

Summary
Mitochondrial diseases comprise a heterogeneous group of genetically inherited disorders that cause failures in energetic and metabolic function. Boosting residual oxidative phosphorylation (OXPHOS) activity can partially correct these failures. Herein, using a high-throughput chemical screen, we identified a bromodomain inhibitor I-BET 525762A as one of the top hits that augments COX5a protein levels in complex I (CI)-mutant cybrid cells. In parallel, Bromodomain-containing protein 4 (Brd4), a target of I-BET 525762A, was identified using a genome-wide CRISPR screen to search for genes whose loss-of-function rescue death of CI-impaired cybrids grown under conditions requiring OXPHOS activity for survival. We show that I-BET525762A or loss-of-Brd4 remodeled the mitochondrial proteome to increase the levels and activity of OXPHOS protein complexes leading to rescue of the bioenergetic defects and cell death caused by mutations or chemical inhibition of CI. These studies show that Brd4 inhibition may have therapeutic implications for the treatment of mitochondrial diseases.
Graphical abstract
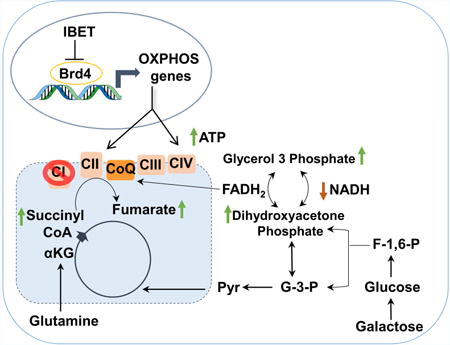
Introduction
Mutations in mitochondrial or nuclear DNA that compromise OXPHOS system lead to a spectrum of debilitating or even fatal human disorders known as mitochondrial diseases (Koopman et al., 2012). Among them, mitochondrial complex I (CI) deficiency is the most common OXPHOS defect observed in patients and to date no cure is available (Pfeffer et al., 2013; Swalwell et al., 2011). The impairment of oxidative phosphorylation due to dysfunction in the electron transport chain largely compromise ATP production (Nunnari and Suomalainen, 2012) and depending on the mutation and/or insult, increase the generation of reactive oxygen species (ROS) (Lin et al., 2012; Vafai and Mootha, 2012) and unbalance the NAD+/NADH ratio due to NADH accumulation (Karamanlidis et al., 2013). Proposed metabolic strategies to correct mitochondrial CI deficiencies include mitochondria-targeted antioxidant molecules (Koopman et al., 2016) or biochemical bypass of the defective complex, for example using succinate (Pfeffer et al., 2013) or short-chain quinones (idebenone or CoQ1) (Haefeli et al., 2011) that can feed electrons into the ETC downstream of CI.
Attempts to boost residual mitochondrial activity to overcome bioenergetics defects have been recently strengthened by several studies reporting that, overexpressing the transcriptional coactivator PGC-1α (a known central regulator of mitochondrial biogenesis) partially corrects pathological phenotypes and extends survival in mouse models with electron transport chain deficiencies (Dillon et al., 2012; Srivastava et al., 2007; St-Pierre et al., 2006). Based on these findings, a possible approach to overcome ETC deficiencies is to enhance the functional OXPHOS capacity which is the failing hallmark of these diseases.
Bromodomain-containing protein 4 (Brd4) is a member of the bromodomain and extraterminal domain (BET) family of proteins that is comprised of Brd2-4 and BrdT (Nicodeme et al., 2010). BET proteins contain two tandem bromodomains (protein module that binds to acetyl-lysines) and an extraterminal domain (ETD) that mediates protein-protein interactions (Dhalluin et al., 1999). Brd4 binds to acetylated histones and coordinately recruits additional proteins via its ETD to promoters and distal enhancers to modulate gene expression (Liu et al., 2013). Chemical inhibitors to the BET family such as I-BET 525762A and JQ1 which occupies the epsilon acetyl lysine binding pocket of Brd4 and prevents its association to acetylated histones at the chromatin have been effective in treating several cancer types (Dawson et al., 2011; Delmore et al., 2011; Filippakopoulos et al., 2010). However, it is unknown whether Brd4 can control genes linked to energy metabolism and impact ETC deficiencies.
Here we have identified Brd4 using a mitochondrial-based high-throughput chemical screen and tandem genome wide-CRISPR screen in human CI mutant cybrid cells. Brd4 inhibition, either chemically or genetically, rescues mitochondrial bioenergetics protecting against cell death caused by CI defects. Deletion or inhibition of Brd4 enhances oxidative phosphorylation genes, proteins, and activity increasing FADH2 levels to bypass defective complex I. These studies show that Brd4 inhibition corrects mitochondrial CI deficiencies and may have therapeutic implications for the treatment of mitochondrial diseases.
Results
Identification of Bromodomain Inhibitor and Brd4 in High-Throughput Chemical and Genome-Wide CRISPR Screens
To discover chemical compounds that rescue bioenergetic defects caused by mitochondrial disease mutations through increases of mitochondrial proteins, we designed and developed a high-throughput in-cell enzyme-linked immunoassay using human cybrid cells carrying a mutation (3796 A>G, found in adult onset dystonia) in the mitochondrial-encoded protein ND1—an integral component of the NADH dehydrogenase CI subunit (Simon et al., 2003) (Figure 1A). A diverse library of 10,015 chemical compounds were screened in duplicate and values were normalized to cells expressing PGC-1α—a transcriptional regulator of mitochondrial biogenesis (Puigserver et al., 1998; Wu et al., 1999) as a positive control (Figure 1B). CIV was the most responsive to PGC-1α-stimulation therefore the quantitative measurement of the CIV subunit Cox5a served as the readout. A 70% threshold was established to select top hits for re-test using the same assay. Interestingly, the compound with the highest score was I-BET 525762A, a pan bromodomain and extraterminal domain (BET) inhibitor that targets BET family of proteins including Brd2-4 and BrdT (Nicodeme et al., 2010) (Figure 1C-D). In parallel, and to complement this chemical screen, we performed a genome-wide editing CRISPR screen. We used the standard cell death-based clinical assay to culture ND1-mutant cybrid cells in media with galactose instead of glucose. Galactose is unable to provide sufficient levels of ATP from glycolysis and cells are forced to depend on oxidative phosphorylation (Aguer et al., 2011; Robinson et al., 1992). Thus, ND1-mutant cells in galactose media die within 72 hours while ND1-control cells survive (Figure 1E and Figure S1A). The genome-wide CRISPR screen was performed with ND1-mutant cybrids to discover genes whose loss-of-function would confer cell viability under galactose conditions presumably by boosting mitochondrial oxidative phosphorylation capacity. The screen employed a validated pooled lentiviral sgRNA library targeting 18,675 genes in the human genome with coverage of four sgRNA per gene (Doench et al., 2016) (Figure 1E). We scored sgRNAs for enrichment within the pool following growth in galactose media (Figure 1F). Remarkably, in alignment with the chemical screen, one of the highest scoring genes with high fold enrichment and statistical significance was the Bromodomain-containing protein 4 (Brd4)—a target that is inhibited by I-BET 525762A. To further confirm these results, we repeated the screen using more stringent conditions challenging the cells with a second round of growth in galactose media, to select persistently galactose-resistant cells. Again, Brd4 emerged as one of the most potent scoring genes (Figure S1B-C). Together, the data from these two independent and unbiased screens (chemical and genome-wide CRISPR library) converging to Brd4 strongly points toward Brd4 inhibition as a mechanism to increase OXPHOS proteins (COX5a) and rescue bioenergetic defects caused by mitochondrial CI mutations.
Figure 1. Brd4 is identified as a target by both high-throughput chemical and CRISPR genetic screens to enhance OXPHOS proteins and rescue mitochondrial bioenergetic defects.
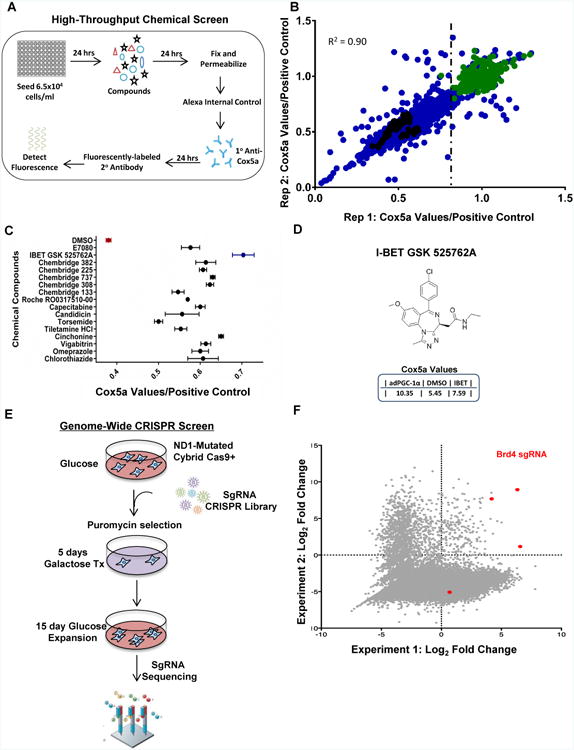
A, Schematic of high-throughput chemical screen. B, Scatterplot of chemicals tested (blue) plotted with first test (x-axis) and repeated test (y-axis) of Cox5a fluorescent values relative to the average PGC-1α positive control (green). DMSO baseline fluorescent values (black) are indicated. Vertical dotted line indicates compounds selected at the 70% cutoff. C, Cox5a fluorescent values from selected compounds tested in secondary screen plotted relative to PGC-1α positive control set to 1.0. Values represent the mean ± s.e.m. n=3. D, Chemical structure of the I-BET 525762A compound with Cox5a fluorescent values below. E, Workflow of genome-wide CRISPR Screen. f, Scatterplot of two replicate CRISPR screens showing enrichment of sgRNAs after galactose treatment. Points highlighted in red represent the different sgRNAs for Brd4. See also Figure S1.
Bromodomain Inhibition Enhances OXPHOS Protein and Activity and Protects Against Cell Death in CI-Defective Cells
To validate and extend the impact of bromodomain inhibition on mitochondrial energetic function, we tested its effects on OXPHOS protein levels in ND1-mutant cybrids. I-BET 525762A increased COX5a and an array of other OXPHOS protein levels in a dose-responsive manner (Figure 2A-B) with increases persisting to the whole complex level (Figure 2C and Figure S2A). Additional Brd4 inhibitors with diverse chemical structures such as JQ1 (Filippakopoulos et al., 2010), GSK1210151A (Dawson et al., 2011), and MS436 (Zhang et al., 2013) also increased COX5a protein levels (Figure 2D). Indeed, comprehensive proteomics analysis indicated that I-BET treatment significantly enhanced proteins involved in mitochondrial oxidative phosphorylation, translation, and organization (Figure 2E and Table S1). The increase in protein levels proved to be functional as treatment with the I-BET 525762A compound enhanced total oxygen consumption as well as the activity of ETC complexes II (CII) and IV (CIV) (Figure 3A-C). I-BET 525762A treatment did not alter glycolysis as measured by ECAR and mitochondrial complex activity for complex III (CIII) and complex V (CV) were unchanged (Figure S2B-D). To assess whether the increase in mitochondrial oxidative capacity from bromodomain inhibitors was sufficient to rescue cell death caused by mitochondrial bioenergetic deficits, ND1-mutant cells were cultured in galactose instead of glucose. Vehicle-treated cells died when exposed to galactose for 72 h, but strikingly, treatment with I-BET 525762A or other bromodomain inhibitors rescued galactose-induced cell death and cells proliferated for several passages (Figure 3D-E and Figure S1A and S2E). Interestingly, the bromodomain inhibitor also prevented galactose-induced cell death and enhanced oxygen consumption in other mitochondrial CI-deficient human cybrid cells such as LHON (Leber Hereditary Optic Neuropathy), carrying the 14459 G>A mutation in ND6 (Jun et al., 1994) (Figure 3F-G) and knock-down of two different CI subunits, Ndufs3 and Ndufv2 (Figure 3H). The energetic rescue by the bromodomain inhibitor was not only observed in CI-mutant cybrid cells, but also in fibroblasts derived from patients harboring a mutation in ACAD9 -a chaperone protein necessary for CI activity (Nouws et al., 2010) (Figure 3I). I-BET 525762A also displayed rescue in cells that were chemically inhibited by rotenone (Figure 3J). I-BET-mediated rescue was only observed in CI-deficient cells as other complex-deficient cells such as MELAS (Mitochondrial Encephalomyopathy and Stroke-Like Episodes/A3243G-Leucine tRNA), MERRF (Myoclonic Epilepsy and Ragged Red Fibers/A8344G-Lysine tRNA), Cox10 KO (CIV Cytochrome C Oxidase assembly protein), and Reiske KO (CIII assembly protein) displayed no rescue when cultured in galactose media despite comparable basal oxygen consumption levels (Figure S2F-G). These results indicate that bromodomain inhibitors remodels the mitochondrial proteome increasing oxidative activity and readjusting bioenergetic defects caused by CI deficiency.
Figure 2. Bromodomain inhibition increases OXPHOS proteins and remodels the mitochondrial proteome.
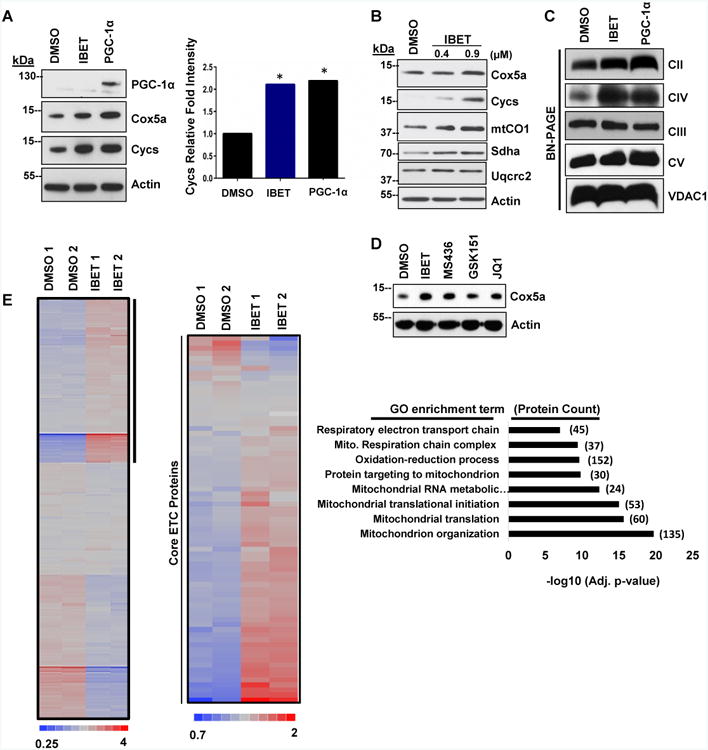
A, I-BET 525762A increases OXPHOS proteins (left). Cycs densitometry of IBET-treated ND1-mutated cybrids is comparable to PGC-1α positive control (right). B, I-BET 525762A increases OXPHOS proteins in a dose-responsive manner. C, Blue Native Page- I-BET 525762A treatment increases OXPHOS complex levels. D, Diverse Brd4 inhibitors increases OXPHOS proteins. Immunoblots shown are representative of greater than 3 independent experiments. E, Proteomic heat map of the hierarchical clustering of all proteins (∼4,800) in duplicate (left). I-BET 525762A-upregulated proteins (> 1.4 fold) are indicated (vertical black bar). Proteomic heat map indicating that I-BET treatment in ND1-mutated cybrids increases core ETC proteins compared to control (middle). Gene Ontology (GO) categories from upregulated proteins enriched after Benjamin-Hochberg correction are indicated to the right. Corresponding −log10 p-values (x axis) and the number of proteins (in parentheses) are listed. See also Figure S2 and Table S1.
Figure 3. Bromodomain inhibition increases mitochondrial bioenergetics and protects against galactose-induced cell death.
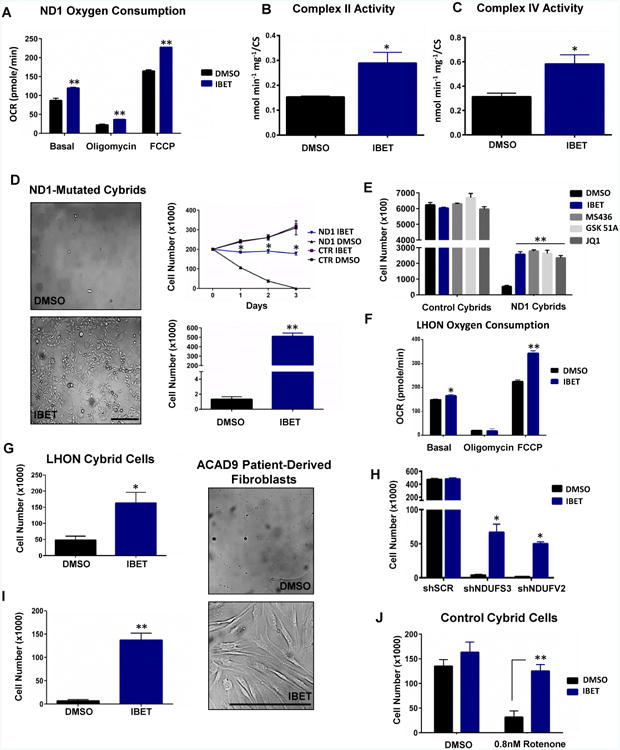
A-C, Total oxygen consumption (A) as well as CII (B) and CIV (C) activities are increased with I-BET 525762A. D, I-BET 525762A protects against galactose-induced cell death. Microscopy image (left) is representative of at least 3 independent experiments and time course (top right) and cell number quantification (bottom right) are the mean ± s.e.m., n=3. Asterisks denote *p<0.05 or **p<0.01 via Student's t-test. Horizontal black scale bar = 200 μm. E, Different Brd4 inhibitors rescue ND1-mutated cybrids from galactose-induced cell death. F-G, I-BET 525762A enhances oxygen consumption in ND6-mutated (LHON) cybrids (F) and protects against galactose-induced cell death (G). H, I-BET 525762A rescues galactose-induced cell death in CI knock-down control cells. i, I-BET 525762A protects against galactose-induced cell death in ACAD9 human patient-derived fibroblasts. Microscopy image (right) is representative of at least 3 independent experiments and cell number quantification (left) is the mean ± s.e.m., n=3. Horizontal black scale bar = 200 μm. J, I-BET 525762A protects against galactose-induced cell death in control human cybrids with rotenone-induced CI deficiency. Data represents the mean ± s.e.m., n=3. Asterisks denote *p<0.05 or **p<0.01 via Student's t-test. All I-BET and Brd4 inhibitor treatments are delivered at final concentration of 0.9 μM. See also Figure S2.
Brd 4 Controls the Mitochondrial OXPHOS Program
We next investigated whether abrogation of Brd4 could increase mitochondrial function in ND1-mutant cells. Brd4 protein was ablated using different sgRNAs and shRNAs and consistent with the I-BET 525762A treatment, sgBrd4 expressing guides and shBrd4 constructs enhanced OXPHOS protein levels (Figure 4A and Figure S3A). In addition, total oxygen consumption and respiratory CII and CIV activities were increased (Figure 4B-D and Figure S3B). Consistent with I-BET treatment, CIII and CV activities were unaltered in sgBrd4 guides (Figure S3C-D). As predicted based on the CRISPR screen, Brd4-ablated cells were protected from galactose-induced cell death (Figure 4E). These results were confirmed using selective competition between sgNeg (GFP+) and sgBrd4 (GFP-) expressing ND1-mutant cells. After 72 h with galactose, 98% of the surviving cells were GFP- indicating that Brd4 ablation conferred a selective advantage under these energy-restricted conditions (Figure 4F). To further support the Brd4 function in mitochondrial energetics, we over-expressed Brd4 in control human cybrid cells. Ectopic expression of Brd4, conversely to its ablation or inhibition, reduced OXPHOS protein levels and decreased total oxygen consumption compared to controls (Figure 4G-H). In addition, increased expression of Brd4 in control cybrids impaired cell growth under galactose conditions (Figure 4I). These findings indicate that changes in expression of Brd4 protein alter mitochondrial energetic capacity and cell survival under nutrient conditions that require oxidative phosphorylation.
Figure 4. Loss-of-function of Brd4 enhances mitochondrial bioenergetics while gain-of-function impairs OXPHOS capacity.
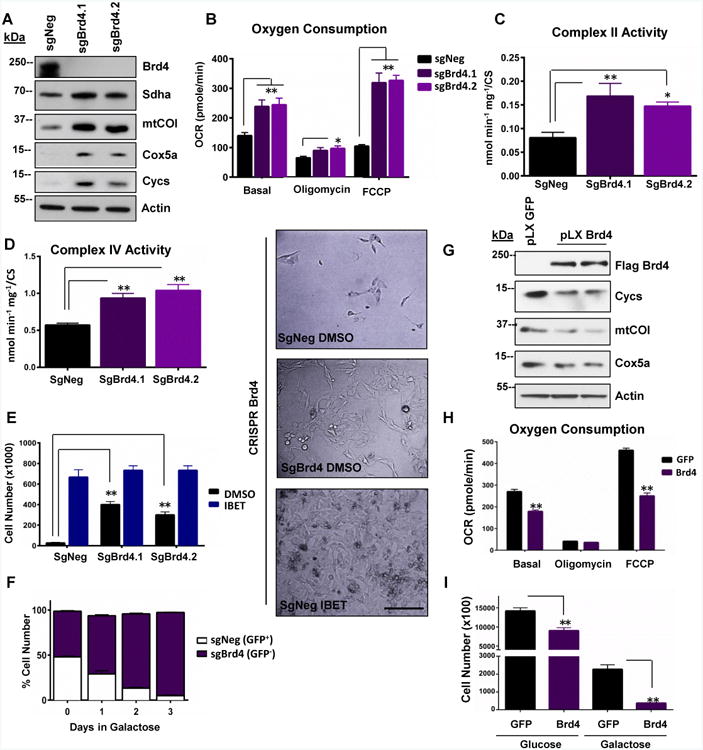
A, CRISPR ablation of Brd4 enhances OXPHOS proteins. B, Total oxygen consumption rates are increased in sgBrd4 ND1-human cybrids compared to sgNeg control. C-D, Complex II (C) and complex IV (D) activities are increased in sgBrd4 ND1-human cybrids compared to sgNeg controls. E, Representative microscopy image (right) of sgNeg, sgBrd4, and IBET-treated sgNeg ND1-mutant cybrids cultured in galactose for 72 hours. Cell quantification after 72 hours in galactose media is indicated to the left. Horizontal black scale bar = 200μm. F, Competition assay. GFP-negative sgBrd4 is more abundant that GFP-positive sgNeg ND1-mutated cybrids. G, Brd4 overexpression in control human cybrids reduces OXPHOS proteins. H, Total oxygen consumption is reduced with Brd4 overexpression in control human cybrids. I, Control human cybrids are sensitized to galactose-induced cell death with Brd4 overexpression. Immunoblots shown are representative of greater than 3 independent experiments and all other experiments are the mean ± s.e.m., n=3. Asterisks denote *p<0.05 or **p<0.01 via Student's t-test. All I-BET treatments are delivered at final concentration of 0.9 μM. See also Figure S3.
Displacing Brd4 Protein from Nuclear-Encoded OXPHOS Promoters Increases OXPHOS Genes
We next evaluated the mechanisms whereby Brd4 inhibition increases mitochondrial oxidative function. Bromodomain proteins bind to chromatin through interaction with histone acetylated lysines (Dhalluin et al., 1999), and I-BET-525762A disrupts this interaction by occupying the epsilon aminolysine pocket leading to direct bromodomain protein displacement from chromatin (Nicodeme et al., 2010). To assess whether Brd4 localizes at promoters of nuclear-encoded mitochondrial genes, we analyzed published data sets containing Brd4 ChIP sequence results (Anders et al., 2014). Genes containing Brd4 binding peaks within 1000 bp upstream and downstream of the transcription start site (TSS) were identified using BETA-minus algorithm and were functionally classified using gene-ontology/Panther algorithm (Wang et al., 2013; Zhang et al., 2008). Interestingly, these data displayed Brd4 occupancy at promoters of genes linked to the oxidative phosphorylation pathway comparable to the control gene CCND2 (Anders et al., 2014) (Figure S4A-B). These findings were validated in ND1–mutant cells by analyzing Brd4 occupancy at promoters of nuclear-encoded mitochondrial genes including SDHD, CYCS, and COX5A. As expected, Brd4 inhibition by I-BET-525762A decreased Brd4 occupancy at these promoters (Figure 5A and Figure S4C). Intriguingly, displacement of Brd4 by the I-BET compound led to enhanced occupancy of PGC-1α—a transcriptional coactivator that binds to promoters and activates expression of mitochondrial genes (Charos et al., 2012) (Figure 5B). Similarly to the Brd4 inhibitor, ectopic expression of PGC-1α strongly displaced Brd4 from these promoters (Figure 5C). Consistent with these results, expression of mitochondrial genes were increased in ND1-mutant cells when Brd4 was displaced from OXPHOS gene promoters by I-BET 525762A (Figure 5D and Figure S4D) or depleted with CRISPR or shRNA (Figure 5E and Figure S4E). This is in contrast to the Brd4 target CCND2, a gene repressed by the Brd4 inhibitor (Figure S4D). Conversely, ectopic expression of Brd4 decreased the expression of OXPHOS genes (Figure 5F). To further confirm these findings, PGC-1α was depleted from ND1-mutated cybrids. PGC-1α knock-down prevented the I-BET-mediated increase in mitochondrial transcripts and partially blunted the rescue from galactose-induced cell death (Figure 5G-H). This data suggests that Brd4 occupancy at nuclear-encoded promoters regulates the expression of mitochondrial genes and prevents PGC-1α and likely other activators from binding.
Figure 5. IBET 525762A treatment displaces Brd4 from OXPHOS promoters and increases transcription in complex I-deficient cells.
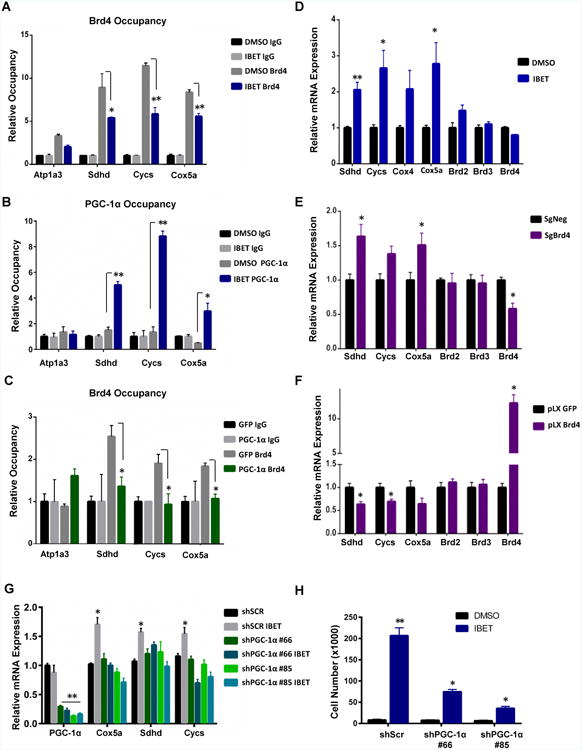
A, ChIP- I-BET 525762A displaces Brd4 from nuclear-encoded mitochondrial promoters. Negative control promoter region Atp1A3 is unaltered. B, ChIP- PGC-1α occupancy increases upon I-BET 525762A-mediated Brd4 displacement. C, ChIP- Brd4 is displaced upon adPGC-1α over-expression. Data is representative of mean ± s.e.m., n=3. Asterisks denote *p<0.05 or **p<0.01 via Student's t-test. D-F, I-BET 525762A-mediated displacement of Brd4 (D) and ablation of Brd4 by CRISPR (E) increases expression of OXPHOS genes while over-expression of Brd4 decreases OXPHOS gene expression (F). Data represents mean ± s.e.m., n=3. Asterisks denote *p<0.05 or **p<0.01 via Student's t-test. G-H, ShPGC-1α KD in ND1-mutated cybrids fail to increase OXPHOS transcripts (G) and partially blunts rescue from galactose-induced cell death (H) when treated with I-BET 525762A. See also Figure S4.
Inhibition of Brd4 Rewires Bioenergetic Metabolism through Complex II to Overcome Complex I Deficiency
To gain additional insight into the mechanism of I-BET-mediated enhancement of OXPHOS bioenergetic capacity and protection from galactose-induced cell death, a comprehensive metabolomics analysis was performed (Figure. 6A). Intriguingly, metabolites such as dihydroxyacetone phosphate (product of mitochondrial glycerol 3 phosphate dehydrogenase), and fumarate (product of succinate dehydrogenase) were significantly enhanced in I-BET-treated ND1 cybrids compared to DMSO controls (Figure 6A-C). Interestingly, these two enzymes are able to bypass the defective CI in the ND1-mutated cells by supplying electrons via FADH2 to CII and CoQ to maintain ATP levels. Indeed, I-BET 525762A treatment in ND1 human cybrids prevented galactose-induced declines in the energetic metabolites ATP and normalized NADH (which accumulates due CI deficiency (Karamanlidis et al., 2013)) to similar levels as control cells (Figure 6D-E and Figure S5A-B). Consistent with increases in metabolites converging to CII and CoQ, FAD levels were elevated in cells treated with the bromodomain inhibitor (Figure 6F), suggesting increased utilization of FADH2. Given that galactose-derived pyruvate is not an optimal substrate for oxidation due to the defective CI we speculated that the increase in oxygen consumption in I-BET-treated ND1-mutant cybrids was accomplished by oxidizing alternative energy sources such as glutamine, which enters the I-BET-enhanced CII via the TCA cycle. Indeed, I-BET 525762A treatment increased 14C glutamine oxidation and was able to moderately prevent cell death when glutamine was the main carbon source (Figure 6G). In addition, I-BET treatment enhanced genes in the glutamine utilization pathway (Figure S5C). In contrast, I-BET 525762A failed to prevent galactose-induced cell death when glutamine was either removed or the glutaminase enzyme (catalyses glutamine to glutamate conversion) was chemically blocked (Figure 6H-I). Taken together, I-BET 525762A augments glutamine oxidation and metabolites that generate FADH2, which can be oxidized through increased CII and CIV activities to produce ATP and rescue cell death. To assess whether CII is required for I-BET 525762A-induced survival, we genetically abrogated CII function using two shRNAs targeting the Sdha subunit which will disrupt both the ETC and TCA cycle. Interestingly, I-BET 525762A-mediated cell rescue was considerably minimized in knockdown cells when placed under galactose for 72hrs (Figure 6J). To further test whether increases in mitochondrial oxidative activity mediated via Brd4 inhibition is necessary to maintain bioenergetics and cell survival in CI-deficient cells, we used two different approaches to completely disrupt mitochondrial respiration: chloramphenicol (an antibiotic that blocks mitochondrial protein translation), and ethidium bromide (EtBr) (causes mitochondrial DNA depletion). In both cases, the levels of mtCOI, a mtDNA encoded-protein translated by mitochondrial ribosomes and essential for respiration (Tsukihara et al., 1996) were undetectable (Figure S5D-E). I-BET 525762A failed to rescue galactose-induced cell death in chloramphenicol or EtBr treated cells (Figure S5D-E). In summary, these results indicate that I-BET 525762A-induced mitochondrial respiration is necessary to rescue the bioenergetic defects and maintain survival in human cybrids with mitochondrial CI mutations.
Figure 6. I-BET 525762A treatment rewires and enhances metabolic and energetic states of complex I-deficient cells.
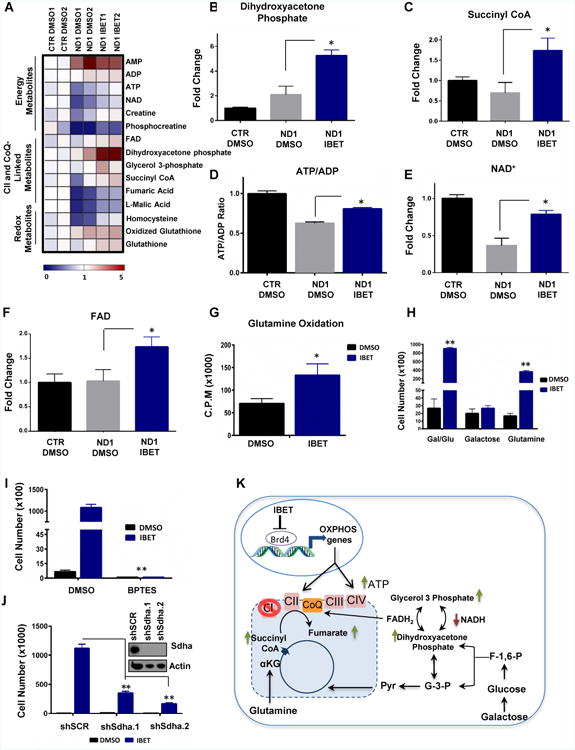
A, Metabolomics Heatmap in control and ND1-mutated cybrids in duplicate. I-BET 525762A-treated ND1-mutated human cybrids increases metabolites linked to energy, CII, and CoQ, and redox metabolism. B-C, I-BET 525762A treatment increases dihydroxyacetone phosphate (B) and succinyl CoA (C) metabolite levels. D-F, I-BET 525762A treatment increases ATP (D), NAD+ (E) and FAD (F) metabolite levels in ND1-mutated cybrids. Data represents mean ± s.e.m., n=2. G, 14C Glutamine oxidation is enhanced by I-BET 525762A treatment in ND1-mutated cybrids. H, I-BET-mediated galactose rescue is blunted when glutamine is removed from culture media in ND1-mutated cybrids. I, Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES) at 25 μM which selectively inhibits Glutaminase (GLS1) to block glutamine to glutamate conversion abolished the positive of effects of I-BET 525762A in ND1-mutated cybrids. J, I-BET 525762A displays minimal rescue upon Sdha knockdown. Immunoblot indicates complete Sdha knockdown at the protein level. Data represent the mean ± s.e.m., n=3. Asterisks denote *p<0.05 or **p<0.01 via Student's t-test. All I-BET treatments are delivered at a final concentration of 0.9 μM. K, Model detailing Brd4 inhibition rescues bioenergetic defects in complex I-deficient cells. Arrows indicate metabolites that were increased (green) or decreased (red) upon I-BET 575762A treatment. αKG, alpha-ketoglutarate, G-3-P, glyceraldehyde-3-phosphate, F-1-6-P, Fructose 1,6 bisphosphate, and CoQ, Coenzyme Q. See also Figure S5.
Discussion
Cells harboring mitochondrial mutations in CI fail to cope with excessive energetic demands and undergo cell death. In principle, a metabolic reprogramming increasing ETC activity, bypassing mutated CI, could rescue the bioenergetic defects and maintain cell survival. Our studies have identified that inhibition of Brd4 constitutes a regulator of this metabolic reprogramming. Brd4 binds to nuclear-encoded mitochondrial genes and its inhibition efficiently rewires cellular energetics caused by complex I defects (Figure 6K). Brd4 inhibition remodels the mitochondrial proteome and causes changes in metabolic routes that increase and utilize FADH2, transferring electrons to the respiratory chain downstream of CI and increasing ATP.
Metabolic strategies to reprogram and bypass ETC defects in mitochondrial disease mutations have been based on CoQ derivatives or NAD+ precursors but the efficacy has been limited, particularly in complex I deficiencies (Pfeffer et al., 2013). Bromodomain inhibitors might overcome these metabolic limitations acting through a broad mitochondrial-based gene expression program that generates energetic efficient mitochondria to bypass complex I defects. Thus, coordinated increases in CII and CIV through Brd4 inactivation would allow oxidation of substrates such as glutamine, fatty acids, or branched chain amino acids increasing FADH2 that will enter the ETC at complex II or CoQ sites. Mounting evidence demonstrates that oxidizing glutamine can serve as the major substrate for energy production (Lu et al., 2010; Reitzer et al., 1979). We show that in addition to OXPHOS genes, transcripts linked to glutamine metabolism were also upregulated by I-BET treatment, supporting the role of glutamine catabolism and ATP production by Brd4 inhibition in CI-deficient mutant cells. Consistent with this energetic and metabolic rescue via Brd4 inhibition, previous studies have corrected CI-deficiency by introducing the Saccharomyces cerevisiae NADH-quinone oxido- reductase (NDI1) protein to bypass the defective complex I and increase OXPHOS and ATP production from CIII and CIV (Bai et al., 2001).
A metabolic hallmark of CI malfunction is NADH accumulation caused by reduction in CI-dependent NADH reductase activity (Karamanlidis et al., 2013). Increased NADH might alter catalytic activity of NAD+ dependent enzymes such as sirtuins, TCA cycle, or glycolytic dehydrogenases (Nunnari and Suomalainen, 2012). We show that NADH levels are partially restored after bromodomain inhibition and may contribute to the bioenergetic rescue in CI deficient cells. Although a complete mechanism of how bromodomain inhibition normalizes NADH is unknown, some putative candidates such as mitochondrial GPD2 (glycerol phosphate dehydrogenase) emerged in the proteomic analysis and was elevated in isolated mitochondria from I-BET treated samples (Figure 2E). GPD2 is a mitochondrial membrane bound protein that forms part of the glycerol phosphate shuttle transferring cytosolic NADH to FADH2 and bypassing CI inhibition (Mracek et al., 2013). The accumulated mitochondrial NADH caused by the defective CI could be transported to the cytosolic compartment utilizing the malate-aspartate pathway to provide the substrate for the glycerol phosphate shuttle (Gut and Verdin, 2013). Furthermore, the metabolomic analysis shows that several metabolites related to cellular redox states and antioxidant response were increased by I-BET treatment. Deleterious phenotypes caused by CI failure have been shown to arise by increased reactive oxygen species (ROS) levels, in addition to compromised ATP generation (Haefeli et al., 2011). Thus, increases in ROS detoxification through bromodomain inhibition might also contribute to the bioenergetic rescue and survival caused by defective CI.
In these studies, I-BET 525762A was extremely effective in increasing the OXPHOS capacity and promoting survival in nutrient-restrictive galactose conditions in cells with CI disruption. I-BET however was unable to rescue cells with other complex mutations such as MELAS and MERRF cybrids. It would appear that I-BET 525762A can only provide rescue in a context when the defective complexes occur at an early point in the ETC such that it can be bypassed to boost residual OXPHOS capacity. Given that CIV levels and activity are also increased upon I-BET treatment, it is conceivable that Brd4 inhibition, similar to PGC-1α (Srivastava et al., 2009), would positively impact mutations where CIV is not fully ablated. A basal threshold of ETC activity seems to be indispensable for the bromodomain inhibitors to rescue the bioenergetic defects and promote survival as shown in the experiments using chloramphenicol or EtBr cells (Figure S5D-E).
Brd4 is a chromatin-bound transcriptional regulator linked to expression of genes associated with different biological processes including tumor progression or inflammation (Baratta et al., 2015; Huang et al., 2009). However, how and whether these programs integrate with mitochondrial energetics and metabolic control is unknown. As a scaffolding protein, Brd4 recruits activator or repressor complexes to target promoters. Its activator function has been extensively characterized through interactions with pTEFb, Mediator and JMJD6 that promotes Pol II elongation (Liu et al., 2013). When displaced from chromatin by bromodomain inhibitors, this corresponds to a decrease of target genes such as myc (Delmore et al., 2011). Brd4 however can also be incorporated into repressor complexes as demonstrated in the human papillomavirus (HPV) system (Wu et al., 2006). Chromatin bound Brd4 recruits and binds to the HPV viral protein E2 and prevents the binding of TFIID and Pol II to the target promoters leading to gene repression. Genome-wide RNA seq analysis of Brd4 abrogation reveals a dual role of Brd4 increasing or decreasing mRNA transcripts (Shu et al., 2016). Brd4 occupies nuclear-encoded mitochondrial promoters and its inhibitor, I-BET 525762A, significantly reduces Brd4 occupancy at the mitochondrial promoters. Our results indicate that I-BET-mediated gene repression occurs almost immediately after IBET treatment, however activation of OXPHOS genes occurs over a longer time point. These time-dependent differences between I-BET-mediated gene repression or activation could be attributed to the difference in dynamic recruitment and composition of transcription factors to these promoters. Increased transcription of OXPHOS genes suggests that Brd4 competes with a potent activator and/or might be associated with a repressor protein. Given our current data that 1) the displacement of Brd4 from nuclear-encoded mitochondrial promoters increased the occupancy of potent transcriptional activators such as PGC-1α (Figure 5B) and 2) that I-BET-mediated increased in OXHPOS genes and cell survival under galactose conditions was greatly impeded with PGC-1α knockdown would suggest a competition model (Jang et al., 2005; Wallberg et al., 2003). The involvement of a repressor protein or an alternate mechanism however, may also be plausible as there was partial rescue, albeit significantly blunted, with ND1-mutated cybrids upon I-BET treatment in the context of PGC-1α knock-down under galactose conditions.
The ability of bromodomain inhibition to correct bioenergetic defects caused by deficient CI activity through different metabolic strategies may be employed as a possible avenue to treat mitochondrial and neurodegenerative diseases where the inhibition of CI has been associated with disease progression.
Methods
High-throughput chemical screening
1.0×106 human ND1-mutant (3796 A>G) cybrid cells were seeded in a 384-well plate (Corning, 3712) and incubated for 24 hours at 37°C with 5% CO2. Positive control wells (column 24) were infected with adenoviral Pgc-1α and negative control lanes (column 23) received 0.3% DMSO. Following a media change, chemical compounds were added to experimental wells via pin transfer and cells were incubated with compound for 24 hours at 37°C with 5% CO2. The In Cell Elisa (ICE) protocol was then performed. Cells were washed with PBS (HyClone) prior to being fixed with 4% paraformaldehyde (Santa Cruz, sc281692) and permeabilized with 0.2% Triton-X-100 (Sigma, X100). Cells were washed twice with PBS and treated with Alexa Fluor 680 succinimidyl ester (Thermo Fisher, A37567) for internal control before blocking with blocking buffer (LI-COR, 927-40000) and incubated with Cox5a antibody (Abcam, ab110262) for 16 h at 4°C. Cells were then washed with 0.2% Triton X-100 in PBS before the addition of the secondary anti-mouse IRDye 800CW antibody (LI-COR, 926-32212) for 1h. Fluorescence was then quantified using the LI-COR Aerius instrument and software. Values in the 800 channel were normalized to the respective internal control (700 channel). Normalized data was then expressed as a ratio to the average positive control (set to 100%). Further details for screening methods are available on request.
Design and creation of the CRISPR library
Human genome-wide lentipool CRISPR library was designed and created as previously described (Doench et al., 2016)
Genome-Wide CRISPR Screen
1.1×108 human ND1-mutant (3796 A>G) cybrid cells, stably expressing Cas9, were seeded in a total of 38- 150 mm × 25 mm dishes (3.0×106 per dish). Cells were infected with the lenti-pooled library to achieve a 30%-50% infection efficiency, corresponding to a multiplicity of infection (MOI) of ∼0.3 -0.5. Media was changed 24 hours later and 0.5 ug/mL puromycin was added 48 h later and selected for 7 additional days. Cells were trypsinized and separated into 150 mm × 25 mm dishes (4.0×106 per dish). Next day, cells were washed twice with PBS and media was changed to non-glucose DMEM with glutamine (4mM) supplemented with 10 mM galactose (Sigma D7050), 10% FBS, and 1% P/S and 1mM pyruvate. Cells were cultured for 5 days in galactose media. After 5 days, galactose media was replaced to high glucose DMEM in order to expand the remaining cells. Cells were maintained and passaged in glucose DMEM for two weeks before genomic DNA was isolated from two biological replicates using QIAamp DNA mini kit (QIAGEN 51304). A third independent experiment was performed as described above but this time cells were challenged with two rounds of 10 mM galactose treatment.
Illumina sequencing
Samples were submitted and sequenced at the Broad Institute using a HiSeq2000 (Illumina) as previously described (Doench et al., 2016)
Screen analysis and statistics
For analysis, the log2-fold-change of each sgRNA was normalized to the starting plasmid DNA (pDNA) pool for each biological replicate unless otherwise stated. Each sgRNA was ranked by log2 fold-change, and this number was then divided by the total number of sgRNAs in the pool to determine a percent-rank. These percent-rank values from two biological replicates were represented as scatter-blots. A third independent experiment using more stringent culture conditions was analyzed equally but represented as a bar graph. For performing RIGER and STARS analysis, the percent-rank values across subpools were merged. STARS allow the calculation of P-values, FDR and q-value (corrected FDR) for hit genes. STARS is written in Python, and is publically available: http://www.broadinstitute.org/rnai/public/software/index.
Cell Culture and Treatments
All cell lines were maintained in DMEM high glucose (HyClone), 10% FBS and 1% P/S at 37°Cand 5% CO2. For compound treatment, cells were seeded at a density of 1.0×105 cells/well in a 6 well plate (Falcon, 353046) and allowed to adhere for 24 hours before treatment with 0.9 μM I-BET 525762A, JQ1, GSK1210151A, or MS436 chemical compounds or vehicle control for 24 hours in DMEM with high glucose. For galactose experiments cells were seeded in 6-well plates grown in DMEM high glucose, 10% FBS and 1% P/S at 37 °Cand 5% CO2 for 24h to allow cells to adhere. Cells were then washed twice with PBS and media was changed to DMEM with no glucose but supplemented with 4mM glutamine (HyClone) and 10 mM galactose (Sigma G0750), 10% FBS, and 1% P/S. Cells were incubated in galactose-containing media ± 0.9 μM I-BET 525762A for 72 hours with daily media changes. 50 μM BPTES (Sigma SML0601) was included in the galactose media for experiments assessing glutamine requirement. Cells were then trypsinized and quantified using a hemocytometer (NanoEnTek, DHC-N01). Generation of RhoO cells involved treating control cybrids with DMEM high glucose supplemented with 50ng/mL of ethidium bromide (Bio-Rad 1610433) and 50 μg/mL of uridine (Sigma U3750) for 2 weeks with media changes every 48 hours. To abrogate mitochondrial protein translation, control cybrids were treated with DMEM high glucose supplemented with 40 μM chloramphenicol (Sigma C0378) and 50 μg/mL uridine for 5 days with daily media changes. Mitochondrial protein depletion for both ethidium bromide and chloramphenicol treatments were assessed by measuring mtCO1 protein level. Cells were then placed in galactose media (described above) in the presence or absence of 0.9 μM I-BET for 72 hours. For rotenone treatments, control cybrid cells were seeded in DMEM with high glucose with 0.8 nM rotenone (Sigma 8875) in the presences or absence of 0.9 μM I-BET 525762A for 5 days with daily media changes. Cell quantity was assessed by cell counting after treatment. Selective competition assay: 5×104 of sgNeg GFP positive and sgBrd4 GFP negative ND1 cybrids cells (1:1) were plated together in a 6 well plate and cultured in galactose media (previously described). Cells were trypsinized and analyzed by FACS (BD FACS Canto II) daily for 3 consecutive days. The ratio between sgNeg (GFP+) and sgBrd4 GFP-) was calculated. Transfections for gain and loss-of-function studies were performed according to the manufacturer's instruction using the polyfect reagent (Qiagen, 301107). The following constructs were used: pLJM1-sgBrd4.1 5′-GAGCAGGTATTGCAGTTGGT-3, pLJM1-eGFP (addgene 19319), pLKO.1 shBrd4 5′-CCGGCCTGGAGATGACATAGTCTTACTCGAGTAAGACTATGTCATCTCCAGGTTTTTG-3′ (Sigma), pLKO.1 shSdha.1 5′-GATTTGCTGATGGAAGCATAA-3′, pLKO.1 shShda.25′-TCGCTATTGCACACCTTATAT-3′, pLKO.1 shNdufs4 5′-TTTGTTCCTGCTCGCAATAAC-3′, pLKO.1 shNdufV2 5′-CCAGTTGGAAAGTATCACATT-3′, pLKO.1 shPGC-1α 66. 5′-CCGGTATGACAGCTACGAGGAATATCTCGAGATATTCCTCGTAGCTGTCATATTTTTG, pLKO.1 shPGC-1α 85. 5′-CCGGCCGTTATACCTGTGATGCTTTCTCGAGAAAGCATCACAGGTATAACGGTTTTT pLKO.1 shCTR, pLX-GFP, pLX-Brd4 (Human ORFeome 71377), and lentiCRISPR v2 (addgene #52951). Guide sequences for Brd4 were as follows: sgBrd4.1: 5′-GAGCAGGTATTGCAGTTGGT-3′ and sgBrd4.2 5′- ACTGCAATACCTGCTCAGAG-3′.
Supplementary Material
Acknowledgments
We thank all the members of the Puigserver lab and Edward Chouchani for discussions regarding this project. MELAS, MERRF, Cox10 and Reiske KO mouse fibroblasts, ND6-mutated LHON cybrids were a generous gift from Carlos Moraes at the University of Miami. ACAD9-mutated patient-derived fibroblasts was obtained from and Michio Hirano from Columbia University. We thank the Institute of Chemistry and Cell Biology at Harvard Medical School and the Broad Institute at M.I.T. for their training, advice and facilities. These studies were supported in part by NIH/NIDDK by RO1DK081418 (P.P.) and R24DK080261 awarded to P.P. J.J.B was supported in part by a diversity supplement award under R01DK081418-06S1 parental grant (P.P). E.B.M was supported in part by Alfonso Martin Escudero fellowship and EMBO postdoctoral fellowship. J.S. is the founding CEO of Khondrion.
Footnotes
Supplemental Information: Supplemental information includes supplemental experimental procedures, figures and associated figure legends which can be found with the online version of the manuscript.
Author Contributions: P.P, E.B.M, and J.J.B conceived the project. P.P and J.J.B designed, optimized, and initiated the high-throughput chemical screen. J.J.B, in collaboration with the ICCB, performed the high-throughput chemical screen, data processing, identified and characterized the I-BET 525762A compound, performed oxygen consumption and ECAR analyses, galactose rescue experiments of MELAS, MERRF, Cox10, Resike, LHON cybrids and ACAD9 patient-derived fibroblasts, Brd4 gain-of-function experiments and metabolomics with assistance from the BIDMC Metabolomics Core. P.P and E.B.M., with advice and expertise from J.D and D.E.R., conceived and designed the galactose-based genome wide CRISPR screen. E.B.M performed the genome-wide CRISPR screen with assistance from L.R.H. Library generation, sequencing and bioinformatics analysis of the CRISPR sequencing data was performed by J.D. and D.E.R. E.B.M performed mitochondrial activity assays, BN-PAGE, glutamine experiments, loss-of-function experiments of CI and CII, galactose rescue of ND1 and CI and CII cybrid cell lines and generation and characterization of the shPGC-1α cell lines. R.V. and J.S. were involved in the initial experimental design of the chemical screen and provided ND1 cybrids. E.B.M and J.J.B generated and performed experiments characterizing CRISPR Brd4. J.J.B, F.V and C.T. performed the ChIP experiments. M.J, J.A.P, and S.P.G performed and analyzed the proteomic experiments. J.J.B., E.B.M and P.P. wrote the manuscript. M.S.S. critically reviewed data and manuscript.
Author Information: The authors declare no competing financial interests. Correspondence and requests for materials should be directed to P.P.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguer C, Gambarotta D, Mailloux RJ, Moffat C, Dent R, McPherson R, Harper ME. Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PloS one. 2011;6:e28536. doi: 10.1371/journal.pone.0028536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders L, Guenther MG, Qi J, Fan ZP, Marineau JJ, Rahl PB, Loven J, Sigova AA, Smith WB, Lee TI, et al. Genome-wide localization of small molecules. Nature biotechnology. 2014;32:92–96. doi: 10.1038/nbt.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Hajek P, Chomyn A, Chan E, Seo BB, Matsuno-Yagi A, Yagi T, Attardi G. Lack of complex I activity in human cells carrying a mutation in MtDNA-encoded ND4 subunit is corrected by the Saccharomyces cerevisiae NADH-quinone oxidoreductase (NDI1) gene. The Journal of biological chemistry. 2001;276:38808–38813. doi: 10.1074/jbc.M106363200. [DOI] [PubMed] [Google Scholar]
- Baratta MG, Schinzel AC, Zwang Y, Bandopadhayay P, Bowman-Colin C, Kutt J, Curtis J, Piao H, Wong LC, Kung AL, et al. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:232–237. doi: 10.1073/pnas.1422165112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charos AE, Reed BD, Raha D, Szekely AM, Weissman SM, Snyder M. A highly integrated and complex PPARGC1A transcription factor binding network in HepG2 cells. Genome research. 2012;22:1668–1679. doi: 10.1101/gr.127761.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- Dillon LM, Williams SL, Hida A, Peacock JD, Prolla TA, Lincoln J, Moraes CT. Increased mitochondrial biogenesis in muscle improves aging phenotypes in the mtDNA mutator mouse. Human molecular genetics. 2012;21:2288–2297. doi: 10.1093/hmg/dds049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature biotechnology. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- Haefeli RH, Erb M, Gemperli AC, Robay D, Courdier Fruh I, Anklin C, Dallmann R, Gueven N. NQO1-dependent redox cycling of idebenone: effects on cellular redox potential and energy levels. PloS one. 2011;6:e17963. doi: 10.1371/journal.pone.0017963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Yang XD, Zhou MM, Ozato K, Chen LF. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Molecular and cellular biology. 2009;29:1375–1387. doi: 10.1128/MCB.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Molecular cell. 2005;19:523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Jun AS, Brown MD, Wallace DC. A mitochondrial DNA mutation at nucleotide pair 14459 of the NADH dehydrogenase subunit 6 gene associated with maternally inherited Leber hereditary optic neuropathy and dystonia. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:6206–6210. doi: 10.1073/pnas.91.13.6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC, Jr, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell metabolism. 2013;18:239–250. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman W, Willems P, Smeitink J. Monogenic mitochondrial disorders. New England Journal of Medicine. 2012;366:1132–1141. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- Koopman W, Beyrath J, Fung CW, Koene S, Rodenburg R, Willems P, Smeitink J. Mitochondrial Disorders in children: toward development of small-molecule treatment strategies. EMBO Molecular Medicine. 2016;8:311–327. doi: 10.15252/emmm.201506131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapuente-Brun E, Moreno-Loshuertos R, Acin-Perez R, Latorre-Pellicer A, Colas C, Balsa E, Perales-Clemente E, Quiros PM, Calvo E, Rodriguez-Hernandez MA, et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science. 2013;340:1567–1570. doi: 10.1126/science.1230381. [DOI] [PubMed] [Google Scholar]
- Lin CS, Sharpley MS, Fan W, Waymire KG, Sadun AA, Carelli V, Ross-Cisneros FN, Baciu P, Sung E, McManus MJ, et al. Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:20065–20070. doi: 10.1073/pnas.1217113109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Ma Q, Wong K, Li W, Ohgi K, Zhang J, Aggarwal AK, Rosenfeld MG. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell. 2013;155:1581–1595. doi: 10.1016/j.cell.2013.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Pelicano H, Huang P. Cancer metabolism: is glutamine sweeter than glucose? Cancer cell. 2010;18:199–200. doi: 10.1016/j.ccr.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mracek T, Drahota Z, Houstek J. The function and the role of the mitochondrial glycerol-3-phosphate dehydrogenase in mammalian tissues. Biochimica et biophysica acta. 2013;1827:401–410. doi: 10.1016/j.bbabio.2012.11.014. [DOI] [PubMed] [Google Scholar]
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouws J, Nijtmans L, Houten SM, van den Brand M, Huynen M, Venselaar H, Hoefs S, Gloerich J, Kronick J, Hutchin T, et al. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell metabolism. 2010;12:283–294. doi: 10.1016/j.cmet.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer G, Horvath R, Klopstock T, Mootha VK, Suomalainen A, Koene S, Hirano M, Zeviani M, Bindoff LA, Yu-Wai-Man P, et al. New treatments for mitochondrial disease-no time to drop our standards. Nature reviews Neurology. 2013;9:474–481. doi: 10.1038/nrneurol.2013.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. The Journal of biological chemistry. 1979;254:2669–2676. [PubMed] [Google Scholar]
- Robinson BH, Petrova-Benedict R, Buncic JR, Wallace DC. Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochemical medicine and metabolic biology. 1992;48:122–126. doi: 10.1016/0885-4505(92)90056-5. [DOI] [PubMed] [Google Scholar]
- Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529:413–417. doi: 10.1038/nature16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DK, Friedman J, Breakefield XO, Jankovic J, Brin MF, Provias J, Bressman SB, Charness ME, Tarsy D, Johns DR, et al. A heteroplasmic mitochondrial complex I gene mutation in adult-onset dystonia. Neurogenetics. 2003;4:199–205. doi: 10.1007/s10048-003-0150-3. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Barrett JN, Moraes CT. PGC-1alpha/beta upregulation is associated with improved oxidative phosphorylation in cells harboring nonsense mtDNA mutations. Human molecular genetics. 2007;16:993–1005. doi: 10.1093/hmg/ddm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Diaz F, Iommarini L, Aure K, Lombes A, Moraes CT. PGC-1alpha/beta induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Human molecular genetics. 2009;18:1805–1812. doi: 10.1093/hmg/ddp093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S, Handschin C, Zheng K, Lin J, Yang W, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Swalwell H, Kirby DM, Blakely EL, Mitchell A, Salemi R, Sugiana C, Compton AG, Tucker EJ, Ke BX, Lamont PJ, et al. Respiratory chain complex I deficiency caused by mitochondrial DNA mutations. European journal of human genetics : EJHG. 2011;19:769–775. doi: 10.1038/ejhg.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–383. doi: 10.1038/nature11707. [DOI] [PubMed] [Google Scholar]
- Wallberg AE, Yamamura S, Malik S, Spiegelman BM, Roeder RG. Coordination of p300-mediated chromatin remodeling and TRAP/mediator function through coactivator PGC-1alpha. Molecular cell. 2003;12:1137–1149. doi: 10.1016/s1097-2765(03)00391-5. [DOI] [PubMed] [Google Scholar]
- Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, Tang Q, Meyer CA, Zhang Y, Liu XS. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nature protocols. 2013;8:2502–2515. doi: 10.1038/nprot.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SY, Lee AY, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, Chiang CM. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes & development. 2006;20:2383–2396. doi: 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Zhang G, Plotnikov AN, Rusinova E, Shen T, Morohashi K, Joshua J, Zeng L, Mujtaba S, Ohlmeyer M, Zhou MM. Structure-guided design of potent diazobenzene inhibitors for the BET bromodomains. Journal of medicinal chemistry. 2013;56:9251–9264. doi: 10.1021/jm401334s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome biology. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.