

ABSTRACT
The ancestral Bacillus subtilis strain 3610 contains an 84-kb plasmid called pBS32 that was lost during domestication of commonly used laboratory derivatives. Here we demonstrate that pBS32, normally present at 1 or 2 copies per cell, increases in copy number nearly 100-fold when cells are treated with the DNA-damaging agent mitomycin C. Mitomycin C treatment also caused cell lysis dependent on pBS32-borne prophage genes. ZpdN, a sigma factor homolog encoded by pBS32, was required for the plasmid response to DNA damage, and artificial expression of ZpdN was sufficient to induce pBS32 hyperreplication and cell death. Plasmid DNA released by cell death was protected by the capsid protein ZpbH, suggesting that the plasmid was packaged into a phagelike particle. The putative particles were further indicated by CsCl sedimentation but were not observed by electron microscopy and were incapable of killing B. subtilis cells extracellularly. We hypothesize that pBS32-mediated cell death releases a phagelike particle that is defective and unstable.
IMPORTANCE Prophages are phage genomes stably integrated into the host bacterium's chromosome and less frequently are maintained as extrachromosomal plasmids. Here we report that the extrachromosomal plasmid pBS32 of Bacillus subtilis encodes a prophage that, when activated, kills the host. pBS32 also encodes both the sigma factor homolog ZpdN that is necessary and sufficient for prophage induction and the protein ComI, which is a potent inhibitor of DNA uptake by natural transformation. We provide evidence that the entire pBS32 sequence may be part of the prophage and thus that competence inhibition may be linked to lysogeny.
INTRODUCTION
Pure-culture propagation of bacteria under artificial laboratory conditions favors rapid growth as individuals over multicellular behaviors, and domestication can drive genetic fixation of selected alleles (1–3). An example of domestication is found in the model organism Bacillus subtilis subsp. subtilis, in which phenotypes of the ancestral strain 3610 were bred out of the commonly used laboratory strain 168 and its derivatives (4). Specifically, the lab strains lack robust biofilm formation, swarming and sliding motility over solid surfaces, and the synthesis of extracellular polymers and antimicrobials found in the ancestor (5–9). Furthermore, the mutations responsible for each lab strain defect have been identified and the corresponding phenotypes can be restored when corrected to the ancestral allele (9–12). Whereas most of the mutations are polymorphisms in the chromosome, the laboratory strains were also cured of an 84-kb plasmid called pBS32 (13–15).
The pBS32 plasmid is highly homologous to the pLS32 plasmid found in the close relative Bacillus subtilis subsp. natto (15, 16). Studies of pLS32 have identified the plasmid origin and shown that it is a low-copy-number theta-replicating plasmid (16, 17). Relatively few proteins encoded by pLS32 have been studied in detail, but the protein RepN is the DNA binding replication initiator protein and the origin-proximally encoded proteins AlfA/AlfB constitute a plasmid segregation system (16, 18, 19). pBS32 encodes at least two proteins shown to alter the physiology of the host: RapP and ComI. RapP is a phosphatase that inhibits biofilm formation by restricting phosphate flow to the sporulation master regulator Spo0A and antagonizing the quorum-sensing response regulator protein ComA (20, 21). ComI is a small transmembrane protein that inhibits the ability of the ancestral strain to take up exogenous DNA, rendering it 100-fold less transformable than its pBS32-deficient descendants (15). The benefit of encoding RapP and ComI on pBS32 is unknown, but RapP homologs are often associated with mobile genetic elements, and some mobile genetic elements have been associated with inhibition of genetic transfer (22–25).
The functions of the remaining products encoded by pBS32 genes are unexplored. Here we confirm that pBS32 is a low-copy-number plasmid but that the copy number increases 100-fold in response to DNA damage by mitomycin C. In addition, mitomycin C treatment caused pBS32-dependent cell lysis, which required a subset of the contiguous prophage genes that constitute nearly one-half of the plasmid genetic sequence. Consistent with the release of a phagelike particle, extracellular DNA was protected from DNase by the phage capsid protein ZpbH. The putative particle is likely defective, however, because intact phage particles were not observed by electron microscopy, nor was the concentrated lysate able to generate plaques or kill cells extracellularly. Finally, we demonstrate that ZpdN, a sigma factor homolog encoded by pBS32, is both necessary and sufficient for increasing plasmid copy number and inducing cell lysis.
MATERIALS AND METHODS
Strains and growth conditions.
B. subtilis strains were grown in lysogeny broth (LB; 10 g tryptone, 5 g yeast extract, 5 g NaCl per liter) or on LB plates fortified with 1.5% Bacto agar at 37°C. When appropriate, antibiotics were included at the following concentrations: 10 μg/ml tetracycline, 100 μg/ml spectinomycin, 5 μg/ml chloramphenicol, 5 μg/ml kanamycin, and 1 μg/ml erythromycin plus 25 μg/ml lincomycin (mls). Mitomycin C (MMC; Santa Cruz Biotech) and isopropyl β-d-thiogalactopyranoside (IPTG; Sigma) were added to the medium at the indicated concentration when appropriate.
Strain construction.
Constructs either were first introduced into the lab strain PY79 or into the pBS32 plasmid-cured strain DS2569 (ΔpBS32) by natural competence and then crossed into the 3610 background using SPP1-mediated generalized phage transduction (26) or were transformed directly into the competent 3610 derivatives DS7187 (ΔcomI) or DK1042 (with a Q-to-L change at position 12 encoded by comI [comIQ12L]) (15). All strains used in this study are listed in Table 1. All plasmids used in this study are listed in Table S1 in the supplemental material. All primers used in this study are listed in Table S2 in the supplemental material.
TABLE 1.
B. subtilis strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| 3610 | Wild type | |
| DS1143 | hag::Tn10 Specr | |
| DS2569 | ΔpBS32 | 15 |
| DS7187 | ΔcomI | 15 |
| DK280 | tetO37 amyE::PftsW-tetR-mCherry Specr | |
| DK297 | ΔSPβ ΔPBSX | |
| DK399 | ΔSPβ ΔpBS32 | |
| DK451 | ΔSPβ ΔPBSX ΔpBS32 | |
| DK1042 | comIQ12L | 15 |
| DK1917 | ΔSPβ ΔPBSX ΔQII amyE::Physpank-gfp Specr | |
| DK1918 | ΔSPβ ΔPBSX ΔQIII amyE::Physpank-gfp Specr | |
| DK1919 | ΔSPβ ΔPBSX ΔQIV amyE::Physpank-gfp Specr | |
| DK1233 | ΔSPβ ΔPBSX amyE::Physpank-gfp Specr | |
| DK1234 | ΔSPβ ΔPBSX ΔpBS32 amyE::Physpank-gfp Specr | |
| DK1551 | ΔSPβ ΔPBSX amyE::Physpank-zpcW Specr | |
| DK1552 | ΔSPβ ΔPBSX amyE::Physpank-zpbW Specr | |
| DK1539 | amyE::Physpank-zpdN Specr comIQ12L | |
| DK1634 | amyE::Physpank-wkRBSzpdN Specr ΔSPβ ΔPBSX ΔcomI | |
| DK1651 | ΔSPβ ΔPBSX ΔpBS32 amyE::Physpank-wkRBSzpdN Specr | |
| DK1687 | ΔPBSX ΔpBS32 | |
| DK2063 | ΔSPβ ΔPBSX amyE::Physpank-wkRBSzpdN Specr | |
| DK3287 | ΔzpdN ΔSPβ ΔPBSX ΔcomI | |
| DK3650 | ΔpBS32 hag::Tn10 Specr | |
| DK3651 | ΔSPβ ΔPBSX hag::Tn10 Specr | |
| DK3652 | ΔpBS32 ΔSPβ ΔPBSX hag::Tn10 Specr | |
| DK3962 | ΔpBS32 amyE::Physpank-wkRBSzpdN Specr | |
| DK4112 | ΔzpbH ΔSPβ ΔPBSX ΔcomI amyE::Physpank-wkRBSzpdN Specr | |
| DK4162 | ΔpBS32 amyE::Km | |
| DK4180 | ΔQII amyE::Physpank-wkRBSzpdN Specr | |
| DK4181 | ΔQIII amyE::Physpank-wkRBSzpdN Specr | |
| DK4182 | ΔQIV amyE::Physpank-wkRBSzpdN Specr | |
| DK4188 | ΔQII ΔSPβ ΔPBSX amyE::Physpank-wkRBSzpdN Specr thrC::Phag-gfp mls | |
| DK4189 | ΔQIV ΔSPβ ΔPBSX amyE::Physpank-wkRBSzpdN Specr thrC::Phag-gfp mls | |
| DK4192 | ΔcomI ΔSPβ ΔPBSX amyE::Physpank-wkRBSzpdN Specr thrC::Phag-gfp mls | |
| DK4193 | ΔpBS32 ΔSPβ ΔPBSX amyE::Physpank-wkRBSzpdN Specr thrC::Phag-gfp mls | |
| DK4196 | ΔQIII ΔSPβ ΔPBSX amyE::Physpank-wkRBSzpdN Specr thrC::Phag-gfp mls | |
| PY79 | sfp0 swrA |
In-frame deletions.
To generate the ΔPBSX in-frame markerless deletion construct, the region upstream of yjpA was PCR amplified using the primer pair 2856/2857 and digested with KpnI and XhoI, and the region downstream of xlyA was PCR amplified using the primer pair 2858/2859 and digested with XhoI and HindIII. The two fragments were then simultaneously ligated into the KpnI and HindIII sites of pMiniMAD2, which carries a temperature-sensitive origin of replication and an erythromycin resistance cassette (27), to generate pMP105. The plasmid pMP105 was introduced to PY79 by transformation at the permissive temperature for plasmid replication (22°C) using mls resistance as a selection. The extrachromosomal plasmid was then transduced into strain 3610 at the nonpermissive temperature of 37°C to force integration. To evict the plasmid, the strain was incubated in 3 ml LB at a permissive temperature for plasmid replication (22°C) for 14 h, diluted 30-fold in fresh LB, and incubated at 22°C for another 8 h. Dilution and outgrowth were repeated 2 more times. Cells were then serially diluted and plated on LB agar at 37°C. Individual colonies were patched on LB plates and on LB plates containing mls to identify mls-sensitive colonies that had evicted the plasmid. Chromosomal DNA from colonies that had excised the plasmid was purified and screened by PCR using primers 2856/2859 to determine which isolate had retained the ΔPBSX allele. Primers 3034/3035 amplify a 1-kb region internal to PBSX and were used as a proxy to confirm loss of PBSX.
To generate the ΔSPβ in-frame markerless deletion construct, the region upstream of yotN was PCR amplified using the primer pair 2981/2982 and digested with EcoRI and XhoI, and the region downstream of yokA was PCR amplified using the primer pair 2983/2984 and digested with XhoI and BamHI. The two fragments were then simultaneously ligated into the EcoRI and BamHI sites of pMiniMAD2 to generate pMP115. The plasmid was integrated and evicted as described above. Chromosomal DNA from colonies that had excised the plasmid was purified and screened by PCR using primers 2981/2984 to determine which isolate had retained the ΔSPβ allele. Primers 3032/3033 amplify a 1-kb region internal to SPβ and were used as a proxy to confirm loss of SPβ.
To generate the ΔzpdN in-frame markerless deletion construct, the region upstream of zpdN was PCR amplified using the primer pair 3811/3812 and the region downstream of zpdN was PCR amplified using the primer pair 3813/3814, inserted into the SmaI site of pMiniMAD2 by Gibson isothermal assembly, and electroporated into Escherichia coli DH5α to generate pMP190. The plasmid was integrated and evicted as described above. Chromosomal DNA from colonies that had excised the plasmid was purified and screened by PCR using primers 3811/3814 to determine which isolate had retained the ΔzpdN allele.
To generate the ΔzpbH in-frame markerless deletion construct, the region upstream of zpbH was PCR amplified using the primer pair 4584/4585 and the region downstream of zpbH was PCR amplified using the primer pair 4586/4587, inserted into the SmaI site of pMiniMAD2 by Gibson isothermal assembly, and electroporated into E. coli DH5α to generate pBM1. The plasmid was integrated and evicted as described above. Chromosomal DNA from colonies that had excised the plasmid was purified and screened by PCR using primers 4584/4587 to determine which isolate had retained the ΔzpbH allele.
The generation of quadrant deletions of pBS32 (ΔQII, ΔQIII, and ΔQIV) was previously described (15).
tetO array integrated in pBS32.
The tetO array was integrated into pBS32 in multiple steps. First, an 800-bp arm containing the 5′ region of zpaJ was PCR amplified using the primer pair 3337/3338 and digested with EcoRI and NheI. An 800-bp arm containing the 3′ region of zpaJ was PCR amplified using the primer pair 3339/3340 and digested with NheI and HindIII. Primer 3339 contained the sequences for BamHI, SphI, and SalI cut sites. The two fragments were then simultaneously ligated into the EcoRI and HindIII sites of pMiniMAD2, which carries a temperature-sensitive origin of replication and an erythromycin resistance cassette to generate pMP159. pLAU39 contains 37 intact repeats of the tetO sequence (tetO37) (a generous gift of David Rudner, Harvard Medical School). The tetO37 sequence was excised from pLAU39 by digestion with NheI and SalI and ligated into the NheI and SalI sites of pMP159 to generate pMP160. The plasmid pMP160 was introduced to DS2569 by transformation at the permissive temperature for plasmid replication (22°C) using mls resistance as a selection. The extrachromosomal plasmid was then transduced into strain 3610 at the nonpermissive temperature of 37°C to force integration. To evict the plasmid, the strain was incubated in 3 ml LB at a permissive temperature for plasmid replication (22°C) for 14 h, diluted 30-fold in fresh LB, and incubated at 22°C for another 8 h. Dilution and outgrowth were repeated 2 more times. Cells were then serially diluted and plated on LB agar at 37°C. Individual colonies were patched on LB plates and on LB plates containing mls to identify mls-sensitive colonies that had evicted the plasmid. Chromosomal DNA from colonies that had excised the plasmid was purified and screened by PCR using primers 3337/3340 to determine which isolate had retained the tetO37 allele.
PftsW-TetR-mCherry translational fusion.
To generate the translational fusion of TetR to mCherry, a fragment containing the PftsW promoter and tetR translationally fused to mCherry was excised from pWX355 (a generous gift of David Rudner, Harvard Medical School) by digestion with EcoRI and BamHI and ligated into the EcoRI and BamHI sites of pAH25 (a generous gift of Amy Camp, Mount Holyoke College) containing a polylinker and spectinomycin (Sp) resistance marker between two arms of the amyE gene to generate pMP162.
Inducible transcriptional fusions.
To generate the inducible amyE::Phypsank-wkRBSzpdN Specr construct pMP200, a PCR product containing zpdN was amplified from strain 3610 chromosomal DNA using primer pair 3959/3951, digested with NheI and SphI, and cloned into the NheI and SphI sites of pDR111 containing a spectinomycin resistance cassette, a polylinker downstream of the Physpank promoter, and the gene encoding the LacI repressor between the two arms of the amyE gene (a generous gift of David Rudner, Harvard Medical School). Primer 3959 replaced the sequence immediately upstream of the zpdN start codon with the TTAGGGGGATAACA ribosome binding site (RBS) sequence from sigD.
The inducible amyE::Physpank-zpbW and amyE::Physpank-zpcW constructs were a generous gift of Kevin Griffith (University of Massachusetts, Amherst).
SPP1 phage transduction.
To 0.2 ml of dense culture grown in TY broth (LB supplemented after autoclaving with 10 mM MgSO4 and 100 μM MnSO4), serial dilutions of SPP1 phage stock were added and statically incubated for 15 min at 37°C. To each mixture, 3 ml TYSA (molten TY supplemented with 0.5% agar) was added and poured atop fresh TY plates, and the plates were incubated at 37°C overnight. Top agar from the plate containing nearly confluent plaques was harvested by scraping into a 50-ml conical tube, vortexed, and centrifuged at 5,000 × g for 10 min. The supernatant was treated with 25 μg/ml (final concentration) DNase before being passed through a 0.45-μm syringe filter and stored at 4°C.
Recipient cells were grown to stationary phase in 2 ml TY broth at 37°C. A 0.9-ml volume of cells was mixed with 5 μl of SPP1 donor phage stock; 9 ml of TY broth was added to the mixture and allowed to stand at 37°C for 30 min. The transduction mixture was then centrifuged at 5,000 × g for 10 min, the supernatant was discarded, and the pellet was resuspended in the remaining volume. One hundred microliters of cell suspension was then plated on TY fortified with 1.5% agar, the appropriate antibiotic, and 10 mM sodium citrate.
Microscopy.
Fluorescence microscopy was performed with a Nikon 80i microscope with a phase-contrast objective Nikon Plan Apo 100× and an Excite 120 metal halide lamp. mCherry was visualized with a C-FL HYQ Texas Red Filter Cube (excitation filter, 532 to 587 nm; barrier filter, >590 nm). Green fluorescent protein (GFP) was visualized using a C-FL HYQ fluorescein isothiocyanate (FITC) Filter Cube (excitation filter, 460 to 500 nm; barrier filter, 515 to 550 nm). TMA-DPH (N,N,N-trimethyl-4-(6-phenyl-1,3,5-hexatrien-1-yl)phenylammonium (p-toluenesulfonate) fluorescent signals were visualized using a UV-2E/C DAPI (4′,6-diamidino-2-phenylindole) Filter Cube (excitation filter, 340 to 380 nm; barrier filter, 435 to 485 nm). Images were captured with a Photometrics Coolsnap HQ2 camera in black and white, false colored, and superimposed using Metamorph image software.
For detection of mCherry-TetR binding to the tetO array, cells were grown to mid-log phase at 37°C in TY medium. Cells (1.0 ml) were pelleted and resuspended in 40 μl phosphate-buffered saline (PBS) containing 100 μg ml−1 TMA-DPH (Molecular Probes) and incubated for 5 min at room temperature. Samples were observed by spotting 4 μl of the suspension on a glass microscope slide and were immobilized with a poly-l-lysine-treated coverslip.
For fluorescence microscopy of the lysis time course using Physpank-gfp transcriptional fusions, strains were grown overnight at 22°C in TY medium. Cultures were subcultured to an optical density at 600 nm (OD600) of ∼0.01 and grown at 37°C in TY. When appropriate, mitomycin C was added to a final concentration of 1 μg/ml and IPTG was added to a final concentration of 1 mM. A 1.0-ml volume of each culture was harvested by centrifugation and resuspended in PBS. Samples were observed by spotting 4 μl of the suspension on a glass microscope slide and were immobilized with a poly-l-lysine-treated coverslip.
qPCR.
For quantitative PCR (qPCR), strains were grown overnight at 22°C in TY medium. Cultures were subcultured to an OD600 of 0.01 and grown at 37°C in TY. When appropriate, mitomycin C was added to a final concentration of 0.3 μg/ml. After 30 min, 1 ml of culture was harvested by centrifugation at mid-log phase and genomic and plasmid DNA was isolated. qPCR was performed with diluted DNA template and specific primer pairs (1 μM) using SYBR green Supermix (Quanta Biosciences) on the Stratagene MX3500 Pro thermocycler. Data were analyzed using the MXPro Stratagene software package. Primer pair 3106/3107 (sigA) was used to measure chromosomal DNA, and primer pairs 3102/3103 (zpaB) and 3108/3109 (zpbI) were used to measure pBS32 DNA. Plasmid copy number was calculated as described previously (28).
DNase protection assay.
Strain DK1539 was grown overnight at 22°C in TY medium. The culture was then subcultured to an OD600 of ∼0.01 in 25 ml TY medium and grown with shaking at 37°C. After 90 min of incubation, IPTG was added to a final concentration of 1 mM when appropriate. Cultures were allowed to grow an additional 3.5 h at 37°C. After 5 h total incubation, 20 ml of each culture was harvested by centrifugation. Supernatants were passed through a 0.45-μm syringe filter and stored at 4°C. One-milliliter samples were aliquoted from each supernatant and were either left untreated, treated with 0.01 μg/ml DNase for 30 min at 37°C, or heated at 80°C for 30 min before being treated with 0.01 μg/ml DNase for 30 min at 37°C. One hundred microliters of these samples was then phenol-chloroform extracted. Primer pairs 4709/4710, 4762/4763, and 4764/4765 were used to PCR amplify pBS32 loci zpcJ, zpaB, and zpbI, respectively, and primer pair 3879/3880 was used to PCR amplify chromosomal locus fliG.
Phage particle isolation and protein analysis.
Strains DS1143, DK3650, DK3651, and DK3652 were grown overnight at 22°C in TY medium, subcultured the next morning to an OD600 of ∼0.01 in 500 ml TY medium, and grown with shaking at 37°C. When the cultures reached an OD600 of 0.15, mitomycin C was added to a final concentration of 0.5 μg/ml. Cultures were allowed to grow for an additional 3.5 h at 37°C, and 1 M NaCl was added to the culture to dissociate materials from the cell exterior. The NaCl was dissolved by gentle swirling. After a 10-min incubation at 4°C, the culture was centrifuged at 5,000 × g for 5 min, and the supernatant was harvested and centrifuged a second time at 5,000 × g for 5 min. To precipitate potential phage particles, polyethylene glycol (PEG) 8000 was added at 10%, wt/vol, to the supernatant, and the mixture was stirred overnight at 4°C. Samples were centrifuged at 4,400 × g for 60 min at 4°C, and the pellets were resuspended in 10 mM Tris (pH 7.4), 10 mM MgSO4, 5 mM CaCl2 overnight with gentle agitation at 4°C. Resuspended pellets were clarified by centrifugation for 10 min at 5,000 × g. Note that the strains used for the PEG precipitation were each mutated for the hag gene, encoding the flagellar filament protein Hag, such that flagellar filaments would not complicate electron microscopy (EM) analysis of any potential phage particles.
The PEG-precipitated supernatants were further purified by CsCl step gradient (1.35 g/ml, 1.45 g/ml, 1.55 g/ml, and 1.65 g/ml) ultracentrifugation in an SW-41 rotor 3 h at 154,000 × g. In the pBS32-only sample, a band was extracted from the 1.45:1.55 g/ml interface, the interface typically enriched for phage particles with roughly equal amounts of proteins and DNA. The isolated band was dialyzed against 10 mM Tris (pH 7.4), 10 mM MgSO4, and 5 mM CaCl2 with a molecular weight cutoff (MWCO) of 10,000 to remove the CsCl.
Transmission electron microscopy.
Samples were applied to 300-mesh copper grids coated with Formvar and carbon (Ted Pella, Inc.) after glow discharging. The grids were then stained with 0.75% uranyl formate and imaged on a JEOL JEM 3200FS 300-kV electron microscope. Images were obtained using a Gatan Ultrascan 4000 charge-coupled-device (CCD) camera.
Mass spectrometry.
Samples were resolved on a 12% SDS-PAGE and were Coomassie blue- or silver-stained to detect bands. Predominant bands and regions of the gel were treated with trypsin and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) on a Thermo Scientific LTQ Velos Pro by the IU Laboratory for Biological Mass Spectrometry.
RESULTS
pBS32 plasmid copy number increases after mitomycin C treatment.
Plasmid pBS32 is genetically similar to plasmid pLS32, and previous work using a cloned pLS32 origin of replication indicated that the resulting derivative was maintained at 2 or 3 copies per B. subtilis cell (15, 16). To determine the copy number of pBS32, two complementary approaches were used: quantitative PCR and a fluorescent reporter-operator system (FROS). First, qPCR was used to compare the amplification of one chromosomally encoded locus (sigA) to two plasmid-borne loci (zpaB and zpbI) (Fig. 1A). Both the zpaB and zpbI loci were found to be at roughly the same relative copy number as sigA by qPCR (Fig. 1B, WT Ø). Second, a FROS strain in which a tandem repeat of 37 copies of the Tet operator (tetO37) was inserted near the pBS32 origin was generated, and in the same strain, a translational fusion between the Tet repressor (TetR) and mCherry was placed under the control of the constitutive ftsW promoter and integrated at an ectopic locus (amyE::PftsW-tetR-mCherry). Thus, TetR-mCherry bound to the plasmid-borne operators, and plasmid copy number could be determined according to the number of fluorescent puncta observed (29). In the pBS32 FROS strain, one or two red puncta were observed per cell (Fig. 1C). We conclude that like pLS32, pBS32 is a low-copy-number plasmid with 1 or 2 copies per cell.
FIG 1.

pBS32 copy number increases in response to mitomycin C treatment. (A) Map of the pBS32 plasmid (open circle) divided into conceptual quadrants (QI, QII, QIII, and QIV). Arrows indicate open reading frames, and internal annotations indicate the identity of the adjacent genes. Origin (oriZ) is indicated by a solid circle. The region of putative prophage genes is outlined in orange. Genes mentioned in the text are indicated by triangles. (B) Plasmid copy number as determined by quantitative PCR (qPCR). WT strain 3610 and a ΔzpdN mutant (DK3287) at an OD600 of 0.1 were grown for 90 min in the presence (+MMC) or absence (Ø) of 0.3 μg/ml mitomycin C prior to being harvested for DNA isolation. A strain containing a Physpank-zpdN construct (DK1634) at an OD600 of 0.1 was grown for 60 min in the presence (+IPTG) or absence (Ø) of 1 mM IPTG prior to being harvested for DNA isolation. qPCR was performed on the isolated DNA using primer pairs specific for two different plasmid loci, zpaB and zpbI, and compared to qPCR performed using primer pairs specific for a chromosomal locus, sigA. Plasmid copy number was calculated as described previously (13). (C) Fluorescence microscopy in which TetR-mCherry foci (false colored red) and membrane stained with TMA-DPH (false colored blue) are indicated for strain DK280, which encodes a constitutively expressed TetR-mCherry construct, and an array of 37 copies of the tetO operator sequence were integrated into pBS32.
pBS32 contains genes encoding a putative prophage. Between genes encoding two putative phagelike integrases (ZpaO and ZpbV) are genes predicted to encode phage structural genes, including large and small phage terminase subunits (ZpbL and ZpbK), a portal protein (ZpbJ), a capsid protein (ZpbH), a DNA packaging protein (ZpbF), a head-tail adaptor protein (ZpbE), a tail tape measure protein (ZpaZ), and tail proteins (ZpbB, ZpaY, ZpaW) (Fig. 1A) (15, 30–32). As the region with genes encoding phagelike proteins constituted nearly one-half of the pBS32 plasmid sequence, we wondered if the plasmid copy number would be affected by treating the cells with mitomycin C, a DNA-alkylating agent that can induce prophage expression, excision, and amplification (33–36). Compared to the untreated culture, the copies of the plasmid-borne zpaB and zpbI genes increased 20- and 100-fold, respectively, relative to sigA when treated with 0.3 μg/ml mitomycin C (MMC) and measured by qPCR (Fig. 1B, WT +MMC). We attempted to determine the copy number in parallel using the FROS, but when the culture was treated with mitomycin C, extensive cell lysis prevented the interpretation of any potential punctate localization.
pBS32 is sufficient to lyse B. subtilis when treated with mitomycin C.
To better understand the lysis phenotype, mitomycin C was added to a culture of the wild-type B. subtilis and optical density (OD) measurements were taken over time. Cells began to lyse 30 min after the addition of mitomycin C, and the OD was reduced nearly 10-fold relative to the peak (Fig. 2A). The ancestral strain harbors multiple prophages in the chromosome, including SPβ and PBSX, which could be responsible for cell lysis above and beyond any contribution of pBS32 (37–39). To determine the relative effects of SPβ, PBSX, and pBS32 on mitomycin C-induced cell lysis, strains were constructed in which combinations of two of the prophages were eliminated so that only one of the three remained (14). Following mitomycin C treatment, a decrease in OD was observed when either PBSX (Fig. 2B) or pBS32 (Fig. 2C) was the only prophage of the three present. In contrast, when SPβ was the only prophage of the three present (Fig. 2D) or all three prophages were deleted (Fig. 2E), growth was arrested but only a slight decrease in OD was observed after mitomycin C addition. We conclude that although B. subtilis encodes multiple prophagelike elements (even beyond those tested here), only PBSX and pBS32 are sufficient to cause a mitomycin C-dependent decrease in optical density.
FIG 2.

pBS32 induction inhibits growth. Growth curves of strains either treated at time zero with 1 μg/ml MMC or 1 mM IPTG as indicated (solid symbols) or left untreated (open symbols). The following strains were used: WT (3610) (A); DK399 (B); DK297 (C); DK1687 (D); DK451 (E); DK1552 (F); DK1551 (G); DK2063 (H); DK3952 (I); DK3287 (J); DK1634 (Physpank-wkRBSzpdN in an otherwise wild-type strain; gray circles), DK4180 (ΔQII), DK4181 (ΔQIII), and DK4182 (ΔQIV) (K); and DK4112 (L). Shorthand strain features are given in parentheses to aid the reader.
The mitomycin C-dependent decrease in optical density was likely due to cell lysis. To cytologically monitor cell lysis, the gene encoding GFP was transcriptionally fused to an IPTG-inducible Physpank promoter and inserted at the amyE ectopic locus (amyE::Physpank-gfp) such that growth in the presence of IPTG induces cytoplasmic fluorescence in living cells. Cells in which pBS32 was present but both SPβ and PBSX were deleted lost GFP fluorescence 1.5 h after the addition of mitomycin C and showed signs of lysis by the accumulation of empty sacculi (Fig. 3A). In comparison, when SPβ, PBSX, and pBS32 were simultaneously deleted, addition of mitomycin C caused cells to elongate within 1 h of treatment, but the cells did not begin to lose fluorescence or lyse until the 3.5 h time point (Fig. 3B). We conclude that pBS32 is sufficient to lyse B. subtilis cells in the presence of mitomycin C.
FIG 3.
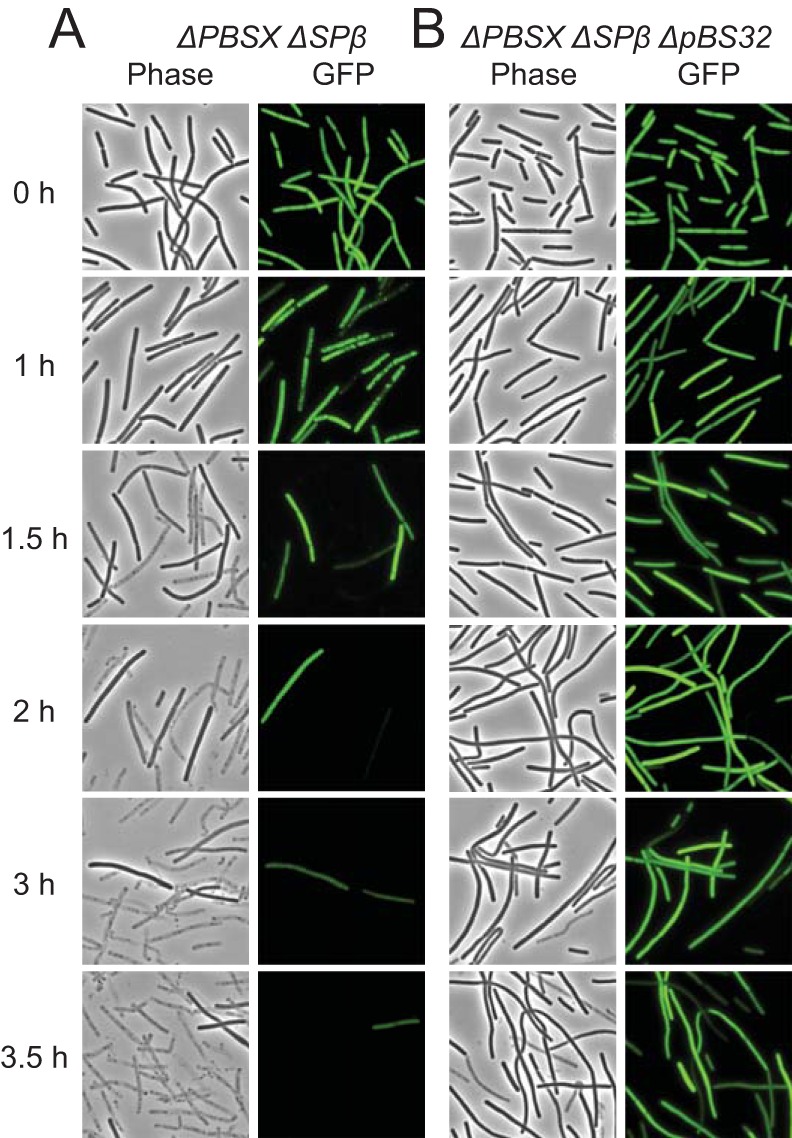
pBS32 induction causes cell lysis. Phase-contrast and epifluorescence microscopy of GFP-expressing strains DK1233 (A) and DK1234 (B) at the indicated time points following treatment with 1 μg/ml mitomycin C at an OD600 of 0.1 (0 h). Strains were grown in the presence of 1 mM IPTG to induce GFP fluorescence. GFP fluorescence was false colored in green.
ZpdN, a sigma factor homolog, is necessary and sufficient for pBS32-mediated cell lysis.
Given that cell lysis was being induced by the presence of mitomycin C-mediated DNA damage, we next wondered if pBS32 encoded a regulator to activate genes required for lysis. Plasmid pBS32 has two genes that encode putative transcriptional regulators, zpbW and zpcW, encoding ZpbW and ZpcW, each containing a helix-turn-helix DNA binding motif. In addition, pBS32 has the gene zpdN, encoding ZpdN, a sigma factor homolog (see Fig. S1 in the supplemental material). To test whether zpbW, zpcW, or zpdN expression was sufficient to activate lysis in the absence of mitomycin C, each gene was transcriptionally fused to an IPTG-inducible Physpank promoter and integrated at the amyE chromosomal ectopic locus in a strain harboring pBS32. Induction of zpbW or zpcW with IPTG was not sufficient to cause a decrease in optical density (Fig. 2F and G). The strain expressing Physpank-zpdN, however, exhibited a growth defect even in the absence of IPTG induction (data not shown), and we inferred that the growth defect was due to low-level expression from the Physpank promoter in the absence of inducer. We conclude that, of the three genes tested, ectopic expression of zpdN had the most severe effect on cell growth.
To better control zpdN expression, the zpdN ribosome binding site in the inducible construct was weakened by fusing the open reading frame to an RBS far from consensus (amyE::Physpank-wkRBSzpdN). Cells with the weakened RBS expression construct did not exhibit a growth defect in the absence of inducer, but the optical density decreased 60 min after IPTG addition (Fig. 2H). The IPTG-dependent decrease in optical density did not occur when zpdN was expressed in a strain lacking pBS32, indicating that cell death was pBS32 dependent and not due to spurious induction of PBSX or other chromosomally borne genes (Fig. 2I). Furthermore, treatment of cells with deletions of zpdN, Spβ, and PBSX with mitomycin C caused a cessation of cell growth but did not cause a decrease in optical density, phenocopying the deletion of pBS32 (compare Fig. 2J and E). Finally, mutation of zpdN abolished pBS32 overreplication in the presence of mitomycin C, and artificial IPTG-dependent expression of zpdN induced overreplication of pBS32 (Fig. 1B). Thus, we conclude that the product of the zpdN gene is both necessary and sufficient to activate pBS32 overreplication and pBS32-dependent cell lysis in response to mitomycin C-induced DNA damage.
In previous work, pBS32 was divided into 4 conceptual quadrants (Fig. 1A), in which quadrant I (QI) contained the plasmid origin of replication and partitioning functions, quadrants II and III (QII and QIII) contained many genes annotated as potential prophage genes, and quadrant IV (QIV) contained genes of unknown function (15, 16, 19). To determine which genes on pBS32 were required for ZpdN-mediated cell lysis, deletions of QII, QIII, and QIV were introduced into a genetic background containing the IPTG-inducible zpdN construct (Physpank-wkRBSzpdN). Deletion of QII resulted in an optical density decrease comparable to that observed in the wild type when IPTG was added (Fig. 2K). Deletions of QIII and QIV, however, caused an arrest of growth, but optical density did not decrease when IPTG was added (Fig. 2K). Furthermore, cells in which QII was deleted lost GFP fluorescence when induced for ZpdN comparable to what was observed for the wild type, whereas cells in which QIII or QIV were deleted did not (see Fig. S2 in the supplemental material). We conclude that QII of pBS32 is dispensable for ZpdN-induced cell lysis. We infer that one or more genes within quadrants QIII and QIV are required for ZpdN-induced cell lysis. Finally, we infer that pBS32-mediated growth arrest and cell lysis are separable phenotypes.
pBS32-dependent lysis by ZpdN induction releases defective phage particles.
One way in which ZpdN induction could cause pBS32-dependent cell lysis is by causing the putative pBS32 prophage to enter a lytic cycle. Thus, we wondered whether we could detect evidence of phage packaging and lysis indirectly by release of pBS32 DNA. To detect released DNA, cells containing the IPTG-inducible zpdN construct were induced with 1 mM IPTG, and supernatants were harvested after 60 min, phenol-chloroform extracted, and used as a template for PCR with primers to plasmid (zpaB, zpbI, and zpcJ) and chromosomal (fliG) loci. Products were observed from each locus, suggesting that plasmid and chromosomal DNA had been released into the supernatant (Fig. 4A, left lane). Treatment of the supernatant with DNase prior to extraction and amplification abolished product from the chromosomal locus but not the three plasmid loci, suggesting that the plasmid loci were being protected (Fig. 4A, middle lane). When the supernatants were heat treated prior to DNase addition, the amount of PCR product detected from plasmid loci was reduced, suggesting that the factor that protected the plasmid loci was heat labile (Fig. 4A, right lane). Further, a strain in which the zpbH gene, encoding the putative phage capsid protein ZpbH, was deleted lysed upon expression of ZpdN (Fig. 2L), but DNase protection of extracellular plasmid loci in the supernatant was abolished (Fig. 4B). We conclude that induction of ZpdN released plasmid DNA that was protected by the ZpbH capsid protein, perhaps due to the formation of a phagelike particle.
FIG 4.

pBS32 produces a defective phage that protects plasmid DNA. (A) DNase protection assay. Strain DK1539 (Physpank-wkRBSzpdN) was induced to lyse by the addition of 1 mM IPTG, and the resulting supernatant was harvested. Left lane, extracellular DNA was phenol-chloroform extracted and used as a PCR template to amplify the chromosomal locus fliG and the plasmid loci zpaB, zpbI, and zpcJ (Fig. 1A). Middle lane, extracellular DNA was treated with DNase prior to extraction and PCR. Right lane, extracellular DNA was heat treated and then DNase treated prior to extraction and PCR. Panels were cropped from the same gel (see Fig. S5A in the supplemental material). (B) DNase protection assay. Strain DK4112 (ΔzpbH ΔSPβ ΔPBSX Physpank-wkRBSzpdN) was induced to lyse by the addition of 1 mM IPTG, and the resulting supernatant was harvested, phenol-chloroform extracted, and used as a PCR template to amplify the plasmid loci zpaB, zpbI, and zpcJ. Middle lane, extracellular DNA was treated with DNase prior to extraction and PCR. Right lane, extracellular DNA was heat treated and then DNase treated prior to extraction and PCR. Panels were cropped from the same gel (see Fig. S5B in the supplemental material). (C and D) EM images of PEG-precipitated supernatants of a hag mutant (DS1143) (C) and a hag ΔpBS32 mutant (DK3650) (D) induced with mitomycin C. Scale bar, 100 nm. Inset, enlarged images of PBSX particles. (E and F) EM images of PEG-precipitated supernatants of a hag ΔpBS32 ΔSPβ ΔPBSX mutant (DK3652) (E) and a hag ΔSPβ ΔPBSX mutant (DK3651) (F) induced with mitomycin C. Scale bar, 100 nm. Inset, enlarged images. (G) Silver-stained fraction taken from CsCl gradient resolved at the interface between 1.45 and 1.55 g/ml of PEG-precipitated supernatant of DK1634 (hag ΔSPβ ΔPBSX) induced with mitomycin C. The triangle indicates the location of a band that was excised and determined to be the ZpbH capsid protein by mass spectrometry analysis.
Next, we attempted to detect phage activity by a plaque assay similar to that established for the generalized transducing phage SPP1 (26). Cells containing pBS32 were induced for lysis by an ectopically integrated Physpank-wkRBSzpdN construct for 1 h with 1 mM IPTG, and supernatants were collected and concentrated by PEG precipitation. Uninoculated medium was concentrated by PEG precipitation as a control. Both concentrates were spotted on a lawn of recipient cells deleted for pBS32 to minimize the possibility of preexistent superinfection resistance systems. No zone of clearing was observed for either the mock or experimental lysate after the lawn had grown. We infer that the particles released upon pBS32-mediated cell death either were incapable of establishing a lytic infection or were somehow inactivated during concentration.
To further examine whether a killing activity was released upon pBS32-mediated cell death with minimal manipulation, we developed a mixed-culture assay. Cells containing the Physpank-wkRBSzpdN construct and in which the chromosomal prophages SPβ and PBSX were deleted (“donor”) were mixed 10:1 with cells in which pBS32 was deleted (“recipient”). Each cell type also contained a different antibiotic resistance cassette, so the abundance of each could be assessed by dilution plating on their respective antibiotics. In the absence of IPTG, the coculture exhibited exponential growth (Fig. 5A) and each subpopulation accumulated proportionally (Fig. 5B). In contrast, the optical density of the coculture plateaued soon after IPTG induction (Fig. 5C) due to the loss of CFU of the donor and later increased as the recipient grew to predominance (Fig. 5D). Importantly, the CFU of the recipient increased unabated in the presence of the dying donor (Fig. 5D). We conclude that pBS32-dependent cell lysis releases particles capable of protecting plasmid DNA but that the particles are defective and incapable of infecting or killing other B. subtilis cells extracellularly.
FIG 5.
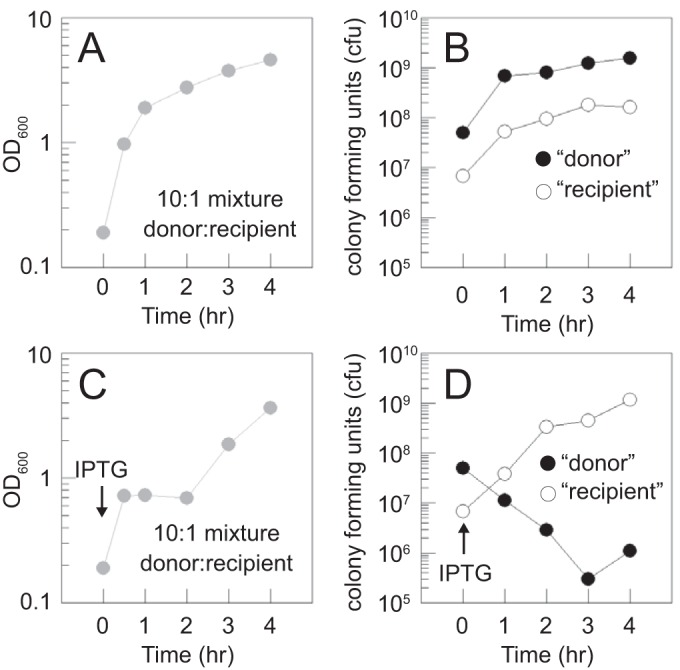
pBS32-dependent cell lysis does not release particles capable of killing cells in trans. (A) Growth curve by OD600 of a culture mixed at T0 with 10 parts “donor” (DK1634 [ΔSPβ ΔPBSX amyE::Physpank-wkRBSsigD Specr]) cells to 1 part “recipient” (DK4162 [ΔpBS32 amyE::Km]) cells in LB medium at 37°C. (B) Growth curve of CFU by dilution plating on LB containing spectinomycin (to permit growth of the donor cells) and separately on LB containing kanamycin (to permit growth of the recipient cells) taken in parallel with the optical density measurements as described for panel A. (C and D) Optical density and CFU measurements, respectively, of the same strains used to generate data in panels A and B, mixed at the same ratio as for data in panels A and B, but inoculated into LB medium containing 1 mM IPTG (to induce zpdN expression in the donor) at 37°C.
Transmission electron microscopy (TEM) was conducted on the MMC-treated PEG-precipitated supernatants to detect pBS32-derived particles. As positive controls, particles consistent with PBSX were isolated from the wild type (Fig. 4C) and a strain in which pBS32 was deleted (Fig. 4D) (33). No particles were observed when SPβ, PBSX, and pBS32 were deleted (Fig. 4E) or when pBS32 was the only prophage of the three present (Fig. 4F). Thus, we were unable to observe phagelike particles that originated from pBS32 by EM. As an alternative approach to detect particles, the PEG-precipitated supernatant was resolved on a CsCl step gradient, and a band was recovered at the interface between 1.45 g/ml and 1.55 g/ml CsCl, indicative of a protein-nucleic acid complex consistent with a phagelike particle. When the CsCl fraction was resolved on SDS-PAGE, a dominant band at 27 kDa was excised and through protease treatment and LC-MS/MS was determined to contain fragments consistent with the ZpbH putative capsid protein (Fig. 4G). Thus, DNase-protected plasmid DNA and the resolution of the ZpbH capsid protein on a CsCl gradient suggest that the pBS32 prophage forms a DNA-protein complex. We infer that the putative phage is assembly defective, which results in the formation of a labile nucleoprotein complex that is lost during EM preparations.
DISCUSSION
Prophages are common in bacterial chromosomes, and here we provide evidence that the large, low-copy-number plasmid pBS32 of B. subtilis encodes a defective prophage. Addition of the DNA-damaging agent MMC caused cell death that was dependent on the presence of the plasmid. Following cell lysis, DNA sequences from the plasmid were protected from DNase treatment and protection was dependent on the production of the ZpbH capsid protein. The released particles, however, were defective, as they were unable to kill other B. subtilis cells when provided extracellularly and were sufficiently unstable that we were unable to observe them after concentration and preparation for electron microscopy. In sum, the observations are consistent with a model in which MMC-induced DNA damage triggers the pBS32 prophage to enter a lytic cycle that kills the donor cell and releases unstable, defective particles that contain plasmid DNA.
The nature of the pBS32 prophage structural and plaque-forming defects is unknown, but we may speculate by drawing comparisons to other phage systems. The pBS32 capsid protein ZpbH shows structural similarity (HHpred, E value = e−47), but not sequence similarity, to the capsid from the lambdoid bacteriophage HK97 (40, 41). The HK97 capsid protein is translated as a 42-kDa protein, and with the portal protein and protease (corresponding to ZpbJ and ZpbI, respectively), self-assembles into a structural stage called prohead I (42, 43). Next, the protease cleaves the capsid protein at residue 104, the N terminus is released, and the smaller 30.7-kDa protein remains in the particle to form the structure called prohead II (42, 44). Similar to the HK97 capsid, the 41-kDa ZpbH was identified as a 27-kDa band by mass spectrometry when resolved by SDS-PAGE. Furthermore, mass spectrometry analysis indicated that all fragments detected mapped to the C terminus of the protein, consistent with an N-terminal cleavage event (see Table S3 and Fig. S3 in the supplemental material). Thus, ZpbH appeared to have been proteolytically processed and may have formed a structure analogous to that of prohead II, perhaps consistent with the HK97 paradigm.
Following the formation of prohead II in HK97, DNA is packaged (45). Consistent with the formation of a prohead II-like state, DNA from the pBS32 plasmid but not the chromosome was protected from DNase treatment by the ZpbH capsid protein, suggesting that the plasmid DNA may have been packaged within a phagelike particle. The packaging size and the boundaries of what constitutes the prophage genome, however, are unclear. In one model, pBS32 could be a plasmid into which a prophage has secondarily integrated. Nearly all genes on pBS32 are colinear and oriented clockwise from the origin except for the region of putative prophage structural genes, which are colinear and oriented in a counterclockwise direction, perhaps suggesting a secondary prophage integration event (Fig. 1A). Prophage insertion can sometimes be detected as a region of GC content that differs from the surrounding DNA, but the GC content appears to be regular throughout the pBS32 sequence (see Fig. S4 in the supplemental material). In another model, pBS32 in its entirety could constitute a horizontally transferred prophage, and consistent with horizontal transfer, the 35% GC content of pBS32 is lower than the 43% GC content of the chromosome (46). Packaging of the entire plasmid was supported when at least three positions well separated on the pBS32 sequence were equally protected from DNase following cell lysis. If the entire plasmid is indeed packaged, pBS32 would represent the first example of a P1-like plasmid prophage reported for the Gram-positive bacteria (45).
Perhaps consistent with the entire plasmid being a single packaged unit is that the zpdN gene is both necessary and sufficient for plasmid prophage-mediated cell lysis but is remote from the prophage structural genes. ZpdN is a sigma factor homolog, but how it induces cell lysis is unknown. Sigma factors activate gene expression by recruiting RNA polymerase core to their cognate promoter sequences. ZpdN exhibits homology to regions 2 and 4 of sigma factors responsible for binding the −10 and −35 promoter elements, respectively (47–49). ZpdN lacks a sigma factor region called 3.1, but the absence of this region does not rule out sigma factor activity as the region is similarly lacking in another B. subtilis sigma factor, SigH (see Fig. S2 in the supplemental material) (50). Thus, ZpdN could induce cell lysis as an alternative sigma factor that directs gene expression of one or more prophage promoters. Alternatively, ZpdN is also required for increasing pBS32 plasmid copy number 100-fold, and amplification of the gene copy number could indirectly titrate a phage repressor. Whatever the case, genes in quadrants III and IV of pBS32 are required for cell lysis but still arrest growth when ZpdN is induced. We note that the B. subtilis chromosomal prophage PBSX encodes the sigma factor homolog Xpf, which is necessary for inducing PBSX-dependent cell lysis, the mechanism of which is also poorly understood (51).
Precisely how and why pBS32 was cured is unknown, but it happened sometime between early work in the B. subtilis ancestral strain 3610 and the events that created the B. subtilis laboratory strain descendant 168 (4). pBS32 may have been cured naturally by serial transfer in the absence of positive environmental selection (whatever that may be), but the low copy number and a high-fidelity partitioning system to ensure its inheritance suggest otherwise. Alternatively, plasmid loss may have been artificial, as scientists mutagenized and specifically selected strains competent for DNA uptake, thereby selecting against the pBS32-encoded competence inhibitor protein ComI (52, 53). Precisely why pBS32 encodes an inhibitor of competence is unknown. The strain cured of pBS32 was generated by selecting for maintenance of an unstable plasmid with an incompatible origin, and ComI may thus enhance the stability of pBS32 in the cell by excluding foreign plasmid uptake (15). As another possibility, we note that DNA taken up by competence systems enters the cell as single-stranded DNA and integration requires homologous recombination such that some bacteria coregulate competence with the DNA damage response (54–56). Thus, perhaps ComI protects the host by inhibiting transformation and the associated DNA repair systems that could trigger spurious entry into the pBS32 lytic cycle.
Supplementary Material
ACKNOWLEDGMENTS
We thank Cassie Xu for her help with qPCR, Aisha Burton for sequence analysis, and David Rudner and Kevin Griffith for generously providing several plasmids. Thanks go to Irene Newton for her help with GC content analysis. We thank Bob Duda, Ben Fane, Ian Molineux, Kristen Parent, and Ry Young for intellectual and technical support and advice.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00213-16.
REFERENCES
- 1.Velicer GJ, Kroos L, Lenski RE. 1998. Loss of social behaviors by Myxococcus xanthus during evolution in an unstructured habitat. Proc Natl Acad Sci U S A 95:12376–12380. doi: 10.1073/pnas.95.21.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Winogradsky S. 1937. The doctrine of pleomorphism in bacteriology. Soil Sci 43:327–339. doi: 10.1097/00010694-193705000-00001. [DOI] [Google Scholar]
- 3.Douglas GL, Klaenhammer TR. 2010. Genomic evolution of domesticated microorganisms. Annu Rev Food Sci Technol 1:397–414. doi: 10.1146/annurev.food.102308.124134. [DOI] [PubMed] [Google Scholar]
- 4.Zeigler DR, Prágai Z, Rodriguez S, Chevreux B, Muffler A, Albert T, Bai R, Wyss M, Perkins JB. 2008. The origins of 168, W23, and other Bacillus subtilis legacy strains. J Bacteriol 190:6983–6995. doi: 10.1128/JB.00722-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Branda SS, Gonzalez-Pastor JE, Ben-Yehuda S, Losick R, Kolter R. 2001. Fruiting body formation by Bacillus subtilis. Proc Natl Acad Sci U S A 98:11621–11626. doi: 10.1073/pnas.191384198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arima K, Kakinuma A, Tamura G. 1968. Surfactin, a crystalline peptidelipid surfactant produced by Bacillus subtilis: isolation, characterization and its inhibition of fibrin clot formation. Biochem Biophys Res Commun 31:488–494. doi: 10.1016/0006-291X(68)90503-2. [DOI] [PubMed] [Google Scholar]
- 7.Butcher RA, Schroeder FC, Fischbach MA, Straight PD, Kolter R, Walsh CT, Clardy J. 2007. The identification of bacillaene, the product of the PksX megacomplex in Bacillus subtilis. Proc Natl Acad Sci U S A 104:1506–1509. doi: 10.1073/pnas.0610503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kearns DB, Losick R. 2003. Swarming motility in undomesticated Bacillus subtilis. Mol Microbiol 49:581–590. [DOI] [PubMed] [Google Scholar]
- 9.Stanley NR, Lazazzera BA. 2005. Defining the genetic differences between wild and domestic strains of Bacillus subtilis that affect poly-γ-DL-glutamic acid production and biofilm formation. Mol Microbiol 57:1143–1158. doi: 10.1111/j.1365-2958.2005.04746.x. [DOI] [PubMed] [Google Scholar]
- 10.Kearns DB, Chu F, Rudner R, Losick R. 2004. Genes governing swarming in Bacillus subtilis and evidence for a phase variation mechanism controlling surface motility. Mol Microbiol 52:357–369. doi: 10.1111/j.1365-2958.2004.03996.x. [DOI] [PubMed] [Google Scholar]
- 11.McLoon AL, Guttenplan SB, Kearns DB, Kolter R, Losick R. 2011. Tracing the domestication of a biofilm-forming bacterium. J Bacteriol 193:2027–2034. doi: 10.1128/JB.01542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakano MM, Corbell N, Besson J, Zuber P. 1992. Isolation and characterization of sfp: a gene that functions in the production of the lipopeptide biosurfactant, surfactin, in Bacillus subtilis. Mol Gen Genet 232:313–321. [DOI] [PubMed] [Google Scholar]
- 13.Earl AM, Losick R, Kolter R. 2007. Bacillus subtilis genome diversity. J Bacteriol 189:1163–1170. doi: 10.1128/JB.01343-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Srivatsan A, Han Y, Peng J, Tehranchi AK, Gibbs R, Wang JD, Chen R. 2008. High-precision, whole-genome sequencing of laboratory strains facilitates genetic studies. PLoS Genet 4:e1000139. doi: 10.1371/journal.pgen.1000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konkol MA, Blair KM, Kearns DB. 2013. Plasmid-encoded ComI inhibits competence in the ancestral 3610 strain of Bacillus subtilis. J Bacteriol 195:4085–4093. doi: 10.1128/JB.00696-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka T, Ogura M. 1998. A novel Bacillus natto plasmid pLS32 capable of replication in Bacillus subtilis. FEBS Lett 422:243–246. doi: 10.1016/S0014-5793(98)00015-5. [DOI] [PubMed] [Google Scholar]
- 17.Hassan AKM, Moriya S, Ogura M, Tanaka T, Kawamura F, Ogasawara N. 1997. Suppression of initiation defects of chromosome replication in Bacillus subtilis dnaA and oriC-deleted mutants by integration of a plasmid replicon into the chromosomes. J Bacteriol 179:2494–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becker E, Herrara NC, Gunderson FQ, Derman AI, Dance AL, Sims J, Larsen RA, Pogliano J. 2006. DNA segregation by the bacterial actin AlfA during Bacillus subtilis growth and development. EMBO J 25:5919–5931. doi: 10.1038/sj.emboj.7601443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanaka T, Ishida H, Maehara T. 2005. Characterization of the replication region of plasmid pLS32 from the natto strain of Bacillus subtilis. J Bacteriol 187:4315–4326. doi: 10.1128/JB.187.13.4315-4326.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parashar V, Konkol MA, Kearns DB, Neiditch MB. 2013. A plasmid-encoded phosphatase regulates Bacillus subtilis biofilm architecture, sporulation, and genetic competence. J Bacteriol 195:2437–2448. doi: 10.1128/JB.02030-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Omer Bendori S, Pollak S, Hizi D, Eldar A. 2015. The RapP-PhrP quorum-sensing system of Bacillus subtilis strain NCIB3610 affects biofilm formation through multiple targets, due to an atypical signal-insensitive allele of RapP. J Bacteriol 197:592–602. doi: 10.1128/JB.02382-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Auchtung JM, Lee CA, Monson RE, Lehman AP, Grossman AD. 2005. Regulation of a Bacillus subtilis mobile genetic element by intercellular signaling and the global DNA damage response. Proc Natl Acad Sci U S A 102:12554–12559. doi: 10.1073/pnas.0505835102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pottathil M, Lazazzera BA. 2003. The extracellular Phr peptide-Rap phosphatase signaling circuit of Bacillus subtilis. Front Biosci 8:d32–d45. doi: 10.2741/913. [DOI] [PubMed] [Google Scholar]
- 24.Singh PK, Ramachandran G, Durán-Alcalde L, Alonso C, Wu LJ, Meijer WJJ. 2012. Inhibition of Bacillus subtilis natural competence by a native, conjugative plasmid-encoded comK repressor protein. Environ Microbiol 14:2812–2825. doi: 10.1111/j.1462-2920.2012.02819.x. [DOI] [PubMed] [Google Scholar]
- 25.Dalia AB, Seed KD, Calderwood SB, Camilli A. 2015. A globally distributed mobile genetic element inhibits natural transformation of Vibrio cholerae. Proc Natl Acad Sci U S A 112:10485–10490. doi: 10.1073/pnas.1509097112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yasbin RE, Young FE. 1974. Transduction in Bacillus subtilis by bacteriophage SPP1. J Virol 14:1343–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patrick JE, Kearns DB. 2008. MinJ (YvjD) is a topological determinant of cell division in Bacillus subtilis. Mol Microbiol 70:1166–1179. doi: 10.1111/j.1365-2958.2008.06469.x. [DOI] [PubMed] [Google Scholar]
- 28.Škulj M, Okršlar V, Jalen Š, Jevševar S, Slanc P, Štrukelj B, Menart V. 2008. Improved determination of plasmid copy number using quantitative real-time PCR for monitoring fermentation processes. Microb Cell Fact 7:6. doi: 10.1186/1475-2859-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gordon GS, Sitnikov D, Webb CD, Teleman A, Straight A, Losick R, Murray AW, Wright A. 1997. Chromosome and low copy plasmid segregation in E. coli: visual evidence for distinct mechanisms. Cell 90:1113–1121. doi: 10.1016/S0092-8674(00)80377-3. [DOI] [PubMed] [Google Scholar]
- 30.Aksyuk AA, Rossman MG. 2011. Bacteriophage assembly. Viruses 3:172–203. doi: 10.3390/v3030172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leiman PG, Arisaka F, van Raaij MJ, Kostyuchenko VA, Aksyuk AA, Kanamaru S, Rossman MG. 2010. Morphogenesis of the T4 tail and tail fibers. Virol J 7:355. doi: 10.1186/1743-422X-7-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rao VB, Black LW. 2010. Structure and assembly of bacteriophage T4 head. Virol J 7:356. doi: 10.1186/1743-422X-7-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okamoto K, Mudd JA, Mangan J, Huang WM, Subbaiah TV, Marmur J. 1968. Properties of the defective phage of Bacillus subtilis. J Mol Biol 34:413–428. doi: 10.1016/0022-2836(68)90169-1. [DOI] [PubMed] [Google Scholar]
- 34.Warner FD, Kitos GA, Romano MP, Hemphill HE. 1977. Characterization of SPβ: a temperate bacteriophage from Bacillus subtilis 168M. J Bacteriol 23:45–51. [Google Scholar]
- 35.Mauël C, Karamata D. 1984. Characterization of proteins induced by mitomycin C treatment of Bacillus subtilis. J Virol 49:806–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goranov AI, Kuester-Schoek E, Wang JD, Grossman AD. 2006. Characterization of the global transcriptional response to different types of DNA damage and disruption of replication in Bacillus subtilis. J Bacteriol 188:5595–5605. doi: 10.1128/JB.00342-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lazarevic V, Düsterhoft A, Soldo B, Hilbert H, Mauël C, Karamata D. 1999. Nucleotide sequence of the Bacillus subtilis temperate bacteriophage SPβc2. Microbiology 145:1055–1067. doi: 10.1099/13500872-145-5-1055. [DOI] [PubMed] [Google Scholar]
- 38.Wood HE, Dawson MT, Devine KM, McConnell DJ. 1990. Characterization of PBSX, a defective prophage of Bacillus subtilis. J Bacteriol 172:2667–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Westers H, Dorenbos R, van Dijl JM, Kabel J, Flanagan T, Devine KM, Jude F, Séror SJ, Beekman AC, Darmon E, Eschevins C, de Jong A, Bron S, Kuipers OP, Albertini AM, Antelmann H, Hecker M, Zamboni N, Sauer U, Bruand C, Ehrlich DS, Alonso JC, Sala M, Quax WJ. 2003. Genome engineering reveals large dispensible regions in Bacillus subtilis. Mol Biol Evol 20:2076–2090. doi: 10.1093/molbev/msg219. [DOI] [PubMed] [Google Scholar]
- 40.Söding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suhanovsky MM, Teschke CM. 2015. Nature's favorite building block: deciphering folding and capsid assembly of proteins with the HK97-fold. Virology 479-480:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duda RL, Hempel J, Michel H, Sabanowitz J, Hunt D, Hendrix RW. 1995. Structural transitions during bacteriophage HK97 head assembly. J Mol Biol 247:618–635. [DOI] [PubMed] [Google Scholar]
- 43.Xie Z, Hendrix RW. 1995. Assembly in vitro of bacteriophage HK97 proheads. J Mol Biol 253:74–85. doi: 10.1006/jmbi.1995.0537. [DOI] [PubMed] [Google Scholar]
- 44.Duda RL, Martincic K, Hendrix RW. 1995. Genetic basis of bacteriophage HK97 prohead assembly. J Mol Biol 247:636–647. [DOI] [PubMed] [Google Scholar]
- 45.Ikeda H, Tomizawa J. 1968. Prophage P1, an extrachromosomal replication unit. Cold Spring Harbor Symp Quant Biol 33:791–798. doi: 10.1101/SQB.1968.033.01.091. [DOI] [PubMed] [Google Scholar]
- 46.Priest FG. 1993. Systematics and ecology of Bacillus, p 3–16. In Sonenshein AL, Hoch JA, Losick R (ed), Bacillus subtilis and other Gram-positive bacteria: biochemistry, physiology, and molecular genetics. American Society for Microbiology, Washington, DC. [Google Scholar]
- 47.Feklístov A, Sharon BD, Darst SA, Cross CA. 2014. Bacterial sigma factors: a historical, structural, and genomic perspective. Annu Rev Microbiol 68:357–376. doi: 10.1146/annurev-micro-092412-155737. [DOI] [PubMed] [Google Scholar]
- 48.Borukhov S, Nudler E. 2003. RNA polymerase holoenzyme: structure, function and biological implications. Curr Opin Microbiol 6:93–100. doi: 10.1016/S1369-5274(03)00036-5. [DOI] [PubMed] [Google Scholar]
- 49.Young BA, Gruber TM, Gross CA. 2004. Minimal machinery of RNA polymerase holoenzyme sufficient for promoter melting. Science 303:1382–1384. doi: 10.1126/science.1092462. [DOI] [PubMed] [Google Scholar]
- 50.Tatti KM, Carter HL III, Moir A, Moran CP Jr. 1989. Sigma H-directed transcription of citG in Bacillus subtilis. J Bacteriol 171:5928–5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McDonnell GE, Wood H, Devine KM, McConnell DJ. 1994. Genetic control of bacterial suicide: regulation of the induction of PBSX in Bacillus subtilis. J Bacteriol 176:5820–5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burkholder PR, Giles NH Jr. 1947. Induced biochemical mutations in Bacillus subtilis. Am J Botany 34:345–348. doi: 10.2307/2437147. [DOI] [PubMed] [Google Scholar]
- 53.Spizizen J. 1958. Transformation of biochemically deficient strains of Bacillus subtilis by deoxyribonucleate. Proc Natl Acad Sci U S A 44:1072–1078. doi: 10.1073/pnas.44.10.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prudhomme M, Attaiech L, Sanchez G, Martin B, Claverys J-P. 2006. Antibiotic stress induces genetic transformability in the human pathogen Streptococcus pneumoniae. Science 313:89–92. doi: 10.1126/science.1127912. [DOI] [PubMed] [Google Scholar]
- 55.Charpentier X, Kay E, Schneider D, Shuman HA. 2011. Antibiotics and UV radiation induce competence for natural transformation in Legionella pneumophila. J Bacteriol 193:1114–1121. doi: 10.1128/JB.01146-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yasbin RE, Wilson GA, Young FE. 1975. Transformation and transfection in lysogenic strains of Bacillus subtilis: evidence for selective induction of prophage in competent cells. J Bacteriol 121:296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.