

ABSTRACT
Cyclic AMP (cAMP) and the cAMP receptor protein (cAMP-CRP) and CsrA are the principal regulators of the catabolite repression and carbon storage global regulatory systems, respectively. cAMP-CRP controls the transcription of genes for carbohydrate metabolism and other processes in response to carbon nutritional status, while CsrA binds to diverse mRNAs and regulates translation, RNA stability, and/or transcription elongation. CsrA also binds to the regulatory small RNAs (sRNAs) CsrB and CsrC, which antagonize its activity. The BarA-UvrY two-component signal transduction system (TCS) directly activates csrB and csrC (csrB/C) transcription, while CsrA does so indirectly. We show that cAMP-CRP inhibits csrB/C transcription without negatively regulating phosphorylated UvrY (P-UvrY) or CsrA levels. A crp deletion caused an elevation in CsrB/C levels in the stationary phase of growth and increased the expression of csrB-lacZ and csrC-lacZ transcriptional fusions, although modest stimulation of CsrB/C turnover by the crp deletion partially masked the former effects. DNase I footprinting and other studies demonstrated that cAMP-CRP bound specifically to three sites located upstream from the csrC promoter, two of which overlapped the P-UvrY binding site. These two proteins competed for binding at the overlapping sites. In vitro transcription-translation experiments confirmed direct repression of csrC-lacZ expression by cAMP-CRP. In contrast, cAMP-CRP effects on csrB transcription may be mediated indirectly, as it bound nonspecifically to csrB DNA. In the reciprocal direction, CsrA bound to crp mRNA with high affinity and specificity and yet exhibited only modest, conditional effects on expression. Our findings are incorporated into an emerging model for the response of Csr circuitry to carbon nutritional status.
IMPORTANCE Csr (Rsm) noncoding small RNAs (sRNAs) CsrB and CsrC of Escherichia coli use molecular mimicry to sequester the RNA binding protein CsrA (RsmA) away from lower-affinity mRNA targets, thus eliciting major shifts in the bacterial lifestyle. CsrB/C transcription and turnover are activated by carbon metabolism products (e.g., formate and acetate) and by a preferred carbon source (glucose), respectively. We show that cAMP-CRP, a mediator of classical catabolite repression, inhibits csrC transcription by binding to the upstream region of this gene and also inhibits csrB transcription, apparently indirectly. We propose that glucose availability activates pathways for both synthesis and turnover of CsrB/C, thus shaping the dynamics of global signaling in response to the nutritional environment by poising CsrB/C sRNA levels for rapid response.
INTRODUCTION
The Csr (carbon storage regulator) or Rsm (repressor of stationary-phase metabolites) system is a widely conserved bacterial posttranscriptional regulatory system (1–3). Its components, their functions, and the mechanisms by which the central regulator of this system, CsrA or RsmA, affects gene expression have been studied primarily in Gammaproteobacteria (4–6). The sequence-specific RNA binding protein CsrA regulates translation, stability, and/or transcription or elongation of numerous target mRNAs. CsrA regulates the expression of genes involved in lifestyle transitions. In Escherichia coli, CsrA activates glycolysis and central carbon pathways (7–13) and motility (14, 15). Conversely, it represses gluconeogenesis (7), glycogen biosynthesis (16–20), biofilm formation (21–24), the stringent response (25), and expression of genes involved in other stress resistance and stationary-phase processes, e.g., cstA, hfq, cel, sdiA, and nhaR (24, 26–30). Its effects on pathogenesis are complex. For example, CsrA both positively and negatively affects expression of enteropathogenic E. coli (EPEC) pathogenicity island genes (31, 32). CsrA was found to copurify with over 700 different mRNAs in E. coli K-12 (25) and to affect the expression of hundreds of genes (33).
Consistent with its extensive regulatory role, CsrA activity is tightly controlled. In E. coli, csrA is transcribed from five promoters using two different sigma factors (34). Furthermore, CsrA directly represses its own translation while indirectly activating its transcription (34). CsrA activity is antagonized by the noncoding small RNAs (sRNAs) CsrB and CsrC, which contain multiple CsrA binding sites that allow them to sequester this protein (35, 36). Fluctuations in the levels of these RNAs regulate CsrA activity in response to the environment. Transcription of both csrB and csrC (csrB/C) is activated by the BarA-UvrY two-component signal transduction system (TCS) in response to carboxylic acids such as formate and acetate (3, 36–41). CsrA indirectly activates transcription of CsrB and CsrC through its effects on BarA-UvrY, creating a negative-feedback loop within the Csr circuitry (37, 38, 42). CsrB/C turnover requires the GGDEF-EAL domain protein CsrD, which is necessary for cleavage by RNase E and turnover (42, 43). Therefore, CsrD affects the expression of CsrA-regulated genes and processes. Recent studies showed that glucose availability activates CsrB/C decay. The unphosphorylated form of EIIAGlc of the phosphoenolpyruvate:carbohydrate phosphotransferase system (PTS), which predominates during glucose transport, binds to the EAL domain of CsrD (44). We previously proposed a model for the influence of carbon nutrition on the workings of the Csr system based on these observations. The elimination of a preferred carbon source and the buildup of carboxylic acid products of metabolism together facilitate CsrB/C sRNA accumulation, inhibit CsrA activity, and promote the physiological switch from the exponential phase to the stationary phase of growth and a stress-resistant phenotype (43, 44).
Noteworthy among the many E. coli mRNAs that copurified with CsrA were transcripts for global regulatory factors such as relA and dksA of the stringent response system and crp and cyaA of the catabolite repression system (25). Reciprocal regulatory interactions between the Csr and stringent response systems permit the Csr system to posttranscriptionally reinforce the transcriptional effects of DksA and (p)ppGpp on the expression of genes that are coregulated by these systems (25). Details of the interactions between the Csr and catabolite repression regulatory systems were not previously determined and are the subject of the present study.
The genes crp and cyaA encode the cyclic AMP (cAMP) receptor protein (CRP) and the enzyme that synthesizes cAMP, adenylate cyclase, respectively. The cAMP-CRP complex regulates transcription in response to the availability of a preferred carbon source, e.g., glucose (45–47). Under conditions of carbon limitation, the PTS proteins, including the glucose-specific protein EIIAGlc, are predominantly phosphorylated. In this form, P-EIIAGlc binds to adenylate cyclase and activates cAMP synthesis (48). Transport and phosphorylation of glucose or other PTS sugars leads to dephosphorylation of EIIAGlc and loss of its ability to activate cAMP synthesis. The cAMP-CRP complex mediates hierarchical utilization of nonpreferred carbon sources, referred to as carbon catabolite repression (CCR), by activating the expression of genes required for the transport and utilization of alternative carbon sources (49). cAMP-CRP also influences the expression of genes not directly involved in carbon metabolism such as those encoding ribosomal proteins, tRNAs, amino acid biosynthesis enzymes, heat shock proteins, sRNAs, and perhaps as many as 70 transcription factors (45–47, 50–55). cAMP levels and cAMP-CRP regulatory functions have been suggested to respond to both the carbon status and the nitrogen status of the cell, leading to reorganization of the proteome (56).
cAMP-CRP is a bifunctional protein that can activate or repress transcription (57). As a transcriptional activator, cAMP-CRP binds to a sequence located upstream from (class I activation) or close to (class II activation) promoter DNA and participates in protein-protein interactions leading to transcription initiation by RNA polymerase (58). Activation using CRP binding sites positioned farther upstream from the promoter requires cAMP-CRP to work in conjunction with other regulatory proteins and may involve protein-protein interactions and/or DNA bending. DNA binding mechanisms similar to those observed in activation are employed when cAMP-CRP acts as a repressor (57).
Bioinformatics analysis for potential cAMP-CRP binding sites in the E. coli genome identified the coding region of syd, the gene immediately upstream from csrB, as a possible target (47). In addition, an online tool (Virtual Footprint [www.prodoric.de/vfp]) for predicting the binding sequences of regulatory proteins identified potential CRP binding sites in csrB, csrC, csrA, csrD, and uvrY. Also, possible reciprocal regulatory interactions between cAMP-CRP and the Csr system prompted us to undertake the present study. We provide evidence that cAMP-CRP inhibits the transcription of csrC directly and that of csrB indirectly, while CsrA modestly and conditionally activates crp expression. The implications of this new circuitry for determining the complex global regulatory response of E. coli to its carbon nutritional environment are discussed.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The strains, plasmids, and bacteriophage used in this study are listed in Table 1. Oligonucleotides are listed in Table 2. Unless otherwise indicated, bacteria were grown at 37°C, with shaking at 250 rpm, in Luria-Bertani (LB) medium (59), LB medium buffered with 0.1 M MOPS (3-morpholinopropane-1-sulfonic acid) (LB-MOPS) and with or without an added carbon source, or Kornberg (KB) medium (1.1% K2HPO4, 0.85% KH2PO4, 0.6% yeast extract containing 0.5% glucose for liquid medium). Media were supplemented with antibiotics at the following concentrations or as indicated otherwise: kanamycin at 100 μg/ml; ampicillin at 100 μg/ml; chloramphenicol at 25 μg/ml, and tetracycline at 10 μg/ml. P1vir transductions were performed as previously described (59).
TABLE 1.
List of the strains, plasmids, and bacteriophage used in this studya
| Strain, plasmid, or bacteriophage | Genotype or description | Source or reference |
|---|---|---|
| E. coli strains | ||
| MG1655 | F− λ− rph-1 | CGSC (no. 6300) |
| AP379 | MG1655 ΔlacZ | This study |
| AP455 | MG1655 crp::cam | This study |
| AP1000 | MG1655 cyaA::kan | This study |
| AP779 | MG1655 ΔcsrD::kan | This study |
| AP792 | MG1655 ΔcsrD::kan crp::cam | This study |
| AP461 | MG1655 ΔlacZ/pLFXcsrB-lacZ | This study |
| AP482 | MG1655 ΔlacZ/pLFXcsrB-lacZ crp::cam | This study |
| AP858 | MG1655 ΔlacZ/pLFXcsrC-lacZ | This study |
| AP864 | MG1655 ΔlacZ/pLFXcsrC-lacZ crp::cam | This study |
| AP724 | MG1655 ΔlacZ/pLFTcrp′-′lacZ | This study |
| AP736 | MG1655 ΔlacZ/pLFTcrp′-′lacZ csrA::kan | This study |
| AP804 | MG1655 uvrY::3× FLAG | This study |
| AP854 | MG1655 uvrY::3× FLAG crp::cam | This study |
| CF7789 | MG1655 ΔlacI-Z (MluI) | Michael Cashel |
| PLB2286 | CF7789 crp′-′lacZ | This study |
| PLB2287 | CF7789 crp′-′lacZ csrA::kan | This study |
| PLB2289 | CF7789 cyaA′-′lacZ | This study |
| PLB2290 | CF7789 cyaA′-′lacZ csrA::kan | This study |
| BL21(λDE3) | F− lon-11 Δ(ompT-nfrA)885 Δ(galM-ybhJ)884 λDE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5] Δ46 [mal+]K-12 (λs) hsdS10 | Novagen |
| Plasmids | ||
| pLFX | Plasmid used for constructing transcriptional fusions | 25 |
| pLFT | Plasmid used for constructing translational fusions | 25 |
| pPFINT | Helper plasmid used for integrating lacZ fusions into the chromosome | 25 |
| pET24a | Plasmid used for constructing crp clone for expressing the CRP protein | Novagen |
| pLFXcsrC-lacZ | csrC upstream region cloned into pLFX | This study |
| Bacteriophage P1vir | Strictly lytic P1 | Carol Gross |
Strains harboring crp::cam, cyaA::kan, csrD::kan, and csrA::kan mutations were obtained by P1vir transduction. CGSC, E. coli Genetic Stock Center.
TABLE 2.
List of oligonucleotides used in this study
| Oligonucleotide | Sequence | Purpose |
|---|---|---|
| csrB lacZ Fwd | 5′-GTCCTGCAGCCGGGGATATGCACGCGCAGTTTGT-3′ | Forward primer for constructing csrB-lacZ transcriptional fusion |
| csrB lacZ Rev | 5′-GTCGGTACCACGAAGATAGAATCGTCTTTTTCG-3′ | Reverse primer for constructing csrB-lacZ transcriptional fusion |
| csrC lacZ Fwd | 5′-GTCCTGCAGAATGCGTCTGTTGATAATTCAAATTAGTC-3′ | Forward primer for constructing csrC-lacZ transcriptional fusion |
| csrC lacZ Rev | 5′-GTCGGTACCTATGGGTGCTACTTTACGCCTTTGC-3′ | Reverse primer for constructing csrC-lacZ transcriptional fusion |
| crp trsln Fwd | 5′-GCATGACTGCAGCGCTTTTTCCAGCATCAACGCCACTG-3′ | Forward primer for constructing crp′-′lacZ translational fusion |
| crp trsln Rev | 5′-GTCGTCGAATTCCAAGCATGCGCGGTTATCCTCTG-3′ | Reverse primer for constructing crp′-′lacZ translational fusion |
| cyaA trsln Fwd | 5′-TATACTGCAGAGAGTGCAAGTGGGCTTTG-3′ | Forward primer for constructing cyaA′-′lacZ translational fusion |
| cyaA trsln Rev | 5′-TATAGGATCCACACGCAATTGATTTATGGC-3′ | Reverse primer for constructing cyaA′-′lacZ translational fusion |
| crp WT Fwd SP6 | 5′-ATTTAGGTGACACTATAGAAGATGCTACAGTAATACATTGATGTACTGCATGTATGC-3′ | Forward primer for generating template for crp WT RNA |
| crp P1 Rev | 5′-CCAAGCACCATGCGCGGTTATCCTCTG-3′ | Reverse primer for generating crp template for WT RNA |
| cyaA WT Fwd T7 | 5′-TAATACGACTCACTATAGGGTTTTAGACCATTT-3′ | Forward primer for generating template for cyaA WT RNA |
| cyaA WT Rev T7 | 5′-TTTATGGCATCCAGTCTCTGT-3′ | Reverse primer for generating template for cyaA WT RNA |
| crp Fwd Exp | 5′-GTCGTCGGATCCATGGTGCTTGGCAAACCGCAAACAG-3′ | Forward primer for cloning crp coding region into pET24a vector |
| crp Rev Exp | 5′-GTAGTACTCGAGTTAACGAGTGCCGTAAACGACGATG-3′ | Reverse primer for cloning crp coding region into pET24a vector |
| UvrY-His-F | 5′-TAGCTACTACTCGAGCTGACTTGATAATGTCTCCGCATTACA CAG-3′ | Forward primer for cloning uvrY coding region into pET24a vector |
| UvrY-His-R | 5′-CGAGTTCTTCATATGATCAACGTTCTACTTGTTGATGACCACGAA-3′ | Reverse primer for cloning uvrY coding region into pET24a vector |
| csrB DBA Fwd | 5′-TCTGGTGACTCAGAAAAGGCTAAAACTGCCG-3′ | Forward primer for generating csrB DNA for binding assays |
| csrB DBA Rev | 5′-GAAGATAGAATCGTCTTTTTCGCGAAGTCTTACAAGG-3′ | Reverse primer for generating csrB DNA for binding assays |
| csrC DBA Fwd | 5′-GCGTGAGCGTTGTAAGTAAAAGCCATACGC-3′ | Forward primer for generating csrC DNA for binding assays |
| csrC DBA Rev | 5′-GGGTGCTACTTTACGCCTTTGCTTTAAACAAACAATCAACC-3′ | Reverse primer for generating csrC DNA for binding assays |
| csrB DFP Fwd | 5′-CAGGAAAATCTGATTGGTCATCTGGTGAC-3′ | Forward primer for generating csrB template for footprinting |
| csrB DFP Rev | 5′-GTGTCATCATCCTGATGTTCACTTCGTTG-3′ | Reverse primer for generating csrB template for footprinting |
| csrC DFP Fwd | 5′-CAGGCGCACTCATCACAAAATGCGTCTG-3′ | Forward primer for generating csrC template for footprinting |
| csrC DFP Rev | 5′-GTCTCCGGACGTTTGTCTTCCTGAC-3′ | Reverse primer for generating csrC template for footprinting |
| csrB (Ec) probe Rev T7 | 5′-TAATACGACTCACTATAGGGTTCGTTTCGCAGCATTCCAG-3′ | Reverse primer for generating T7 template for CsrB riboprobe in E. coli |
| csrB (Ec) probe Fwd | 5′-GCGTTAAAGGACACCTCCAGG-3′ | Forward primer for generating T7 template for CsrB riboprobe in E. coli |
| csrC (Ec) probe Rev T7 | 5′-TAATACGACTCACTATAGGGTCTTACAATCCTTGCAGGC-3′ | Reverse primer for generating T7 template for CsrC riboprobe in E. coli |
| csrC (Ec) probe Fwd | 5′-GAGGACGCTAACAGGAACAATG-3′ | Forward primer for generating T7 template for CsrC riboprobe in E. coli |
| uvrY 3× FLAG Fwd | 5′-TCGCCATGGTCTGTGTAATGCGGAGACATTATCAAGTCAGGACTACAAAGACCATGACGG-3′ | Forward primer for constructing uvrY::3× FLAG |
| uvrY 3× FLAG Rev | 5′-GTTACGGTTTTTAAAAACGCTTTTGCGTCAAACTGATCACCATATGAATATCCTCCTTAG-3′ | Reverse primer for constructing uvrY::3× FLAG |
Construction of lacZ reporter fusions.
Single-copy, chromosomally integrated transcriptional and translational fusions to lacZ were constructed using the CRIM system (60) with plasmid vectors pLFX and pLFT, derived from pAH125 (25), and integrated into the chromosome at the λ att site, and single integrants were confirmed by PCR, as described previously (60).
For constructing csrB-lacZ and csrC-lacZ transcriptional fusions, 502 (−500 to +2 with respect to the csrB transcription initiation site)-nucleotide (nt) and 304 (−301 to +3 with respect to the csrC transcription initiation site)-nt regions of csrB and csrC were amplified by PCR from E. coli MG1655 genomic DNA using primer pairs csrB lacZ Fwd/csrB lacZ Rev and csrC lacZ Fwd/csrC lacZ Rev. PCR products were gel purified, digested with PstI and KpnI, ligated to PstI- and KpnI-digested and dephosphorylated plasmid pLFX, and electroporated into DH5αλpir cells. Sequence-verified plasmids pLFXcsrB-lacZ and pLFXcsrC-lacZ were integrated into the λ att site of E. coli MG1655 ΔlacZ, using helper plasmid pPFINT.
For constructing the crp′-′lacZ translational fusion, the primer pair crp trsln Fwd/crp trsln Rev was used to amplify a 677-nucleotide region (nucleotides −667 to +10 with respect to the translational start site) of crp from MG1655. The resulting PCR product was gel purified, digested with PstI and EcoRI, ligated to PstI- and EcoRI-digested, dephosphorylated plasmid pLFT, and electroporated into DH5αλpir cells. Primer pair cyaA trsln Fwd/cyaA trsln Rev was used to amplify a 497-nucleotide region (nucleotides −438 to +59 with respect to the cyaA translational start site) of cyaA from MG1655, gel purified, digested with PstI and BamHI, ligated to PstI- and BamHI-digested, dephosphorylated plasmid pLFT, and electroporated into DH5αλpir cells. The sequence-verified plasmids, pLFTcrp′-′lacZ and pLFTcyaA′-′lacZ, were integrated into the λ att site of E. coli MG1655 ΔlacZ using helper plasmid pPFINT.
β-Galactosidase and protein assays.
Assays to examine the effects of csrA on expression of cyaA′-′lacZ and crp′-′lacZ translational fusions were performed as described previously (19). Assays to examine the effects of cAMP-CRP on csrB-lacZ and csrC-lacZ transcriptional fusions were conducted as described previously (61) with minor modifications (25). Total cell protein was measured after precipitation with 10% trichloroacetic acid, using the bicinchoninic acid assay (Pierce Biotechnology) with bovine serum albumin as a protein standard. Purified proteins were quantified similarly but without trichloroacetic acid precipitation.
Northern blotting.
Total RNA was isolated using a RiboPure-Bacteria kit (Ambion) or by phenol-chloroform extraction. Phenol-chloroform extraction was performed following the Gross Lab protocol (http://derisilab.ucsf.edu/microarray/pdfs/Total_RNA_from_Ecoli.pdf) for isolation of total RNA from E. coli. For Northern blotting, 2 μg of total RNA was mixed with 2 volumes of loading buffer (50% [vol/vol] deionized formamide; 6% [vol/vol] formaldehyde; 1× MOPS [20 mM]; 5 mM sodium acetate [NaOAc]; 2 mM EDTA [pH 7.0]; 10% [vol/vol] glycerol; 0.05% [wt/vol] bromophenol blue; 0.01% [wt/vol] ethidium bromide), denatured by heating at 75°C for 5 min, chilled on ice, and separated by electrophoresis on a 7 M urea–5% polyacrylamide gel. RNA was transferred overnight to a positively charged nylon membrane (Roche) in 1× Tris-borate-EDTA (TBE) buffer and fixed to the membrane by UV cross-linking. The rRNA that was transferred was stained with methylene blue and imaged using a Gel-Doc, and its signal intensity was quantified using Quantity One software. CsrB or CsrC RNAs were detected with DIG-labeled RNA probes according to a digoxigenin (DIG) Northern starter kit manual (Roche), and the signals were captured with a ChemiDoc XRS+ system (Bio-Rad, Hercules, CA).
Construction of a carboxy-terminal FLAG-tagged UvrY protein.
A strain expressing a recombinant UvrY protein containing a 3× FLAG tag at the carboxy terminus from the native uvrY locus was constructed as described earlier (62). The recombinant UvrYFLAG protein appeared to be fully functional, as determined by its ability to activate the synthesis of CsrB and CsrC RNAs relative to the results seen with the wild-type (WT) UvrY protein (data not shown).
Western blotting of CsrA and UvrY-FLAG.
For Western blotting, cultures were grown with shaking at 37°C at 250 rpm and harvested throughout the growth curve. Cells were mixed with 2× sample buffer (4% [wt/vol] SDS; 0.16 M Tris; 1.5% [vol/vol] β-mercaptoethanol; 20% [vol/vol] glycerol; 0.02% [wt/vol] bromophenol blue, pH 6.0) and lysed by sonication and then by boiling. Samples (1 to 5 μg of total cellular protein) were subjected to SDS-PAGE, transferred to 0.2 μM polyvinylidene difluoride (PVDF) membranes, and detected using polyclonal anti-CsrA, monoclonal anti-FLAG (for UvrY-FLAG), or anti-RpoB (for RpoB) antibodies as described previously (62). Unphosphorylated UvrY was resolved from phosphorylated UvrY (P-UvrY) on SDS gels containing Phos-Tag reagent, as described previously (62).
Electrophoretic gel mobility shift assays (EMSA) for RNA binding.
Binding of CsrA to crp and cyaA transcripts was determined by EMSA with in vitro-synthesized crp and cyaA transcripts (MAXIscript SP6/MEGAshortscript kit; Ambion) and recombinant CsrA-His6 (2). The template DNA for in vitro transcription of crp and cyaA was generated by PCR from MG1655 genomic DNA, using oligonucleotide pairs crp WT Fwd SP6/crp P1 Rev (crp WT) and cyaA WT Fwd T7/cyaA WT Rev T7 (cyaA WT). WT crp transcripts (178 nt, consisting of 167 nt of the noncoding mRNA leader and 11 nt of the coding region) and cyaA transcripts (201 nt, consisting of 154 nt of the noncoding mRNA leader and 47 nt of the coding region) were gel purified, treated with Antarctic phosphatase (NEB), and radiolabeled at the 5′ end using [γ-32P]ATP and T4 polynucleotide kinase. Binding reaction mixtures contained 0.6 nM RNA, 10 mM MgCl2, 100 mM KCl, 32.5 ng total yeast RNA, 20 mM dithiothreitol (DTT], 7.5% glycerol, 4 U SUPERasin (Ambion), and various concentrations of CsrA (0 to 640 nM) and were incubated at 37°C for 30 min. Reaction mixtures were separated on 9% native polyacrylamide gels using 1× TBE buffer as the electrophoresis buffer, and labeled RNA was analyzed using a phosphorimager equipped with Quantity One software, as described previously (25).
Purification of native CRP protein.
The E. coli crp coding region was PCR amplified from MG1655 genomic DNA using the primer pair crp Fwd Exp/crp Rev Exp. The resulting PCR product was gel purified, digested with EcoRI and XhoI, ligated to EcoRI and XhoI digested, dephosphorylated pET24a(+), and transformed into DH5α cells. The resulting clone pET24a(+) crp was verified by sequencing and transformed into the expression host, BL21(λDE3). Shaking cultures of BL21(λDE3) pET24a(+) crp were grown in LB (300 ml) containing kanamycin at 37°C for 3 h, and expression of crp was induced for 3 h with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Cells were collected by centrifugation, resuspended in 30 ml of binding buffer (20 mM Tris-HCl [pH 7.9]; 500 mM NaCl; 20 mM imidazole), lysed using a French press, and centrifuged to remove cell debris from the cell lysate. The native CRP protein was fractionated using HisTrap column chromatography (63). The cell lysate was loaded onto a His-Trap column (HisTrap HP; GE Healthcare), rinsed with binding buffer (20 mM Tris-HCl [pH 7.9]; 500 mM NaCl; 20 mM imidazole), and eluted with a gradient of binding buffer and elution buffer (20 mM Tris-HCl [pH 7.9]; 500 mM NaCl; 500 mM imidazole). CRP-containing fractions were pooled and dialyzed against a buffer containing 50 mM Tris-HCl (pH 7.6], 200 mM KCl, 10 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, and 10% glycerol. The final CRP solution was adjusted to 50% glycerol and stored at −20°C. Purity was estimated to be ≥98% by SDS-PAGE.
Purification of carboxy-terminal His-tagged UvrY protein.
His6-tagged UvrY protein was purified from a strain expressing the recombinant protein as described previously (3).
In vitro phosphorylation of UvrY.
UvrY protein was phosphorylated by incubation with 100 mM lithium potassium acetyl-phosphate (Sigma-Aldrich) for 60 min at room temperature in a buffer containing 50 mM HEPES, 100 mM NaCl, and 10 mM MgCl2 as described previously (40).
Electrophoretic gel mobility shift assays for DNA binding.
For DNA gel shift assays, the regions from nt −400 to −1 and nt −200 to −1, with respect to the transcriptional start sites of csrB and csrC, respectively, were amplified by PCR from MG1655 genomic DNA and subjected to end labeling with [γ-32P]ATP using T4 polynucleotide kinase. Binding reaction mixtures (10 μl) contained 0.5 nM end-labeled DNA, 20 mM Tris HCl (pH 7.5), 10% (vol/vol) glycerol, 50 mM KCl, 3 mM MgCl2, 1 mM dithiothreitol, 100 μg/ml bovine serum albumin, and, as indicated, cAMP, CRP, and/or P-UvrY. Reaction mixtures were incubated for 30 min at 37°C degrees, and then 1 μl xylene cyanol was added and samples were separated by electrophoresis on 6% native polyacrylamide gels with 0.5× TBE buffer as the running buffer. The gels were dried, and radioactive signals were captured by phosphorimaging and analyzed using Quantity One software.
DNase I footprinting.
DNA of csrB and csrC regions, extending from nt −420 to +46 and nt −319 to +100 relative to the respective transcriptional start sites, was amplified by PCR from MG1655 to generate the csrB and csrC templates for footprinting. To label the 5′ end of the nontemplate or template strand, a 32P end-labeled forward or reverse primer, respectively, was used in the PCR and the resulting PCR product was gel purified. Binding reaction mixtures (10 μl) contained labeled DNA (0.5 nM), 20 mM Tris HCl (pH 7.5), 10% (vol/vol) glycerol, 50 mM KCl, 3 mM MgCl2, 1 mM dithiothreitol, 100 μg/ml BSA, 200 μM cAMP, and CRP or P-UvrY. Reaction mixtures were incubated for 30 min at 37°C and cooled on ice. Then, a solution containing 0.025 U of DNase I (Roche) and CaCl2 (1 mM final concentration) was added, and the contents were gently mixed by pipetting and incubated in a 37°C water bath for 1 min. Thereafter, DNase I was heat inactivated at 75°C for 10 min, samples were chilled on ice, and 2 vol of loading buffer was added. The DNA was denatured by heating at 95°C for 5 min and separated by electrophoresis on a 7 M urea–6% polyacrylamide gel. After electrophoresis, the gel was dried and radioactive signals were collected by phosphorimaging and quantified using Quantity One software. Sequencing ladders were prepared with the use of a ThermoSequenase cycle sequencing kit (Affymetrix, USB; catalog no. 78500), as recommended by the manufacturer.
In vitro coupled transcription-translation.
Coupled transcription-translational assays for expression of pLFXcsrC-lacZ were performed with S-30 extracts prepared from a uvrY-deficient strain (CF7789 uvrY::cam) as described previously (38, 61), except that the reaction mixtures were assembled to reach 32 μl and contained 0.5 U E. coli RNA polymerase holoenzyme and 3 μl of [35S]methionine (1,175 Ci/mmol). Radiolabeled proteins were separated by electrophoresis through Bis-Tris SDS-PAGE. Gels were stained, destained, and dried, and radioactive signals were detected by phosphorimaging and quantified using Quantity One Software.
RESULTS
CRP represses csrB and csrC expression.
To examine the in vivo effects of CRP on the expression of CsrB and CsrC RNAs, we first monitored its effects on csrB-lacZ and csrC-lacZ transcriptional fusions. While a crp mutant grows more slowly than the isogenic WT strain in media lacking a preferred carbon source, including LB, we found that the two strains grew equally well on LB-MOPS buffered medium containing 0.2% fructose (Fig. 1A and C). Furthermore, relative to glucose transport, fructose favors the phosphorylated form of EIIAGlc (P-EIIAGlc) (64), which activates cAMP synthesis. In this medium, csrB-lacZ expression in isogenic WT and isogenic crp mutant strains increased during the exponential phase of growth and decreased as the cultures approached the stationary phase of growth (Fig. 1A). Expression of the csrB-lacZ fusion ranged from 2-fold to 15-fold greater in the crp mutant than in the WT strain in the exponential phase of growth (2 to 5 h) and decreased to similar levels in both strains thereafter. Expression of csrC-lacZ increased in the exponential phase up to the late exponential phase and decreased slightly thereafter in both the WT and crp mutant strains (Fig. 1C). Expression was ∼2-fold greater in the crp mutant throughout the growth curve. To determine if the effect of the crp mutation on csrB-lacZ and csrC-lacZ expression was due to an inability to form the cAMP-CRP complex, we examined the effects of crp and cyaA mutations in the presence and absence of exogenous cAMP. The addition of cAMP was expected to restore cAMP-CRP formation in the cyaA mutant but not in the crp strain. As seen before, at 3 h (Fig. 1A), we observed that csrB-lacZ expression was substantially greater in the crp mutant as well as in the cyaA mutant (Fig. 1B). Addition of cAMP (10 mM) to the cyaA mutant culture caused a dramatic decrease in csrB-lacZ expression, whereas only a small decrease was observed in the crp mutant (Fig. 1B). The cyaA and crp mutations both caused a modest (∼2-fold to 3-fold) increase in csrC-lacZ expression, while cAMP restored expression to normal WT levels only in the cyaA mutant (Fig. 1D). These results indicated that transcription of csrB and csrC is negatively influenced by cAMP-CRP.
FIG 1.

cAMP-CRP represses expression of csrB-lacZ (A and B) and csrC-lacZ (C and D) transcriptional fusions. E. coli MG1655 ΔlacZ (WT) and its isogenic crp or cyaA mutant, harboring chromosomal csrB-lacZ or csrC-lacZ fusions, were cultured in LB-MOPS buffered medium with 0.2% fructose, and β-galactosidase levels were determined. (A and C) Enzyme-specific activity (main panels) and growth (insets) of the WT strain and crp mutant. (B and D) cAMP was included in the growth medium, as indicated, at a final concentration of 10 mM. Error bars show means ± standard deviations (SD) of results of experiments that were conducted twice.
We next determined the effect of CRP on CsrB and CsrC RNA levels in strains grown in LB-MOPS with 0.2% fructose using Northern blotting, which would reflect the effects of CRP on csrB/C transcription as well as on CsrB/C RNA decay. CsrB levels in the WT strain were relatively constant through the exponential phase of growth in this medium and decreased in the stationary phase (Fig. 2A). CsrC levels also decreased as the culture entered the stationary phase. While the CsrB/C RNA levels were expected to differ between the WT and the crp mutant in the exponential phase of growth, based on the behavior of the corresponding lacZ fusions (Fig. 1A and C), they were not substantially altered by the crp mutation. On the other hand, in the stationary phase of growth (8 and 10 h), levels of both CsrB and CsrC were elevated in the crp mutant, although by 24 h this difference was much less pronounced.
FIG 2.
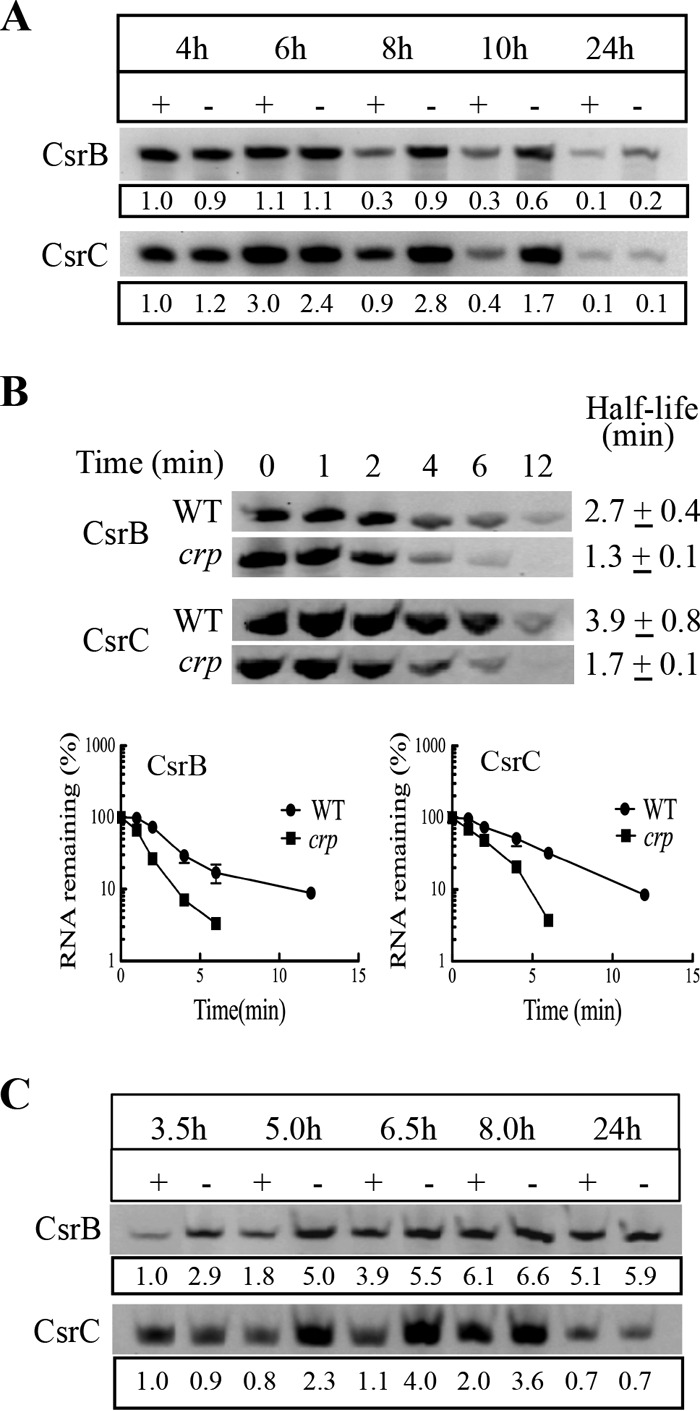
Effects of cAMP-CRP on CsrB and CsrC levels and decay rates. (A) Relative steady-state CsrB and CsrC levels in MG1655 (+) and its isogenic crp mutant (−). (B) Turnover of CsrB and CsrC sRNAs in the mid-exponential phase (3.5 h) of growth, following addition of rifampin. (C) CsrB and CsrC levels in a ΔcsrD genetic background, which compromises their turnover. All E. coli strains were grown in LB-MOPS buffered medium with 0.2% fructose. For panels A and C, CsrB or CsrC levels are indicated with respect to the crp WT strain (+) at 3.5 h, with normalization against 16S rRNA. Each experiment was repeated twice with essentially the same results, and a representative blot is shown. Error bars represent standard deviations.
A possible explanation for the finding that CsrB and CsrC RNA levels were lower than expected in the crp mutant during exponential phase is that their decay rates may be increased in this strain. To test this idea, we determined the decay rates of CsrB and CsrC in WT (MG1655) and crp mutant strains in the mid-exponential phase, where crp disruption increased the expression of csrB-lacZ and csrC-lacZ reporter fusions (Fig. 1A and C) without substantially affecting the steady-state levels of the small RNAs (Fig. 2A). Total RNA was isolated from cells at several times following rifampin addition to block transcription, and CsrB and CsrC sRNAs were detected by Northern blotting. The half-lives of Csr small RNAs in the WT and crp mutant, respectively, were 2.7 and 1.3 min for CsrB and 3.9 and 1.7 min for CsrC (Fig. 2B). Thus, the decay of CsrB and CsrC was accelerated approximately 2-fold in the crp mutant, explaining the distinct behavior of the reporter fusions versus the steady-state levels of these RNAs in response to CRP.
Degradation of CsrB and CsrC requires the protein CsrD, which facilitates endonucleolytic cleavage of these sRNAs by RNase E (42, 43). Thus, the effect of CRP on synthesis of CsrB and CsrC under conditions of greatly reduced turnover was examined in a ΔcsrD strain (Fig. 2C). In exponential phase (3.5 and 5 h), CsrB levels were approximately 3-fold higher in the csrD crp double mutant than in the csrD mutant background (Fig. 2C), while the effect was minimal thereafter. The CsrC levels were 2.9-, 3.6-, and 1.8-fold higher in the csrD crp double mutant at 5, 6.5, and 8 h of growth, respectively (Fig. 2C). Thus, in the relative absence of their decay, CsrB and CsrC RNA levels responded to CRP similarly to those seen with the csrB-lacZ and csrC-lacZ transcriptional fusions (Fig. 1). We also examined the effect of crp and cyaA mutations on CsrB and CsrC levels in LB-MOPS medium under a growth condition with minimal availability of a preferred source of carbon. Recall that under these conditions, P-EIIAGlc is unable to bind to CsrD, which decreases the decay rates of CsrB/C (44). Under these conditions, the steady-state levels of CsrB and CsrC in both the cyaA and the crp mutant were higher than in the isogenic WT strain (Fig. 3). Furthermore, addition of 10 mM cAMP caused a substantial decrease in CsrB/C levels in the cyaA mutant but not in a crp mutant, confirming that CRP effects on CsrB/C levels require the cAMP-CRP complex.
FIG 3.
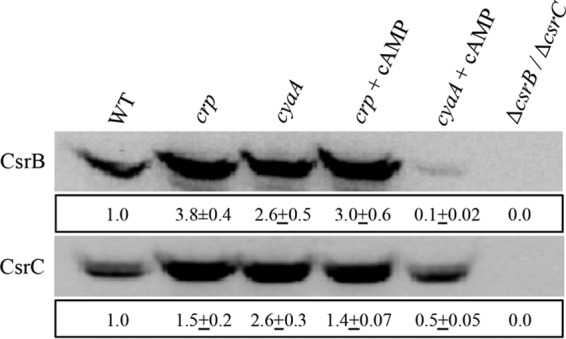
Effect of cAMP (10 mM) addition on CsrB and CsrC levels in LB-MOPS medium. The CsrB or CsrC levels were expressed, after rRNA normalization, with respect to WT (MG1655) after 3 h of growth at 37°C. This experiment was repeated twice with essentially identical results. The means ± ranges are shown for all values. A representative blot is shown.
CRP does not repress CsrB/C expression via effects on CsrA or P-UvrY levels.
Normal transcription of both csrB and csrC depends on P-UvrY, although csrB transcription is more highly dependent than that of csrC (3, 36, 38). In addition, the RNA helicase DeaD activates UvrY translation, which then activates csrB/C transcription (62). To test the possibility that CRP functions by altering the level or phosphorylation state of UvrY, we monitored UvrYFLAG protein expressed from the native uvrY locus in WT and crp mutant strains by Western blotting, as described previously (62). This experiment indicated that the unphosphorylated form of FLAG-tagged UvrY protein was the more abundant form under our experimental conditions (LB-MOPS with 0.2% fructose) (Fig. 4A), as previously observed in LB medium (62). P-UvrY levels were unaltered or slightly lower in the crp mutant in the exponential phase and were less than half of the WT levels in the stationary phase of growth. These effects were not consistent with the observed increases in csrB-lacZ and csrC-lacZ expression (Fig. 1A and C) and in CsrB/CsrC RNA levels (Fig. 2C) seen in the crp mutant. Thus, CRP does not negatively regulate CsrB/C levels via effects on UvrY levels or its phosphorylation status. CsrA protein levels were equivalent in the WT and crp mutant strains (Fig. 4B), indicating that CRP does not repress CsrB/C through effects on CsrA levels.
FIG 4.
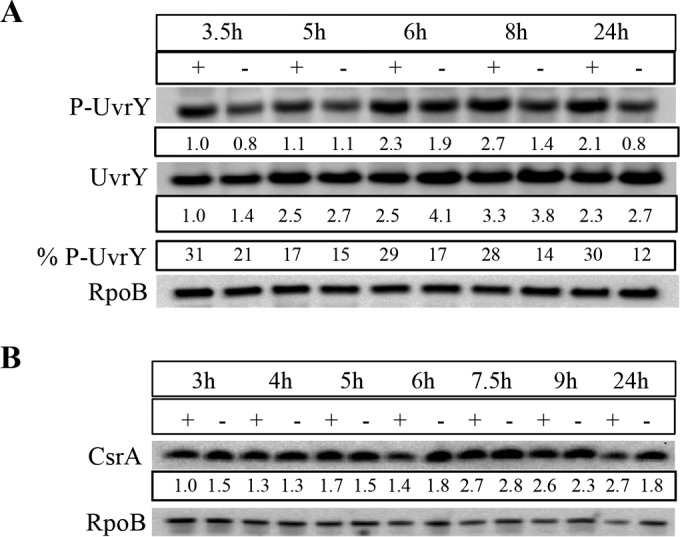
Effect of crp mutation on levels of phosphorylated and unphosphorylated UvrYFLAG and CsrA protein. (A) Cultures were grown in LB-MOPS medium with 0.2% fructose and harvested at the indicated time points for preparation of lysates. Proteins (5 μg) were separated by electrophoresis on Phos-Tag gels, transferred to nitrocellulose, and detected with anti-FLAG and anti-RpoB antibodies. % P-UvrY, percentage of P-UvrY with respect to total UvrY protein (P-UvrY plus UvrY after RpoB normalization). (B) Cell lysates were prepared from cultures grown in LB-MOPS medium with 0.2% fructose, and protein (2 μg) was separated on a 12% acrylamide gel, transferred to a PVDF membrane, and detected with anti-CsrA and anti-RpoB antibodies. Proteins from WT (+) and crp mutant (−) strains are shown. Each experiment was repeated twice with essentially the same results, and a representative blot is shown.
cAMP-CRP binds to csrC DNA in a region overlapping the P-UvrY binding site.
Because P-UvrY and CsrA do not appear to mediate cAMP-CRP effects on csrB and csrC expression, we hypothesized that cAMP-CRP might directly repress transcription by binding to csrB and/or csrC DNA. P-UvrY activates transcription of csrB and csrC by binding to 18-nt inverted repeat (IR) DNA sequences, TGTGAGAGATCTCTTACA and TGTGAGACATTGCCGATA, respectively, centered at nt −183 and −159 from the transcriptional start site (3). CRP binds to a conserved 22-bp DNA sequence (5′-AAATGTGATCTAGATCACATTT-3′) in the regulatory regions of its target genes (57, 58). Possible CRP binding sequences in the upstream regulatory regions of csrB and csrC were identified using an online tool, Virtual Footprint (www.prodoric.de/vfp). This analysis yielded two potential binding sites for csrB (CRP1 and CRP2) and a single site for csrC (CRP1) (Fig. 5A). In the case of csrB, CRP1 overlaps the P-UvrY binding site, while in the case of csrC, CRP1 is immediately downstream of the P-UvrY binding site (Fig. 6) (3).
FIG 5.

Binding of cAMP-CRP to csrB and csrC DNA. (A) Consensus sequence for cAMP-CRP binding and predicted binding sites in the upstream flanking regions of csrB and csrC. Nucleotides in uppercase letters in the crp consensus sequence denote the conserved residues, and the size of the nucleotide letters indicates the relative degree of conservation of the nucleotide residue (47). Nucleotides with the asterisk indicate the second most highly conserved residue in comparison to the nucleotide under which they are placed. Numbers indicate the position of each nucleotide relative to the 5′ end of the consensus. Numbers beneath the csrB and csrC sequences are indicative of location with respect to the transcription initiation site. (B) Electrophoretic gel mobility shift analysis of cAMP-CRP binding to csrB and csrC DNA. A 32P end-labeled PCR product (0.5 nM) was used in binding reactions performed with or without cAMP (0.2 mM) and CRP, as shown. Positions of free (F) and bound (B) DNA are shown. The apparent Kd for binding of cAMP-CRP to csrC DNA was 18 ± 2 nM. Each experiment was conducted twice, and a representative image is shown.
FIG 6.

DNase I footprinting of csrC DNA in the presence of cAMP-CRP and P-UvrY. (A and B) Lanes G, A, T, and C depict the sequencing ladder. The numbers to the left of the images indicate distance from the transcription start site. Protection by cAMP-CRP and P-UvrY is shown by filled and open bars, respectively. Sites hypersensitive to cleavage in the presence of cAMP-CRP and P-UvrY are indicated by asterisks and a white star, respectively. (A) Footprinting of csrC in the presence of CRP or P-UvrY. Reaction mixtures in lanes 3 and 4 contained 100 and 250 nM CRP with 0.2 mM cAMP. Lanes 6, 7, and 8, 250, 500, and 1,000 nM P-UvrY. (B) Footprinting of csrC DNA in the presence of cAMP-CRP or P-UvrY or both. Lanes 3 to 6, 100 nM CRP and 0.2 mM cAMP; lanes 4, 5, and 6, 250, 500, and 700 nM P-UvrY; lanes 7 to 10, 500 nM P-UvrY; lanes 8, 9, and 10, 50, 100, and 200 nM CRP with 0.2 mM cAMP. The filled and open arrowheads and the arrow mark the nucleotide residues that were used to assess competition of cAMP-CRP and P-UvrY (the hypersensitive cleavage site caused by P-UvrY binding, a site protected specifically by P-UvrY, and a site protected specifically by cAMP-CRP, respectively). (C) The csrC sequence from −230 to −80 with respect to transcription initiation, displaying regions protected by cAMP-CRP and P-UvrY. The regions protected by cAMP-CRP and P-UvrY are indicated by filled and open bars, respectively. Nucleotides marked with an asterisk or a white star represent sites hypersensitive to cleavage in the presence of cAMP-CRP or P-UvrY, respectively. The arrowhead and arrow indicate the single nucleotides used to assess competition. The three regions protected by cAMP-CRP in the DNase I footprinting are indicated by R-I, R-II, and R-III. Sequences underlined with dashed lines are related to the consensus CRP binding sequence (Fig. 5A).
To determine whether cAMP-CRP binds specifically and with high affinity to the upstream regulatory regions of csrB and csrC, we first performed electrophoretic gel mobility shift assays (EMSA). Reactions without cAMP were used as a means to address binding specificity. csrB DNA showed a shifted complex at a relatively high CRP concentration of ∼180 nM (Fig. 5B, panel i). However, the shifted complex was diffuse and there was no difference in the binding of CRP (200 nM) in the presence or absence of cAMP, indicating that binding to CsrB DNA was nonspecific. In contrast, csrC DNA formed a distinct shifted complex at a CRP concentration of as low as 10 nM (Fig. 5B, panel ii). Increasing the concentration of CRP resulted in an increasing signal intensity of the shifted complex, with nearly complete binding seen at ∼50 nM. In the absence of cAMP, CRP failed to form the distinct complex, confirming that cAMP-CRP bound specifically to csrC DNA. The apparent dissociation constant (Kd value) for cAMP-CRP binding to csrC DNA was 18 ± 2 nM, similar to that of the lactose and galactose promoters (5 to 10 nM), which are known targets of cAMP-CRP binding (65). EMSA of CRP binding to csrA, uvrY, and csrD DNA in all cases resulted in the formation of diffuse complexes at high CRP concentrations (≥100 nM), similarly to the results seen with csrB DNA, indicative of nonspecific binding (data not shown).
To identify the cAMP-CRP and P-UvrY binding site(s) in csrC, we performed DNase I footprinting reactions. cAMP-CRP protected three prominent regions (R-I, R-II, and R-III) on csrC DNA (Fig. 6A, lanes 3 and 4, and C [marked with filled bars]). R-I included the site predicted by Virtual Footprint (www.prodoric.de/vfp), consisting of the region from nt −95 to −121 relative to the csrC transcription start site (Fig. 5A and 6A [lanes 3 and 4] and C). R-I appeared to represent a high-affinity site for binding of cAMP-CRP to csrC, since protection occurred at a low concentration of CRP (50 nM) and in the presence of P-UvrY (Fig. 6B, lane 8). cAMP-CRP binding to the two other sites, nt −137 to −168 (R-II) and nt −174 to −199 (R-III), required a higher concentration of CRP (Fig. 6B). Inspection of the nucleotide sequences at R-II and R-III revealed sequences that were related to the conserved nucleotides in the CRP consensus (Fig. 5A and 6C). The binding of cAMP-CRP also led to hypersensitive cleavage at several sites (−93A, −105T, −115G, −149T, −173C, −180A, −205T, and −215A) (Fig. 6, asterisks). DNase I footprinting of csrC DNA with P-UvrY showed a protected region from nt −125 to −193 (Fig. 6A, lane 7 [indicated with open bars]), as well as a pronounced hypersensitive cleavage site at −149T. Importantly, the region protected by P-UvrY overlapped the R-II and R-III regions protected by cAMP-CRP.
The results of the DNase I protection studies performed with individual proteins suggested that cAMP-CRP and P-UvrY may compete with each other for binding to csrC DNA. To test for binding competition, we first conducted DNase I protection assays in the presence of a constant concentration of cAMP-CRP and increasing concentrations of P-UvrY. In this experiment, the protection pattern of cAMP-CRP at R-II (nt −137 to −168) and R-III (nt −174 to −199) was increasingly replaced by the pattern observed for P-UvrY, including the strong hypersensitive cleavage at −149T (Fig. 6B; compare lanes 3 to 6). In contrast, protection of the R-I site (nt −95 to −121) by CRP was unaffected by the presence of P-UvrY (Fig. 6B, lanes 3 to 6). Alternatively, when increasing amounts of cAMP-CRP were added to reaction mixtures containing a constant amount of P-UvrY, we observed that the lowest concentration of CRP that was tested (50 nM) showed strong protection at the R-I site (nt −95 to −121) (Fig. 6B; compare lanes 7 and 8). Increasing the concentration of CRP (Fig. 6B, lanes 7 to 10) led to the appearance of CRP protection at the R-II site (nt −137 to 168) and the R-III site (nt −174 to −199) and a concomitant decrease in signal intensity of the P-UvrY hypersensitive site at −149T.
We also examined competition at two nucleotide residues (−159T and −196G) which were differentially sensitive to DNase I in the presence of cAMP-CRP versus P-UvrY. Residue −159T was protected by P-UvrY but not by cAMP-CRP. An increase in the concentration of P-UvrY led to increasing protection of this residue (Fig. 6B, lanes 3 to 6), while increasing the concentration of cAMP-CRP resulted in loss of protection at this residue (Fig. 6B, lanes 7 to 10). Similarly, −196G was protected by cAMP-CRP but not by P-UvrY. Increasing the concentration of P-UvrY resulted in decreasing protection of this residue (Fig. 6B, lanes 3 to 6), while increasing the concentration of cAMP-CRP resulted in increasing protection of this residue (Fig. 6B, lanes 7 to 10). These results strengthened the evidence suggesting that cAMP-CRP competes with P-UvrY for binding to csrC DNA.
We also conducted DNase I footprinting studies of csrB DNA in the presence of P-UvrY and cAMP-CRP (data not shown). As reported previously, P-UvrY protected a region from nt −138 to bp −210 upstream from the csrB transcriptional start site (3). cAMP-CRP did not generate a distinct footprint with csrB DNA, confirming the findings from EMSA (Fig. 5B) showing that cAMP-CRP does not bind specifically to csrB DNA.
cAMP-CRP inhibits in vitro expression of csrC.
Although we were unable to detect expression of csrB or csrC using in vitro transcription reactions with purified components (data not shown), P-UvrY was previously found to directly activate the expression of csrB-lacZ and csrC-lacZ transcriptional fusions in S-30 transcription-translation assays (36, 38). Thus, we used the latter approach to determine if cAMP-CRP directly represses expression from the csrC-lacZ transcriptional fusion in the plasmid pLFXcsrC-lacZ. Addition of phosphorylated UvrY (P-UvrY) alone stimulated β-galactosidase synthesis, as expected (Fig. 7A, lanes 1 and 2). Addition of cAMP alone to the P-UvrY reaction caused a slight (∼20%) relative decrease in β-galactosidase synthesis, which was likely due to the presence of endogenous CRP in the S-30 extract (Fig. 7A, lanes 2 and 4, and B). In the presence of cAMP, the addition of increasing CRP concentrations led to decreasing β-galactosidase synthesis relative to the results seen with the β-lactamase gene product (Bla) encoded by pLFXcsrC-lacZ, which served as an internal control (Fig. 7A, lanes 4 to 8). The inhibitory effect began to saturate at a concentration of 1.0 μM CRP, resulting in ∼55% inhibition (Fig. 7). This is a minimal estimation of CRP inhibition because it does not include the (∼20%) inhibition caused by the endogenous CRP from the S-30 extract (Fig. 7A, lane 2 versus lane 4, and B).
FIG 7.
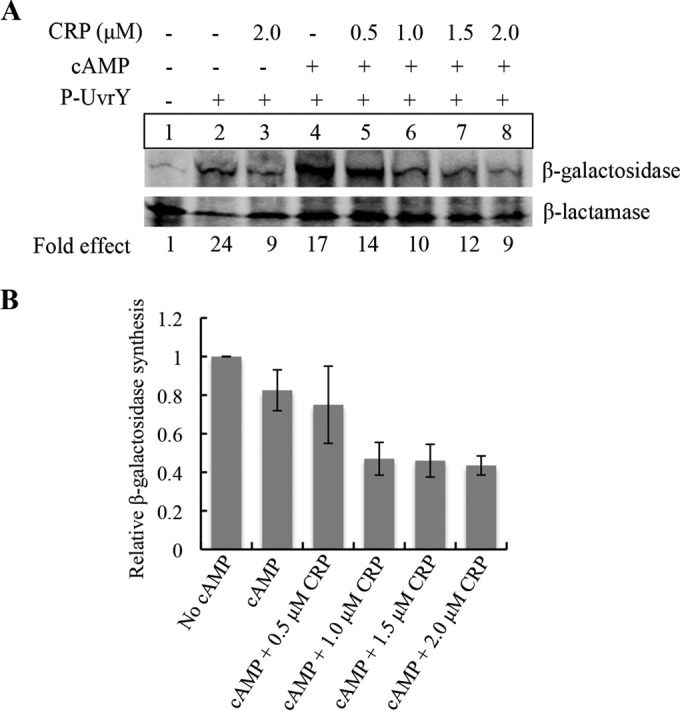
Effect of cAMP-CRP on in vitro expression of csrC-lacZ in S-30 extracts. (A) An in vitro coupled transcription-translation assay was performed using plasmid pLFXcsrC-lacZ (4 μg), P-UvrY (2.3 μM), cAMP (0.2 mM), and CRP as indicated. 35S-methionine-labeled proteins were detected by phosphorimaging, and signal intensity was quantified using Quantity One software. Specific β-galactosidase synthesis values were determined from the β-galactosidase/β-lactamase ratio for each reaction, and fold effects were determined relative to the reaction mixture that lacked CRP, cAMP, and P-UvrY (lane 1). (B) Effect of CRP and cAMP on P-UvrY-dependent csrC-lacZ expression. Relative levels of β-galactosidase synthesis were determined by comparing the specific β-galactosidase synthesis values to that of a control reaction (panel A, lane 2). Results depict average values from the results of two independent experiments ± SD.
In vitro binding of CsrA to crp and cyaA transcripts.
Transcripts for crp and cyaA previously copurified with CsrA protein (25). This finding suggested that CsrA might directly regulate crp and cyaA expression at a posttranscriptional level, resulting in reciprocal interactions among these two global regulatory systems. A position-weight matrix (PWM) analysis for potential CsrA binding sequences in the E. coli transcriptome (27) suggested that there may be as many as four such sites in the 167-nucleotide untranslated leader of crp mRNA (BS1 to BS4) (Fig. 8A). The cyaA mRNA leader contains a single potential binding sequence (BS1) in the early coding region (Fig. 8C). Electrophoretic gel mobility shift assays (EMSA) were used to first examine the binding interaction of CsrA with crp mRNA. Although major and minor RNA conformers were present in the absence of CsrA, the intensity of a band that ran at a position similar to that of the minor conformer began to increase at ∼5 nM CsrA, which is indicative of a CsrA-crp RNA complex. This complex became more prominent as the CsrA concentration was increased further (Fig. 8B). A nonlinear least-squares analysis of the data for this binding interaction yielded an apparent dissociation constant (Kd) of 25 nM. At a CsrA concentration of 40 nM, a second shifted complex was observed in addition to the first complex, and little of the RNA remained unbound. At a CsrA concentration of 80 nM, a third shifted complex was observed along with the second complex. A further increase in CsrA concentration to 160 nM led to formation of only the third shifted complex. This series of reactions strongly suggested the binding of multiple CsrA proteins to the crp RNA, although this possibility was not further investigated. Unlabeled crp RNA was able to compete for the formation of CsrA complexes with the labeled crp RNA, while unlabeled phoB RNA, which does not bind to CsrA (25), did not compete with crp RNA. These findings confirmed that CsrA bound specifically to crp RNA (Fig. 8B).
FIG 8.

Binding of CsrA to crp and cyaA transcripts analyzed by EMSA. (A and C) Nucleotide sequences of the untranslated leader and initial coding region of crp (A) and cyaA (C) RNAs and the predicted sites (underlined) for CsrA binding. The first nucleotide of each sequence (•) is numbered with respect to the translation initiation site. Scores next to each predicted binding site (BS) are based on a position-weight-matrix analysis of CsrA binding sequences (27). The ATG and TTG initiation codons are bolded. (B and D) Binding and competition assays of CsrA with crp (B) and cyaA (D) transcripts. Positions of free (F) and bound (B) RNA are indicated.
EMSA of cyaA RNA showed two RNA species in the absence of CsrA (Fig. 8D), the faster migrating species being the more abundant. CsrA bound similarly to the two species, indicating that they are two conformers of a single RNA. A shifted complex began to appear at a CsrA concentration of 40 nM and became more prominent as the CsrA concentration was increased further. A nonlinear least-squares analysis of the binding data for this complex yielded an apparent dissociation constant (Kd) of 133 ± 7 nM, indicating that the affinity of CsrA for cyaA mRNA was much weaker than for crp mRNA. Unlabeled cyaA RNA was able to compete for the formation of the CsrA complex with labeled cyaA RNA, but unlabeled nonspecific RNA, Bacillus subtilis trp mRNA, also competed (Fig. 8D). These results suggested that the binding interaction of CsrA with cyaA mRNA might not be specific.
CsrA conditionally affects crp and cyaA expression at low temperatures.
The binding affinity of CsrA for crp mRNA (25 nM) was similar to that of several authenticated mRNA targets, e.g., glgCAP (39 nM, 4 CsrA binding sites), pgaABCD (22 nM, 6 binding sites), hfq (38 nM, 1 binding site), cstA (40 nM, 4 binding sites), relA (17 nM, 6 binding sites), and csrA (27 nM, 4 binding sites). Therefore, we decided to examine the effect of CsrA on the in vivo expression of a chromosomally integrated crp′-′lacZ translational fusion. Although the binding affinity of CsrA to cyaA mRNA was weak and presumably nonspecific, we also tested for CsrA effects on a chromosomally integrated cyaA′-′lacZ translational fusion. Both fusions were monitored in LB and KB media when cells were grown at 22°C and 37°C. Studies were conducted at 22°C because we found that CsrA activates expression of another E. coli gene at 22°C but not at 37°C (L. C. McGibbon, T. Romeo, and P. Babitzke, unpublished results). Growth levels of WT and csrA mutant strains in LB and KB media were comparable. At 22°C, expression of the crp′-′lacZ fusion in KB medium was lower in the csrA mutant at all stages of growth except for early exponential phase, when the level was slightly higher in the csrA mutant (Fig. 9A). CsrA had little or no effect on the expression of this fusion in LB medium (Fig. 9B). Expression of the cyaA′-′lacZ fusion was moderately lower in the csrA mutant in both KB and LB media, but a stronger effect was observed in KB medium at early exponential phase (Fig. 9E and F). The csrA::kan mutation had negligible effects on the expression of another reporter fusion (dppA-lacZ) in KB medium at 22°C (data not shown), suggesting that the observed effects of CsrA on crp′-′lacZ and cyaA′-′lacZ fusions were specific. No significant effects of CsrA on the expression of either reporter fusion at 37°C in either medium were observed (Fig. 9C, D, G, and H). These findings indicated that CsrA positively affects crp and cyaA expression in a temperature-dependent fashion.
FIG 9.

Effect of csrA::kan disruption on in vivo expression of crp′-′lacZ and cyaA′-′lacZ translational fusions in KB (A, C, E, and G) and LB medium (B, D, F, and H) at 22°C or 37°C, as shown. The results represent the averages of the results of two independent experiments, and error bars depict standard deviations. Growth levels of CF7789 (WT) and its isogenic csrA::kan strain were similar in KB medium and LB medium under the growth conditions employed for the assay.
DISCUSSION
These studies were inspired by observations suggestive of regulatory connections between the catabolite repression and Csr systems. For example, (i) crp and cya mRNAs copurified with the CsrA protein (25), (ii) potential cAMP-CRP binding sites were identified upstream of the csrB gene by bioinformatics analysis (47), and (iii) a number of genes and processes have been reported to be regulated by both CsrA and cAMP-CRP (14, 18, 21, 66) (Fig. 10). In addition, the phosphorylation state of the PTS protein EIIAGlc serves as the key sensory mechanism for cAMP synthesis and catabolite repression control and for the turnover pathway of CsrB/C sRNAs (43, 44, 48). Our present data establish direct and apparently indirect connections between these two important global regulatory systems.
FIG 10.
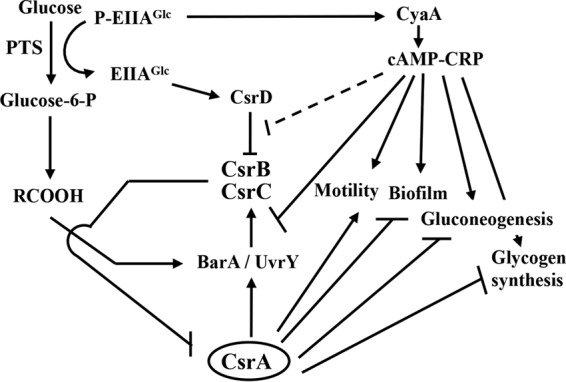
A model for the impact of carbon nutrition on Csr circuitry. Availability of carbon nutrition and end product accumulation impact the workings of the Csr system. During glucose transport, EIIAGlc of the PTS becomes relatively dephosphorylated and is able to bind to CsrD only in this form, activating CsrB/C decay (42–44). End products of metabolism (RCOOH) such as formate and acetate (RCOOH) activate csrB/C transcription via the BarA-UvrY TCS (39). Thus, the depletion of carbon substrate and accumulation of end products cause CsrB/C accumulation and subsequent inhibition of CsrA activity. In turn, CsrA indirectly activates csrB/C transcription, creating a negative-feedback loop that supports rapid response dynamics of the system (3, 36–39, 71). Inhibition of CsrA decreases expression of glycolytic genes and increases expression of genes for gluconeogenesis, glycogen biosynthesis, biofilm formation, and adaptation to stationary-phase and stress conditions (7, 14, 18, 21, 63). In the absence of a preferred carbon source, P-EIIAGlc binds to adenylate cyclase (CyaA), activating cAMP synthesis and cAMP-CRP formation (48), which represses transcription of csrC directly (Fig. 1 to 3 and 5 to 7) and of csrB indirectly (Fig. 1 to 3 and 5). The latter distinct effects of CsrA on csrB and csrC are shown as a combined inhibitory interaction. The results of our present studies imply that a futile cycle of CsrB/C synthesis and turnover may occur when glucose is abundant and end products begin to accumulate, with the potential to poise the Csr system for rapid response to shifting regulatory cues. A broken line indicates that cAMP-CRP has weak (2-fold) effects on CsrB/C decay. Additional details of this circuitry are discussed in the text.
We determined that cAMP-CRP represses the synthesis of E. coli CsrB and CsrC sRNAs using a combination of molecular genetics and biochemical evidence. Levels of these sRNAs and csrB-lacZ and csrC-lacZ expression were elevated in strains unable to produce cAMP-CRP (Fig. 1 to 3). While both CsrB and CsrC responded positively to cAMP-CRP in vivo, only csrC DNA was a target of specific, high-affinity in vitro binding by cAMP-CRP (Fig. 5 to 8). Thus, cAMP-CRP uses distinct mechanisms for regulating csrB versus csrC expression. Consistent with this finding, cAMP-CRP had little or no effect on the cellular levels of P-UvrY and CsrA, which are known to activate both csrB and csrC expression (Fig. 4). Integration host factor IHF is the only factor known to differentially activate csrB transcription without affecting csrC (3, 41). However, the ihfA and ihfB genes, which encode the IHF subunits, were not among the E. coli genes found to contain cAMP-CRP binding sites by genomic SELEX analyses (47). Thus, IHF seems unlikely to mediate the effects of cAMP-CRP on csrB.
The Csr regulon is broad in scope and includes many genes involved in carbon and energy metabolism. Not surprisingly, the carbon nutritional status influences the workings of the Csr system. The BarA-UvrY (-SirA) TCS of E. coli and Salmonella activates csrB expression in response to end products of bacterial carbon metabolism that accumulate in the mammalian large intestine, such as formate, acetate, and propionate (39, 67). Furthermore, chromatin immunoprecipitation-exo (ChIP-exo) studies have shown that P-UvrY (P-SirA) binds primarily to csrB and csrC DNA in vivo in these bacteria, indicating that activation of csrB and csrC transcription is the main function of BarA-UvrY (3). In contrast, citrate accumulation in Vibrio fischeri (68) and other tricarboxylic acid cycle intermediates in Pseudomonas fluorescens (69) are correlated with the function of this TCS, also referred to as GacS-GacA. The biochemical mechanisms involved in these sensory processes remain to be determined. In E. coli, CsrA positively regulates several enzymes of glycolysis, in particular, the enzyme phosphofructokinase A, which drives metabolic flux beyond the upper trunk of the glycolysis pathway (7, 13). By inference, products of carbon metabolism downregulate CsrA activity and glycolytic flux through the Embeden-Meyerhof-Parnas pathway, while they activate gluconeogenesis, glycogen synthesis, synthesis of the biofilm exopolysaccharide dPNAG, and pathways and processes favoring stress resistance and survival, which are repressed by CsrA (Fig. 10) (7, 17, 18, 22, 24, 33).
The Csr system is also regulated in complex ways by the availability of preferred carbon substrate for growth (Fig. 10). Transport of glucose by the PTS pathway leads to dephosphorylation of EIIAGlc, which binds to CsrD and activates the decay pathway for CsrB/C in E. coli (43, 44). This kind of regulatory pathway may function in most Enterobacteriaceae, Vibrionaceae, and Shewanellaceae species and yet is absent in the majority of gammaproteobacterial families, the members of which lack a csrD homolog (42, 43). Furthermore, cAMP-CRP modestly inhibits CsrB/C decay (Fig. 2). Therefore, EIIAGlc and P-EIIAGlc relay complementary information to CsrD and adenylate cyclase, respectively, favoring CsrB/C decay when glucose is present. Together, csrB/C transcription, which is stimulated by end products of carbon metabolism, and CsrB/C decay, which is activated by glucose, have the potential to reinforce each other's effects on CsrB/C. Both pathways should drive CsrB/C accumulation when preferred carbon resources have been expended and end products have accumulated, promoting the physiological switch from glycolytic growth to stationary-phase metabolism (9, 44).
Our new observations present a twist on the role of carbon substrate in the Csr system. cAMP-CRP formation, which is inhibited by the effect of glucose on EIIAGlc phosphorylation, leads to repression of csrB/C transcription (Fig. 1). Thus, the presence of glucose has the potential to activate both the synthesis and turnover of CsrB/C, through its effects on the phosphorylation state of EIIAGlc (Fig. 10). How might these conflicting effects of glucose be of benefit to E. coli? When a preferred carbon source is present and metabolic end products such as formate or acetate are accumulating, both the synthesis and turnover of CsrB/C should occur. We propose that this may lead to accelerated responses to cues or stimuli affecting the Csr system, as described in general for the behavior of incoherent feedback loops (53, 70). In support of this hypothesis, results of modeling studies with genes of the Csr system suggest that the CsrD-dependent decay pathway for CsrB/C sRNAs enhances rates of Csr response to signals, although the involvement of glucose or carbon metabolites in this process has not been demonstrated (71). In view of the hundreds of genes and numerous pathways and processes that are controlled by CsrA (13, 25, 33), the proposed operation of a futile cycle of CsrB/C synthesis and turnover when a preferred carbon source is available may be a small price to pay to poise the Csr system for rapid response.
We demonstrated that cAMP-CRP directly represses csrC expression by binding to csrC DNA (Fig. 5, 6, and 8), while csrB appears to be repressed indirectly (Fig. 5 and 7). Repression by cAMP-CRP can be accomplished in a number of ways. For example, CRP can bind at a location close to the promoter and directly interfere with transcription initiation or elongation (57, 72). Alternatively, CRP can prevent binding of an activator (52) or can activate transcription from a promoter and indirectly lead to repression of transcription from an overlapping divergent promoter (73, 74). Our results suggest that repression of csrC expression by cAMP-CRP acts in conjunction with P-UvrY-dependent activation. DNase I footprinting experiments show that cAMP-CRP and P-UvrY compete for binding in a region far upstream of the csrC promoter (Fig. 6). We should emphasize that cAMP-CRP binds even more tightly at a location immediately downstream of the P-UvrY binding site (Fig. 6). Whether repression is mediated by direct competition of cAMP-CRP with P-UvrY for binding to csrC DNA or is a consequence of cAMP-CRP binding to the downstream site and inhibiting the productive interaction of bound P-UvrY with RNA polymerase or other regulatory elements for csrC transcription remains to be seen.
The Csr system appears to be conserved in all Gammaproteobacteria species, but details such as the number of CsrA paralogs and Csr sRNAs produced by a given species can differ (6, 44, 75, 76). Not surprisingly, even among closely related Enterobacteriaceae species, the links between Csr and catabolite repression circuitry differ. In Yersinia pseudotuberculosis, cAMP-CRP exerts indirect and opposite regulatory effects on csrB and csrC (77). Furthermore, while csrB expression in Y. pseudotuberculosis depends on BarA-UvrY, csrC expression is directly activated by the PhoP-PhoQ TCS (78). Salmonella enterica was reported to somehow activate expression of the uvrY ortholog, sirA, via cAMP-CRP, with positive downstream effects on csrB-lacZ and csrC-lacZ gene fusions (79). In addition, CLIP-seq studies in Salmonella revealed binding of CsrA to crp (cap) leader mRNA at two locations (80), one of which is related in sequence to E. coli BS2 (Fig. 8A). Additional studies will be required to unravel the biological significance of such variations in the Csr and catabolite repression networks.
ACKNOWLEDGMENTS
This study was supported by grants to T.R. and P.B. (NIH GM059969) and D.G. (CONACyT 178033).
REFERENCES
- 1.White D, Hart ME, Romeo T. 1996. Phylogenetic distribution of the global regulatory gene csrA among eubacteria. Gene 182:221–223. doi: 10.1016/S0378-1119(96)00547-1. [DOI] [PubMed] [Google Scholar]
- 2.Mercante J, Suzuki K, Cheng X, Babitzke P, Romeo T. 2006. Comprehensive alanine-scanning mutagenesis of Escherichia coli CsrA defines two subdomains of critical functional importance. J Biol Chem 281:31832–31842. doi: 10.1074/jbc.M606057200. [DOI] [PubMed] [Google Scholar]
- 3.Zere TR, Vakulskas CA, Leng Y, Pannuri A, Potts AH, Dias R, Tang D, Kolaczkowski B, Georgellis D, Ahmer BM, Romeo T. 2015. Genomic targets and features of BarA-UvrY (-SirA) signal transduction systems. PLoS One 10:e0145035. doi: 10.1371/journal.pone.0145035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romeo T, Vakulskas CA, Babitzke P. 2013. Post-transcriptional regulation on a global scale: form and function of Csr/Rsm systems. Environ Microbiol 15:313–324. doi: 10.1111/j.1462-2920.2012.02794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babitzke P, Baker CS, Romeo T. 2009. Regulation of translation initiation by RNA binding proteins. Annu Rev Microbiol 63:27–44. doi: 10.1146/annurev.micro.091208.073514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vakulskas CA, Potts AH, Babitzke P, Ahmer BM, Romeo T. 2015. Regulation of bacterial virulence by Csr (Rsm) systems. Microbiol Mol Biol Rev 79:193–224. doi: 10.1128/MMBR.00052-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sabnis NA, Yang H, Romeo T. 1995. Pleiotropic regulation of central carbohydrate metabolism in Escherichia coli via the gene csrA. J Biol Chem 270:29096–29104. doi: 10.1074/jbc.270.49.29096. [DOI] [PubMed] [Google Scholar]
- 8.Revelles O, Millard P, Nougayrede JP, Dobrindt U, Oswald E, Letisse F, Portais JC. 2013. The carbon storage regulator (Csr) system exerts a nutrient-specific control over central metabolism in Escherichia coli strain Nissle 1917. PLoS One 8:e66386. doi: 10.1371/journal.pone.0066386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pernestig AK, Georgellis D, Romeo T, Suzuki K, Tomenius H, Normark S, Melefors O. 2003. The Escherichia coli BarA-UvrY two-component system is needed for efficient switching between glycolytic and gluconeogenic carbon sources. J Bacteriol 185:843–853. doi: 10.1128/JB.185.3.843-853.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murray EL, Conway T. 2005. Multiple regulators control expression of the Entner-Doudoroff aldolase (Eda) of Escherichia coli. J Bacteriol 187:991–1000. doi: 10.1128/JB.187.3.991-1000.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei B, Shin S, LaPorte D, Wolfe AJ, Romeo T. 2000. Global regulatory mutations in csrA and rpoS cause severe central carbon stress in Escherichia coli in the presence of acetate. J Bacteriol 182:1632–1640. doi: 10.1128/JB.182.6.1632-1640.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patterson-Fortin LM, Vakulskas CA, Yakhnin H, Babitzke P, Romeo T. 2013. Dual posttranscriptional regulation via a cofactor-responsive mRNA leader. J Mol Biol 425:3662–3677. doi: 10.1016/j.jmb.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morin M, Ropers D, Letisse F, Laguerre S, Portais JC, Cocaign-Bousquet M, Enjalbert B. 2016. The post-transcriptional regulatory system CSR controls the balance of metabolic pools in upper glycolysis of Escherichia coli. Mol Microbiol 100:686–700. doi: 10.1111/mmi.13343. [DOI] [PubMed] [Google Scholar]
- 14.Wei BL, Brun-Zinkernagel AM, Simecka JW, Pruss BM, Babitzke P, Romeo T. 2001. Positive regulation of motility and flhDC expression by the RNA-binding protein CsrA of Escherichia coli. Mol Microbiol 40:245–256. doi: 10.1046/j.1365-2958.2001.02380.x. [DOI] [PubMed] [Google Scholar]
- 15.Yakhnin AV, Baker CS, Vakulskas CA, Yakhnin H, Berezin I, Romeo T, Babitzke P. 2013. CsrA activates flhDC expression by protecting flhDC mRNA from RNase E-mediated cleavage. Mol Microbiol 87:851–866. doi: 10.1111/mmi.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu MY, Yang H, Romeo T. 1995. The product of the pleiotropic Escherichia coli gene csrA modulates glycogen biosynthesis via effects on mRNA stability. J Bacteriol 177:2663–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang H, Liu MY, Romeo T. 1996. Coordinate genetic regulation of glycogen catabolism and biosynthesis in Escherichia coli via the CsrA gene product. J Bacteriol 178:1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romeo T, Gong M, Liu MY, Brun-Zinkernagel AM. 1993. Identification and molecular characterization of csrA, a pleiotropic gene from Escherichia coli that affects glycogen biosynthesis, gluconeogenesis, cell size, and surface properties. J Bacteriol 175:4744–4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker CS, Morozov I, Suzuki K, Romeo T, Babitzke P. 2002. CsrA regulates glycogen biosynthesis by preventing translation of glgC in Escherichia coli. Mol Microbiol 44:1599–1610. doi: 10.1046/j.1365-2958.2002.02982.x. [DOI] [PubMed] [Google Scholar]
- 20.Mercante J, Edwards AN, Dubey AK, Babitzke P, Romeo T. 2009. Molecular geometry of CsrA (RsmA) binding to RNA and its implications for regulated expression. J Mol Biol 392:511–528. doi: 10.1016/j.jmb.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackson DW, Suzuki K, Oakford L, Simecka JW, Hart ME, Romeo T. 2002. Biofilm formation and dispersal under the influence of the global regulator CsrA of Escherichia coli. J Bacteriol 184:290–301. doi: 10.1128/JB.184.1.290-301.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang X, Dubey AK, Suzuki K, Baker CS, Babitzke P, Romeo T. 2005. CsrA post-transcriptionally represses pgaABCD, responsible for synthesis of a biofilm polysaccharide adhesin of Escherichia coli. Mol Microbiol 56:1648–1663. doi: 10.1111/j.1365-2958.2005.04648.x. [DOI] [PubMed] [Google Scholar]
- 23.Jonas K, Edwards AN, Simm R, Romeo T, Romling U, Melefors O. 2008. The RNA binding protein CsrA controls cyclic di-GMP metabolism by directly regulating the expression of GGDEF proteins. Mol Microbiol 70:236–257. doi: 10.1111/j.1365-2958.2008.06411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pannuri A, Yakhnin H, Vakulskas CA, Edwards AN, Babitzke P, Romeo T. 2012. Translational repression of NhaR, a novel pathway for multi-tier regulation of biofilm circuitry by CsrA. J Bacteriol 194:79–89. doi: 10.1128/JB.06209-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edwards AN, Patterson-Fortin LM, Vakulskas CA, Mercante JW, Potrykus K, Vinella D, Camacho MI, Fields JA, Thompson SA, Georgellis D, Cashel M, Babitzke P, Romeo T. 2011. Circuitry linking the Csr and stringent response global regulatory systems. Mol Microbiol 80:1561–1580. doi: 10.1111/j.1365-2958.2011.07663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dubey AK, Baker CS, Suzuki K, Jones AD, Pandit P, Romeo T, Babitzke P. 2003. CsrA regulates translation of the Escherichia coli carbon starvation gene, cstA, by blocking ribosome access to the cstA transcript. J Bacteriol 185:4450–4460. doi: 10.1128/JB.185.15.4450-4460.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker CS, Eory LA, Yakhnin H, Mercante J, Romeo T, Babitzke P. 2007. CsrA inhibits translation initiation of Escherichia coli hfq by binding to a single site overlapping the Shine-Dalgarno sequence. J Bacteriol 189:5472–5481. doi: 10.1128/JB.00529-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang TY, Sung YM, Lei GS, Romeo T, Chak KF. 2010. Posttranscriptional repression of the cel gene of the ColE7 operon by the RNA-binding protein CsrA of Escherichia coli. Nucleic Acids Res 38:3936–3951. doi: 10.1093/nar/gkq177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yakhnin H, Baker CS, Berezin I, Evangelista MA, Rassin A, Romeo T, Babitzke P. 2011. CsrA represses translation of sdiA, which encodes the N-acylhomoserine-l-lactone receptor of Escherichia coli, by binding exclusively within the coding region of sdiA mRNA. J Bacteriol 193:6162–6170. doi: 10.1128/JB.05975-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Figueroa-Bossi N, Schwartz A, Guillemardet B, D'Heygere F, Bossi L, Boudvillain M. 2014. RNA remodeling by bacterial global regulator CsrA promotes Rho-dependent transcription termination. Genes Dev 28:1239–1251. doi: 10.1101/gad.240192.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhatt S, Edwards AN, Nguyen HT, Merlin D, Romeo T, Kalman D. 2009. The RNA binding protein CsrA is a pleiotropic regulator of the locus of enterocyte effacement pathogenicity island of enteropathogenic Escherichia coli. Infect Immun 77:3552–3568. doi: 10.1128/IAI.00418-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhatt S, Romeo T, Kalman D. 2011. Honing the message: post-transcriptional and post-translational control in attaching and effacing pathogens. Trends Microbiol 19:217–224. doi: 10.1016/j.tim.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esquerré T, Bouvier M, Turlan C, Carpousis AJ, Girbal L, Cocaign-Bousquet M. 2016. The Csr system regulates genome-wide mRNA stability and transcription and thus gene expression in Escherichia coli. Sci Rep 6:25057. doi: 10.1038/srep25057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yakhnin H, Yakhnin AV, Baker CS, Sineva E, Berezin I, Romeo T, Babitzke P. 2011. Complex regulation of the global regulatory gene csrA: CsrA-mediated translational repression, transcription from five promoters by Eσ70 and Eσ(S), and indirect transcriptional activation by CsrA. Mol Microbiol 81:689–704. doi: 10.1111/j.1365-2958.2011.07723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu MY, Romeo T. 1997. The global regulator CsrA of Escherichia coli is a specific mRNA-binding protein. J Bacteriol 179:4639–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weilbacher T, Suzuki K, Dubey AK, Wang X, Gudapaty S, Morozov I, Baker CS, Georgellis D, Babitzke P, Romeo T. 2003. A novel sRNA component of the carbon storage regulatory system of Escherichia coli. Mol Microbiol 48:657–670. doi: 10.1046/j.1365-2958.2003.03459.x. [DOI] [PubMed] [Google Scholar]
- 37.Camacho MI, Alvarez AF, Chavez RG, Romeo T, Merino E, Georgellis D. 2015. Effects of the global regulator CsrA on the BarA/UvrY two-component signaling system. J Bacteriol 197:983–991. doi: 10.1128/JB.02325-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suzuki K, Wang X, Weilbacher T, Pernestig AK, Melefors O, Georgellis D, Babitzke P, Romeo T. 2002. Regulatory circuitry of the CsrA/CsrB and BarA/UvrY systems of Escherichia coli. J Bacteriol 184:5130–5140. doi: 10.1128/JB.184.18.5130-5140.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chavez RG, Alvarez AF, Romeo T, Georgellis D. 2010. The physiological stimulus for the BarA sensor kinase. J Bacteriol 192:2009–2012. doi: 10.1128/JB.01685-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martínez LC, Yakhnin H, Camacho MI, Georgellis D, Babitzke P, Puente JL, Bustamante VH. 2011. Integration of a complex regulatory cascade involving the SirA/BarA and Csr global regulatory systems that controls expression of the Salmonella SPI-1 and SPI-2 virulence regulons through HilD. Mol Microbiol 80:1637–1656. doi: 10.1111/j.1365-2958.2011.07674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martínez LC, Martínez-Flores I, Salgado H, Fernández-Mora M, Medina-Rivera A, Puente JL, Collado-Vides J, Bustamante VH. 2014. In silico identification and experimental characterization of regulatory elements controlling the expression of the Salmonella csrB and csrC genes. J Bacteriol 196:325–336. doi: 10.1128/JB.00806-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Suzuki K, Babitzke P, Kushner SR, Romeo T. 2006. Identification of a novel regulatory protein (CsrD) that targets the global regulatory RNAs CsrB and CsrC for degradation by RNase E. Genes Dev 20:2605–2617. doi: 10.1101/gad.1461606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vakulskas CA, Leng Y, Abe H, Amaki T, Okayama A, Babitzke P, Suzuki K, Romeo T. 2016. Antagonistic control of the turnover pathway for the global regulatory sRNA CsrB by the CsrA and CsrD proteins. Nucleic Acids Res doi: 10.1093/nar/gkw484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leng Y, Vakulskas CA, Zere TR, Pickering BS, Watnick PI, Babitzke P, Romeo T. 2016. Regulation of CsrB/C sRNA decay by EIIAGlc of the phosphoenolpyruvate:carbohydrate phosphotransferase system. Mol Microbiol 99:627–639. doi: 10.1111/mmi.13259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gosset G, Zhang Z, Nayyar S, Cuevas WA, Saier MH Jr. 2004. Transcriptome analysis of CRP-dependent catabolite control of gene expression in Escherichia coli. J Bacteriol 186:3516–3524. doi: 10.1128/JB.186.11.3516-3524.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gutierrez-Ríos RM, Freyre-Gonzalez JA, Resendis O, Collado-Vides J, Saier M, Gosset G. 2007. Identification of regulatory network topological units coordinating the genome-wide transcriptional response to glucose in Escherichia coli. BMC Microbiol 7:53. doi: 10.1186/1471-2180-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shimada T, Fujita N, Yamamoto K, Ishihama A. 2011. Novel roles of cAMP receptor protein (CRP) in regulation of transport and metabolism of carbon sources. PLoS One 6:e20081. doi: 10.1371/journal.pone.0020081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deutscher J, Ake FM, Derkaoui M, Zebre AC, Cao TN, Bouraoui H, Kentache T, Mokhtari A, Milohanic E, Joyet P. 2014. The bacterial phosphoenolpyruvate:carbohydrate phosphotransferase system: regulation by protein phosphorylation and phosphorylation-dependent protein-protein interactions. Microbiol Mol Biol Rev 78:231–256. doi: 10.1128/MMBR.00001-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Görke B, Stülke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol 6:613–624. doi: 10.1038/nrmicro1932. [DOI] [PubMed] [Google Scholar]
- 50.De Lay N, Gottesman S. 2009. The CRP-activated small noncoding regulatory RNA CyaR (RyeE) links nutritional status to group behavior. J Bacteriol 191:461–476. doi: 10.1128/JB.01157-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Papenfort K, Pfeiffer V, Lucchini S, Sonawane A, Hinton JC, Vogel J. 2008. Systematic deletion of Salmonella small RNA genes identifies CyaR, a conserved CRP-dependent riboregulator of OmpX synthesis. Mol Microbiol 68:890–906. doi: 10.1111/j.1365-2958.2008.06189.x. [DOI] [PubMed] [Google Scholar]
- 52.Polayes DA, Rice PW, Garner MM, Dahlberg JE. 1988. Cyclic AMP-cyclic AMP receptor protein as a repressor of transcription of the spf gene of Escherichia coli. J Bacteriol 170:3110–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beisel CL, Storz G. 2011. The base-pairing RNA spot 42 participates in a multioutput feedforward loop to help enact catabolite repression in Escherichia coli. Mol Cell 41:286–297. doi: 10.1016/j.molcel.2010.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomason MK, Fontaine F, De Lay N, Storz G. 2012. A small RNA that regulates motility and biofilm formation in response to changes in nutrient availability in Escherichia coli. Mol Microbiol 84:17–35. doi: 10.1111/j.1365-2958.2012.07965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grainger DC, Hurd D, Harrison M, Holdstock J, Busby SJ. 2005. Studies of the distribution of Escherichia coli cAMP-receptor protein and RNA polymerase along the E. coli chromosome. Proc Natl Acad Sci U S A 102:17693–17698. doi: 10.1073/pnas.0506687102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.You C, Okano H, Hui S, Zhang Z, Kim M, Gunderson CW, Wang YP, Lenz P, Yan D, Hwa T. 2013. Coordination of bacterial proteome with metabolism by cyclic AMP signalling. Nature 500:301–306. doi: 10.1038/nature12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kolb A, Busby S, Buc H, Garges S, Adhya S. 1993. Transcriptional regulation by cAMP and its receptor protein. Annu Rev Biochem 62:749–795. doi: 10.1146/annurev.bi.62.070193.003533. [DOI] [PubMed] [Google Scholar]
- 58.Busby S, Ebright RH. 1999. Transcription activation by catabolic activator protein (CAP). J Mol Biol 293:199–213. doi: 10.1006/jmbi.1999.3161. [DOI] [PubMed] [Google Scholar]
- 59.Miller J. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 60.Haldimann A, Wanner BL. 2001. Conditional-replication, excision and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol 183:6384–6393. doi: 10.1128/JB.183.21.6384-6393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Romeo T, Black J, Preiss J. 1990. Genetic regulation of glycogen synthesis in Escherichia coli: in vivo effects of the catabolic repression and stringent response systems in glg gene expression. Curr Microbiol 21:131–137. doi: 10.1007/BF02091831. [DOI] [Google Scholar]
- 62.Vakulskas CA, Pannuri A, Cortes-Selva D, Zere TR, Ahmer BM, Babitzke P, Romeo T. 2014. Global effects of the DEAD-box RNA helicase DeaD (CsdA) on gene expression over a broad range of temperatures. Mol Microbiol 92:945–958. doi: 10.1111/mmi.12606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wickstrum JR, Egan SM. 2002. Ni+-affinity purification of untagged cAMP receptor protein. Biotechniques 33:728–730. [DOI] [PubMed] [Google Scholar]
- 64.Hogema BM, Arents JC, Bader R, Eijkemans K, Yoshida H, Takahashi H, Aiba H, Postma PW. 1998. Inducer exclusion in Escherichia coli by non-PTS substrates: the role of the PEP to pyruvate ratio in determining the phosphorylation state of enzyme IIAGlc. Mol Microbiol 30:487–498. doi: 10.1046/j.1365-2958.1998.01053.x. [DOI] [PubMed] [Google Scholar]
- 65.Kolb A, Busby S, Herbert M, Kotlarz D, Buc H. 1983. Comparison of the binding sites for the Escherichia coli cAMP receptor protein at the lactose and galactose promoters. EMBO J 2:217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jackson DW, Simecka JW, Romeo T. 2002. Catabolite repression of Escherichia coli biofilm formation. J Bacteriol 184:3406–3410. doi: 10.1128/JB.184.12.3406-3410.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lawhon SD, Maurer R, Suyemoto M, Altier C. 2002. Intestinal short-chain fatty acids alter Salmonella typhimurium invasion gene expression and virulence through BarA/SirA. Mol Microbiol 46:1451–1464. doi: 10.1046/j.1365-2958.2002.03268.x. [DOI] [PubMed] [Google Scholar]
- 68.Septer AN, Bose JL, Lipzen A, Martin J, Whistler C, Stabb EV. 2015. Bright luminescence of Vibrio fischeri aconitase mutants reveals a connection between citrate and the Gac/Csr regulatory system. Mol Microbiol 95:283–296. doi: 10.1111/mmi.12864. [DOI] [PubMed] [Google Scholar]
- 69.Takeuchi K, Kiefer P, Reimmann C, Keel C, Dubuis C, Rotli J, Vorholt JA, Haas D. 2009. Small RNA dependent expression of secondary metabolism is controlled by Krebs cycle function in Pseudomonas fluorescens. J Biol Chem 284:34976–34985. doi: 10.1074/jbc.M109.052571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mangan S, Alon U. 2003. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci U S A 100:11980–11985. doi: 10.1073/pnas.2133841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Adamson DN, Lim HN. 2013. Rapid and robust signaling in the CsrA cascade via RNA-protein interactions and feedback regulation. Proc Natl Acad Sci U S A 110:13120–13125. doi: 10.1073/pnas.1308476110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nakano M, Ogasawara H, Shimada T, Yamamoto K, Ishihama A. 2014. Involvement of cAMP-CRP in transcription activation and repression of the pck gene encoding PEP carboxykinase, the key enzyme of gluconeogenesis. FEMS Microbiol Lett 355:93–99. doi: 10.1111/1574-6968.12466. [DOI] [PubMed] [Google Scholar]
- 73.Hanamura A, Aiba H. 1991. Molecular mechanism of negative autoregulation of Escherichia coli crp gene. Nucleic Acids Res 19:4413–4419. doi: 10.1093/nar/19.16.4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Osuna R, Boylan SA, Bender RA. 1991. In vitro transcription of the histidine utilization (hutUH) operon from Klebsiella aerogenes. J Bacteriol 173:116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abbott ZD, Yakhnin H, Babitzke P, Swanson MS. 2015. csrR, a paralog and direct target of CsrA, promotes Legionella pneumophila resilience in water. mBio 6:e00595. doi: 10.1128/mBio.00595-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lenz DH, Miller MB, Zhu J, Kulkarni RV, Bassler BL. 2005. CsrA and three redundant small RNAs regulate quorum sensing in Vibrio cholerae. Mol Microbiol 58:1186–1202. doi: 10.1111/j.1365-2958.2005.04902.x. [DOI] [PubMed] [Google Scholar]
- 77.Heroven AK, Sest M, Pisano F, Scheb-Wetzel M, Steinmann R, Bohme K, Klein J, Munch R, Schomburg D, Dersch P. 2012. CRP induces switching of the CsrB and CsrC RNAs in Yersinia pseudotuberculosis and links nutritional status to virulence. Front Cell Infect Microbiol 2:158. doi: 10.3389/fcimb.2012.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nuss AM, Schuster F, Heroven AK, Heine W, Pisano F, Dersch P. 2014. A direct link between the global regulator PhoP and the Csr regulon in Yersinia pseudotuberculosis through the small regulatory RNA CsrC. RNA Biol 11:580–593. doi: 10.4161/rna.28676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Teplitski M, Goodier RI, Ahmer BMM. 2006. Catabolite repression of the SirA regulatory cascade in Salmonella enterica. Int J Med Microbiol 296:449–466. doi: 10.1016/j.ijmm.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 80.Holmqvist E, Wright PR, Li L, Bischler T, Barquist L, Reinhardt R, Backofen R, Vogel J. 2016. Global RNA recognition patterns of post-transcriptional regulators Hfq and CsrA revealed by UV crosslinking in vivo. EMBO J 35:991–1011. doi: 10.15252/embj.201593360. [DOI] [PMC free article] [PubMed] [Google Scholar]