

The Polycomb complex PRC1 is a gatekeeper of leukemic cell identity in the presence of distinct oncogenic signals.
Keywords: Polycomb, leukemia, MLL-AF9, histone modifications, PRC1
Abstract
Leukemia is a complex heterogeneous disease often driven by the expression of oncogenic fusion proteins with different molecular and biochemical properties. Whereas several fusion proteins induce leukemogenesis by activating Hox gene expression (Hox-activating fusions), others impinge on different pathways that do not involve the activation of Hox genes (non–Hox-activating fusions). It has been postulated that one of the main oncogenic properties of the HOXA9 transcription factor is its ability to control the expression of the p16/p19 tumor suppressor locus (Cdkn2a), thereby compensating Polycomb-mediated repression, which is dispensable for leukemias induced by Hox-activating fusions. We show, by genetically depleting the H2A ubiquitin ligase subunits of the Polycomb repressive complex 1 (PRC1), Ring1a and Ring1b, that Hoxa9 activation cannot repress Cdkn2a expression in the absence of PRC1 and its dependent deposition of H2AK119 monoubiquitination (H2AK119Ub). This demonstrates the essential role of PRC1 activity in supporting the oncogenic potential of Hox-activating fusion proteins. By combining genetic tools with genome-wide location and transcription analyses, we further show that PRC1 activity is required for the leukemogenic potential of both Hox-activating and non–Hox-activating fusions, thus preventing the differentiation of leukemic cells independently of the expression of the Cdkn2a locus. Overall, our results genetically demonstrate that PRC1 activity and the deposition of H2AK119Ub are critical factors that maintain the undifferentiated identity of cancer cells, positively sustaining the progression of different types of leukemia.
INTRODUCTION
Leukemia is a heterogeneous tumor type sustained by the presence of cancer stem cells (1) and characterized by diverse genetic lesions and rearrangements (2, 3). Acute and chronic myeloid leukemia are frequently characterized by the expression of aberrant oncogenic fusion proteins that are essential to initiate and maintain malignant transformation (4, 5). Gene loci encoding for chromatin remodelers (also referred to as epigenetic factors) are often involved in chromosomal translocations, suggesting a crucial role for these proteins in different types of leukemic transformations (6). In addition, several chromatin modifiers have been found extensively involved in the development of different types of hematopoietic disorders and leukemia (7). For these reasons, potential druggable targets have been proposed for specific types of leukemia, and therapeutic approaches that target different mechanisms of epigenetic regulation are currently under investigation for the treatment of these tumors (6). These include the inhibition of the histone lysine (K) demethylases LSD1 or JMJD3 for treating acute myeloid or lymphoid leukemia, respectively (8, 9), and the inhibition of the Polycomb repressive complex 2 (PRC2), which acts as the specific histone H3K27 methyltransferase (10, 11), for treating leukemia driven by the MLL-AF9 oncogenic fusion protein.
Polycomb group (PcG) proteins are present in two different transcriptional repressive complexes: the aforementioned PRC2 and PRC1, which mediates histone H2AK119 monoubiquitination (H2AK119Ub) (12, 13). PRC2 has been described as having both tumor suppressor and oncogenic functions, depending on cellular context. In mice, genetic inactivation of PRC2 activity induces myelodysplastic syndrome and T cell acute lymphoid leukemia (14, 15). Whereas it has been established that inactivation or pharmacological inhibition of EZH2/EZH1 (the catalytic subunit of PRC2) and Eed (an essential component of the PRC2 core complex) compromises MLL-AF9 leukemic growth through a multifactorial mechanism not entirely dependent on the Cdkn2a locus (16), the roles of PRC1 activity and H2AK119Ub deposition in the leukemic processes have not yet been fully elucidated.
PRC1 was recently shown to have a degree of variation in its subcomplexes. In all of them, the essential E3 ligases, Ring1a and Ring1b components, which both contribute to the deposition of H2AK119Ub, interact with biochemically distinct subunits whose properties and selective functions still remain to be addressed (17). Whereas the so-called canonical PRC1 depends on the activity of PRC2, the other PRC1 variants, generally referred to as noncanonical PRC1 complexes, do not (18). BMI1, a critical component for canonical PRC1 activity (19), was identified as a Myc-cooperative oncogene in lymphomagenesis and has already been implicated in leukemia pathogenesis (20, 21). BMI1 can interact with PLZF-RARα and modulates its oncogenic activity through the transcriptional repression of the well-known tumor-suppressive locus Cdkn2a (also known as Ink4a/Arf) (22). More recently, Bmi1 was found to be indispensable for PML-RARα–dependent leukemia but dispensable for MLL-AF9–driven leukemogenesis (20). The MLL-AF9 oncogenic properties involve the specific activation of the transcription factor HOXA9 (4), which directly mediates the transcriptional repression of the Cdkn2a locus, favoring the leukemic transformation independently of Bmi1 and canonical PRC1 repression (20). Although the proven main function of Bmi1 in leukemia is to transcriptionally repress the Cdkn2a locus, the overall roles of PRC1 activity and H2AK119Ub deposition and their relationship with Cdkn2a transcriptional repression in leukemic cells remain to be addressed.
By using genetic and molecular approaches, we have now characterized, both ex vivo and in vivo, the overall role of PRC1 activity in leukemogenesis, driven by different oncogenic proteins. We show that PRC1 activity and H2AK119Ub are required to repress Cdkn2a expression and sustain the growth of leukemic cells independently of any ability of fusion proteins to activate Hoxa9 expression or of ectopic HOXA9-driven transformation. We further show that PRC1 activity is essential for the development and maintenance of different types of leukemia by sustaining the undifferentiated state of tumor cells independently of Cdkn2a expression. Overall, our data place PRC1 activity and H2AK119Ub deposition as critical events in the different types of leukemogenesis.
RESULTS
PRC1 activity is essential for leukemogenesis independently of oncogenic Hoxa9 activation
To elucidate the roles of the PRC1 activity and the ensuing whole deposition of H2AK119Ub in the self-renewal of hematopoietic cells and during the development of leukemia, we isolated lineage-negative (Lin−) cells from the bone marrow of C57BL/6 mice with a constitutive Ring1a knockout (KO) allele (Ring1a−/−) and a Cre-dependent conditional Ring1b KO allele (Ring1bf/f; cKO) in the presence of a constitutive CreERT2 expression from the Rosa 26 locus (R26CreERT2). Because Ring1a deficiency is fully compensated by Ring1b expression, we will refer to this model from now on as Ring1a/b cKO. The purified Lin− cells were transduced with retroviruses that express the MLL-AF9, HOXA9, or PML-RARα human leukemic oncogenes. In all three cases, we observed the acquisition of a transformed phenotype, which was determined by measuring the immortal growth of the transduced cells in liquid cultures (Fig. 1A), analyzing the maintenance of an undifferentiated morphology upon expression of the three different oncogenes (Fig. 1B and fig. S1A), and determining the maintenance of the self-renewing capacity upon serial replating of three-dimensional (3D) methylcellulose Lin− cell cultures (Fig. 1C). Thus, we used these models to characterize the role of PRC1 activity in the leukemic transformation induced by different oncogenic signals. To do this, we induced full inactivation of Ring1a/b by adding 500 nM 4-hydroxytamoxifen (4-OHT) to the culture medium. This 4-OHT concentration was sufficient to induce the almost complete loss of Ring1b expression and the global loss of H2AK119Ub deposition (fig. S1, B and C) and did not show any toxicity effects on R26CreERT2 Lin− control cells (fig. S1, D to G). The loss of PRC1 activity induced a rapid arrest of leukemic cell growth independently of the oncogenic stimulus in both liquid cultures (Fig. 1A and fig. S1D) and methylcellulose colony formation assays (Fig. 1C and fig. S1E). The normal Lin− cells and the leukemic blasts acquired a clear differentiated morphology in all cases (Fig. 1B and fig. S1, A and F). The loss of PRC1 activity specifically prevented the growth of leukemic cells without affecting the expression of the transduced oncogenes (Fig. 1D). Expression analyses in the same cells demonstrated that, whereas Ring1b was efficiently inactivated under all conditions (Fig. 1E), the loss of PRC1 transcriptional repression clearly activated Cdkn2a expression independently of the type of oncogenic signal involved (Fig. 1E). This result was further confirmed at the protein level, showing that the efficient loss of Ring1b expression correlated with a global loss of H2AK119Ub deposition and with a strong accumulation of p16 levels (Fig. 1F). Consistent with previous reports (23), leukemic transformation induced by the MLL-AF9 fusion protein or by the human form of HOXA9 correlated with a strong activation of endogenous Hoxa9 (Fig. 1G), which, in part, can repress p16 expression (fig. S1H). However, neither physiological (MLL-AF9) nor ectopic activation of HOXA9 are sufficient to maintain p16 and p19 repression in the absence of PRC1 activity (Fig. 1, E and G). Together, these results demonstrate that Hoxa9 expression is not sufficient to compensate the lack of H2AK119Ub deposition induced by the complete loss of PRC1 activity for the maintenance of Cdkn2a transcriptional silencing during leukemogenesis.
Fig. 1. PRC1 activity is required for leukemic cell growth independently of oncogenic Hoxa9 activation.
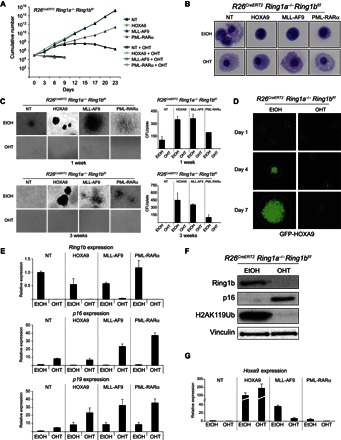
(A) Growth curves of R26CreERT2 Ring1a−/− Ring1bf/f Lin− cells purified from bone marrow and transformed by transduction with lentiviruses expressing the indicated human oncogenic proteins. Full PRC1 inactivation was induced by 4-OHT treatment. Ethanol (EtOH) was used as treatment control. Nontransduced cells were used as nontransformed control (NT). (B) May-Grünwald-Giemsa staining of cells obtained from the experiment presented in (A) at 72 hours after 4-OHT treatment, which shows a differentiated morphology in the absence of PRC1 activity. (C) Methylcellulose colony assays starting from 5000 plated cells, the same presented in (A) at the first and third passages. Quantifications of the number of colonies obtained per plate are presented in the right panels. CFU, colony-forming unit. (D) Green fluorescent protein (GFP) expression in the HOXA9-transformed Lin− cells, demonstrating that the loss of PRC1 activity does not induce a silencing of the transduced oncogene expression construct. (E) Relative expression levels of Ring1b, p16, and p19 determined by quantitative reverse transcription polymerase chain reaction (qRT-PCR) analyses in the Lin− cells shown in (A) and (B), demonstrating both the loss of Ring1b expression upon 72 hours of 4-OHT treatment and the transcriptional activation of the Cdkn2a locus products. Gene expression is normalized to Rpo levels. (F) Western blot analyses with the indicated antibodies on proteins extracted from R26CreERT2 Ring1a−/− Ring1bf/f Lin− cells treated with 4-OHT for 72 hours, showing the loss of Ring1b expression and H2AK119Ub deposition and the concurrent accumulation of p16 protein levels. EtOH was used as treatment control. Vinculin is presented as loading control. (G) Relative expression levels of Hoxa9 determined by qRT-PCR analyses in the R26CreERT2 Ring1a−/− Ring1bf/f Lin− cells expressing the indicated oncogenic proteins and treated for 72 hours with 4-OHT or EtOH (treatment control). Gene expression is normalized to Rpo levels and to the Hoxa9 levels in nontransduced cells (NT).
PRC1 activity sustains leukemogenesis independently of Cdkn2a repression
These results suggest that Cdkn2a activation could have an important role in arresting the growth of leukemic cells. However, we and others (24, 25) have previously reported that PcG proteins can control the growth of normal and tumor cells through Cdkn2a-independent mechanisms. To address this issue, we crossed R26CreERT2 Ring1a−/− Ring1bf/f mice with a constitutive Cdkn2a KO allele [Cdkn2a−/− (26)], from which we isolated Lin− cells with undetectable expression levels of both p16 and p19 (Fig. 2, A and B). The purified cells were transduced and subjected to the same phenotypic analyses performed on R26CreERT2 Ring1a/b cKO Lin− cells (Fig. 2, C to E). Here, the inactivation of Cdkn2a was sufficient to confer an immortal growth and an undifferentiated phenotype on the nontransduced Lin− cells, consistent with the tumor-suppressive properties of p16 and p19 (Fig. 2, C to E, and fig. S2A).
Fig. 2. PRC1 activity is required for leukemic cell growth independently of p16 and p19 expression.
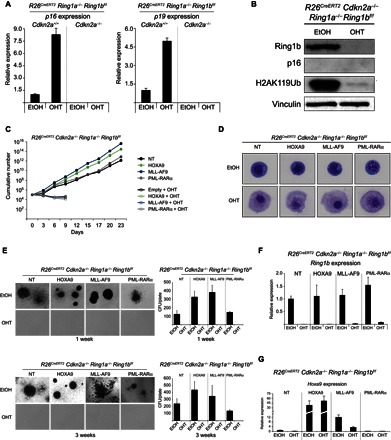
(A) Relative expression levels of p16 and p19 determined by qRT-PCR analyses in the Cdkn2a-proficient and Cdkn2a-deficient Lin− cells treated for 72 hours with 4-OHT or EtOH (treatment control). Gene expression is normalized to Rpo levels. (B) Western blot analyses with the indicated antibodies on proteins extracted from R26CreERT2 Cdkn2a−/− Ring1a−/− Ring1bf/f Lin− cells treated with 4-OHT for 72 hours, demonstrating the loss of Ring1b expression, lack of H2AK119Ub deposition, and absence of p16 expression. EtOH was used as treatment control. Vinculin is presented as loading control. (C) Growth curves of R26CreERT2 Cdkn2a−/− Ring1a−/− Ring1bf/f Lin− cells purified from bone marrow and transformed by transduction with lentiviruses expressing the indicated oncogenic proteins. Full PRC1 inactivation in the absence of p16 and p19 transcriptional activation (Cdkn2a−/−) was induced by 4-OHT treatment. EtOH was used as treatment control. Nontransduced cells were used as nontransformed control. (D) May-Grünwald-Giemsa staining of the cells obtained from the experiment presented in (C) at 72 hours after 4-OHT treatment. (E) Methylcellulose colony assays starting from 5000 plated cells, the same presented in (C) at the first and third passages, displayed the transformed phenotype acquired upon expression of the different oncogenes, highlighting a dependency on PRC1 activity independent of Cdkn2a expression. Quantifications of the number of colonies per plate are presented in the right panels. (F) Relative expression levels of Ring1b determined by qRT-PCR analyses in the Lin− cells shown in (C) and (D), showing efficient loss of Ring1b expression upon 72 hours of 4-OHT treatment. Gene expression is normalized to Rpo levels. (G) Relative expression levels of Hoxa9 determined by qRT-PCR analyses in the R26CreERT2 Cdkn2a−/− Ring1a−/− Ring1bf/f Lin− cells expressing the indicated oncogenic proteins and treated for 72 hours with 4-OHT or EtOH (treatment control). Gene expression is normalized to Rpo levels and used as a nontransformed control on the nontransduced cells.
However, the expression of all oncogenic proteins, particularly MLL-AF9 and HOXA9, conferred a significant growth advantage in both liquid and 3D cultures (Fig. 2, C and E). The efficient inactivation of Ring1b expression (Fig. 2, B and F) induced a marked arrest of leukemic cell growth (Fig. 2, C and E) coupled with the acquisition of a differentiated morphology (Fig. 2D and fig. S2A), independently of the oncogenic stimulus.
Also in this case, Hoxa9 expression was specifically activated in MLL-AF9– and HOXA9-transformed cells (Fig. 2G), without affecting the PRC1 loss-of-function phenotype and suggesting that Hoxa9 oncogenic properties do not involve Cdkn2a repression. Overall, these results demonstrate that PRC1 activity and H2AK119Ub deposition are required to sustain leukemic growth independently of Cdkn2a repression.
PRC1 activity and H2AK119Ub maintain the undifferentiated state of leukemic cells
To characterize the direct molecular effects controlled by PRC1 activity in leukemic cells, we performed chromatin immunoprecipitation–sequencing (ChIP-seq) analyses for H2AK119Ub in Cdkn2a−/− Lin− cells nontransduced or transduced with MLL-AF9, HOXA9, or PML-RARα. The specificity of the H2AK119Ub signal was confirmed by the ChIP-qPCR approach in Ring1a/b-proficient or Ring1a/b-deficient mouse embryonic stem (ES) cells (fig. S3A) and by checking established H2AK119Ub-positive and H2AK119Ub-negative regions on Cdkn2a+/+ or Cdkn2a−/− Lin− cells (fig. S3, B and C). The analysis of the H2AK119Ub genome-wide deposition displayed a largely overlapping profile upon the expression of each different oncogenic protein (Fig. 3A, fig. S3D, and table S1), suggesting that PRC1 activity modifies the same genomic loci independently of the transformation mechanism of Lin− cells. Consistent with the repressive role of H2AK119Ub, RNA sequencing (RNA-seq) analysis performed on the same cells 72 hours after 4-OHT–induced Ring1a/b inactivation revealed a larger number of genes that were transcriptionally activated (up-regulated) compared to the genes that were silenced (down-regulated) by PRC1 loss of function (Fig. 3B, fig. S3E, and table S2).
Fig. 3. Conserved genomic association of PRC1 activity in different leukemic cells.
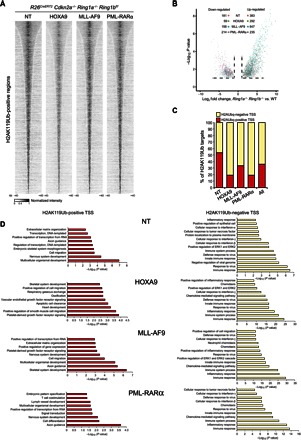
(A) Cumulative H2AK119Ub ChIP-seq signals among all enriched loci in the R26CreERT2 Cdkn2a−/− Ring1a−/− Ring1bf/f nontreated Lin− cells expressing the indicated oncogenic proteins show that H2AK119Ub accumulated to the same genomic loci upon transformation with different oncogenes. Rows are sorted by decreasing normalized intensity centered in the ±40-kb window surrounding the H2AK119Ub peaks. (B) Composite volcano plot showing the significantly differentially regulated genes 72 hours after 4-OHT or EtOH (treatment control) treatment in the R26CreERT2 Cdkn2a−/− Ring1a−/− Ring1bf/f Lin− cells expressing the indicated oncogenic proteins. WT, wild type. (C) Percentage of H2AK119Ub-enriched promoters undergoing transcriptional activation (fold change ≥ 4) after loss of PRC1 activity. Expression was determined by RNA-seq analysis in the R26CreERT2 Cdkn2a−/− Ring1a−/− Ring1bf/f Lin− cells expressing the indicated oncogenic proteins at 72 hours after the 4-OHT treatment. EtOH was used as control treatment. Red bars represent the percentage of H2AK119Ub-positive promoters (PRC1 direct targets); yellow bars represent the H2AK119Ub-negative promoters (PRC1 indirect targets). TSS, transcription start site. (D) P values of the significantly enriched pathways identified by gene ontology interrogation for the activated genes presented in (C). Left panels represent the functional pathways enriched among the PRC1 direct targets; right panels represent the functional pathways enriched among the PRC1 indirect targets.
The combination of RNA-seq and ChIP-seq analyses revealed that a significant number of H2AK119Ub decorated promoters underwent direct transcriptional activation (>30%; red bars in Fig. 3C). Despite the conserved profile of H2AK119Ub deposition among the different leukemic cells (Fig. 3A), the proportion of activated direct targets varied among samples (Fig. 3C), suggesting that the different oncogenic signals or differentiation statuses of the leukemic cells could diversely affect PRC1 direct target reactivation. With gene ontology analysis, it also emerged that under all conditions, the loss of PRC1 directly affects the expression of a set of genes with developmental and differentiation functions, whereas the activation of pathways involved in hematopoietic differentiation in Ring1a/Ring1b double-KO cells is the result of a secondary effect of the primary deregulation (Fig. 3D).
The common differentially regulated genes between the different leukemic cells, which displayed a larger number of up-regulated genes (fig. S3, F and G), showed a general enrichment for ontology annotations that are related to the acquisition of a differentiated phenotype (fig. S3H). Overall, these results reveal that the loss of PRC1 transcriptional control triggers differentiation into multiple hematopoietic lineages.
To further confirm that PRC1 activity is required to sustain the undifferentiated phenotype of leukemic cells, we performed fluorescence-activated cell sorting (FACS) staining using markers that characterize different lineages of hematopoietic differentiation. Consistent with the differentiation block of leukemic cells in the myeloid precursor, cells transformed with MLL-AF9, HOXA9, or PML-RARα showed high levels of macrophage (Mac1) and granulocyte (Gr1) markers (fig. S4) and low levels of markers for different hematopoietic lineages, such as megakaryocytes (CD61), erythrocytes (Ter119), B cells (B220), and T cells (CD3e) (Fig. 4). In agreement with the RNA-seq results, Ring1a/b inactivation induced the activation of several differentiation markers in all types of leukemic cells (Fig. 4 and fig. S4), suggesting that PRC1 activity may sustain leukemic transformation by preventing the activation of differentiation programs.
Fig. 4. Loss of PRC1 activity induces leukemic cell differentiation.
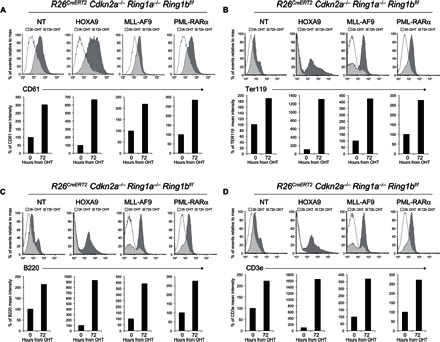
FACS analyses and quantification of the levels of differentiation markers upon loss of PRC1 activity in the different transformed Lin− cells treated with 4-OHT at the indicated time points. CD61 (A) is presented for megakaryocyte differentiation; Ter119 (B) for erythrocytes; B220 (C) for B cells; and CD3e (D) for T cell differentiation.
Loss of PRC1 induces MLL-AF9 leukemic cell growth arrest and transdifferentiation independently of HOXA9 and p53 activation
These results suggest that the global PRC1 activity and the deposition of H2AK119Ub play critical roles in maintaining the undifferentiated state of different leukemic cells. However, these data only demonstrate the requirement of PRC1 activity in the early onset of leukemic transformation without addressing its role in primary tumors. To also address this point, we focused our attention on MLL-AF9 because of the essential role of Hoxa9 activation in the development of this type of leukemia (23). Thus, we generated primary leukemia in vivo by inoculating R26CreERT2 Cdkn2a−/− Ring1a−/− Ring1bf/f Lin− cells transduced with MLL-AF9 into immunocompromised recipient mice (fig. S5A). The occurrence of advanced primary leukemia was confirmed by hematoxylin and eosin (H&E) staining of spleen and liver sections isolated from leukemic mice that showed diffuse, cell-dense infiltrations and disruption of the architecture of both organs (fig. S5B, top panels) and by May-Grünwald-Giemsa staining that highlighted the presence of undifferentiated blasts in the peripheral blood samples (fig. S5B, bottom panels). We isolated primary leukemia cells from both the spleen and the bone marrow of these mice and confirmed their transformed phenotype by indefinite growth in methylcellulose 3D replating assays (Fig. 5A and fig. S5C). Consistent with the essential role of PRC1 in sustaining leukemia development, the depletion of Ring1a/b activity in primary leukemia cells derived from either the spleen or the bone marrow severely impaired their growth and self-renewing capacity (Fig. 5A and fig. S5C).
Fig. 5. Loss of H2AK119Ub in MLL-AF9–induced primary leukemia blocks cell growth.
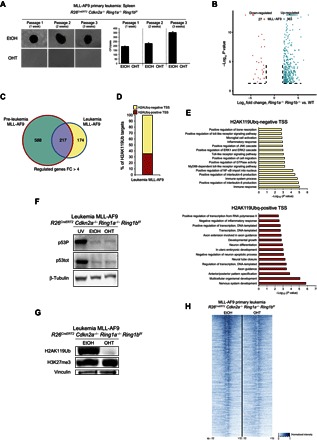
(A) Methylcellulose colony assays starting from 1000 plated cells isolated from the spleen of a mouse that had developed a primary MLL-AF9 leukemia after intravenous inoculation of the MLL-AF9–transduced R26CreERT2 Cdkn2a−/− Ring1a−/− Ring1bf/f Lin− cells. Loss of PRC1 activity was induced by 4-OHT treatment at each methylcellulose passage. Quantifications of the colony number per plate are presented in the bottom panel. (B) Composite volcano plot showing the significantly differentially regulated genes 72 hours after 4-OHT treatment of the R26CreERT2 Cdkn2a−/− Ring1a−/− Ring1bf/f MLL-AF9 primary leukemia cells. EtOH was used as control treatment. (C) Overlaps between the up-regulated genes [fold change (FC) ≥ 4] in the MLL-AF9 preleukemic cells and MLL-AF9 leukemic cells upon loss of PRC1 activity. (D) Percentage of H2AK119Ub-enriched promoters undergoing transcriptional activation (fold change ≥ 4) after loss of PRC1 activity. Expression was determined by RNA-seq analysis in the indicated cells after 72 hours from the 4-OHT treatment. EtOH was used as control treatment. Red bars represent the percentage of H2AK119Ub-positive promoters (PRC1 direct targets); yellow bars represent the H2AK119Ub-negative promoters (PRC1 indirect targets). (E) P values of the significantly enriched pathways identified by gene ontology interrogation among the activated genes presented in (D). Top panel represents the functional pathways enriched among the PRC1 indirect targets; bottom panel represents the functional pathways enriched among the PRC1 direct targets. (F) Western blot analyses for total p53 (p53tot) and p53 phosphorylated (p53P) in the R26CreERT2 Cdkn2a−/− Ring1a−/− Ring1bf/f MLL-AF9 primary leukemia cells 72 hours after EtOH or 4-OHT addition, showing that the loss of PRC1 activity does not activate the p53 pathway. Ultraviolet-irradiated R26CreERT2 Cdkn2a−/− Ring1a−/− Ring1bf/f MLL-AF9 primary leukemia cells were used as p53/p53P-positive control (UV). β-Tubulin is presented as loading control. (G) Western blot analysis for H3K27me3 showing that its global deposition is not affected by the loss of H2AK119Ub in the R26CreERT2 Cdkn2a−/− Ring1a−/− Ring1bf/f MLL-AF9 primary leukemia cells 72 hours after EtOH or 4-OHT addition. Vinculin is presented as loading control. (H) Heat map representing the normalized H3K27me3 ChIP-seq intensities ±10 kb around the summit of H3K27me3-positive promoters identified in R26CreERT2 Cdkn2a−/− Ring1a−/− Ring1bf/f MLL-AF9 primary leukemia cells 72 hours after EtOH or 4-OHT addition.
To gain further insights into the apparent loss of cell identity observed in the Lin− in vitro model, we decided to characterize the transcriptional program controlled by PRC1 in the MLL-AF9 primary leukemia cells. In accordance with our previous results (Fig. 3), loss of global PRC1 activity (fig. S5D) resulted in a larger number of up-regulated genes (363 up-regulated versus 27 down-regulated genes; Fig. 5B and table S3). The comparison of the transcriptional deregulation between the preleukemic MLL-AF9– and leukemic MLL-AF9–expressing cells showed highly similar expression profiles with more than 55% of genes commonly deregulated under both conditions (Fig. 5C). Moreover, the RNA-seq and ChIP-seq analyses revealed that 35% of these up-regulated genes are PRC1 direct targets (Fig. 5D, red bar), enriched in ontology pathways related to tissue development (Fig. 5E, bottom panel), whereas the up-regulated genes harboring H2AK119Ub-negative promoters are associated with the immune response (Fig. 5, D and E, top panel, and tables S3 and S4).
Western blot analyses for total and phosphorylated p53 in the MLL-AF9 primary leukemic cells 72 hours after EtOH or 4-OHT addition revealed that the loss of PRC1 activity does not activate the p53 pathway (Fig. 5F), further supporting a role for PRC1 in maintaining the undifferentiated state of leukemic cells.
Moreover, because the loss of PRC1 function in MLL-AF9 leukemic cells also resulted in a reduced expression of endogenous Hoxa9 (Figs. 1G and 2G), we tested whether HOXA9 overexpression in MLL-AF9 primary leukemia cells was sufficient to rescue this phenotype. Consistent with the requirement of PRC1 to sustain leukemic growth induced by HOXA9 expression alone (Figs. 1 and 2), the ectopic expression of HOXA9 in MLL-AF9 leukemic cells was not sufficient to revert PRC1 essentiality (fig. S5E), further confirming that the PRC1 role in the leukemic cells is Hoxa9- and Cdkn2a-independent.
PRC1 was recently shown to modulate PRC2 activity on chromatin through direct recognition of the H2AK119Ub mark, which globally sustains H3K27me3 deposition in ES cells (27–29). To test whether the global loss of PRC1 affects PRC2 activity even in leukemic cells, we assayed the deposition of H3K27me3 in MLL-AF9 leukemic cells upon Ring1a/Ring1b double KO. Unlike ES cells, the loss of H2AK119Ub deposition did not affect the ability of PRC2 to methylate H3K27 in bulk (Fig. 5G). To rule out the possibility of a redistribution of H3K27me3, we also performed ChIP-seq analyses for H3K27me3 in the same cells upon inactivation of PRC1 activity (Fig. 5H, fig. S5F, and table S4). This confirmed that PRC1-dependent H2AK119Ub does not control PRC2 activity in MLL-AF9 primary leukemia cells, underlining the specific role of PRC1 in sustaining leukemic transformation.
Finally, to address the controversial role of Bmi1 in relation to MLL-AF9 transformation (20, 21) by taking into consideration, for the first time, the impact of the catalytic activity of PRC1, we down-regulated its expression through constitutive short hairpin RNA (shRNA). To do so, we first tested a panel of individual shRNA molecules that specifically target Bmi1 in mouse embryonic fibroblasts (fig. S6, A and B), and the two most effective shRNAs were then transduced in MLL-AF9–transformed leukemic cells. Loss of Bmi1 expression had a minor effect on MLL-AF9 leukemic cell growth in liquid culture, with respect to the global loss of PRC1 activity (fig. S6C). This result was further confirmed by serial replating of Bmi1 knockdown cells in methylcellulose (fig. S6D). Consistent with this, the loss of Bmi1 was not counterselected (fig. S6E) and did not affect the global deposition of H2AK119Ub, suggesting redundant functions among different forms of PRC1 (fig. S6F) in the MLL-AF9 leukemic cells. Overall, the strong correlation between the loss of H2AK119Ub and the cell defects observed in the MLL-AF9 primary leukemia cells (Fig. 5) supports a model by which loss of H2AK119Ub blocks the growth of cells and induces their transdifferentiation in a p16/p19- and p53-independent manner.
PRC1 activity is required to maintain primary leukemia independently of Cdkn2a
Whereas transplantation of primary MLL-AF9 leukemic cells in immunocompromised [nonobese diabetic/severe combined immunodeficient (NOD/SCID)] or immunocompetent (C57BL/6) recipient mice rapidly induced secondary leukemias, the loss of PRC1 activity, driven by a single intraperitoneal injection of tamoxifen, significantly delayed the occurrence of leukemia, considerably increasing the life span of the mice (Fig. 6, A and B). Weekly tamoxifen injections improve the survival rate of the mice (Fig. 6B), suggesting that the leukemic cells that kill the mice could be PRC1-proficient escapee cells.
Fig. 6. PRC1 activity is essential for leukemia development both in vitro and in vivo.
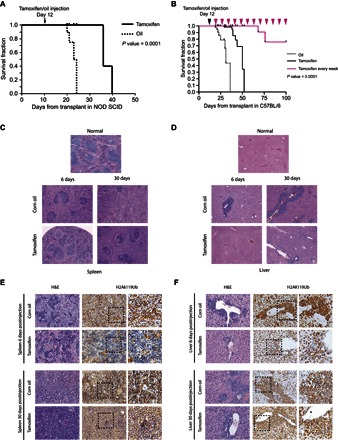
(A) Kaplan-Meier survival curves of NOD/SCID mice intravenously inoculated with 1 × 106 MLL-AF9 primary leukemic cells. Mice were treated with tamoxifen to inactivate the Ring1b allele at 12 days after leukemia transplant (arrow). For mice transplants: oil, n = 4; tamoxifen, n = 4. P value was determined by χ2 test. (B) Kaplan-Meier survival curves of C57BL/6 mice intravenously inoculated with 1 × 106 MLL-AF9 primary leukemic cells. Mice were treated with tamoxifen to inactivate the Ring1b allele at 12 days after leukemia transplant (black arrowhead). For mice transplants: oil, n = 8; tamoxifen, n = 9. The pink arrowheads indicate the weekly tamoxifen injections for the group of mice (n = 5) that show improvement in the survival rate (pink survival curve), demonstrating that the leukemic cells that kill the mice are PRC1-proficient escapee cells. P value was determined by χ2 test. (C and D) H&E staining of spleen (C) and liver tissues (D) collected at day 6 or 30 after injection shows a milder leukemic phenotype in the tamoxifen-treated mice. (E and F) Immunohistochemical analyses of spleen (E) and liver tissues (F) collected at days 6 and 30 after tamoxifen injections, showing the rapid impairment of the active H2AK119Ub deposition in infiltrated leukemic cells compared to the control tissues (6 days after injection; top panels) as a consequence of the acute inactivation of PRC1 activity. Thirty days after tamoxifen injections, the infiltrated leukemic cells display an H2AK119Ub-positive staining (bottom panels), indicating the counterselection of H2AK119Ub-negative cells, with respect to the infiltrated leukemic cells with unexcised Ring1b allele. The far right panels are a magnification of the dashed square areas.
H&E staining of spleen and liver tissues collected at day 6 or 30 after injection shows a milder leukemic phenotype in the tamoxifen-treated mice (Fig. 6, C and D). Immunohistochemical analyses show the rapid impairment of the active H2AK119Ub deposition (which is a consequence of the acute inactivation of PRC1 activity) in infiltrated leukemic cells, as compared to control tissues 6 days after tamoxifen treatment (Fig. 6, E and F). On the contrary, 30 days after the tamoxifen injections, the infiltrated leukemic cells displayed a positive H2AK119Ub staining (Fig. 6, E and F), which demonstrates that the H2AK119Ub-negative cells were strongly counterselected compared to the cells with unexcised Ring1b allele. Together, these results demonstrate that PRC1 activity is essential not only for the development of leukemia but also for maintaining a leukemic phenotype in vivo.
DISCUSSION
All available data that related PRC1 with the oncogenic activity of leukemic fusion proteins focused on the roles of different specific components of the canonical PRC1 complex, such as Bmi1 (20, 21) and Cbx (30) proteins, without analyzing the overall requirement of PRC1-dependent H2AK119Ub deposition in leukemic cells. Our study highlights, for the first time, the critical roles of the PRC1 activity and the global deposition of H2AK119Ub in controlling the undifferentiated state of different types of leukemic cells.
Here, we have shown that the full loss of all PRC1 activities in Lin− cells severely impairs leukemic cell growth and self-renewal capacity, independently of the oncogenic stimulus. Moreover, the inactivation of Ring1a/b E3 ubiquitin ligases results in the complete lack of H2AK119Ub deposition and the transcriptional activation of the Cdkn2a locus in all types of leukemic cells, without any sign of compensation mediated by Hoxa9 activation, as previously reported for specific Bmi1 loss of function (20). Our results extend the previous observations that identified the ability of oncogenes to activate Hox genes, especially Hoxa9, as a molecular determinant for the dependency of tumor growth on PRC1 activity. This discrepancy places PcG and H2AK119Ub as critical determinants for p16 and p19 silencing in all tumor contexts and further suggests that the overall Ring1a/b activity will likely have broader functions in normal hematopoiesis and in leukemic transformation than previously supposed.
In addition to Cdkn2a regulation, our results also show that MLL-AF9 or direct HOXA9 expression transformed Cdkn2a-null Lin− cells, conferring a ~100-fold increased proliferation rate on immortal cells in liquid cultures. This strongly suggests that both oncogenes transform normal Lin− cells through additional mechanisms. Moreover, we established that PRC1 activity is not just required to maintain Cdkn2a repression upon different oncogenic insults, but it is also essential for the leukemic cell growth, in a mechanism that is independent of p16 and p19 expression, which is consistent with our previous findings in different model systems (24, 31). The evaluation of the response of a primary MLL-AF9 Cdkn2a−/− leukemia to the complete loss of Ring1a/Ring1b activity also demonstrates the fundamental role of PRC1 in the maintenance of established leukemia. Furthermore, these data show that neither Cdkn2a repression nor p53 activation is involved in Ring1a/b-dependent control of the leukemic transformation.
By mapping the genome-wide deposition of H2AK119Ub, we established that the genomic loci directly controlled by PRC1 activity are conserved to a high degree in the different leukemic cells. This means that different oncogene stimulations do not perturb the mechanisms that determine PRC1 recruitment to its genomic targets or that regulate its enzymatic activity. Notably, even if the PRC1 targets in Lin− cells are similar independently of the leukemogenic proteins expressed, their reactivation upon the loss of H2AK119Ub deposition seems to be strongly influenced by the oncogenic triggering mechanism. Knowing that the epigenetic regulation of gene expression is a sophisticated process that involves several regulators, different epigenetic settings may influence the activation or repression patterns of the PRC1 target genes. This result is consistent with previous literature showing that only a minor fraction of PcG targets can be derepressed by loss of function (32), stressing the relevance of the cellular context for downstream effects, which is also in agreement with the dual role of oncogenes and tumor suppressors in different types of cancers.
Our transcriptional analyses show that PRC1 repressive activity is required to prevent the activation of direct and indirect lineage differentiation programs, which ensure the cellular identity of leukemic cells. The direct effect of PRC1 activity is strictly linked with the suppression of developmental programs and differentiation-triggering genes, whereas the activation of markers of hematopoietic differentiation and inflammatory response resulted from an indirect consequence of this primary deregulation. However, it is worth noting that Ring1a/b also plays a similar role in normal Lin− cells, suggesting that PRC1 activity counteracts differentiation stimuli to preserve the undifferentiated, high-proliferative state of hematopoietic progenitors. This observation further highlights the dominant “gatekeeper” properties of PRC1 over the activity of different oncogenic stimuli.
All types of leukemic cells start to transdifferentiate, expressing all the hematopoietic differentiation markers as soon as the Ring1a/b activities are abrogated. This observation is in accordance with the well-established role of PRC1 in maintaining the correct lineage identity (31, 33) of the cells and strongly supports our hypothesis about its role in sustaining leukemic transformation by preventing the activation of hematopoietic differentiation programs. Because epigenetic factors represent the master regulators of “lineage switching” observed within acute leukemic patients, defined as the capacity of changing cell fate without altering the genotype (34, 35), anomalous PcG activity may deregulate stem cell plasticity by derepressing lineage-specific genes, allowing the onset and progression of leukemia.
In our cellular context, the loss of H2AK119Ub deposition does not affect the levels and the chromatin localization of H3K27me3, suggesting that PRC2 recruitment and activity in primary MLL-AF9 Cdkn2a−/− leukemia cells are not influenced by the PRC1 enzymatic activity, as observed in other cellular systems (27–29). This emphasizes the dominant role of PRC1 in the maintenance of the Lin− cell–specific transcription program.
In primary MLL-AF9 Cdkn2a−/− leukemia cells, knocking down Bmi1, a central player for the canonical PRC1 complex activity (19), did not affect the bulk deposition of H2AK119Ub and only mildly impaired the proliferative capacity. This possibly depends on the residual expression of Bmi1 protein, which could be sufficient to support leukemogenesis, as well as on its compensation in the canonical PRC1 complex by Mel18. In contrast, the loss of Ring1a/b not only fully abrogates H2AK119Ub deposition but also severely impairs the growth of the cells and their self-renewal capacity. This corroborates the central role of H2AK119Ub deposition in the leukemogenesis process, highlighting a large degree of functional redundancies among the different PRC1 subcomplexes.
On the one hand, our results suggest that PRC1 activity could be targeted to treat different types of leukemia; on the other hand, these findings highlight the essential function of PRC1 activity in the self-renewal of normal hematopoietic Lin− cells. Therefore, our findings underscore the need to develop strategies to directly target Ring1a/b activity as well as the need to dissect, in molecular detail, the mechanisms by which different PRC1 complexes would contribute to normal hematopoiesis and to leukemia development.
MATERIALS AND METHODS
Growth curves and methylcellulose assays
Lin− cells were cultured in Dulbecco’s modified Eagle’s medium (Lonza) supplemented with 10% fetal bovine serum for mouse myeloid colony-forming cells (scFBS, STEMCELL Technologies), stem cell factor (100 ng/ml) (PeproTech), recombinant interleukin-3 (IL-3) (20 ng/ml) (PeproTech), and IL-6 (20 ng/ml) (PeproTech).
Growth curves were performed by plating 1 × 105 Lin− cells per well in a 24-well plate for each day of the growth curve in the presence of 500 nM 4-OHT (Sigma). EtOH (Panreac) was used as a vector control.
For the methylcellulose assay, 5 × 103 cells were plated in a 35-mm dish and mixed with 1.2 ml of MethoCult GF M3434 (STEMCELL Technologies) in the presence of 500 nM 4-OHT (Sigma). EtOH (Panreac) was used as a vector control. Colonies were scored after 7 days of culture and replated every 7 days. Pictures of colonies were taken using the EVOS FL microscope.
Transplantation
Five-week-old NOD/SCID (Charles River) mice were injected intravenously with 1 × 106 MLL-AF9–transduced Lin− cells harvested from the third methylcellulose assay. Ten-week-old NOD/SCID (8 mice) and C57BL/6 (17 mice) were injected intravenously with 1 × 106 MLL-AF9 primary leukemic blasts. Tamoxifen treatment was performed by two intraperitoneal injections with 2 mg of tamoxifen at days 12 and 14 after leukemic cell transplant, and every 7 days after that. Identical volumes of oil were injected into the control cohort of mice.
Additional methods
A detailed description of the mouse models, the retroviral vectors, Lin− purification, transduction and morphological evaluation, real-time qPCRs, Western blots, flow cytometry, and ChIP-seq and RNA-seq sample preparation is available in the Supplementary Materials.
Data set accession
The RNA-seq and ChIP-seq data are deposited at the Gene Expression Omnibus (GEO) database under accession no. GSE67552.
Supplementary Material
Acknowledgments
We thank V. Raker for editing support and H. Koseki and M. Vidal for the Ring1a and Ring1b KO mice. We thank all members of the Pasini group for helpful discussions. We thank M. Rescigno for providing conjugated antibodies and B. Amati and S. Minucci for the lentiviral vectors. Funding: The work in the Pasini laboratory was supported by grants from the Italian Association for Cancer Research (IG-2014-15798), the Italian Ministry of Health (1/GR-2011-02348313), and the Umberto Veronesi Foundation (FUV, bando 2014). A.R., A.P., and A.S. were supported by fellowships from the Italian Foundation for Cancer Research. F.C. was supported by a fellowship from FUV. Author contributions: D.P., A.P., and A.R. conceived the experiments. A.R. and K.J.F. performed the experimental work. F.C. provided support with the experiments. A.S. generated the Ring1b antibody. S.J. performed all the computational analyses. L.M. provided reagents and assistance with the experiments. P.G.P. provided reagents and intellectual contribution. D.P. and A.R. wrote the manuscript. A.R., K.J.F., and S.J. revised the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Next-generation sequencing data sets have been deposited and are available at the GEO database under accession no. GSE67552. Correspondence for reagents should be addressed to D.P. (diego.pasini@ieo.eu).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/10/e1600972/DC1
Supplementary Materials and Methods
fig. S1. Loss of PRC1 induces a differentiated phenotype in Lin− transformed cells.
fig. S2. Loss of PRC1 induces a differentiated phenotype independently of Cdkn2a expression.
fig. S3. Specificity check of H2AK119Ub signal.
fig. S4. Loss of PRC1 activity induces leukemic cell differentiation.
fig. S5. Loss of PRC1 activity negatively affects the growth of primary leukemia both in vitro and in vivo.
fig. S6. Bmi1 knockdown does not recapitulate the Ring1a/b-deficient phenotype.
table S1. ChIP-seq results in Lin− and leukemic cells.
table S2. Genome-wide expression in wild-type, PRC1 Lin−, and leukemic cells.
table S3. Genome-wide expression in primary MLL-AF9 leukemic cells.
table S4. ChIP-seq results in primary MLL-AF9 leukemic cells.
REFERENCES AND NOTES
- 1.Wang J. C. Y., Dick J. E., Cancer stem cells: Lessons from leukemia. Trends Cell Biol. 15, 494–501 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Marcucci G., Haferlach T., Döhner H., Molecular genetics of adult acute myeloid leukemia: Prognostic and therapeutic implications. J. Clin. Oncol. 29, 475–486 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Hehlmann R., Hochhaus A., Baccarani M. European LeukemiaNet , Chronic myeloid leukaemia. Lancet 370, 342–350 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Faber J., Krivtsov A. V., Stubbs M. C., Wright R., Davis T. N., van den Heuvel-Eibrink M., Zwaan C. M., Kung A. L., Armstrong S. A., HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood 113, 2375–2385 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martens J. H. A., Stunnenberg H. G., The molecular signature of oncofusion proteins in acute myeloid leukemia. FEBS Lett. 584, 2662–2669 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Greenblatt S. M., Nimer S. D., Chromatin modifiers and the promise of epigenetic therapy in acute leukemia. Leukemia 28, 1396–1406 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Helin K., Dhanak D., Chromatin proteins and modifications as drug targets. Nature 502, 480–488 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Schenk T., Chen W. C., Göllner S., Howell L., Jin L., Hebestreit K., Klein H.-U., Popescu A. C., Burnett A., Mills K., Casero R. A. Jr, Marton L., Woster P., Minden M. D., Dugas M., Wang J. C. Y., Dick J. E., Müller-Tidow C., Petrie K., Zelent A., Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat. Med. 18, 605–611 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ntziachristos P., Tsirigos A., Welstead G. G., Trimarchi T., Bakogianni S., Xu L., Loizou E., Holmfeldt L., Strikoudis A., King B., Mullenders J., Becksfort J., Nedjic J., Paietta E., Tallman M. S., Rowe J. M., Tonon G., Satoh T., Kruidenier L., Prinjha R., Akira S., Van Vlierberghe P., Ferrando A. A., Jaenisch R., Mullighan C. G., Aifantis I., Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 514, 513–517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neff T., Sinha A. U., Kluk M. J., Zhu N., Khattab M. H., Stein L., Xie H., Orkin S. H., Armstrong S. A., Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc. Natl. Acad. Sci. U.S.A. 109, 5028–5033 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim W., Bird G. H., Neff T., Guo G., Kerenyi M. A., Walensky L. D., Orkin S. H., Targeted disruption of the EZH2–EED complex inhibits EZH2-dependent cancer. Nat. Chem. Biol. 9, 643–650 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aloia L., Di Stefano B., Di Croce L., Polycomb complexes in stem cells and embryonic development. Development 140, 2525–2534 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Stock J. K., Giadrossi S., Casanova M., Brookes E., Vidal M., Koseki H., Brockdorff N., Fisher A. G., Pombo A., Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat. Cell Biol. 9, 1428–1435 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Ntziachristos P., Tsirigos A., Van Vlierberghe P., Nedjic J., Trimarchi T., Flaherty M. S., Ferres-Marco D., da Ros V., Tang Z., Siegle J., Asp P., Hadler M., Rigo I., De Keersmaecker K., Patel J., Huynh T., Utro F., Poglio S., Samon J. B, Paietta E., Racevskis J., Rowe J. M., Rabadan R., Levine R. L., Brown S., Pflumio F., Dominguez M., Ferrando A., Aifantis I., Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 18, 298–303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simon C., Chagraoui J., Krosl J., Gendron P., Wilhelm B., Lemieux S., Boucher G., Chagnon P., Drouin S., Lambert R., Rondeau C., Bilodeau A., Lavallée S., Sauvageau M., Hébert J., Sauvageau G., A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev. 26, 651–656 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danis E., Yamauchi T., Echanique K., Haladyna J., Kalkur R., Riedel S., Zhu N., Xie H., Bernt K. M., Orkin S. H., Armstrong S. A., Neff T., Inactivation of Eed impedes MLL-AF9–mediated leukemogenesis through Cdkn2a-dependent and Cdkn2a-independent mechanisms in a murine model. Exp. Hematol. 43, 930–935 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao Z., Zhang J., Bonasio R., Strino F., Sawai A., Parisi F., Kluger Y., Reinberg D., PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 45, 344–356 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Comet I., Helin K., Revolution in the Polycomb hierarchy. Nat. Struct. Mol. Biol. 21, 573–575 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Di Croce L., Helin K., Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 20, 1147–1155 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Smith L.-L., Yeung J., Zeisig B. B., Popov N., Huijbers I., Barnes J., Wilson A. J., Taskesen E., Delwel R., Gil J., Van Lohuizen M., So C. W. E., Functional crosstalk between Bmi1 and MLL/Hoxa9 axis in establishment of normal hematopoietic and leukemic stem cells. Cell Stem Cell 8, 649–662 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Yuan J., Takeuchi M., Negishi M., Oguro H., Ichikawa H., Iwama A., Bmi1 is essential for leukemic reprogramming of myeloid progenitor cells. Leukemia 25, 1335–1343 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Boukarabila H., Saurin A. J., Batsché E., Mossadegh N., van Lohuizen M., Otte A. P., Pradel J., Muchardt C., Sieweke M., Duprez E., The PRC1 Polycomb group complex interacts with PLZF/RARA to mediate leukemic transformation. Genes Dev. 23, 1195–1206 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thiel A. T., Blessington P., Zou T., Feather D., Wu X., Yan J., Zhang H., Liu Z., Ernst P., Koretzky G. A., Hua X., MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell 17, 148–159 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Piunti A., Rossi A., Cerutti A., Albert M., Jammula S., Scelfo A., Cedrone L., Fragola G., Olsson L., Koseki H., Testa G., Casola S., Helin K., d’Adda di Fagagna F., Pasini D., Polycomb proteins control proliferation and transformation independently of cell cycle checkpoints by regulating DNA replication. Nat. Commun. 5, 3649 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruggeman S. W. M., Hulsman D., Tanger E., Buckle T., Blom M., Zevenhoven J., van Tellingen O., van Lohuizen M., Bmi1 controls tumor development in an Ink4a/Arf-independent manner in a mouse model for glioma. Cancer Cell 12, 328–341 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Serrano M., Lee H.-W., Chin L., Cardo-Carlo C., Beach D., DePinho R. A., Role of the INK4a locus in tumor suppression and cell mortality. Cell 85, 27–37 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Cooper S., Dienstbier M., Hassan R., Schermelleh L., Sharif J., Blackledge N. P., De Marco V., Elderkin S., Koseki H., Klose R., Heger A., Brockdorff N., Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep. 7, 1456–1470 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blackledge N. P., Farcas A. M., Kondo T., King H. W., McGouran J. F., Hanssen L. L. P., Ito S., Cooper S., Kondo K., Koseki Y., Ishikura T., Long H. K., Sheahan T. W., Brockdorff N., Kessler B. M., Koseki H., Klose R. J., Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 157, 1445–1459 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalb R., Latwiel S., Baymaz H. I., Jansen P. W. T. C., Müller C. W., Vermeulen M., Müller J., Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 21, 569–571 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Tan J., Jones M., Koseki H., Nakayama M., Muntean A. G., Maillard I., Hess J. L., CBX8, a polycomb group protein, is essential for MLL-AF9-induced leukemogenesis. Cancer Cell 20, 563–575 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiacchiera F., Rossi A., Jammula S., Piunti A., Scelfo A., Ordóñez-Morán P., Huelsken J., Koseki H., Pasini D., Polycomb complex PRC1 preserves intestinal stem cell identity by sustaining Wnt/β-catenin transcriptional activity. Cell Stem Cell 18, 91–103 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Bracken A. P., Dietrich N., Pasini D., Hansen K. H., Helin K., Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 20, 1123–1136 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boyer L. A., Plath K., Zeitlinger J., Brambrink T., Medeiros L. A., Lee T. I., Levine S. S., Wernig M., Tajonar A., Ray M. K., Bell G. W., Otte A. P., Vidal M., Gifford D. K., Young R. A., Jaenisch R., Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Dorantes-Acosta E., Pelayo R., Lineage switching in acute leukemias: A consequence of stem cell plasticity?. Bone Marrow Res. 2012, 406796 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zardo G., Cimino G., Nervi C., Epigenetic plasticity of chromatin in embryonic and hematopoietic stem/progenitor cells: Therapeutic potential of cell reprogramming. Leukemia 22, 1503–1518 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Blecher-Gonen R., Barnett-Itzhaki Z., Jaitin D., Amann-Zalcenstein D., Lara-Astiaso D., Amit I., High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat. Protoc. 8, 539–554 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S. L., TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anders S., Huber W., Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/10/e1600972/DC1
Supplementary Materials and Methods
fig. S1. Loss of PRC1 induces a differentiated phenotype in Lin− transformed cells.
fig. S2. Loss of PRC1 induces a differentiated phenotype independently of Cdkn2a expression.
fig. S3. Specificity check of H2AK119Ub signal.
fig. S4. Loss of PRC1 activity induces leukemic cell differentiation.
fig. S5. Loss of PRC1 activity negatively affects the growth of primary leukemia both in vitro and in vivo.
fig. S6. Bmi1 knockdown does not recapitulate the Ring1a/b-deficient phenotype.
table S1. ChIP-seq results in Lin− and leukemic cells.
table S2. Genome-wide expression in wild-type, PRC1 Lin−, and leukemic cells.
table S3. Genome-wide expression in primary MLL-AF9 leukemic cells.
table S4. ChIP-seq results in primary MLL-AF9 leukemic cells.