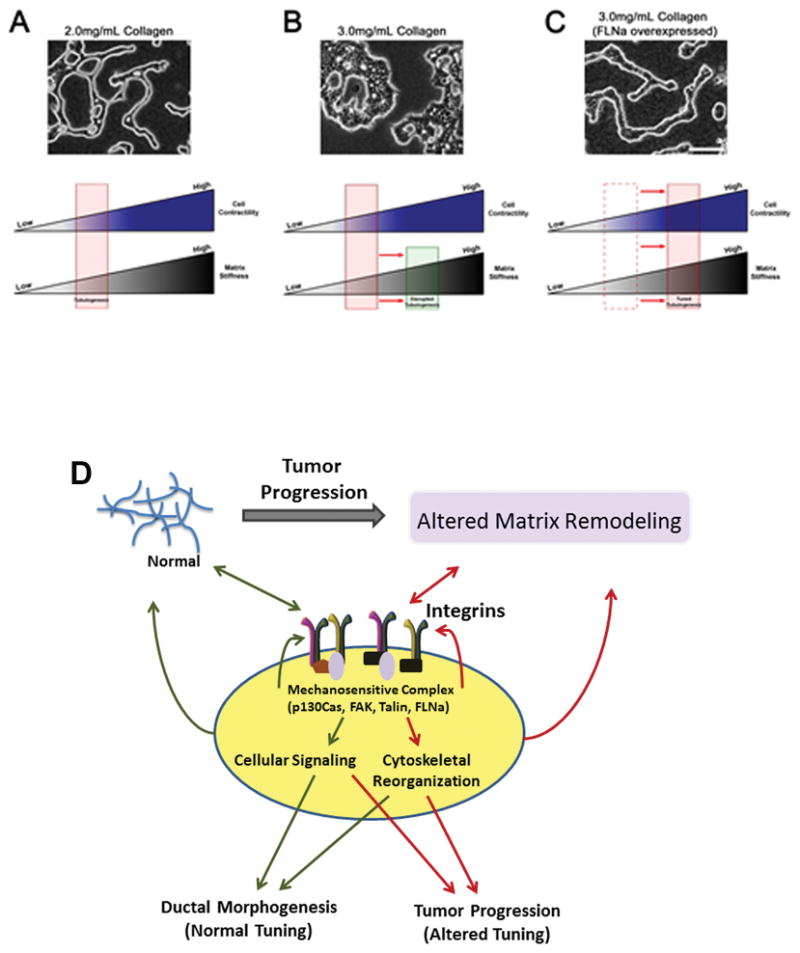
Cell contractility can be tuned to reciprocate matrix stiffness to result in phenotypic reversion. A) T47D cells cultured in a 2.0mg/mL collagen gel exhibits normal tubulogenesis. This suggests cell contractility reciprocates the stiffness of the matrix. B) When matrix stiffness increases (3.0mg/mL collagen gel), cells undergo disrupted tubulogenesis as a result of the inability of the cells to counter the increased matrix tension. C) T47D cells that overexpress FLNa, which exhibit enhanced cell contractility, undergo phenotypic reversion when cultured in a 3.0mg/mL collagen gel. Enhanced FLNa binding with β1 integrin produces similar results. Scale bar = 100μm. D) Matrix-induced changes in integrin-mediated cellular signaling and cytoskeletal organization involve a mechanosensitive complex that can undergo reciprocal signaling to alter matrix remodeling and integrin clustering.
The observation that enhanced FLNa binding to β1 integrin reverts cell phenotype in a stiff matrix suggests FLNa may be part of a mechanosensor that tunes cell contractility to reciprocate the external tension created. In support of this notion, mechanical deformation of F-actin/FLNa networks has been shown to enhanced FLNa binding to the cytoplasmic domain of β7 integrin and Filamin A-binding RhoGTPase-activating protein (FilGAP), which is a regulator of Rac activity and actin assembly (82). This suggests FLNa binding to β1 integrin not only provides a link between the ECM and the actin cytoskeleton, but can also regulate signaling pathways that may be important for actomyosin contractility (Figure 2). Interestingly, Paszek et al. (2005) demonstrated that transformed epithelial cells with elevated levels of Rho activity exhibit disorganized tissue structures (42). However, when Rho-generated contractility is reduced, these cells undergo phenotypic reversion, thus restoring tissue polarity in vitro. Conversely, when collagen crosslinking was reduced, thus decreasing matrix stiffness, tumor cell proliferation and focal adhesion formation was decreased in vivo (55). Although our understanding of mechanotransduction continues to evolve, integrins likely form multimolecular mechanosensitive complexes that regulate both biochemical signaling events and cell-generated contractility through bi-directional mechanisms to respond to matrix stiffness during tumor progression.
Rho Signaling During Tumor Progression
Rho GTPases are one branch of the larger superfamily of Ras-related small GTPases. To date, 22 human genes encoding at least 25 GTPase-containing proteins have been identified, and of these, Rho, Rac and CDC42 have been the most widely studied mainly due to their role in regulating the actin cytoskeleton and coordinating cell migration (88, 89). Like all Ras-related small GTPases, Rho GTPases are activated downstream of integrins and growth factor receptors and they function as molecular switches that cycle between the active GTP-bound form and the inactive GDP-bound form. In their active form they bind to downstream effector molecules which regulate changes in adhesive state, actin cytoskeleton organization, cellular contractility, and cell motility (90–92). The cycling of Rho activity is regulated by three classes of proteins: GAPs (GTP Activating proteins), GEFs (guanine nucleotide exchange factors) and GDIs (GDP dissociation inhibitors). RhoGAPs facilitate the hydrolysis of GTP to GDP, thereby inactivating Rho, whereas activation of Rho by GEFs occurs by phosphorylation of GDP to GTP. RhoGDIs bind and maintain Rho in its inactive form by blocking localization to the plasma membrane and inhibiting the release of GDP. While Rho is activated downstream of growth factor and integrin signaling, the exact mechanisms of regulation involving GAPs, GEFs, and GDIs largely remains under investigation.
Many tumors carry a mutation in RasGTPases leading to overexpression or increased activation of Ras. While no mutations have been identified in the Rho family of GTPases, several groups have reported overexpression of RhoGTPases in many types of cancer including breast cancers. In fact, increased levels of RhoA protein in breast cancer is associated with increased invasiveness (93, 94). Like RhoA, RhoC is also overexpressed in ductal carcinoma and inflammatory breast cancer and it has been identified as a prognostic marker of mammary tumors and metastasis (95). Furthermore, CDC42 and Rac1 have also been found to be overexpressed in breast carcinoma (93, 94). Rac1 has been implicated in cell transformation in vitro and constitutive activation of Rac1 has been suggested to play a role in aggressive breast carcinoma (96). Thus, identifying the mechanisms regulating GTPase activity is important to understanding the progression of breast carcinoma.
Several types of mechanical signals, including the increase in matrix stiffness and collagen alignment (29, 69), are associated with mammary carcinoma, and have been identified as regulators of Rho GTPases (97–100). Rho GTPase activation in response to matrix stiffness plays an important role in altering cell migration within the changing tumor associated microenvironment (29, 92). Subsequently, force generated by Rho-mediated cellular contractility causes a feedback loop to enhance matrix stiffness and alignment by inducing a conformational change in FN, which is necessary for fibrillar collagen deposition and assembly (101, 102). Additionally, enhanced actomyosin contractility, through increased Rho-ROCK activation, increases collagen deposition and matrix stiffness, which also promotes tumor cell proliferation (103). Alternately, inhibition of ROCK or myosin reduced gel contraction and disrupted tubulogenesis (Figure 3). Thus, integrin signaling to RhoGTPases is a key mechanism by which cells tune to changes in the microenvironment during tumor progression (Figure 3).
Figure 3.
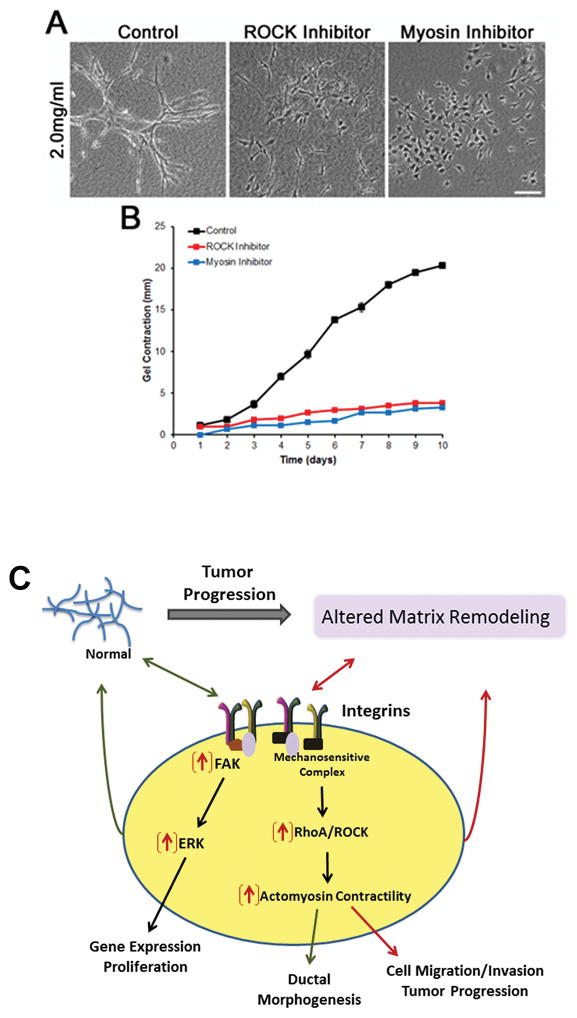
RhoA-mediated cell contractility is essential for branching morphogenesis and gel contraction. Inhibition of ROCK (2.5μM H-1152) or myosin (10μM Blebbistatin) disrupt branching morphogenesis (A) and gel contraction (B) in NMuMG breast epithelial cells cultured in a 2.0mg/mL collagen gel. Scale bar = 100μm. C) Matrix-induced changes in integrin-mediated cell signaling and actomyosin contractility which are important for cell behaviors, such as proliferation and morphogenesis, and reciprocal matrix remodeling. Increased FAK/ERK and RhoA/ROCK signaling (indicated by red arrows) promotes tumor progression through enhanced cell migration/invasion and proliferation.
Rho GTPases have been implicated at all stages of cancer progression. The initial finding of their role in regulating cytoskeletal dynamics implicated their primary function in coordinating cell migration and adhesion, in addition to matrix reorganization. During cell migration within three-dimensional matrices, tumor cells can transition between mesenchymal, amoeboid, and collective cell migration. All three of these migratory phenotypes involve signaling through RhoGTPases (reviewed in (92)). Additionally, activation of the cell cycle via cyclin-D1 and promotion of epithelial to mesenchymal transition (EMT) by increased MMP expression and decreased stability of adherens junctions can be attributed to Rac (104–106). Recent findings have given light to the function of Rho GTPases in other processes relevant to tumor progression including cell survival, protein secretion, vesicle trafficking and gene transcription (104). Thus, elucidating the exact mechanisms of the RhoGTPases in tumor progression remains quite challenging however, their role as potential therapeutic targets is promising.
Physical Cues and Cell Cycle Control during Tumor Progression
Integrin signaling has been linked to cellular proliferation through its activation of the FAK—Ras—ERK pathway under conditions of stiff matrices (65). Activation of focal adhesion kinase (FAK) is one of the dominant integrin-mediated signaling events, and FAK is emerging as a central regulator of mammary phenotype. Targeted loss of FAK in mammary epithelial cells results in a gland that is hypoplastic, and corresponds to diminished activation of ERK and reduced expression of cyclin D1, suggesting the defect is due in part to diminished cell proliferation during lobuloaveolar development (107). The role for FAK in regulating cell proliferation is further supported by the finding that FAK is strongly implicated in tumor formation. Several independent investigators demonstrated that loss of FAK suppresses tumor formation in mouse models (108–111). This is consistent with the finding that FAK is often over-expressed in human breast carcinomas (112). Analysis of genes regulated by FAK loss in mouse mammary tumors link FAK to proliferation genes involved in both G1 and G2/M (109, 113). FAK loss resulted in the down-regulation of several signaling pathways that link to proliferation including ERK, PI3K, and Rho/ROCK (109–111).
FAK levels are increased in many breast carcinoma samples (114), and FAK is associated with increased progression in several tumors including breast and colon carcinoma, and astrocytoma (115–118). FAK transcriptional expression is increased by the action of NF-kB, which is an important survival mediator (119, 120). However, the finding that FAK levels diminish in some liver and cervical carcinomas (121, 122) and that FAK phosphorylation does not always correspond to tumor progression and metastasis (123) suggests that extracellular signals impinge upon FAK and determine its role in tumor suppression or progression.
Several investigators have demonstrated that activation of FAK downstream of integrins signals directly to the Ras/ERK pathway and also regulates growth-factor signaling to this pathway (124–128). In the context of matrix stiffness, a dense or stiff matrix activates ERK in mammary epithelial cells, while a compliant matrix does not (Figure 3)(42, 65). Moreover, a stiff matrix results in dramatic upregulation of a proliferation signature that is found in human breast cancer, which can be reversed by inhibition of MEK/ERK (65). A striking feature of cells that are cultured in compliant 3D collagen matrices is that cell proliferation is markedly attenuated in a compliant matrix compared to stiff 3D or 2D matrices, which is linked to the down-regulation of this proliferation signature (65). Importantly, the regulation of cell cycle progression has been specifically linked to tissue stiffness, rather than integrin ligation per se, demonstrating the ability of the ECM stiffness to regulate how integrins signal to proliferation (64).
Stiff matrices also induce expression of several genes that are pro-invasive, including MMPs, integrin subunits, and migratory chemokine receptors. As with proliferation, inhibition of MEK reverts this gene expression, reverts the invasive phenotype, and promotes normal tubulogenesis in a stiff 3D collagen matrix (65). These findings support the notion that one way a stiff matrix promotes tumor progression to invasiveness is by activating gene expression through the MEK/ERK pathway (Figure 3).
Collagen Alignment during Tumor Progression
Recent findings have demonstrated that not only does the chemical and physical composition of the ECM affect mammary tumor progression, but the re-organization of matrix proteins into aligned fibers is also an important contributor to tumor progression. In mouse mammary tumors, the deposition and organization of collagen fibers changes dramatically as tumors progress. Provenzano et al (2006) described these changes as Tumor Associated Collagen Signatures (TACS) with TACS-3 representing perpendicular alignment of stromal collagen to the tumor in MMTV-PyVT mice (129). In mice with collagen-dense mammary glands, the presence of TACS-3 increases and correlates with increased tumor burden, invasion, and metastasis to the lung (69). These findings strongly implicate that aligned collagen fibers provide “tracks” by which tumor cells can escape from a primary tumor, thus facilitating metastasis. This is supported by observations that collagen aligned perpendicular to a tumor-explant boundary promotes the invasion of mammary epithelial cells in vitro (130). Conklin et al. (2011) corroborated these results in a study of human breast cancer, where TACS-3 fibers surrounding hyperplastic mammary ducts were identified in human breast tumor histological sections, and were associated with poor patient outcome (131). These initial observations of tumor-induced matrix reorganization provided some of the first evidence that matrix topographical cues in developing tumors may direct cell migration away from a primary tumor, and may reveal some insight into potential mechanisms of metastasis. However, the mechanism by which ECM alignment is produced and maintained in vivo, particularly with regard to TACS-3 aligned fibers surrounding breast tumors, is not well understood.
Generation of Collagen Alignment
The ability of cells to create regions of aligned fibers in vitro has been well established in fibroblast-collagen gel cultures and occurs concomitantly with gel contraction by cells (132). In this contraction-induced model, cells exert strain on surrounding matrix fibers such that when matrix stiffness is less than the force imparted by cells, the matrix deforms and undergoes changes in local alignment but the cells do not exhibit substantial levels of migration. However, when matrix stiffness exceeds cell contractile forces, cells are then able to migrate (18). These findings further suggest that matrix stiffness determines the cellular tuning response, and that an appropriate amount of stiffness is required for cells to generate traction and subsequent cell migration (Figure 4).
Figure 4.
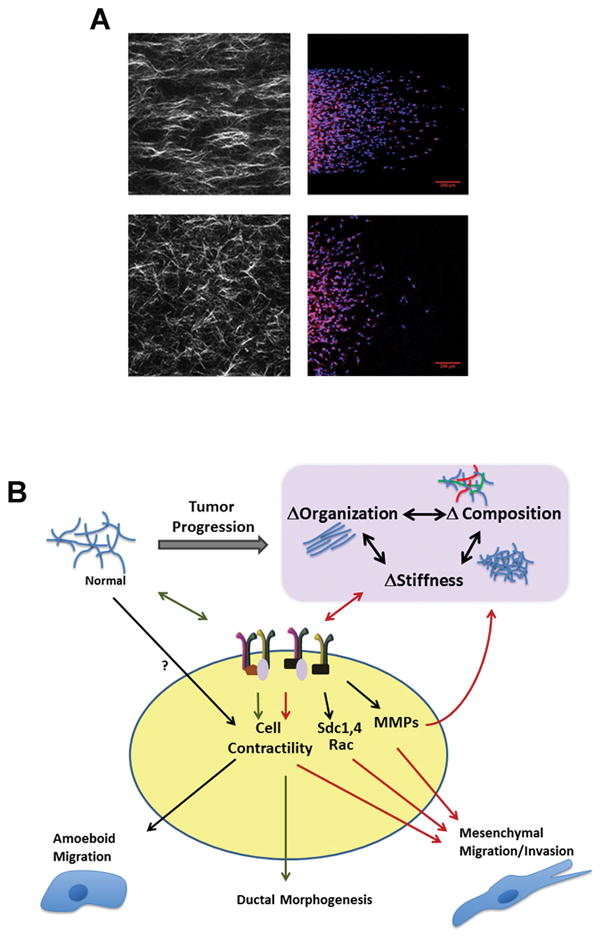
Bi-directional signals from integrins and extracellular matrix composition, organization, and stiffness lead to altered cell behavior in tumor progression. A. MDA-MB-231 cells cultured in microchannels containing aligned or random collagen matrices migrate more when collagen is aligned (top panel). Collagen is visualized via second harmonic generation (left) and cells are stained for nuclei and actin with DAPI and Phalloidin, respectively (right). B. Cells respond to changes in ECM chemical and physical composition by altering signaling through integrins, and in tumor progression, regulate further changes in the ECM by increasing the expression and activation of MMPs or by enhancing Rho-mediated contractility. In so doing, tumor cells rapidly adapt to changes in their environment, and consequently promote invasion and metastasis.
A study of vascular smooth muscle cells cultured in collagen gels showed that integrin linkages to collagen fibers are required for gel contraction by cells. When the α2 and β1 integrin subunits are blocked, contraction is halted, and the contraction-induced matrix tension is lost (133). Similarly, Provenzano et al. (2008) showed that the generation of alignment between tissue explants or cell-seeded collagen gels is dependent on actin-myosin contractility and requires signaling via Rho GTPase and its effector, ROCK. This contractile mechanism, however, is only required to set up the alignment; when cells are confronted with a matrix that is already aligned, they migrate along fibers even when Rho is inhibited, further suggesting that cells must first utilize contractile means downstream of integrin linkages to align fibers, after which may then provide the necessary traction to support migration (130).
It is unclear what cells in the tumor microenvironment are responsible for creating aligned matrices around developing tumors. Although carcinoma cell lines and cells from tumor explants have been shown to align collagen in vitro, multiple cell types may work synergistically to produce matrix alignment in vivo. Additionally, generation of alignment may also result from de novo collagen deposition by stromal fibroblasts. Indeed, fibroblasts can be exploited in vitro to deposit highly aligned collagen matrices, and the orientation of the fibroblasts determines that of the newly deposited aligned matrix. In turn, fibroblast orientation can be controlled by the initial matrix on which they are seeded. For example, when fibroblasts are allowed to adhere to a matrix that is already aligned, the cells will deposit more aligned matrix (134–137).
One mechanism by which stromal cells deposit aligned matrices appears to involve the proteoglycan, syndecan-1. The cell surface proteoglycan, syndecan-1, interacts with integrins among other cell surface proteins to aid in adhesion and migration (138). Yang et al (2011) showed that fibroblasts expressing elevated levels of syndecan-1 generate parallel-organized ECM fibers that allow seeded tumor cells to adhere to and migrate directionally along fibers. This occurs in contrast to cells cultured on randomly organized matrices generated by fibroblasts lacking syndecan-1 expression (135). Furthermore, increased expression of syndecan-1 in human breast carcinomas is associated with the aligned matrix architecture present in tumors (131, 135). These findings indicate syndecan-1 is a key player in the process of generating aligned fibers, which may shed some additional light on other potential signaling pathways; however, more work must be done to further understand this process.
The presence of TACS-3 aligned collagen fibers in breast cancer is strongly correlated with cancer cell invasion and metastasis, however, little is known how cells sense and respond to these changes in ECM topography. When cancer cells are cultured in matrices of pre-aligned collagen, they exhibit more directionally persistent migration compared to cells cultured in random collagen matrices (130). Although the mechanisms of directional sensing are not yet fully understood, several hypotheses exist for how alignment of the ECM guides migrating cells. Clusters of β1 integrin localize along collagen fibers (unpublished data, Keely Lab) and fiber alignment likely guides newly formed integrin clusters along the leading edge of the cell. In addition to the effects of increased integrin signaling on cell motility and invasion, the channels created by parallel-aligned fibers provide less spatial impedance for migrating cells (139). Furthermore, aligned collagen may be stiffer than randomly organized collagen (140), suggesting that the increase in ECM stiffness may provide enhanced traction for motility and restrict motility to the direction of greatest traction, namely along parallel aligned ECM fibers.
In addition to mechanical recognition of aligned collagen, recent findings show that the ability of cells to recognize patterned fibronectin substrates is dependent on syndecan-4. Bass et al (2007) demonstrated that cells lacking syndecan-4 expression fail to migrate directionally when cultured on matrices containing aligned fibronectin. This response was shown to depend on the ability of syndecan-4 to bind to and localize PKCalpha to points of ECM engagement at the leading edge, followed by subsequent rac localization by PKCalpha (141). In this scenario, cells are guided by the organization of the matrix via their points of contact with the matrix. These findings reinforce the concept that cells tune their responses through integrins to changes in matrix mechanics, composition, and organization, allowing them to constantly adapt to and modify their ever-changing environment.
Matrix Metalloproteinases in Breast Cancer Invasion and Metastasis
While matrix remodeling that leads to changes in the mechanical properties of the ECM plays an important role during tumor progression, proteolytic degradation is another key component in this process. Matrix metalloproteinases (MMPs) have broad implications in many cancers and their functions involving tumor promotion and invasion have been extensively studied, although our understanding of this complex regulatory network remains limited. MMPs are most widely known for degrading ECM proteins, however, they have a diverse group of substrates and are implicated in many aspects of cell signaling. MMPs can bind integrins, release surface-bound growth factors and growth factor receptors, as well as cleave cell-cell and cell-matrix adhesion molecules (142). In so doing, MMPs can directly regulate proliferation, migration, and apoptosis.
Expression of MMPs is markedly increased in many cancers including breast carcinomas, and is associated with poor prognosis (143, 144). In mouse xenograft models, elevated expression of MMPs increases tumor burden and metastasis, whereas decreased levels of MMPs in more invasive cell lines reduces tumor malignancy and lung metastases (142). Several in vitro experiments show a positive correlation between MMP expression and invasion of carcinoma cell lines through collagen type I matrices or matrigel (145–147). While basement membrane and to a lesser degree, stromal collagen, largely serve as a barrier to migration, MMPs allow cells to navigate through dense fibrillar networks. In addition to simply severing ECM fibers that may be obstructing a cell’s trajectory, the cleavage of ECM proteins can expose cryptic binding sites that bind integrins and promote migration (142).
Evidence for MMP-dependent migration
Cancer cells cultured in 3D matrices employ a mesenchymal migration phenotype that is dependent on integrin-ECM engagement. In dense collagen matrices or matrigel, where the ECM poses a significant structural barrier, cells will extend pseudopods into the matrix, generate traction force, and locally secrete MMPs to sever fibers adjacent and posterior to the leading edge (148), (149). This allows cells to maintain traction on fibers where integrins are engaged, and subsequently enables translocation of the cell body through the matrix. Cleaved fibers are then displaced and become aligned as cells exert tension and push their way through the matrix (139). The resulting aligned ECM tracks represent a permanent matrix deformation, which can then promote subsequent cancer cell migration in a protease-independent manner (150), (148).
Evidence for MMP-independent migration
There is mounting evidence that tumor cells exhibit remarkable plasticity in their ability to change migration modes. When MMPs are inhibited in vitro, tumor cells can adopt a more amoeboid migration profile that is primarily dependent on Rho-mediated actin-myosin contractility (Figure 4), allowing cells to change shape and squeeze through pores in the matrix (151, 152). Additionally, cells that employ amoeboid migration do not permanently deform matrix fibers, but rather leave the matrix relatively undisturbed (153). For this mode of migration, matrix density plays a much more significant role in that migration is arrested if the matrix pore size is sufficiently small (154, 155).
Conversely, numerous reports have also shown that a mesenchymal-like migration phenotype can prevail independent of MMP action. In nested collagen gel assays, cells retain their ability to mechanically deform matrix fibers via Rho and Rock allowing subsequent migration even when proteases are inhibited (130). In these scenarios, matrix stiffness may play a larger role in allowing cells’ contractile machinery to reorganize matrix fibers, rendering MMPs unnecessary. If, however, the matrix stiffness exceeds the amount of force cells are capable of producing, protease-dependent migration would likely dominate (Figure 4). The presence of aligned collagen fibers is expected to alleviate some of the restrictions that a dense matrix might create, and instead create an open track complete with adhesive ligand along which cells can migrate. Thus, it is not surprising that migration along aligned collagen fibers appears to be protease independent (130).
Further support for protease independent cancer cell migration comes from recent work with mice that have a mutation in the collagenase cleavage site of the col1a1 gene, and results in an overabundance of collagenase-resistant stromal collagen (156). These mice, in a PyMT background, have much more aggressive mammary tumors and more lung metastases, suggesting that proteases are not required for invasion through collagen-rich mammary glands (69). Of interest, in Col1a1tm1jae animals with protease resistant collagen, collagen alignment is augmented (69). These findings highlight an important distinction between the role of proteases and the respective ECM compartments in which their function is necessary for cells to effectively migrate. Conceivably, proteases may only be necessary for invasion through dense basement membrane, or during intra- or extravasation of the vasculature, where a cell cannot penetrate the ECM utilizing any other means. Once cells have crossed these superficial barriers to migration, however, they may then migrate independently of proteases.
Conclusions
Cells interact with their microenvironment through reciprocal interactions that involve both chemical AND mechanical properties of the ECM, such as matrix stiffness, topography, organization, and alignment. Together, these ECM properties regulate integrin signaling to influence cell behavior that promotes growth and differentiation, proliferation, and invasion and metastasis during tumor progression. For instance, cells can tune their response, within a range, to changes in the mechanical properties of the ECM through integrins and adjust the force they exert upon the matrix. This process likely involves numerous signaling molecules that comprise a mechanosensitive complex. This cellular tuning profoundly affects the cellular response, such as proliferation and invasion, to the ECM that can lead to tumor progression. Conversely, cell-mediated activities, such as myosin-generated traction forces, cross-linking, and protease digestion, modify and alter the chemical and structural composition of the ECM that in turn influences cell behavior. While our understanding of the mechanotransduction is incomplete, further elucidating the mechanisms involved in the reciprocal interactions between cells and various properties of the microenvironment will provide valuable insight to how mechanical cues regulate cell behavior.
Acknowledgments
This work is supported by CA114462-01A2 and CA142833-01 to PJK.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. Epub 2011/03/08. [DOI] [PubMed] [Google Scholar]
- 2.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449(7162):557–63. doi: 10.1038/nature06188. Epub 2007/10/05. [DOI] [PubMed] [Google Scholar]
- 3.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677–89. doi: 10.1016/j.cell.2006.06.044. Epub 2006/08/23. [DOI] [PubMed] [Google Scholar]
- 4.Keely PJ. Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J Mammary Gland Biol Neoplasia. 2011;16(3):205–19. doi: 10.1007/s10911-011-9226-0. Epub 2011/08/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jodele S, Blavier L, Yoon JM, DeClerck YA. Modifying the soil to affect the seed: role of stromal-derived matrix metalloproteinases in cancer progression. Cancer Metastasis Rev. 2006;25(1):35–43. doi: 10.1007/s10555-006-7887-8. Epub 2006/05/09. [DOI] [PubMed] [Google Scholar]
- 6.Larsen M, Wei C, Yamada KM. Cell and fibronectin dynamics during branching morphogenesis. J Cell Sci. 2006;119(Pt 16):3376–84. doi: 10.1242/jcs.03079. Epub 2006/08/03. [DOI] [PubMed] [Google Scholar]
- 7.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–87. doi: 10.1016/s0092-8674(02)00971-6. Epub 2002/09/26. [DOI] [PubMed] [Google Scholar]
- 8.van der Flier A, Sonnenberg A. Function and interactions of integrins. Cell and tissue research. 2001;305(3):285–98. doi: 10.1007/s004410100417. Epub 2001/09/27. [DOI] [PubMed] [Google Scholar]
- 9.Barczyk M, Carracedo S, Gullberg D. Integrins. Cell and tissue research. 2010;339(1):269–80. doi: 10.1007/s00441-009-0834-6. Epub 2009/08/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–47. doi: 10.1146/annurev.immunol.25.022106.141618. Epub 2007/01/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geiger B, Yamada KM. Molecular architecture and function of matrix adhesions. Cold Spring Harbor perspectives in biology. 2011;3(5) doi: 10.1101/cshperspect.a005033. Epub 2011/03/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zamir E, Geiger B. Components of cell-matrix adhesions. J Cell Sci. 2001;114(Pt 20):3577–9. doi: 10.1242/jcs.114.20.3577. Epub 2001/11/15. [DOI] [PubMed] [Google Scholar]
- 13.Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285(5430):1028–32. doi: 10.1126/science.285.5430.1028. Epub 1999/08/14. [DOI] [PubMed] [Google Scholar]
- 14.Calderwood DA. Integrin activation. J Cell Sci. 2004;117(Pt 5):657–66. doi: 10.1242/jcs.01014. Epub 2004/02/03. [DOI] [PubMed] [Google Scholar]
- 15.Zamir E, Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. J Cell Sci. 2001;114(Pt 20):3583–90. doi: 10.1242/jcs.114.20.3583. Epub 2001/11/15. [DOI] [PubMed] [Google Scholar]
- 16.Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294(5547):1708–12. doi: 10.1126/science.1064829. Epub 2001/11/27. [DOI] [PubMed] [Google Scholar]
- 17.Cukierman E, Pankov R, Yamada KM. Cell interactions with three-dimensional matrices. Curr Opin Cell Biol. 2002;14(5):633–9. doi: 10.1016/s0955-0674(02)00364-2. Epub 2002/09/17. [DOI] [PubMed] [Google Scholar]
- 18.Miron-Mendoza M, Seemann J, Grinnell F. Collagen fibril flow and tissue translocation coupled to fibroblast migration in 3D collagen matrices. Mol Biol Cell. 2008;19(5):2051–8. doi: 10.1091/mbc.E07-09-0930. Epub 2008/03/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fogerty FJ, Mosher DF. Mechanisms for organization of fibronectin matrix. Cell differentiation and development: the official journal of the International Society of Developmental Biologists. 1990;32(3):439–50. doi: 10.1016/0922-3371(90)90061-z. Epub 1990/12/02. [DOI] [PubMed] [Google Scholar]
- 20.Mao Y, Schwarzbauer JE. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005;24(6):389–99. doi: 10.1016/j.matbio.2005.06.008. Epub 2005/08/03. [DOI] [PubMed] [Google Scholar]
- 21.Yurchenco PD. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harbor perspectives in biology. 2011;3(2) doi: 10.1101/cshperspect.a004911. Epub 2011/03/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ricard-Blum S. The collagen family. Cold Spring Harbor perspectives in biology. 2011;3(1):a004978. doi: 10.1101/cshperspect.a004978. Epub 2011/03/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moschos SJ, Drogowski LM, Reppert SL, Kirkwood JM. Integrins and cancer. Oncology. 2007;21(9 Suppl 3):13–20. Epub 2007/10/12. [PubMed] [Google Scholar]
- 24.Plantefaber LC, Hynes RO. Changes in integrin receptors on oncogenically transformed cells. Cell. 1989;56(2):281–90. doi: 10.1016/0092-8674(89)90902-1. Epub 1989/01/27. [DOI] [PubMed] [Google Scholar]
- 25.Kyutoku M, Taniyama Y, Katsuragi N, Shimizu H, Kunugiza Y, Iekushi K, et al. Role of periostin in cancer progression and metastasis: inhibition of breast cancer progression and metastasis by anti-periostin antibody in a murine model. Int J Mol Med. 2011;28(2):181–6. doi: 10.3892/ijmm.2011.712. Epub 2011/05/28. [DOI] [PubMed] [Google Scholar]
- 26.Dai J, Li B, Shi J, Peng L, Zhang D, Qian W, et al. A humanized anti-osteopontin antibody inhibits breast cancer growth and metastasis in vivo. Cancer immunology, immunotherapy: CII. 2010;59(3):355–66. doi: 10.1007/s00262-009-0754-z. Epub 2009/08/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hancox RA, Allen MD, Holliday DL, Edwards DR, Pennington CJ, Guttery DS, et al. Tumour-associated tenascin-C isoforms promote breast cancer cell invasion and growth by matrix metalloproteinase-dependent and independent mechanisms. Breast Cancer Res. 2009;11(2):R24. doi: 10.1186/bcr2251. Epub 2009/05/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park CC, Zhang H, Pallavicini M, Gray JW, Baehner F, Park CJ, et al. Beta1 integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimensional cultures and in vivo. Cancer Res. 2006;66(3):1526–35. doi: 10.1158/0008-5472.CAN-05-3071. Epub 2006/02/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ. ROCK-generated contractility regulates breast epithelial cell differentiation in response to the physical properties of a three-dimensional collagen matrix. J Cell Biol. 2003;163(3):583–95. doi: 10.1083/jcb.200305010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paszek MJ, Weaver VM. The tension mounts: mechanics meets morphogenesis and malignancy. J Mammary Gland Biol Neoplasia. 2004;9(4):325–42. doi: 10.1007/s10911-004-1404-x. [DOI] [PubMed] [Google Scholar]
- 31.Guo YP, Martin LJ, Hanna W, Banerjee D, Miller N, Fishell E, et al. Growth factors and stromal matrix proteins associated with mammographic densities. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2001;10(3):243–8. [PubMed] [Google Scholar]
- 32.Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310(5751):1139–43. doi: 10.1126/science.1116995. Epub 2005/11/19. [DOI] [PubMed] [Google Scholar]
- 33.Boyd NF, Dite GS, Stone J, Gunasekara A, English DR, McCredie MR, et al. Heritability of mammographic density, a risk factor for breast cancer. N Engl J Med. 2002;347(12):886–94. doi: 10.1056/NEJMoa013390. [DOI] [PubMed] [Google Scholar]
- 34.Boyd NF, Martin LJ, Li Q, Sun L, Chiarelli AM, Hislop G, et al. Mammographic density as a surrogate marker for the effects of hormone therapy on risk of breast cancer. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2006;15(5):961–6. doi: 10.1158/1055-9965.EPI-05-0762. [DOI] [PubMed] [Google Scholar]
- 35.Yaghjyan L, Colditz GA, Rosner B, Tamimi RM. Mammographic breast density and breast cancer risk by menopausal status, postmenopausal hormone use and a family history of breast cancer. Cancer causes & control: CCC. 2012;23(5):785–90. doi: 10.1007/s10552-012-9936-7. Epub 2012/03/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schedin P, O’Brien J, Rudolph M, Stein T, Borges V. Microenvironment of the involuting mammary gland mediates mammary cancer progression. J Mammary Gland Biol Neoplasia. 2007;12(1):71–82. doi: 10.1007/s10911-007-9039-3. Epub 2007/02/24. [DOI] [PubMed] [Google Scholar]
- 37.Boyd NF, Lockwood GA, Martin LJ, Byng JW, Yaffe MJ, Tritchler DL. Mammographic density as a marker of susceptibility to breast cancer: a hypothesis. IARC Sci Publ. 2001;154:163–9. [PubMed] [Google Scholar]
- 38.Martin LJ, Boyd NF. Mammographic density. Potential mechanisms of breast cancer risk associated with mammographic density: hypotheses based on epidemiological evidence. Breast Cancer Res. 2008;10(1):201. doi: 10.1186/bcr1831. Epub 2008/01/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ursin G, Hovanessian-Larsen L, Parisky YR, Pike MC, Wu AH. Greatly increased occurrence of breast cancers in areas of mammographically dense tissue. Breast Cancer Res. 2005;7(5):R605–8. doi: 10.1186/bcr1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lopez JI, Kang I, You WK, McDonald DM, Weaver VM. In situ force mapping of mammary gland transformation. Integr Biol (Camb) 2011;3(9):910–21. doi: 10.1039/c1ib00043h. Epub 2011/08/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Provenzano PP, Eliceiri KW, Keely PJ. Shining new light on 3D cell motility and the metastatic process. Trends Cell Biol. 2009;19(11):638–48. doi: 10.1016/j.tcb.2009.08.009. Epub 2009/10/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8(3):241–54. doi: 10.1016/j.ccr.2005.08.010. Epub 2005/09/20. [DOI] [PubMed] [Google Scholar]
- 43.Walker RA. The complexities of breast cancer desmoplasia. Breast Cancer Res. 2001;3(3):143–5. doi: 10.1186/bcr287. Epub 2001/04/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jahkola T, Toivonen T, Nordling S, von Smitten K, Virtanen I. Expression of tenascin-C in intraductal carcinoma of human breast: relationship to invasion. Eur J Cancer. 1998;34(11):1687–92. doi: 10.1016/s0959-8049(98)00215-9. Epub 1999/01/20. [DOI] [PubMed] [Google Scholar]
- 45.Jahkola T, Toivonen T, Virtanen I, von Smitten K, Nordling S, von Boguslawski K, et al. Tenascin-C expression in invasion border of early breast cancer: a predictor of local and distant recurrence. Br J Cancer. 1998;78(11):1507–13. doi: 10.1038/bjc.1998.714. Epub 1998/12/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jahkola T, Toivonen T, von Smitten K, Virtanen I, Wasenius VM, Blomqvist C. Cathepsin-D, urokinase plasminogen activator and type-1 plasminogen activator inhibitor in early breast cancer: an immunohistochemical study of prognostic value and relations to tenascin-C and other factors. Br J Cancer. 1999;80(1–2):167–74. doi: 10.1038/sj.bjc.6690336. Epub 1999/07/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grigoriadis A, Mackay A, Reis-Filho JS, Steele D, Iseli C, Stevenson BJ, et al. Establishment of the epithelial-specific transcriptome of normal and malignant human breast cells based on MPSS and array expression data. Breast Cancer Res. 2006;8(5):R56. doi: 10.1186/bcr1604. Epub 2006/10/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Puglisi F, Puppin C, Pegolo E, Andreetta C, Pascoletti G, D’Aurizio F, et al. Expression of periostin in human breast cancer. Journal of clinical pathology. 2008;61(4):494–8. doi: 10.1136/jcp.2007.052506. Epub 2007/10/17. [DOI] [PubMed] [Google Scholar]
- 49.Choquet D, Felsenfeld DP, Sheetz MP. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 1997;88(1):39–48. doi: 10.1016/s0092-8674(00)81856-5. Epub 1997/01/10. [DOI] [PubMed] [Google Scholar]
- 50.Riveline D, Zamir E, Balaban NQ, Schwarz US, Ishizaki T, Narumiya S, et al. Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol. 2001;153(6):1175–86. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galbraith CG, Yamada KM, Sheetz MP. The relationship between force and focal complex development. J Cell Biol. 2002;159(4):695–705. doi: 10.1083/jcb.200204153. Epub 2002/11/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pelham RJ, Jr, Wang Y. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc Natl Acad Sci U S A. 1997;94(25):13661–5. doi: 10.1073/pnas.94.25.13661. Epub 1998/02/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giannone G, Dubin-Thaler BJ, Dobereiner HG, Kieffer N, Bresnick AR, Sheetz MP. Periodic lamellipodial contractions correlate with rearward actin waves. Cell. 2004;116(3):431–43. doi: 10.1016/s0092-8674(04)00058-3. Epub 2004/03/16. [DOI] [PubMed] [Google Scholar]
- 54.Sawada S, Iwasaki Y, Nakabayashi N, Ishihara K. Stress response of adherent cells on a polymer blend surface composed of a segmented polyurethane and MPC copolymers. J Biomed Mater Res A. 2006;79(3):476–84. doi: 10.1002/jbm.a.30820. Epub 2006/06/08. [DOI] [PubMed] [Google Scholar]
- 55.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906. doi: 10.1016/j.cell.2009.10.027. Epub 2009/11/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nelson CM, Bissell MJ. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Butcher DT, Alliston T, Weaver VM. A tense situation: forcing tumour progression. Nature reviews Cancer. 2009;9(2):108–22. doi: 10.1038/nrc2544. Epub 2009/01/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692(2–3):103–19. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 59.Geiger B, Bershadsky A. Assembly and mechanosensory function of focal contacts. Curr Opin Cell Biol. 2001;13(5):584–92. doi: 10.1016/s0955-0674(00)00255-6. [DOI] [PubMed] [Google Scholar]
- 60.Beacham DA, Cukierman E. Stromagenesis: the changing face of fibroblastic microenvironments during tumor progression. Seminars in cancer biology. 2005;15(5):329–41. doi: 10.1016/j.semcancer.2005.05.003. Epub 2005/06/23. [DOI] [PubMed] [Google Scholar]
- 61.Berrier AL, Yamada KM. Cell-matrix adhesion. J Cell Physiol. 2007;213(3):565–73. doi: 10.1002/jcp.21237. Epub 2007/08/08. [DOI] [PubMed] [Google Scholar]
- 62.Giannone G, Sheetz MP. Substrate rigidity and force define form through tyrosine phosphatase and kinase pathways. Trends Cell Biol. 2006;16(4):213–23. doi: 10.1016/j.tcb.2006.02.005. Epub 2006/03/15. [DOI] [PubMed] [Google Scholar]
- 63.Miranti CK, Brugge JS. Sensing the environment: a historical perspective on integrin signal transduction. Nat Cell Biol. 2002;4(4):E83–90. doi: 10.1038/ncb0402-e83. Epub 2002/04/11. [DOI] [PubMed] [Google Scholar]
- 64.Klein EA, Yin L, Kothapalli D, Castagnino P, Byfield FJ, Xu T, et al. Cell-cycle control by physiological matrix elasticity and in vivo tissue stiffening. Curr Biol. 2009;19(18):1511–8. doi: 10.1016/j.cub.2009.07.069. Epub 2009/09/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28(49):4326–43. doi: 10.1038/onc.2009.299. Epub 2009/10/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ulrich TA, de Juan Pardo EM, Kumar S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res. 2009;69(10):4167–74. doi: 10.1158/0008-5472.CAN-08-4859. Epub 2009/05/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Williams CM, Engler AJ, Slone RD, Galante LL, Schwarzbauer JE. Fibronectin expression modulates mammary epithelial cell proliferation during acinar differentiation. Cancer Res. 2008;68(9):3185–92. doi: 10.1158/0008-5472.CAN-07-2673. Epub 2008/05/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gehler S, Baldassarre M, Lad Y, Leight JL, Wozniak MA, Riching KM, et al. Filamin A-beta1 integrin complex tunes epithelial cell response to matrix tension. Mol Biol Cell. 2009;20(14):3224–38. doi: 10.1091/mbc.E08-12-1186. Epub 2009/05/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, et al. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. Epub 2008/04/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137(1):231–45. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.White DE, Kurpios NA, Zuo D, Hassell JA, Blaess S, Mueller U, et al. Targeted disruption of beta1-integrin in a transgenic mouse model of human breast cancer reveals an essential role in mammary tumor induction. Cancer Cell. 2004;6(2):159–70. doi: 10.1016/j.ccr.2004.06.025. [DOI] [PubMed] [Google Scholar]
- 72.Guo WH, Frey MT, Burnham NA, Wang YL. Substrate rigidity regulates the formation and maintenance of tissues. Biophys J. 2006;90(6):2213–20. doi: 10.1529/biophysj.105.070144. Epub 2006/01/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lo CM, Wang HB, Dembo M, Wang YL. Cell movement is guided by the rigidity of the substrate. Biophys J. 2000;79(1):144–52. doi: 10.1016/S0006-3495(00)76279-5. Epub 2000/06/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Menon S, Beningo KA. Cancer cell invasion is enhanced by applied mechanical stimulation. PLoS One. 2011;6(2):e17277. doi: 10.1371/journal.pone.0017277. Epub 2011/03/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kostic A, Lynch CD, Sheetz MP. Differential matrix rigidity response in breast cancer cell lines correlates with the tissue tropism. PLoS One. 2009;4(7):e6361. doi: 10.1371/journal.pone.0006361. Epub 2009/07/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morini M, Mottolese M, Ferrari N, Ghiorzo F, Buglioni S, Mortarini R, et al. The alpha 3 beta 1 integrin is associated with mammary carcinoma cell metastasis, invasion, and gelatinase B (MMP-9) activity. Int J Cancer. 2000;87(3):336–42. Epub 2000/07/18. [PubMed] [Google Scholar]
- 77.Goetz JG, Minguet S, Navarro-Lerida I, Lazcano JJ, Samaniego R, Calvo E, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011;146(1):148–63. doi: 10.1016/j.cell.2011.05.040. Epub 2011/07/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saez A, Buguin A, Silberzan P, Ladoux B. Is the mechanical activity of epithelial cells controlled by deformations or forces? Biophys J. 2005;89(6):L52–4. doi: 10.1529/biophysj.105.071217. Epub 2005/10/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Solon J, Levental I, Sengupta K, Georges PC, Janmey PA. Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys J. 2007;93(12):4453–61. doi: 10.1529/biophysj.106.101386. Epub 2007/11/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Na S, Collin O, Chowdhury F, Tay B, Ouyang M, Wang Y, et al. Rapid signal transduction in living cells is a unique feature of mechanotransduction. Proc Natl Acad Sci U S A. 2008;105(18):6626–31. doi: 10.1073/pnas.0711704105. Epub 2008/05/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Friedland JC, Lee MH, Boettiger D. Mechanically activated integrin switch controls alpha5beta1 function. Science. 2009;323(5914):642–4. doi: 10.1126/science.1168441. Epub 2009/01/31. [DOI] [PubMed] [Google Scholar]
- 82.Ehrlicher AJ, Nakamura F, Hartwig JH, Weitz DA, Stossel TP. Mechanical strain in actin networks regulates FilGAP and integrin binding to filamin A. Nature. 2011;478(7368):260–3. doi: 10.1038/nature10430. Epub 2011/09/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tamada M, Sheetz MP, Sawada Y. Activation of a signaling cascade by cytoskeleton stretch. Dev Cell. 2004;7(5):709–18. doi: 10.1016/j.devcel.2004.08.021. Epub 2004/11/05. [DOI] [PubMed] [Google Scholar]
- 84.Giannone G, Jiang G, Sutton DH, Critchley DR, Sheetz MP. Talin1 is critical for force-dependent reinforcement of initial integrin-cytoskeleton bonds but not tyrosine kinase activation. J Cell Biol. 2003;163(2):409–19. doi: 10.1083/jcb.200302001. Epub 2003/10/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP. Stretching single talin rod molecules activates vinculin binding. Science. 2009;323(5914):638–41. doi: 10.1126/science.1162912. Epub 2009/01/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yoshigi M, Hoffman LM, Jensen CC, Yost HJ, Beckerle MC. Mechanical force mobilizes zyxin from focal adhesions to actin filaments and regulates cytoskeletal reinforcement. J Cell Biol. 2005;171(2):209–15. doi: 10.1083/jcb.200505018. Epub 2005/10/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Razinia Z, Makela T, Ylanne J, Calderwood DA. Filamins in mechanosensing and signaling. Annual review of biophysics. 2012;41:227–46. doi: 10.1146/annurev-biophys-050511-102252. Epub 2012/03/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tang Y, Olufemi L, Wang MT, Nie D. Role of Rho GTPases in breast cancer. Front Biosci. 2008;13:759–76. doi: 10.2741/2718. Epub 2007/11/06. [DOI] [PubMed] [Google Scholar]
- 89.Guilluy C, Garcia-Mata R, Burridge K. Rho protein crosstalk: another social network? Trends Cell Biol. 2011;21(12):718–26. doi: 10.1016/j.tcb.2011.08.002. Epub 2011/09/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348(Pt 2):241–55. Epub 2000/05/19. [PMC free article] [PubMed] [Google Scholar]
- 91.Hall A, Nobes CD. Rho GTPases: molecular switches that control the organization and dynamics of the actin cytoskeleton. Philos Trans R Soc Lond B Biol Sci. 2000;355(1399):965–70. doi: 10.1098/rstb.2000.0632. Epub 2000/12/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell communication and signaling: CCS. 2010;8:23. doi: 10.1186/1478-811X-8-23. Epub 2010/09/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fritz G, Just I, Kaina B. Rho GTPases are over-expressed in human tumors. Int J Cancer. 1999;81(5):682–7. doi: 10.1002/(sici)1097-0215(19990531)81:5<682::aid-ijc2>3.0.co;2-b. Epub 1999/05/18. [DOI] [PubMed] [Google Scholar]
- 94.Fritz G, Brachetti C, Bahlmann F, Schmidt M, Kaina B. Rho GTPases in human breast tumours: expression and mutation analyses and correlation with clinical parameters. Br J Cancer. 2002;87(6):635–44. doi: 10.1038/sj.bjc.6600510. Epub 2002/09/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kleer CG, van Golen KL, Zhang Y, Wu ZF, Rubin MA, Merajver SD. Characterization of RhoC expression in benign and malignant breast disease: a potential new marker for small breast carcinomas with metastatic ability. Am J Pathol. 2002;160(2):579–84. doi: 10.1016/S0002-9440(10)64877-8. Epub 2002/02/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H, et al. Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene. 2000;19(26):3013–20. doi: 10.1038/sj.onc.1203621. Epub 2000/06/29. [DOI] [PubMed] [Google Scholar]
- 97.Smith PG, Roy C, Zhang YN, Chauduri S. Mechanical stress increases RhoA activation in airway smooth muscle cells. American journal of respiratory cell and molecular biology. 2003;28(4):436–42. doi: 10.1165/rcmb.4754. Epub 2003/03/26. [DOI] [PubMed] [Google Scholar]
- 98.Kawamura S, Miyamoto S, Brown JH. Initiation and transduction of stretch-induced RhoA and Rac1 activation through caveolae: cytoskeletal regulation of ERK translocation. J Biol Chem. 2003;278(33):31111–7. doi: 10.1074/jbc.M300725200. Epub 2003/06/05. [DOI] [PubMed] [Google Scholar]
- 99.Wojciak-Stothard B, Ridley AJ. Shear stress-induced endothelial cell polarization is mediated by Rho and Rac but not Cdc42 or PI 3-kinases. J Cell Biol. 2003;161(2):429–39. doi: 10.1083/jcb.200210135. Epub 2003/04/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tzima E, del Pozo MA, Shattil SJ, Chien S, Schwartz MA. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J. 2001;20(17):4639–47. doi: 10.1093/emboj/20.17.4639. Epub 2001/09/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol. 2008;20(5):495–501. doi: 10.1016/j.ceb.2008.06.008. Epub 2008/07/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang Q, Magnusson MK, Mosher DF. Lysophosphatidic acid and microtubule-destabilizing agents stimulate fibronectin matrix assembly through Rho-dependent actin stress fiber formation and cell contraction. Mol Biol Cell. 1997;8(8):1415–25. doi: 10.1091/mbc.8.8.1415. Epub 1997/08/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P, et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell. 2011;19(6):776–91. doi: 10.1016/j.ccr.2011.05.008. Epub 2011/06/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Benitah SA, Valeron PF, van Aelst L, Marshall CJ, Lacal JC. Rho GTPases in human cancer: an unresolved link to upstream and downstream transcriptional regulation. Biochim Biophys Acta. 2004;1705(2):121–32. doi: 10.1016/j.bbcan.2004.10.002. Epub 2004/12/14. [DOI] [PubMed] [Google Scholar]
- 105.Sander EE, van Delft S, ten Klooster JP, Reid T, van der Kammen RA, Michiels F, et al. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J Cell Biol. 1998;143(5):1385–98. doi: 10.1083/jcb.143.5.1385. Epub 1998/12/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lozano E, Betson M, Braga VM. Tumor progression: Small GTPases and loss of cell-cell adhesion. Bioessays. 2003;25(5):452–63. doi: 10.1002/bies.10262. Epub 2003/04/30. [DOI] [PubMed] [Google Scholar]
- 107.Nagy T, Wei H, Shen TL, Peng X, Liang CC, Gan B, et al. Mammary epithelial-specific deletion of the focal adhesion kinase gene leads to severe lobulo-alveolar hypoplasia and secretory immaturity of the murine mammary gland. J Biol Chem. 2007;282(43):31766–76. doi: 10.1074/jbc.M705403200. Epub 2007/08/25. [DOI] [PubMed] [Google Scholar]
- 108.Lahlou H, Sanguin-Gendreau V, Zuo D, Cardiff RD, McLean GW, Frame MC, et al. Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumor progression. Proc Natl Acad Sci U S A. 2007;104(51):20302–7. doi: 10.1073/pnas.0710091104. Epub 2007/12/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Provenzano PP, Inman DR, Eliceiri KW, Beggs HE, Keely PJ. Mammary epithelial-specific disruption of focal adhesion kinase retards tumor formation and metastasis in a transgenic mouse model of human breast cancer. Am J Pathol. 2008;173(5):1551–65. doi: 10.2353/ajpath.2008.080308. Epub 2008/10/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG. Ras- and PI3K-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J Clin Invest. 2009;119(2):252–66. doi: 10.1172/JCI37160. Epub 2009/01/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Luo M, Fan H, Nagy T, Wei H, Wang C, Liu S, et al. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res. 2009;69(2):466–74. doi: 10.1158/0008-5472.CAN-08-3078. Epub 2009/01/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cance WG, Harris JE, Iacocca MV, Roche E, Yang X, Chang J, et al. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin Cancer Res. 2000;6(6):2417–23. Epub 2000/06/29. [PubMed] [Google Scholar]
- 113.Zhao JH, Reiske H, Guan JL. Regulation of the cell cycle by focal adhesion kinase. J Cell Biol. 1998;143(7):1997–2008. doi: 10.1083/jcb.143.7.1997. Epub 1998/12/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Owens LV, Xu L, Craven RJ, Dent GA, Weiner TM, Kornberg L, et al. Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 1995;55(13):2752–5. Epub 1995/07/01. [PubMed] [Google Scholar]
- 115.Haskell H, Natarajan M, Hecker TP, Ding Q, Stewart J, Jr, Grammer JR, et al. Focal adhesion kinase is expressed in the angiogenic blood vessels of malignant astrocytic tumors in vivo and promotes capillary tube formation of brain microvascular endothelial cells. Clin Cancer Res. 2003;9(6):2157–65. Epub 2003/06/11. [PubMed] [Google Scholar]
- 116.Lark AL, Livasy CA, Dressler L, Moore DT, Millikan RC, Geradts J, et al. High focal adhesion kinase expression in invasive breast carcinomas is associated with an aggressive phenotype. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2005;18(10):1289–94. doi: 10.1038/modpathol.3800424. Epub 2005/04/30. [DOI] [PubMed] [Google Scholar]
- 117.Matkowskyj KA, Keller K, Glover S, Kornberg L, Tran-Son-Tay R, Benya RV. Expression of GRP and its receptor in well-differentiated colon cancer cells correlates with the presence of focal adhesion kinase phosphorylated at tyrosines 397 and 407. J Histochem Cytochem. 2003;51(8):1041–8. doi: 10.1177/002215540305100807. Epub 2003/07/23. [DOI] [PubMed] [Google Scholar]
- 118.Oktay MH, Oktay K, Hamele-Bena D, Buyuk A, Koss LG. Focal adhesion kinase as a marker of malignant phenotype in breast and cervical carcinomas. Human pathology. 2003;34(3):240–5. doi: 10.1053/hupa.2003.40. Epub 2003/04/04. [DOI] [PubMed] [Google Scholar]
- 119.Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta. 2004;1678(2–3):111–25. doi: 10.1016/j.bbaexp.2004.03.002. Epub 2004/05/26. [DOI] [PubMed] [Google Scholar]
- 120.Golubovskaya VM, Conway-Dorsey K, Edmiston SN, Tse CK, Lark AA, Livasy CA, et al. FAK overexpression and p53 mutations are highly correlated in human breast cancer. Int J Cancer. 2009;125(7):1735–8. doi: 10.1002/ijc.24486. Epub 2009/06/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ayaki M, Komatsu K, Mukai M, Murata K, Kameyama M, Ishiguro S, et al. Reduced expression of focal adhesion kinase in liver metastases compared with matched primary human colorectal adenocarcinomas. Clin Cancer Res. 2001;7(10):3106–12. Epub 2001/10/12. [PubMed] [Google Scholar]
- 122.Gabriel B, zur Hausen A, Stickeler E, Dietz C, Gitsch G, Fischer DC, et al. Weak expression of focal adhesion kinase (pp125FAK) in patients with cervical cancer is associated with poor disease outcome. Clin Cancer Res. 2006;12(8):2476–83. doi: 10.1158/1078-0432.CCR-05-1867. Epub 2006/04/28. [DOI] [PubMed] [Google Scholar]
- 123.Lu Z, Jiang G, Blume-Jensen P, Hunter T. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol Cell Biol. 2001;21(12):4016–31. doi: 10.1128/MCB.21.12.4016-4031.2001. Epub 2001/05/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Howe AK, Juliano RL. Distinct mechanisms mediate the initial and sustained phases of integrin-mediated activation of the Raf/MEK/mitogen-activated protein kinase cascade. J Biol Chem. 1998;273(42):27268–74. doi: 10.1074/jbc.273.42.27268. Epub 1998/10/09. [DOI] [PubMed] [Google Scholar]
- 125.Lin TH, Aplin AE, Shen Y, Chen Q, Schaller M, Romer L, et al. Integrin-mediated activation of MAP kinase is independent of FAK: evidence for dual integrin signaling pathways in fibroblasts. J Cell Biol. 1997;136(6):1385–95. doi: 10.1083/jcb.136.6.1385. Epub 1997/03/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schlaepfer DD, Hunter T. Focal adhesion kinase overexpression enhances ras-dependent integrin signaling to ERK2/mitogen-activated protein kinase through interactions with and activation of c-Src. J Biol Chem. 1997;272(20):13189–95. doi: 10.1074/jbc.272.20.13189. Epub 1997/05/16. [DOI] [PubMed] [Google Scholar]
- 127.Schlaepfer DD, Jones KC, Hunter T. Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinase: summation of both c-Src- and focal adhesion kinase-initiated tyrosine phosphorylation events. Mol Cell Biol. 1998;18(5):2571–85. doi: 10.1128/mcb.18.5.2571. Epub 1998/05/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2(5):249–56. doi: 10.1038/35010517. Epub 2000/05/12. [DOI] [PubMed] [Google Scholar]
- 129.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4(1):38. doi: 10.1186/1741-7015-4-38. Epub 2006/12/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Provenzano PP, Inman DR, Eliceiri KW, Trier SM, Keely PJ. Contact guidance mediated three-dimensional cell migration is regulated by Rho/ROCK-dependent matrix reorganization. Biophys J. 2008;95(11):5374–84. doi: 10.1529/biophysj.108.133116. Epub 2008/09/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. The American journal of pathology. 2011;178(3):1221–32. doi: 10.1016/j.ajpath.2010.11.076. Epub 2011/03/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Guilak F. Functional tissue engineering. New York: Springer; 2003. p. xvi.p. 426. [Google Scholar]
- 133.Ogle BM, Mooradian DL. The role of vascular smooth muscle cell integrins in the compaction and mechanical strengthening of a tissue-engineered blood vessel. Tissue engineering. 1999;5(4):387–402. doi: 10.1089/ten.1999.5.387. Epub 1999/09/09. [DOI] [PubMed] [Google Scholar]
- 134.Isenberg BC, Backman DE, Kinahan ME, Jesudason R, Suki B, Stone PJ, et al. Micropatterned cell sheets with defined cell and extracellular matrix orientation exhibit anisotropic mechanical properties. Journal of biomechanics. 2012;45(5):756–61. doi: 10.1016/j.jbiomech.2011.11.015. Epub 2011/12/20. [DOI] [PubMed] [Google Scholar]
- 135.Yang N, Mosher R, Seo S, Beebe D, Friedl A. Syndecan-1 in breast cancer stroma fibroblasts regulates extracellular matrix fiber organization and carcinoma cell motility. Am J Pathol. 2011;178(1):325–35. doi: 10.1016/j.ajpath.2010.11.039. Epub 2011/01/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sander EA, Barocas VH, Tranquillo RT. Initial fiber alignment pattern alters extracellular matrix synthesis in fibroblast-populated fibrin gel cruciforms and correlates with predicted tension. Ann Biomed Eng. 2011;39(2):714–29. doi: 10.1007/s10439-010-0192-2. Epub 2010/11/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang JH, Jia F, Gilbert TW, Woo SL. Cell orientation determines the alignment of cell-produced collagenous matrix. Journal of biomechanics. 2003;36(1):97–102. doi: 10.1016/s0021-9290(02)00233-6. Epub 2002/12/18. [DOI] [PubMed] [Google Scholar]
- 138.Beauvais DM, Rapraeger AC. Syndecans in tumor cell adhesion and signaling. Reproductive biology and endocrinology: RB&E. 2004;2:3. doi: 10.1186/1477-7827-2-3. Epub 2004/01/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Friedl P, Wolf K. Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer research. 2008;68(18):7247–9. doi: 10.1158/0008-5472.CAN-08-0784. Epub 2008/09/17. [DOI] [PubMed] [Google Scholar]
- 140.Xu B, Chow MJ, Zhang Y. Experimental and modeling study of collagen scaffolds with the effects of crosslinking and fiber alignment. Int J Biomater. 2011;2011:172389. doi: 10.1155/2011/172389. Epub 2011/08/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bass MD, Roach KA, Morgan MR, Mostafavi-Pour Z, Schoen T, Muramatsu T, et al. Syndecan-4-dependent Rac1 regulation determines directional migration in response to the extracellular matrix. The Journal of cell biology. 2007;177(3):527–38. doi: 10.1083/jcb.200610076. Epub 2007/05/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nature reviews Cancer. 2002;2(3):161–74. doi: 10.1038/nrc745. Epub 2002/05/07. [DOI] [PubMed] [Google Scholar]
- 143.Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer metastasis reviews. 2006;25(1):9–34. doi: 10.1007/s10555-006-7886-9. Epub 2006/05/09. [DOI] [PubMed] [Google Scholar]
- 144.Kohrmann A, Kammerer U, Kapp M, Dietl J, Anacker J. Expression of matrix metalloproteinases (MMPs) in primary human breast cancer and breast cancer cell lines: New findings and review of the literature. BMC cancer. 2009;9:188. doi: 10.1186/1471-2407-9-188. Epub 2009/06/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ahonen M, Baker AH, Kahari VM. Adenovirus-mediated gene delivery of tissue inhibitor of metalloproteinases-3 inhibits invasion and induces apoptosis in melanoma cells. Cancer research. 1998;58(11):2310–5. Epub 1998/06/11. [PubMed] [Google Scholar]
- 146.Deryugina EI, Luo GX, Reisfeld RA, Bourdon MA, Strongin A. Tumor cell invasion through matrigel is regulated by activated matrix metalloproteinase-2. Anticancer research. 1997;17(5A):3201–10. Epub 1997/12/31. [PubMed] [Google Scholar]
- 147.Ala-Aho R, Johansson N, Baker AH, Kahari VM. Expression of collagenase-3 (MMP-13) enhances invasion of human fibrosarcoma HT-1080 cells. International journal of cancer Journal international du cancer. 2002;97(3):283–9. doi: 10.1002/ijc.1619. Epub 2002/01/05. [DOI] [PubMed] [Google Scholar]
- 148.Friedl P, Wolf K. Proteolytic interstitial cell migration: a five-step process. Cancer metastasis reviews. 2009;28(1–2):129–35. doi: 10.1007/s10555-008-9174-3. Epub 2009/01/21. [DOI] [PubMed] [Google Scholar]
- 149.Sabeh F, Shimizu-Hirota R, Weiss SJ. Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. The Journal of cell biology. 2009;185(1):11–9. doi: 10.1083/jcb.200807195. Epub 2009/04/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ilina O, Bakker GJ, Vasaturo A, Hofmann RM, Friedl P. Two-photon laser-generated microtracks in 3D collagen lattices: principles of MMP-dependent and -independent collective cancer cell invasion. Physical biology. 2011;8(1):015010. doi: 10.1088/1478-3975/8/1/015010. Epub 2011/02/09. [DOI] [PubMed] [Google Scholar]
- 151.Friedl P. Prespecification and plasticity: shifting mechanisms of cell migration. Current opinion in cell biology. 2004;16(1):14–23. doi: 10.1016/j.ceb.2003.11.001. Epub 2004/03/24. [DOI] [PubMed] [Google Scholar]
- 152.Wyckoff JB, Pinner SE, Gschmeissner S, Condeelis JS, Sahai E. ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Current biology: CB. 2006;16(15):1515–23. doi: 10.1016/j.cub.2006.05.065. Epub 2006/08/08. [DOI] [PubMed] [Google Scholar]
- 153.Friedl P, Wolf K. Proteolytic and non-proteolytic migration of tumour cells and leucocytes. Biochemical Society symposium; 2003. pp. 277–85. Epub 2003/11/01. [DOI] [PubMed] [Google Scholar]
- 154.Haston WS, Shields JM, Wilkinson PC. Lymphocyte locomotion and attachment on two-dimensional surfaces and in three-dimensional matrices. The Journal of cell biology. 1982;92(3):747–52. doi: 10.1083/jcb.92.3.747. Epub 1982/03/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Harley BA, Kim HD, Zaman MH, Yannas IV, Lauffenburger DA, Gibson LJ. Microarchitecture of three-dimensional scaffolds influences cell migration behavior via junction interactions. Biophysical journal. 2008;95(8):4013–24. doi: 10.1529/biophysj.107.122598. Epub 2008/07/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liu X, Wu H, Byrne M, Jeffrey J, Krane S, Jaenisch R. A targeted mutation at the known collagenase cleavage site in mouse type I collagen impairs tissue remodeling. The Journal of cell biology. 1995;130(1):227–37. doi: 10.1083/jcb.130.1.227. Epub 1995/07/01. [DOI] [PMC free article] [PubMed] [Google Scholar]