

Abstract
Several species of β-amyloid peptides (Aβ) exist as a result of differential cleavage from amyloid precursor protein (APP) to yield various C-terminal Aβ peptides. Several N-terminal modified Aβ peptides have also been identified in Alzheimer’s disease (AD) brains, the most common of which is pyroglutamate-modified Aβ (AβpE3-42). AβpE3-42 peptide has an increased propensity to aggregate, appears to accumulate in the brain before the appearance of clinical symptoms of AD, and precedes Aβ1-42 deposition. Moreover, in vitro studies have shown that AβpE3-42 can act as a seed for full length Aβ1-42. In this study, we characterized the Drosophila model of AβpE3-42 toxicity by expressing the peptide in specific sets of neurons using the GAL4-UAS system, and measuring different phenotypic outcomes. We found that AβpE3-42 peptide had an increased propensity to aggregate. Expression of AβpE3-42 in the neurons of adult flies led to behavioural dysfunction and shortened lifespan. Expression of AβpE3-42 constitutively in the eyes led to disorganised ommatidia, and activation of the c-Jun N-terminal kinase (JNK) signaling pathway. The eye disruption was almost completely rescued by co-expressing a candidate Aβ degrading enzyme, neprilysin2. Furthermore, we found that neprilysin2 was capable of degrading AβpE3-42. Also, we tested the seeding hypothesis for AβpE3-42 in vivo, and measured its effect on Aβ1-42 levels. We found that Aβ1-42 levels were significantly increased when Aβ1-42 and AβpE3-42 peptides were co-expressed. Furthermore, we found that AβpE3-42 enhanced Aβ1-42 toxicity in vivo. Our findings implicate AβpE3-42 as an important source of toxicity in AD, and suggest that its specific degradation could be therapeutic.
Electronic supplementary material
The online version of this article (doi:10.1186/s40478-016-0380-x) contains supplementary material, which is available to authorized users.
Keywords: Neurodegeneration, Alzheimer’s disease, pyroglutamate Abeta, Drosophila
Introduction
Alzheimer’s Disease (AD) is a neurodegenerative disorder characterized by amyloid beta (Aβ) deposits and neurofibrillary hyperphosphorylated tau tangles [1]. The amyloid cascade, which has undergone some revision in recent years, is the leading hypothesis for the pathology associated with AD, and states that amyloidogenic Aβ is the trigger of the pathogenic process leading to neuronal cell death [2, 3]. Aβ induces several stressors, which could lead to neuronal cell death [4]. The c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) signaling pathway which influences cell death is activated in response to many forms of stress, such as oxidative stress and endoplasmic reticulum (ER) stress [5, 6]. In particular, Aβ is able to activate the JNK/SAPK pathway, and increased phosphorylation of JNK/SAPK has been observed in post-mortem AD brain tissue in comparison to control cases [5, 7].
Aβ peptides form as a cleavage product from the amyloid precursor protein (APP) [8]. Several species of Aβ peptides exist, as a result of differential cleavage from APP to yield various C-terminal Aβ peptides. Aβ40 and Aβ42 are the most abundant, with Aβ42 being the more toxic form [9]. More recently, Aβ43 has also been identified as a pathogenic species [10, 11]. There are also several N-terminal truncated/modified Aβ peptides that have been identified in AD brains, the most common of which is pyroglutamate-modified Aβ [12–14].
Pyroglutamic (pGlu) acid is generated from N-terminal glutamine during pro-hormone maturation in the secretory pathway; the enzyme glutaminyl cyclase (QC) is directed to the secretory pathway and catalyses the conversion from N-terminal glutamic to pGlu acid [15, 16]. Interestingly, Aβ undergoes this post-translational modification at its amino terminus, also catalyzed by QC [17], which is up-regulated in the cortex of patients with AD [18]. Of particular interest is the highly abundant AβpE3-42, which is generated by cleavage of the first 2 amino acids of the Aβ peptide, followed by pGlu-modification of glutamate in the third amino acid position, which is thought to stabilize it and/or promote its aggregation propensity [14].
Interestingly, water soluble Aβ, which appear before plaques is made up predominantly of AβpE3-42 [19], and indeed AβpE3-42 accumulate early on in the brain before the appearance of clinical symptoms and Aβ1-42 deposition [20–23]. Moreover, in vitro studies have shown that AβpE3-42 has an increased propensity to aggregate in comparison to Aβ1-42 and can act as a seed/primer for full length Aβ1-42 [14, 24, 25]. AβpE3-42 behaves in a prion-like manner, whereby only small quantities of AβpE3-42 are able to increase the amount of metastable low-n Aβ1-42 oligomers in vitro [25]. These attributes have created a lot of interest in AβpE3-42 peptides, and several groups have suggested that they are important in initiating the pathological cascade of AD [14, 20].
Interestingly, AβpE3-42 plaque load has been observed in brain autopsies of familial, sporadic cases and controls although, importantly, oligomeric AβpE3-42 was only found in the familial and sporadic cases [26]. There is also a likely role for AβpE3-42 in intra-neuronal AD toxicity. A study that expressed AβQ3-42 under the Thy-1 promoter in mice (glutamine was used instead of glutamate because it is a better substrate for pyroglutamate conversion, [15]) showed increased levels of intra-neuronal AβpE3-42, severe neurological impairment and loss of Purkinje cells [27]. Furthermore, AβpE3-42 expressing mice which display neuronal loss [28], when crossed into a tau KO background were almost completely protected against neuronal loss, establishing a functional connection between pGluAβ and tau [25]. Moreover, transgenic mouse models of AD that develop more severe pathology, as measured by the appearance of early neurological phenotypes and amyloid plaque deposition, tend to have high levels of AβpE3-42 [14].
Over-expression or reduction of QC has also been shown to exacerbate or rescue behavioural phenotypes and plaque pathology in an AD mouse model [29]. Interestingly, QC KO mice showed a reduction in both AβpE3-42 and Aβ1-42 levels, again supporting the idea that AβpE3-42 plays a role in seeding Aβ1-42 [29]. The data also demonstrate the importance of QC, and suggest that a reduction of QC might be a promising therapeutic strategy.
Many signaling pathways/molecules are conserved between flies and humans, and QC is 1 of them. Drosophila has 2 QCs – Drome QC and isoDrome QC, which have different subcellular locations [30]. IsoDrome QC more closely resembles the mammalian homologue [30]. Interestingly, treatment of AβQ3-42 transgenic flies with a QC inhibitor led to reduced AβpE3-42 levels [18], highlighting the usefulness of Drosophila to investigate the molecular pathogenicity of AβpE3-42. Several labs have generated fly models that express various Aβ peptides [31–33]. AβQ3-42 fly models are available, but have not been fully characterized or utilized to test the “seeding hypothesis”.
In this study, we characterized a Drosophila model of AβpE3-42 toxicity in the fruit-fly. Expression specifically in adult fly neurons led to behavioural dysfunction and shortened lifespan. Expression of the AβpE3-42 constitutively in the eyes led to disorganised ommatidia, which was ameliorated by neprilysin2. Furthermore, we show for the first time that neprilysin2 was able to degrade pyroglutamate Aβ.
Several recent studies have suggested that AβpE3-42 can act as a seed for Aβ1-42, and such a role has been demonstrated in vitro. AβpE3-42 has been shown to increase the amount of metastable low-n Aβ1-42 oligomers in vitro [25]. Furthermore, peri-hippocampal injection of AβpE3-42 into APPswe/NOS2-/- AD mice led to the presence of both AβpE3-42 and conventional Aβ plaques, which the authors mention was hardly seen in sham injected AD mice or WT mice injected with AβpE3-42 [25]. However, the direct effect of AβpE3-42 on Aβ1-42 in vivo remains to be assessed, because the genetic background of the AD mouse lines are mutant for several genes that may well be affected by AβpE3-42. Furthermore, to control for the specificity of the AβpE3-42 species in causing enhanced plaque formation, an important control of peri-hippocampal injection of Aβ1-42 into the AD mice is missing from their studies.
We have utilized the Drosophila model to our advantage by expressing multiple transgenes at the same time to test this seeding hypothesis in vivo, and thus determine whether AβpE3-42 could be a target for therapeutic intervention, and/or a diagnostic marker. We found that total Aβ1-42 levels and toxicity are greatly increased when AβpE3-42 is co-expressed. These data suggest that AβpE3-42 is able to stabilise Aβ1-42 in vivo.
Materials and methods
Fly stocks and maintenance
All fly stocks were maintained either at 25 °C or 28 °C on a 12:12-h light:dark cycle at constant humidity on a standard sugar-yeast (SY) medium (15gl-1 agar, 50 gl-1 sugar, 100 gl-1 autolysed yeast, 100gl-1 nipagin and 3 ml l-1 propionic acid). Adult-onset, neuronal-specific expression of Aβ peptide was achieved by using the elav GeneSwitch (elavGS)-UAS system. ElavGS was derived from the original elavGS 301.2 line [34] and obtained as a generous gift from Dr H. Tricoire (CNRS, France), GMR driver was from Bloomington stock centre. UAS-Aβ1-42 line was obtained from Dr D. Crowther [35]. UAS- AβQ3-42 line has been previously described [36], briefly the rat pre pro-enkephalin signaling sequence was cloned upstream AβQ3-42 and put into the EcoR1 site of the pUAST vector, the construct then gets processed via prohormone convertases and glutaminyl cyclase to generate AβpE3-42 [17, 37] (Additional file 1: Figure S1). GMR, elavGS and UAS-lines used in all experiments were backcrossed six times into the w 1118 genetic background. Expression by elavGS was induced by treatment with mifepristone (RU486; 200 μM) added to the standard SY medium. In the absence of mifepristone (RU486; -RU), the transgene remains transcriptionally silent. Following treatment with RU486, AβQ3-42 peptide is expressed.
Lifespan analyses
For all experiments, flies were raised at a standard density on standard SY medium in 200 mL bottles. Two days after eclosion once-mated females were transferred to experimental vials containing SY medium with or without RU486 (200 μM) at a density of 10 flies per vial (120 flies per genotype were used in Fig. 2, and 150 flies per genotype were used in Fig. 4). Deaths were scored almost every other day and flies were transferred to fresh food. Data are presented as survival curves and statistical analysis was performed using log-rank tests to compare survival of groups.
Fig. 2.
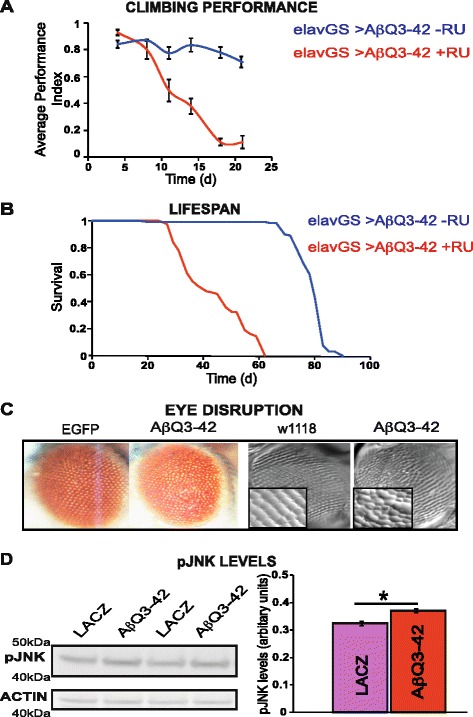
Expression of AβpE3-42 causes locomotor dysfunction, shortened lifespan, eye disruption, and JNK activation in Drosophila. a Climbing ability of elavGS/UAS-AβQ3-42 flies on + and – RU486 SY medium were assessed at the indicated time-points. Data are presented as the average performance index (PI) ± SEM and were compared using 2-way ANOVA (number of independent tests (n) = 3 *P < 0.001 (b) Expression of AβpE3-42 in adult neurons shortens lifespan. Survival curves are depicted and data were compared using the log-rank test, *P < 0.001 comparing elavGS/UAS-AβQ3-42 + RU flies to -RU flies. c Expression of AβpE3-42 causes a neurodegenerative eye phenotype. First 2 images from left to right are light microscopy images and latter 2 images are nail varnish imprints of eyes (magnification is 25×, and 40× objectives for close up images). From left to right, GMR-GAL4/UAS-EGFP, GMR-GAL4/+;UAS-AβQ3-42/+, GMR-GAL4/+, GMR-GAL4/+;UAS-AβQ3-42/+. Note compressed and fused ommatidia in AβQ3-42 expressing flies in comparison to control flies (GMR-GAL4/UAS-EGFP or GMR-GAL4/+). Flies were grown at 28 °C. d Expression of AβpE3-42 increases the levels of phosphorylated JNK. Data are presented as means ± SEM and were analysed by Student t test, *P < 0.05 comparing GMR-GAL4/+;UAS-AβQ3-42/+ flies to control GMR-GAL4/UAS-LACZ flies
Fig. 4.
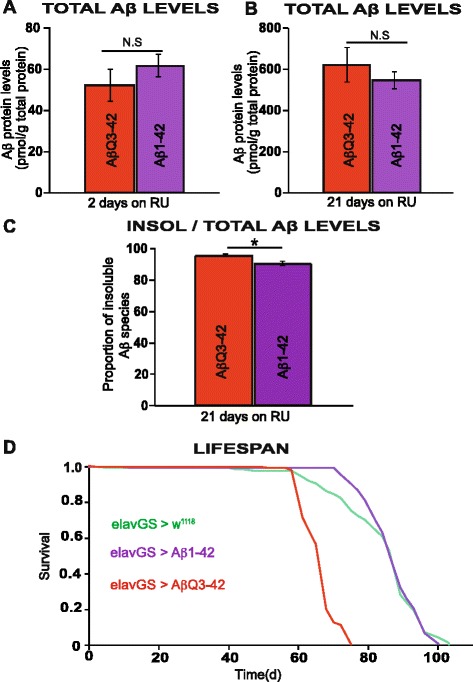
AβpE3-42 is more toxic than Aβ1-42 in Drosophila. Following treatment with RU486, Aβ1-42 and AβpE3-42 peptides were expressed at similar levels, quantified at 2 days and 21 days post-RU486 treatment (a) and (b) respectively. c A significant increase in the amount of insoluble to total Aβ protein levels in AβpE3-42 expressing flies in comparison to Aβ1-42 expressing flies was observed when quantified at 21 days post-RU486 treatment. Data are presented as means ± SEM and were analysed by Student’s t test, P < 0.01. d Expression of AβpE3-42 specifically in adult neurons shortens lifespan significantly relative to both Aβ1-42 and w1118 control. Survival curves are depicted and data were compared using the log-rank test. *P < 0.001 comparing elavGS/UAS-AβQ3-42 + RU flies to elavGS/+ and UAS-Aβ1-42 /+;elavGS/+ +RU flies
Negative geotaxis assays
Climbing assays were performed at 25 °C according to previously published methods [31]. Climbing was analysed every 2–3 days post-RU486 treatment. Fifteen adult flies were placed in a vertical column, then their rate of climb to the top of the column was analysed. Flies reaching the top (12 cm) and flies remaining at the bottom of the column after a 30 s period were counted separately, and 3 trials were performed for each experiment. Scores recorded were the mean number of flies at the top (ntop), the mean number of flies at the bottom (nbottom) and the total number of flies assessed (ntot). A performance index (PI) defined as ½(ntot + ntop - nbottom)/ ntot) was calculated. Data are presented as the mean PI ± SEM obtained in 3 independent experiments for each group, and analyses of variances (ANOVA) were performed using JMP software.
Quantification of Aβ peptide by ELISA
Quantification of Aβ was carried out as previously described [38]. To extract total Aβ, 5 Drosophila heads were homogenised in 50 μl GnHCl extraction buffer (5 M Guanidine HCl, 50 mM Hepes pH 7.3, protease inhibitor cocktail (Sigma, P8340) and 5 mM EDTA), centrifuged at 21,000 g for 5 min at 4 °C, and cleared supernatant retained as the total fly Aβ sample. Alternatively, for soluble and insoluble pools of Aβ, 25 fly heads were homogenised in 50 μl tissue homogenisation buffer (250 mM sucrose, 20 mM Tris base, 1 mM EDTA, 1 mM EGTA, protease inhibitor cocktail (Sigma) then mixed further with 50 μl diethyl acetate (DEA) buffer (0.4 % DEA, 100 mM NaCl and protease inhibitor cocktail). Samples were centrifuged at 135,000 g for one hour at 4 °C (Beckman OptimaTM Max centrifuge, TLA120.1 rotor), and supernatant retained as the cytosolic, soluble Aβ fraction. Pellets were resuspended in 200 μls ice-cold formic acid (FA; 70 %), and sonicated. Samples were re-centrifuged at 135,000 g for one hour at 4 °C, then 100 μl of supernatant diluted with 1 ml FA neutralisation buffer (1 M Tris base, 0.5 M Na2HPO4, 0.05 % NaN3) and retained as the insoluble, formic acid-extractable Aβ fraction. Aβ content was measured using the hAmyloid β42 ELISA kits, X-42 and N3pE-42 (IBL INTERNATIONAL). N of 3 or 4 individual samples were diluted in sample/standard dilution buffer and ELISA performed according to the manufacturers’ instructions. Protein extracts were quantified using the Bradford protein assay (Bio-Rad protein assay reagent; Bio-Rad laboratories (UK) Ltd) and the amount of Aβ in each sample expressed as a ratio of the total protein content (pmol/g total protein).
Western blotting
For total Aβ extraction, we used a procedure previously described [11]. 20 heads per biological replicate were homogenized in 100 μL of 70 % formic acid. Samples were centrifuged at 16,000 g for 20 min at room temperature. The supernatant was collected and evaporated using a SpeedVac. The pellet was resuspended in 100 μL 2× LDS containing reducing agent (Invitrogen) and homogenized by sonication (10 pulses). Samples were then boiled at 100 °C for 10 min and 15 μL of each sample were used for western blotting to determine total Aβ levels. For LDS/SDS oligomer Aβ extraction, 20 heads per biological replicate were homogenized in 100 μL 2× LDS containing reducing agent (Invitrogen). Samples were incubated on ice for 30 min and then boiled at 100 °C for 10 min. 15 μL per sample were used for western blotting to evaluate LDS/SDS-stable Aβ oligomers. Proteins were separated on 16.5 % Tris-Tricine Criterion gels (Biorad) blotted onto nitrocellulose membranes. Membranes were incubated in a blocking solution containing 5 % milk proteins in TBST for 1 h at room temperature, then probed with primary antibody diluted in TBST + 5 % BSA overnight at 4 °C. 82E1 Aβ1-42 Antibody was from Takara, used at 1 in 100 dilution.
For pJNK western blot analyses, total protein was extracted from 5 fly heads in 30 μl 2 × LDS buffer containing reducing agent (Invitrogen). Membranes were incubated in a blocking solution containing 5 % BSA in TBST for 1 h at room temperature, then probed with primary antibody diluted in TBST + 5 % BSA overnight at 4 °C. mouse monoclonal phospho-SAPK/JNK (T183/Y185) antibody was from cell signaling, used at 1 in 1000 dilution. Rabbit polyclonal actin antibody was from abcam and used at 1 in 1000 dilution.
Quantitative RT-PCR
Total RNA was extracted from 20 to 25 fly heads using TRIzol (GIBCO) according to the manufacturers’ instructions. The concentration of total RNA purified for each sample was measured using an Eppendorf biophotometer. 1 μg of total RNA was then subjected to DNA digestion using DNAse I (Ambion), immediately followed by reverse transcription using the Superscript II system (Invitrogen) with oligo(dT) primers. Quantitative PCR was performed using the PRISM 7000 sequence-detection system (Applied Biosystems), SYBR Green (Molecular Probes), ROX Reference Dye (Invitrogen), and Hot StarTaq (Qiagen, Valencia, CA) by following manufacturers’ instructions. Each sample was analysed at a minimum in triplicate with both target gene (AβQ3-42 or NEP2) and control gene (RP49) primers in parallel. The primers for the Aβ transgenes were directed to the 5’ end and 3’ end of the Aβ coding sequence: forward CGACATGACTCAGGTTATGAAGTT; reverse GACAACGCCCACCAT Neprilysin2 primers are, forward ACGAGGTCAACTGGATGGAC and reverse GTCGAGCTTGGCGTAGTAGG. RP49 primers were as follows: forward ATGACCATCCGCCCAGCATCAGG; reverse ATCTCGCCGCAGTAAACG.
Eye phenotype
Eye images of 2/3-day-old female flies expressing AβpE3-42 under the control of the GMR-Gal4 driver at 28 °C were taken. Nail polish imprint of the external eye was carried out as previously described. For adult eye transverse sections, adult heads were fixed, dehydrated, sectioned (10 microns thick) and stained with Harry’s hematoxylin. To investigate the eye phenotype of double transgenic Aβ1-42; AβQ3-42 flies, we kept the flies at 25 °C to minimize AβpE3-42 eye phenotype. Images were taken with ZEISS Axioskop2 plus microscope. The eye phenotype was quantified by assigning numbers, from zero to 2 to individual flies chosen at random. Normal looking eyes were given zero, flies with moderate eye phenotype were assigned 1, and flies with strong eye phenotype were given 2, (N = 5 − 6 flies per genotype). The scoring was carried out blind by 2 independent researchers.
Statistical analyses
For lifespan analyses, log-rank tests were used to assess for statistical differences. Eye phenotype was presented as means ± SEM, and statistically assessed by Student’s t test. Other data are presented as means ± SEM obtained in at least 3 independent experiments, and differences between means were assessed by either Student’s t test or 2-way analysis of variance (ANOVA) using JMP (version 12.0) software (SAS Institute, Cary, NC, USA).
Results
Pyroglutamate Aβ (AβpE3-42) expression can be induced in the adult Drosophila nervous system
A fly model that expresses AβE3-42 has been described [39], however, we are utilising a previously generated AβQ3-42 transgenic fly model for this study, since glutamine is a better substrate for pyroglutamate conversion than glutamate [14]. AβQ3-42 fly models have been generated but little characterized [18, 36]. To ensure that these flies could express AβQ3-42, we drove expression of the AβQ3-42 transgene in adult neurons with the inducible pan-neuronal driver, elav GeneSwitch (elavGS) [31, 34]. We measured RNA levels of AβQ3-42 flies in adult neurons, after treating elavGS;UAS-AβQ3-42 flies with the activator mifepristone (RU486) for 7 days, starting at 2 days post-eclosion (Fig. 1a). We found a significant increase in AβQ3-42 transcripts in RU486-treated elavGS;UAS-AβQ3-42 flies in comparison to untreated flies (Fig. 1a). Furthermore, we confirmed that the flies generate pyroglutamate-modified Aβ by measuring AβpE3-42 protein levels specifically. AβpE3-42 protein levels in adult neurons of elavGS;UAS-AβQ3-42 flies treated with RU486 for 21 days, starting at 2 days post-eclosion were significantly increased in comparison to untreated flies (Fig. 1b). These data demonstrate that AβpE3-42 can be successfully generated in the flies.
Fig. 1.
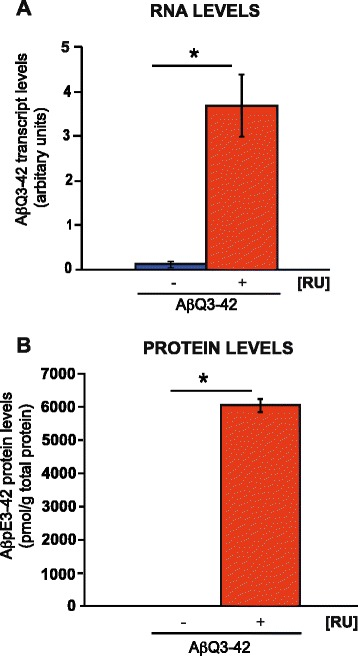
Pyroglutamate Aβ expression in the Drosophila nervous system. a RNA levels were quantified at 7 days post-RU486 treatment and (b) AβpE3-42 protein levels were quantified at 21 days post-RU486 treatment. Data are presented as means ± SEM and were analysed by Student t test, *P < 0.001 comparing Aβ RNA and protein expression in RU486-treated elavGS/UAS-AβQ3-42 flies to their –RU486 controls
Expression of AβpE3-42 causes shortened lifespan, neuronal dysfunction, disorganised eye phenotype, and activates JNK in Drosophila
To determine whether expression of AβpE3-42 in neurons is toxic, we used the elavGS driver to express AβpE3-42 peptide in adult neurons, and measured the effects on behaviour. Impaired geotaxis is a behavioural measure of neuronal dysfunction and can be assessed using a climbing assay [31]. elavGS;UAS-AβQ3-42 flies were treated with RU486 starting at 2 days post-eclosion, and their climbing ability was subsequently recorded. Flies expressing AβpE3-42 displayed substantially increased rate of decline in negative geotaxis with age in comparison to the –RU or driver (elavGS) alone control flies (Fig 2a and Additional file 2: Figure S2).
We also measured the effects of AβpE3-42 on lifespan in comparison to the –RU control flies, by treating elavGS,UAS-AβQ3-42 flies with RU486 starting at 2 days post-eclosion and recording their subsequent survival. Expression of AβpE3-42 in adult neurons significantly shortened median (54 %) and maximum lifespan (31 %) in comparison to control elavGS,UAS-AβQ3-42 -RU flies (Fig. 2b).
To assess the effects of AβpE3-42 on neurodegeneration, we expressed AβpE3-42 constitutively in the fly eye using the GMR-GAL4 driver and observed the effect on the organization of the ommatidia, an assay that has been used extensively to characterise fly models of neurodegenerative diseases [35]. Expression of AβpE3-42 caused disorganisation of the ommatidia in these flies, presenting with eye roughness and fused ommatidia in comparison to flies expressing EGFP, and w1118 control flies (Fig. 2c).
The JNK/SAPK signaling pathway which influences cell death is activated by Aβ, and has been suggested to contribute to Aβ mediated cell death in fly models expressing Aβ1-42 [40, 41]. To determine whether AβpE3-42 expressing flies are also capable of activating JNK, we measured the levels of phosphorylated JNK as a read-out in the flies, by western blot analyses. We found a significant increase in the level of phosphorylated JNK in flies expressing AβpE3-42 (GMR-GAL4/+; AβQ3-42/+) in comparison to control flies expressing LACZ (GMR-GAL4/LACZ), 6 days post-eclosion (Fig. 2d).
Co-expression of neprilysin2 suppresses the toxicity of AβpE3-42 expressing flies
Neprilysin (NEP) and its close homologue Neprilysin2 (Nep2) are candidate Aβ degrading enzymes, and regulate amyloid protein levels in AD [42–44]. We next determined whether the fly orthologue, nep2, which has been shown previously to significantly reduce Aβ1-42 levels and toxicity, is able to similarly ameliorate AβpE3-42 induced toxicity. We made use of the rough eye/disorganised ommatidia phenotype, and found that co-expression of NEP2 using the EP(3)3549 Drosophila strain, with the GMR-GAL4 driver almost completely suppressed both the external disorganised ommatidia and internal retinal degeneration of the AβpE3-42 expressing flies (Fig. 3a). We confirmed that the EP(3)3549 strain had a significant expression of NEP2 levels by RTPCR (Additional file 3: Figure S3A).
Fig. 3.
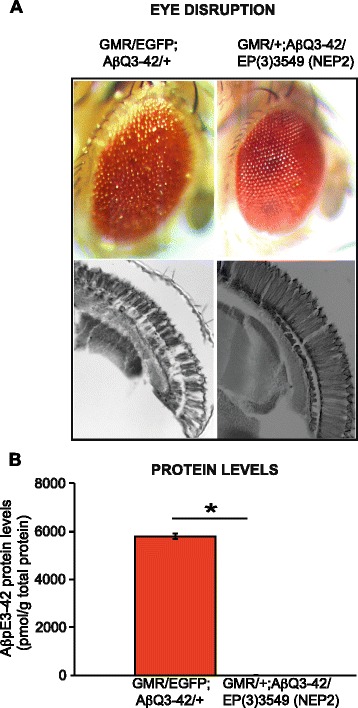
a Co-expression of neprilysin2 suppresses the toxicity of AβpE3-42 expressing flies. Top, light microscopy images and bottom, transverse sections. From left to right, GMR-GAL4/UAS-EGFP;UAS-AβQ3-42/+ and GMR-GAL4/UAS-EGFP;UAS-AβQ3-42/NEP2 (EP(3)3549). Flies were grown at 28 °C. b Neprilysin2 significantly reduces AβpE3-42 protein levels, *P < 0.001. AβpE3-42 levels were quantified by ELISA. Data are presented as means ± SEM and were analysed by Student t test
Co-expression of neprilysin2 reduces AβpE3-42 protein levels
To understand the mechanism by which NEP2 ameliorates the AβpE3-42 eye phenotype, we measured total Aβ load, and AβpE3-42 protein levels specifically in the flies, 7 days post-eclosion. Interestingly, we found by ELISA analyses that Aβ load and importantly, AβpE3-42 levels were significantly reduced in flies co-expressing AβpE3-42 and NEP2 in comparison to flies co-expressing AβpE3-42 and EGFP as a control (Additional file 3: Figure S3B and Fig. 3b). Furthermore, the reduction we see at the protein level is not due to reduced RNA levels, since this reduction was not observed in the RNA by RTPCR analyses, 7 days post eclosion (Additional file 3: Figure S3C). These data demonstrate a major role of NEP2 in ameliorating AβpE3-42 induced toxicity, by reducing AβpE3-42 protein levels.
AβpE3-42 is more toxic than Aβ1-42 in Drosophila
Data from mouse models have indicated that the appearance of AβpE3-42 correlates with increased pathogenicity [45]. To determine whether AβpE3-42 peptide was more toxic in comparison to Aβ1-42, we needed 2 lines with comparable levels of Aβ peptide. We expressed the peptides with the elavGS driver line, by treating AβQ3-42 and Aβ1-42 transgenic flies independently with RU486 starting at 2 days post-eclosion, for 2 days and 21 days, and measured Aβ protein levels in adult neurons, taking advantage of an ELISA kit that recognizes an epitope in the middle of both AβpE3-42 and Aβ1-42 peptides. We found similar levels of total Aβ protein in the AβpE3-42 and Aβ1-42 expressing flies (Fig. 4a and b). However, the solubility/aggregation propensity of Aβ differed between the AβpE3-42 and Aβ1-42 expressing flies, with AβpE3-42 expressing flies having a significantly increased ratio of insoluble to total Aβ in comparison to Aβ1-42 expressing flies (Fig. 4c). AβpE3-42 expressing flies also suffered increased toxicity, because expression of AβpE3-42 in adult neurons significantly shortened median (27 %) and maximum (23 %) lifespan in comparison both to control w1118;;elavGS and Aβ1-42 expressing flies (Fig. 4d), demonstrating a more toxic effect of the AβpE3-42 peptide.
pGluAβ increases accumulation of Aβ in vivo
AβpE3-42 has been shown to increase the amount of metastable low-n Aβ1-42 oligomers in vitro [25]. To study in vivo, the seeding effect of AβpE3-42, we co-expressed AβpE3-42 and Aβ1-42 peptides directly and compared the effects to those seen in flies expressing Aβ1-42. We expressed Aβ1-42 or AβpE3-42 constitutively with the GMR-GAL4 driver line, and measured at 2−3 days post-eclosion total protein levels of flies expressing either peptide. We found a substantial increase in insoluble Aβ levels and a significant decrease in soluble Aβ detected in AβpE3-42 expressing flies in comparison to Aβ1-42 expressing flies with ELISA (Fig. 5a). There was also a significant increase in total Aβ levels in Aβ1-42; AβpE3-42 expressing flies in comparison to Aβ1-42;Aβ1-42 expressing flies (Fig. 5a and Additional file 4: Figure S4). Furthermore, we found a shift in Aβ solubility in Aβ1-42; AβpE3-42 expressing flies, with a significant decrease in soluble Aβ and a substantial increase in insoluble Aβ in comparison to Aβ1-42;Aβ1-42 expressing flies (Fig. 5a).
Fig. 5.
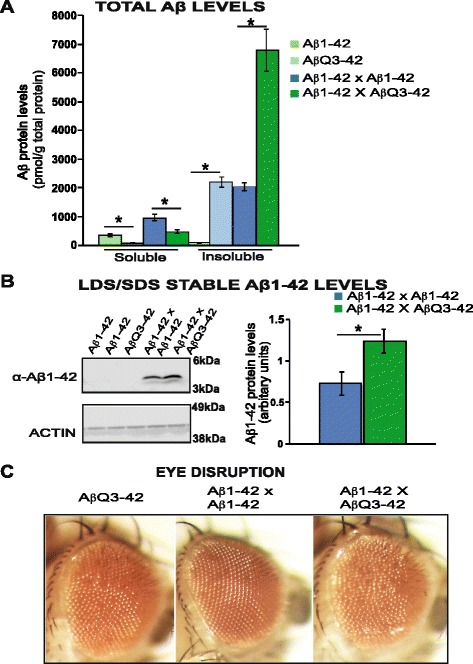
pGluAβ increases accumulation of Aβ in vivo, and exacerbates toxicity (a). Flies expressing Aβ1-42 had significantly more soluble Aβ, P <0.05 but less insoluble Aβ, P <0.0001 in comparison to flies expressing AβpE3-42. Flies co-expressing Aβ1-42 and AβpE3-42 had significantly more insoluble Aβ, P <0.01 but less soluble Aβ P <0.05 Aβ in comparison to flies co-expressing Aβ1-42 and Aβ1-42. Data are presented as means ± SEM and were analysed by Student’s t-test. b There was a significant increase in Aβ1-42 accumulation in flies co-expressing Aβ1-42;AβpE3-42 in comparison to flies expressing either a single copy of Aβ1-42 (expressing one single copy of Aβ1-42 does not accumulate enough protein levels for quantification by western blot analyses), or flies expressing 2 copies of Aβ1-42, P < 0.05. Data are presented as means ± SEM and were analysed by Student’s t test. GMR-GAL4/+;UAS-Aβ1-42/+, GMR-GAL4/+;UAS-AβQ3-42/+, GMR-GAL4/UAS-Aβ1-42;UAS-Aβ1-42/+ and GMR-GAL4/+;UAS-Aβ1-42/UAS-AβQ3-42 flies were used for (a) and (b). c. Co-expression of Aβ1-42 and AβpE3-42 led to a disorganized eye phenotype that was absent in flies co-expressing Aβ1-42 and Aβ1-42, and worse than in AβpE3-42 expressing flies. From left to right, GMR-GAL4/+;UAS-AβQ3-42/+, GMR-GAL4/UAS-Aβ1-42;UAS-Aβ1-42/+ and GMR-GAL4/+;UAS-Aβ1-42/UAS-AβQ3-42 flies. Flies were grown at 25 °C
We next investigated the effect of co-expressing Aβ1-42; AβpE3-42 specifically on Aβ1-42 stability, and whether this contributed to the increased total Aβ levels. We measured Aβ1-42 levels by western blot, by selecting an antibody that detects Aβ1-42, but not AβpE3-42 (Fig. 5b and Additional file 5: Figure S5). Flies co-expressing Aβ1-42; AβpE3-42 had increased levels of Aβ1-42 levels in comparison to flies expressing a single copy of Aβ1-42, which do not accumulate enough protein levels for quantification by western blot analyses (Fig. 5a and Additional file 5: Figure S5). However, Aβ1-42 was specifically detected in flies containing 2 copies of the Aβ1-42 transgene (Aβ1-42;Aβ1-42), confirming protein expression of this line with the antibody (Fig. 5b). Interestingly, the flies co-expressing Aβ1-42;Aβ1-42 significantly expressed lower levels of Aβ1-42 in comparison to flies co-expressing Aβ1-42; AβpE3-42 (Fig. 5b).
pGluAβ enhances Aβ toxicity
Since AβpE3-42 increased the stability of Aβ1-42, we assessed whether it also increased the toxicity of Aβ1-42, using the rough eye/disorganised ommatidia phenotype. Flies co-expressing Aβ1-42; AβpE3-42, but not those expressing Aβ1-42;Aβ1-42 presented with disorganised ommatidia, 2 days post eclosion, and this phenotype was stronger than in flies expressing AβQ3-42 alone (Fig. 5c and Additional file 6: Figure S6) suggesting that AβpE3-42 enhances the toxicity of Aβ1-42.
Collectively, these data suggest that AβpE3-42 is able to increase the stability of the Aβ1-42 peptide and exacerbate its toxicity in vivo.
Discussion
AβpE3-42 is increasingly thought to play a pivotal role in the pathogenesis of Alzheimer’s disease [14]. Although previous studies in vitro have suggested that AβpE3-42 acts as a seed for Aβ stability, and some correlative work has been done in vivo [25], this seeding behaviour and its consequences have not been examined in vivo. Our study demonstrates that AβpE3-42 increases the levels of Aβ1-42, presumably by increasing its stability, and that it enhances toxicity of the Aβ1-42 peptide, as observed in flies co-expressing AβpE3-42 and Aβ1-42 in comparison to flies expressing 2 copies of Aβ1-42.
First, we characterized the model. Expression of the AβpE3-42 peptide specifically in adult fly neurons led to behavioural dysfunction and shortened lifespan, and constitutive expression in the eyes led to disorganised ommatida. Furthermore, we found that AβpE3-42 was able to activate the JNK signaling pathway, suggesting a role for this cell death activating pathway in AβpE3-42 mediated toxicity.
Interestingly, we found that we could ameliorate the AβpE3-42 toxicity by over expressing Neprilsyin2. Increasing the expression of several candidate in vivo Aβ degrading enzymes, such as NEP or Insulin degrading enzyme (IDE) have been shown to reduce the cerebral amyloid plaque burden observed in APP over-expressing mice [46]. However, direct interactions between AβpE3-42 and Neprilysin have not been investigated. We found that over-expression of Drosophila NEP2 was able to reduce AβpE3-42 levels and improve considerably the disorganised ommatidia, demonstrating for the first time that, although AβpE3-42 may aggregate more than Aβ1-42, NEP2 is capable of degrading pyroglutamate-modified Aβ.
Also, we found that these flies had an increase in the ratio of insoluble to total Aβ levels in comparison to flies expressing Aβ1-42, confirming the propensity of AβpE3-42 to aggregate as previously described [14]. Furthermore, we found that AβpE3-42 was more toxic than Aβ1-42 peptide.
Another interesting finding from our analyses is the increase in Aβ levels observed in response to AβpE3-42. We found a general increase in total Aβ levels in flies co-expressing AβpE3-42 and Aβ1-42, and there was also an increase in the ratio of insoluble Aβ to total Aβ levels in these flies. To determine what role Aβ1-42 might have in this, we measured Aβ1-42 levels specifically by western blot analyses. Interestingly, we found that total Aβ1-42 levels were increased when AβpE3-42 was co-expressed. Nussbaum et al. showed that Aβ peptides oligomerise by different pathways, and that the low-n oligomers of AβpE3-42 are structurally distinct from Aβ1-42, and far more cytotoxic, in the order of AβpE3-42 /Aβ1-42 > AβpE3-42 > Aβ1-42 [25]. We found that co-expressing 1 copy each of Aβ1-42 and AβpE3-42 increased the accumulation of Aβ1-42, and was more toxic than expressing 2 copies of Aβ1-42, indicating that AβpE3-42 is able to stabilise Aβ1-42 in a different manner to over expressing the Aβ1-42 peptide, perhaps by affecting its structure.
Conclusions
We have tested and validated the AβpE3-42 seeding hypothesis and shown that indeed AβpE3-42 increases the levels of Aβ, and that AβpE3-42 enhances pathology in this AD model. These results raise AβpE3-42 as both a potential biomarker and new therapeutic target in AD. Furthermore, because Drosophila does not inherently express Aβ, the observation that expression of AβpE3-42 is able to cause toxicity independent of its effect on Aβ1-42 suggests that it is capable of initiating toxicity in other ways. It would be interesting to uncover downstream pathways that are modulated specifically by AβpE3-42, and the fly provides a powerful context for pursuing this question with genetic screens.
Acknowledgements
We thank Dr D Crowther and Dr H Tricoire for their kind donation of UAS-Aβ42 and elavGS fly stocks respectively. We thank Dr Kerr and Dr Woodling for help with blind scoring of eye data.
Funding
This work was supported by grants from the Max Planck Institute for the Biology of Ageing (MK, IS, LP), Alzheimer’s Society (OS-A) and the Wellcome Trust (LP).
Availability of supporting data
Supporting data have been included in the publication as supplementary figures.
Authors’ contributions
OS-A conceived experiments, performed experiments, analysed the data and wrote the manuscript. MK and IS performed experiments, and analyzed the data. LT generated UAS-AβQ3-42 transgenic flies. LP contributed to supervision of the project and writing of the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interest.
Consent for publication
All authors have given consent for publication.
Ethical approval and consent to participate
Not applicable.
Authors’ information
Not applicable.
Abbreviations
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- Aβ
Amyloid beta
- elavGS
elav GeneSwitch
- ER
Endoplasmic reticulum
- IDE
Insulin degrading enzyme
- JNK/SAPK
c-Jun N-terminal kinase/stress-activated protein kinase
- Nep2
Neprilysin2
- pGlu
Pyroglutamic
- QC
glutaminyl cyclase
- RU486, RU
mifepristone
Additional files
Generation of AβpE3-42. The proenkephalin signaling peptide upstream of AβQ3-42 is cleaved by prohormone convertases, AβQ3-42 is then released, and glutaminyl cyclase catalyses its conversion to AβpE3-42. (PDF 271 kb)
Expression of AβpE3-42 causes locomotor dysfunction. Climbing ability of elavGS/UAS-AβQ3-42and elavGS flies on + RU486 SY medium was assessed at the indicated time-points (see Materials & Methods). Expression of AβpE3-42 in adult neurons reduced climbing ability of the flies in comparison to control elavGS driver flies. Data are presented as the average performance index (PI) ± SEM and were compared using 2-way ANOVA (number of independent tests (n) = 3 P < 0.01. (PDF 306 kb)
(A). Confirming the expression of Neprilysin 2 in the EP(3)3549 fly strain. There was a significant increase in nep2 transcript levels in the flies expressing the EP(3)3549 EP element, in comparison to the control fly lines expressing LACZ. Data are presented as means ± SEM and were analysed by student’s t-test, P < 0.001. (B) Neprilysin2 significantly reduces Aβ protein levels, *P < 0.001. AβX-42 levels were quantified by ELISA. Data are presented as means ± SEM and were analysed by Student t test. (C) Neprilysin2 does not reduce AβQ3-42 RNA levels. There was a significant increase in AβQ3-42 RNA levels in flies co-expressing AβQ3-42 and NEP2 in comparison to flies expressing AβQ3-42 and EGFP, by quantitative RTPCR, P < 0.01, student’s t-test. GMR-GAL4 was used to drive expression of AβQ3-42 transgenic flies. (PDF 369 kb)
pGluAβ increases accumulation of Aβ in vivo. Flies co-expressing Aβ1-42 and AβpE3-42 peptide, had significantly higher Aβ levels than flies co-expressing Aβ1-42 and Aβ1-42. Data are presented as means ± SEM and were analysed by student’s t-test, P < 0.01. GMR-GAL4 was used to drive expression of Aβ1-42 and AβQ3-42 transgenic flies. (PDF 417 kb)
pGluAβ increases accumulation of Aβ1-42 in vivo. (A). Flies co-expressing Aβ1-42 and AβpE3-42 peptide, had substantially more Aβ1-42 levels than flies expressing Aβ1-42 alone. Aβ was not detected in flies expressing a single copy of Aβ1-42 because it does not accumulate enough protein. However, Aβ1-42 was detected in flies expressing 2 copies of Aβ1-42 (B), confirming protein expression in these flies with the antibody. Furthermore, to validate specificity, Aβ1-42 was not detected in flies expressing either a single copy or double copy of AβpE3-42. GMR-GAL4 was used to drive expression of Aβ1-42 and AβQ3-42 transgenic flies. (PDF 10727 kb)
Blind scoring of disorganised eye phenotype. The data demonstrate a significant difference in the degree of severity of eye roughness in flies co-expressing Aβ1-42 and AβpE3-42 in comparison to flies expressing either 2 copies of Aβ1-42 (A), or AβpE3-42 only (B), *P < 0.001 for both. Flies were grown at 25 °C. Data are presented as means ± SEM and were analysed by Student’s t test. GMR-GAL4 was used to drive expression of Aβ1-42 and AβQ3-42 transgenic flies. (PDF 303 kb)
Contributor Information
Oyinkan Sofola-Adesakin, Email: o.sofola@ucl.ac.uk.
Mobina Khericha, Email: m.khericha@ucl.ac.uk.
Inge Snoeren, Email: i.snoeren@ucl.ac.uk.
Leo Tsuda, Email: ltsuda@ncgg.go.jp.
Linda Partridge, Email: l.partridge@ucl.ac.uk.
References
- 1.Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 3.Wirths O, Multhaup G, Bayer TA. A modified beta-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide--the first step of a fatal cascade. J Neurochem. 2004;91:513–520. doi: 10.1111/j.1471-4159.2004.02737.x. [DOI] [PubMed] [Google Scholar]
- 4.Mao P, Reddy PH. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: implications for early intervention and therapeutics. Biochim Biophys Acta. 1812;2011:1359–1370. doi: 10.1016/j.bbadis.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu X, Raina AK, Rottkamp CA, Aliev G, Perry G, Boux H, et al. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J Neurochem. 2001;76:435–441. doi: 10.1046/j.1471-4159.2001.00046.x. [DOI] [PubMed] [Google Scholar]
- 6.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 7.Yarza R, Vela S, Solas M, Ramirez MJ. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front Pharmacol. 2015;6:321. doi: 10.3389/fphar.2015.00321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selkoe DJ, Schenk D. Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- 10.Saito T, Suemoto T, Brouwers N, Sleegers K, Funamoto S, Mihira N, et al. Potent amyloidogenicity and pathogenicity of Aβ43. Nat Neurosci. 2011;14:1023–1032. doi: 10.1038/nn.2858. [DOI] [PubMed] [Google Scholar]
- 11.Burnouf S, Gorsky MK, Dols J, Grönke S, Partridge L. Aβ43 is neurotoxic and primes aggregation of Aβ40 in vivo. Acta Neuropathol. 2015;130:35–47. doi: 10.1007/s00401-015-1419-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mori H, Takio K, Ogawara M, Selkoel DJ. Mass spectrometry of purified amyloid beta protein in Alzheimer’s disease. J Biol Chem. 1992;267:17082–17086. [PubMed] [Google Scholar]
- 13.Saido TC, Yamao-Harigaya W, Iwatsubo T, Kawashima S. Amino- and carboxyl-terminal heterogeneity of beta-amyloid peptides deposited in human brain. Neurosci Lett. 1996;215:173–176. doi: 10.1016/0304-3940(96)12970-0. [DOI] [PubMed] [Google Scholar]
- 14.Jawhar S, Wirths O, Bayer TA. Pyroglutamate amyloid-β (Aβ): a hatchet man in Alzheimer disease. J Biol Chem. 2011;286:38825–38832. doi: 10.1074/jbc.R111.288308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schilling S, Hoffmann T, Manhart S, Hoffmann M, Demuth H-U. Glutaminyl cyclases unfold glutamyl cyclase activity under mild acid conditions. FEBS Lett. 2004;563:191–196. doi: 10.1016/S0014-5793(04)00300-X. [DOI] [PubMed] [Google Scholar]
- 16.Cynis H, Schilling S, Bodnár M, Hoffmann T, Heiser U, Saido TC, et al. Inhibition of glutaminyl cyclase alters pyroglutamate formation in mammalian cells. Biochim Biophys Acta. 2006;1764:1618–1625. doi: 10.1016/j.bbapap.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 17.Schilling S, Appl T, Hoffmann T, Cynis H, Schulz K, Jagla W, et al. Inhibition of glutaminyl cyclase prevents pGlu-Abeta formation after intracortical/hippocampal microinjection in vivo/in situ. J Neurochem. 2008;106:1225–1236. doi: 10.1111/j.1471-4159.2008.05471.x. [DOI] [PubMed] [Google Scholar]
- 18.Schilling S, Zeitschel U, Hoffmann T, Heiser U, Francke M, Kehlen A, et al. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer’s disease-like pathology. Nat Med. 2008;14:1106–1111. doi: 10.1038/nm.1872. [DOI] [PubMed] [Google Scholar]
- 19.Russo C, Saido TC, DeBusk LM, Tabaton M, Gambetti P, Teller JK. Heterogeneity of water-soluble amyloid β-peptide in Alzheimer’s disease and Down’s syndrome brains. FEBS Lett. 1997;409:411–416. doi: 10.1016/S0014-5793(97)00564-4. [DOI] [PubMed] [Google Scholar]
- 20.Perez-Garmendia R, Gevorkian G. Pyroglutamate-Modified Amyloid Beta Peptides: Emerging Targets for Alzheimer’s Disease Immunotherapy. Curr Neuropharmacol. 2013;11:491–498. doi: 10.2174/1570159X11311050004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S. Dominant and differential deposition of distinct β-amyloid peptide species, AβN3(pE), in senile plaques. Neuron. 1995;14:457–466. doi: 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- 22.Sergeant N, Bombois S, Ghestem A, Drobecq H, Kostanjevecki V, Missiaen C, et al. Truncated beta-amyloid peptide species in pre-clinical Alzheimer’s disease as new targets for the vaccination approach. J Neurochem. 2003;85:1581–1591. doi: 10.1046/j.1471-4159.2003.01818.x. [DOI] [PubMed] [Google Scholar]
- 23.Iwatsubo T, Saido TC, Mann DM, Lee VM, Trojanowski JQ. Full-length amyloid-beta (1-42(43)) and amino-terminally modified and truncated amyloid-beta 42(43) deposit in diffuse plaques. Am J Pathol. 1996;149:1823–1830. [PMC free article] [PubMed] [Google Scholar]
- 24.He W, Barrow CJ. The Aß 3-pyroglutamyl and 11-pyroglutamyl peptides found in senile plaque have greater ß-sheet forming and aggregation propensities in vitro than full-length Aß. Biochemistry. 1999;38:10871–10877. doi: 10.1021/bi990563r. [DOI] [PubMed] [Google Scholar]
- 25.Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature. 2012;485:651–655. doi: 10.1038/nature11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wirths O, Erck C, Martens H, Harmeier A, Geumann C, Jawhar S, et al. Identification of Low Molecular Weight Pyroglutamate Aβ Oligomers in Alzheimer Disease. J Biol Chem. 2010;285:41517–41524. doi: 10.1074/jbc.M110.178707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wirths O, Breyhan H, Cynis H, Schilling S, Demuth H-U, Bayer TA. Intraneuronal pyroglutamate-Abeta 3-42 triggers neurodegeneration and lethal neurological deficits in a transgenic mouse model. Acta Neuropathol. 2009;118:487–496. doi: 10.1007/s00401-009-0557-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexandru A, Jagla W, Graubner S, Becker A, Bauscher C, Kohlmann S, et al. Selective hippocampal neurodegeneration in transgenic mice expressing small amounts of truncated Abeta is induced by pyroglutamate-Abeta formation. J Neurosci. 2011;31:12790–12801. doi: 10.1523/JNEUROSCI.1794-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jawhar S, Wirths O, Schilling S, Graubner S, Demuth H-U, Bayer TA. Overexpression of glutaminyl cyclase, the enzyme responsible for pyroglutamate A{beta} formation, induces behavioral deficits, and glutaminyl cyclase knock-out rescues the behavioral phenotype in 5XFAD mice. J Biol Chem. 2011;286:4454–4460. doi: 10.1074/jbc.M110.185819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schilling S, Lindner C, Koch B, Wermann M, Rahfeld J-U, von Bohlen A, et al. Isolation and characterization of glutaminyl cyclases from Drosophila: evidence for enzyme forms with different subcellular localization. Biochemistry. 2007;46:10921–10930. doi: 10.1021/bi701043x. [DOI] [PubMed] [Google Scholar]
- 31.Sofola O, Kerr F, Rogers I, Killick R, Augustin H, Gandy C, et al. Inhibition of GSK-3 ameliorates Abeta pathology in an adult-onset Drosophila model of Alzheimer’s disease. PLoS Genet. 2010;6:e1001087. doi: 10.1371/journal.pgen.1001087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crowther D, Page R, Chandraratna D, Lomas D. A Drosophila model of Alzheimer’s disease. Methods Enzymol. 2006;412:234–255. doi: 10.1016/S0076-6879(06)12015-7. [DOI] [PubMed] [Google Scholar]
- 33.Iijima K, Liu H-P, Chiang A-S, Hearn SA, Konsolaki M, Zhong Y. Dissecting the pathological effects of human Aβ40 and Aβ42 in Drosophila: A potential model for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:6623–6628. doi: 10.1073/pnas.0400895101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Osterwalder T, Yoon KS, White BH, Keshishian H. A conditional tissue-specific transgene expression system using inducible GAL4. Proc Natl Acad Sci U S A. 2001;98:12596–12601. doi: 10.1073/pnas.221303298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie FAI, et al. Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience. 2005;132:123–135. doi: 10.1016/j.neuroscience.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 36.Omata Y, Lim Y-M, Akao Y, Tsuda L. Age-induced reduction of autophagy-related gene expression is associated with onset of Alzheimer’s disease. Am J Neurodegener Dis. 2014;3:134–142. [PMC free article] [PubMed] [Google Scholar]
- 37.Johanning K, Juliano MA, Juliano L, Lazure C, Lamango NS, Steiner DF, et al. Specificity of prohormone convertase 2 on proenkephalin and proenkephalin-related substrates. J Biol Chem. 1998;273:22672–22680. doi: 10.1074/jbc.273.35.22672. [DOI] [PubMed] [Google Scholar]
- 38.Rogers I, Kerr F, Martinez P, Hardy J, Lovestone S, Partridge L. Ageing increases vulnerability to aβ42 toxicity in Drosophila. PLoS ONE. 2012;7:e40569. doi: 10.1371/journal.pone.0040569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jonson M, Pokrzywa M, Starkenberg A, Hammarstrom P, Thor S. Systematic Aβ Analysis in Drosophila Reveals High Toxicity for the 1-42, 3-42 and 11-42 Peptides, and Emphasizes N- and C-Terminal Residues. PLoS ONE. 2015;10:e0133272. doi: 10.1371/journal.pone.0133272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tare M, Modi RM, Nainaparampil JJ, Puli OR, Bedi S, Fernandez-Funez P, et al. Activation of JNK signaling mediates amyloid-ß-dependent cell death. PLoS ONE. 2011;6:e24361. doi: 10.1371/journal.pone.0024361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hong YK, Lee S, Park SH, Lee JH, Han SY, Kim ST, et al. Inhibition of JNK/dFOXO pathway and caspases rescues neurological impairments in Drosophila Alzheimer’s disease model. Biochem Biophys Res Commun. 2012;419:49–53. doi: 10.1016/j.bbrc.2012.01.122. [DOI] [PubMed] [Google Scholar]
- 42.Finelli A, Kelkar A, Song HJ, Yang H, Konsolaki M. A model for studying Alzheimer’s Aβ42-induced toxicity in Drosophila melanogaster. Mol Cell Neurosci. 2004;26:365–375. doi: 10.1016/j.mcn.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 43.Marr RA, Guan H, Rockenstein E, Kindy M, Gage FH, Verma I, et al. Neprilysin regulates amyloid Beta peptide levels. J Mol Neurosci. 2004;22:5–11. doi: 10.1385/JMN:22:1-2:5. [DOI] [PubMed] [Google Scholar]
- 44.Huang JY, Hafez DM, James BD, Bennett DA, Marr RA. Altered NEP2 expression and activity in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis. 2012;28:433–441. doi: 10.3233/JAD-2011-111307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wirths O, Bethge T, Marcello A, Harmeier A, Jawhar S, Lucassen PJ, et al. Pyroglutamate Abeta pathology in APP/PS1KI mice, sporadic and familial Alzheimer’s disease cases. J Neural Transm. 2010;117:85–96. doi: 10.1007/s00702-009-0314-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, et al. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–1093. doi: 10.1016/S0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]