

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal cancers. Cyclopamine (CPA), a potent inhibitor for sonic hedgehog pathway (SHH), shows great promises in PDAC treatment, including the disruption of tumor-associated stroma, and enhancement of radiation therapy. However, CPA is insoluble in water; therefore requires a nanometric delivery platform to achieve satisfactory performance. We herein encapsulated CPA in a core-crosslinked polymeric micelle system (M-CPA). M-CPA was combined with Cs-137 radiation and evaluated in vitro in PDAC cell lines and a human pancreatic stellate cell line. The results showed M-CPA had higher cytotoxicity than CPA, abolished Gli-1 expression (a key component of SHH), and enhanced the radiation therapy of Cs-137. M-CPA radio-sensitization correlated with its ability to disrupt the repair of radiation-induced DNA damage. These findings indicate that the combination therapy of M-CPA and radiation is an effective strategy to simultaneously treat pancreatic tumors and tumor-associated stroma.
Keywords: Cyclopamine, radiation sensitization, pancreatic cancer, stroma disruption, DNA damage repair
Graphical abstract
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth-leading cause of cancer-related death in the United States, with an overall 5-year survival rate below 5% 1. About 40% of pancreatic cancer patients have locally advanced disease that cannot be surgically resected 2. This cohort of patients is usually treated with concurrent chemoradiation therapy (CCRT) 3. Still, the majority of patients die of local or distant tumor recurrence within several years following CCRT. Therefore, there is an urgent need for more effective adjuvant therapies to further improve the efficacy of radiation therapy. On the other hand, PDAC is accompanied by abundant desmoplastic stroma, an excessive fibrotic deposition produced by pancreatic stellate cells. Stromal tissue presents a unique challenge to PDAC therapy by protecting tumor cells against radiation or chemotherapy, promoting tumor progression, and inducing metastasis. Importantly, the stroma tissue creates a hypoxic tumor microenvironment that could negate the efficacy of radiation therapy 4. Therefore, disruption of stromal tissue is a critical step in treating PDAC 5.
The sonic hedgehog (SHH) pathway is a key pathway active in PDAC progression. SHH regulates tissue development, differentiation, and homeostasis in normal tissues 6. While mostly quiescent in the normal pancreas, SHH pathway is aberrantly activated in PDAC primary tumors or precursor lesions. In a typical scenario of activation, Shh ligands bind to Patched (PTCH) and release its inhibition of Smoothened (SMO). SMO then activates the Gli-family of transcription factors and their downstream targets. In PDAC and other malignancies, the SHH pathway has been shown to function in a paracrine manner 7. Specifically, the pancreatic stellate cells (PSCs) in the tumor associated stroma of PDAC have been shown to express the SMO receptor that becomes activated by hedgehog ligands produced by the PDAC cells 7b. Over-expression of factors in the SHH signaling pathway such as PTCH1, SMO, and Gli-1 are related to poor prognosis of PDAC 8. Blockage of SHH not only suppressed the proliferation of tumor cells but also disrupted stroma and facilitated the delivery of other chemotherapy drugs 6, 9.
Cyclopamine (CPA) is a potent SMO inhibitor with promising therapeutic effects against PDAC, such as depleting cancer stem cells, preventing metastasis, and disrupting stroma 10. In particular, CPA enhances the response of PDAC to ionizing radiation (IR) 10b. However, CPA is insoluble in water and has high systemic toxicity; therefore, it cannot be used directly in humans. The bioavailability of CPA can be improved using nanometric drug delivery systems 11. Polymeric micelles are attractive candidates for drug delivery 12. They can prolong the blood circulation of an encapsulated drug, minimize drug degradation, prevent undesirable drug deposition in healthy organs, and increase drug concentrations in the tumor region. Once tagged with active targeting moieties, polymeric micelles can preferentially accumulate in tumors and thus further improve the efficacy of drug delivery.
In the present study, we developed a core-crosslinked polymeric micelle system as a drug-delivery platform for CPA. This micellar formulation (M-CPA) was combined with ionizing radiation (IR) and tested against pancreatic cancer cell lines as well as a pancreatic stellate cell line. We hypothesized that M-CPA could enhance radiation response in SMO-expressing pancreatic cancer cells and pancreatic stellate cells.
2. Experimental Sections
2.1. Synthesis of Block Copolymer Precursor
This procedure is described in detail in the supplementary materials section. As shown in Fig. 1, the first step was to synthesize poly[oligo(ethylene glycol) monomethyl ether methacrylate)31-b-poly(2-hydroxyethyl methacrylate)26 (compound 2), abbreviated as P(OEGMA)31-b-P(HEMA)26, using atom transfer radical polymerization. Next, a short chain of hydrophobic oligo(ε-caprolactone) was grafted onto the P(HEMA) block, using controlled ring-opening polymerization, to make the amphiphilic polymer (compound 3) abbreviated as P(OEGMA)31-b-P(HEMA-CL5)26. Triethoxysilane function groups were then tethered to the hydrophobic block as crosslinkers to yield compound 4, abbreviated as P(OEGMA)31-b-P(HEMA-CL5-Si)26. The structures of the polymers were monitored using proton nuclear magnetic resonance spectroscopy (1H-NMR) (Bruker Avance 600 spectrometer, Billerica, MA).
Figure 1.
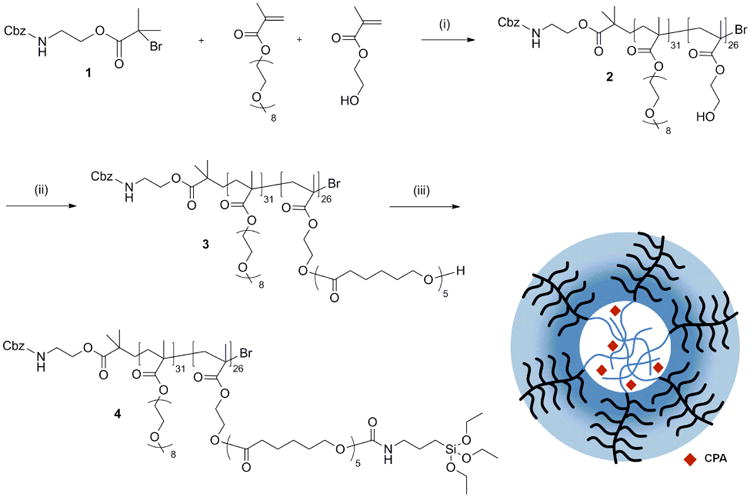
Synthesis of polymer precursor for the core-crosslinked polymeric micelles: (1) 2-(benzyloxycarbonyl amino)ethyl 2-bromo-isobutyrate; (2) P(OEGMA)31-b-P(HEMA)26; (3) P(OEGMA)31-b-P(HEMA-CL5)26; and (4) P(OEGMA)31-b-P(HEMA-CL5-Si)26. Reaction conditions: (i) cuprous bromide, bipyridine, 50 °C in methanol; (ii) tin(II) 2-ethylhexanoate, 120 °C in toluene; (iii) 3-isocyanatopropyl triethoxysilane, dibutyltin dilaurate, 40 °C in toluene. CPA: cyclopamine.
2.2. Formation and Characterization of CPA-loaded Micelles (M-CPA)
CPA (6.0 mg) and copolymer 4 (200 mg) were dissolved in 1 ml tetrahydrofuran. Double-distilled water (2 ml) was then added to the solution under vigorous vortexing, followed by dialysis against double-distilled water at 5°C for 24 hours. The mixture was passed through a 0.22-μm filter to remove un-encapsulated CPA. Drug-release
The size and size distribution of M-CPA were measured by dynamic light scattering (Brookhaven 90Plus, Holtsville, NY). To determine CPA loading, micelles were first dissolved in ethanol to release encapsulated CPA. The ethanol solution was analyzed using an Agilent 1100 Series high-performance liquid chromatography system (Santa Clara, CA). The mobile phase was a mixture of 0.1% trifluoroacetic acid in water (phase A) and acetonitrile (phase B), running over 30 minutes in gradient from 5%B to 95%B (v/v). The column was an Agilent C18 (4.6 × 250 mm) with 5-μm particles. The flow rate was 1.0 ml/min, and the detection wavelength was 210 nm.
For drug releasing profile, micelles were added to a micro-dialyzer (MWCO ∼ 3000), and incubated in PBS (pH7.4) or sodium acetate buffer (pH5.2) at 37 °C with agitation. At pre-determined time points, 20 μL of the solution inside dialyzer was retrieved and centrifuged at 5,000 RPM for 5 min. 10 μL of the supernatant was analyzed using HPLC for the concentrations of CPA.
2.3. Cell Lines
We selected pancreatic cancer cell lines with variant sensitivities to CPA according to previous reports 13. Two pancreatic cancer cell lines were obtained from ATCC: Miapaca-2 (CPA-sensitive), L3.6pl (moderate CPA-sensitive) 13 and Panc-1 (CPA-resistant) 10a. An SMO-expressing human pancreatic stellate cell line (HPSC) was kindly provided by Dr. Rosa F. Hwang (MD Anderson Cancer Center).
2.4. Cytotoxicity
Cytotoxicity was evaluated with a (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) assay. Cells were treated with M-CPA (dispersed in water) or free CPA (dissolved in DMSO) for 96 hrs before test. Cell viability was normalized to un-treated cells. For the combination therapy, cells were treated with M-CPA for 1 hr and exposed to pre-determined doses of IR using a MARK I Cesium-137 Irradiator (JL Shepherd & Associates, San Fernando, CA) at 309 cGy/min. MTS assay was performed 96 hrs later.
2.5. Potential Lethal Damage Repair (PLDR)
The PLDR assay was performed according to previous reports 14. Cells were grown to full confluency, irradiated, and plated following three schedules: (1) cells were plated right away without recovery; (2) cells were further cultured in growth medium at 37 °C for 24 hrs; and (3) cells were further incubated in the growth medium containing 10 μM M-CPA (in equivalent CPA concentration) at 37°C for 24 hrs. Cells were then re-plated in single-cell suspensions for colony formation. Such delayed plating was used to control the exposure of cells to M-CPA treatment, and to give cells sufficient time for repair of DNA damages. Colony survival curves were fitted using the linear-quadratic equation; mean inactivation dose was calculated according to previous reports 15. The formula used for calculating PLDR is given in Supplemental Materials.
2.6. Detection of DNA Damage by Comet Assay and 53BP1 staining
DNA single-strand breaks were analyzed using the single-cell gel electrophoresis assay (comet assay) under alkali lysing conditions (17). DNA double-strand breaks were analyzed using 53BP1 staining assay using rabbit-anti-human 53BP1 antibody and visualized using anti-rabbit IgG (H+L), F(ab')2 Fragment (Alexa Fluor® 488 Conjugate) (Cell Signaling, Beverly, MA). Fluorescence microscopic images were acquired using a Zeiss Axio Observer.Z1 fluorescence microscope (Carl Zeiss MicroImaging GmbH, Thornwood, NY).
2.7. Western Blot Analysis
After treatment, cell lysate was fractioned and transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, MA). Membranes were blotted with rabbit-anti-human monoclonal Gli-1 antibody (ab134906, Abcam Inc., Cambridge, MA, 1:1000 dilution) and visualized using fluorescent IRDye 680RD goat anti-rabbit IgG (H+L) (LI-COR, Lincoln, NE 1:5000 dilution). β-Actin (Sigma-Aldrich) was used as an internal control to ensure equal protein loading.
2.8. Statistical analysis
All values are expressed as mean ± standard error of mean (SEM). Either Student's t test or a one-way analysis of variance (ANOVA) was used to evaluate the data. A p-value less than 0.05 was considered to be statistically significant.
3. Results
3.1. Synthesis and Characterization of Polymers and M-CPA
Block copolymers were prepared using atom transfer radical polymerization (ATRP), followed by controlled ring-opening polymerization. The ATRP initiator, 2-(benzyloxycarbonyl amino)ethyl 2-bromo-isobutyrate, abbreviated as Z-BIBB, was first synthesized (1H-NMR spectrum shown in Fig. S1A). The peaks around 7.3 ppm (benzene H of Z-BIBB) were used as internal standards to calculate polymer molecular weight. P(OEGMA) and P(HEMA) blocks were sequentially built using ATRP (2, Fig. S1B). The peak at 3.25 ppm was assigned to -OCH3 at the end of the OEG chain; the peak at 4.8 ppm was assigned to -OH of HEMA. By comparing peak integrations, we concluded that polymer 2 had 31 units of OEGMA and 26 units of HEMA. The -OH groups of HEMA were then extended with short oligo(ε-caprolactone) (oligo-CL) side chains (3, Fig. S1C). The average number of CL units on each side chain was calculated from Fig. S1C by comparing the peak at 3.25 ppm (-OCH3 of OEG) with the peak at 1.54 ppm (β and δ -CH2- of the CL unit). There were about 5 units of CL per side chain. The oligo-CL chains were then tethered with triethoxysilanes (4, Fig. S1D) that could crosslink the micelle core in an aqueous medium. The number of grafted Si(OEt)3 units was determined by integrating the peak at 0.5 ppm (-CH2-Si) (Fig. S1D). Each copolymer had 23 units of Si(OEt)3. CPA was then encapsulated in micelles at 1.4% by weight. Micelles were spontaneously crosslinked through the hydrolysis of triethoxysilane groups in aqueous solution. The formed micelles (M-CPA) were 69.5 ± 6.4 nm in diameter. The release of CPA from micelles was faster at pH 5.2 than at pH 7.4 (Fig. 2A). After 24 hr of incubation at 37 °C, 21.4 ± 3.4 % of CPA was released at pH 7.4; while 77.2 ± 0.7% was released at pH 5.2.
Figure 2.
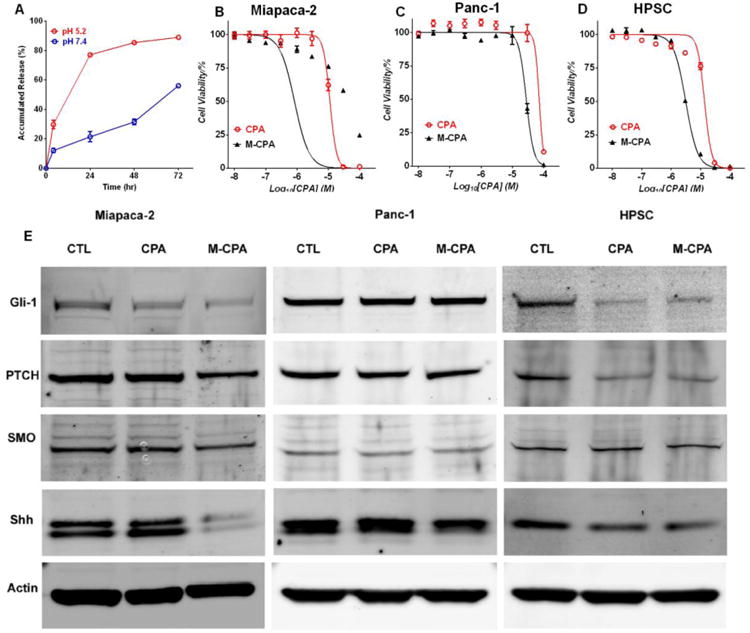
Effects of CPA and M-CPA on cell viability and SHH pathway in Miapaca-2, Panc-1 and HPSC cells. (A) CPA release profiles at 37 °C in pH7.4 and pH 5.2 buffers. (B-D) Cell viability after treatment with free CPA and M-CPA. Data points are presented as mean ± standard error of mean (N = 6) (E) Expression of SHH pathway proteins in Miapaca-2, Panc-1 and HPSC cells after treatment with CPA and M-CPA (both at 10 M CPA equivalent concentration). 30 μg protein was loaded into each lane for immunoblotting.
3.2. M-CPA Had Higher Cytotoxicity than CPA, Disrupted SHH Pathway in CPA-Sensitive Cell Lines
M-CPA was more potent than free CPA to Miapaca-2, Panc-1, and HPSC cells (Fig. 2B-C). The IC50 values of M-CPA were 0.8, 29.0, and 3.1 μM, while those of free CPA were 11.3, 72.1, and 13.7 μM against the Miapaca-2, Panc-1, and HPSC cells, respectively, representing 14.1-, 2.5-, and 4.4-fold decrease in IC50 values, respectively (p < 0.05), with M-CPA. Similarly, the IC50 values of M-CPA and CPA against L3.6pl cells were 6.0 μM and 21.6 μM, respectively (Fig. S2). L3.6pl cells were moderately sensitive to CPA treatment. In order to exclude the effect of blank micelles, the same cytotoxicity assay was performed on Miapaca-2 using both blank micelles and M-CPA. The IC50 value for blank micelles was at least 2-orders of magnitude higher than that of M-CPA, indicating that the blank micelles contributed minimal effect to the cytotoxicity of M-CPA (Fig. S3).
To evaluate the treatment response in SHH pathway, immunoblotting was performed to analyze the expression of Gli-1, PTCH, SMO, and Shh in the three cell lines (Fig. 2E). In Miapaca-2 and HPSC cells, CPA or M-CPA treatment reduced the expression of Gli-1, PTCH, and Shh; while M-CPA was more potent than CPA in Miapaca-2 cells. Neither CPA nor M-CPA decreased the expression of Gli-1 in Panc-1 cells. We also compared the effects of CPA and M-CPA (10 μM for 48 hrs) on cell apoptosis and cell cycles. M-CPA induced more cell apoptosis and cell death than CPA did in Miapaca-2, L3.6pl and HPSC cells but not in Panc-1 cells (Fig. S4). Both CPA and M-CPA induced only slight re-distribution of cell cycles in all tested cell lines (Fig. S5).
3.3. M-CPA Enhanced IR Response in Miapaca-2 and HPSC Cells
We first measured the cytotoxicity of IR and M-CPA mono-therapy (Fig. 3A). The doses that caused about 50% cell death in the IR and M-CPA monotherapies were then chosen for the combination therapy: 2 Gy + 1 μM M-CPA for Miapaca-2, 2 Gy + 30 μM M-CPA for Panc-1, and 5 Gy + 3 μM M-CPA for HPSC. The same doses of free CPA were used for combined treatment of CPA and IR: 1μM CPA for Miapaca-2, 30 μM CPA for Panc-1 and 3 μM CPA for HPSC. The results are shown in Fig. 3 (B-D). These results show that with the exception of Pan-1 cells, M-CPA plus IR has significantly better cell killing effect than CPA plus IR. We further measured cell viability after treating the cell lines with fixed M-CPA concentrations but varying IR doses (Fig. S6). Cell viability for Panc-1 cells that were treated with 30 μM M-CPA remained unchanged after IR exposures at doses of 0, 1, 2 and 5 Gy, indicating that M-CPA could not sensitize CPA-resistant Panc-1 cells (Fig. S6B).
Figure 3.
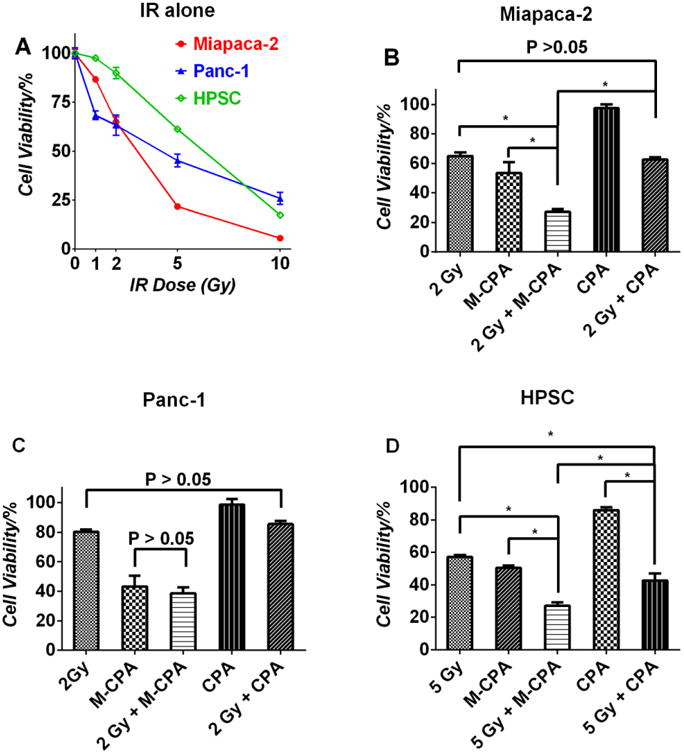
Effects of radiation and M-CPA on cell viability. (A) Effect of radiation doses on the viability of three cell lines. (B) Combination effects of 2 Gy IR and 1 μM CPA or M-CPA on the viability of Miapaca-2 (B); effects of 2Gy IR and 30 μM CPA or M-CPA on the viability of Panc-1 (C); and effects of 5Gy IR and 3 μM CPA or M-CPA on the viability of HPSC (D). Significant differences were marked by * (p < 0.05).
3.4. M-CPA Reduced Post-IR Clonogenicity in Miapaca-2, L3.6pl, and HPSC Cells
We then examined the effect of M-CPA on post-IR clonogenicity via PLDR assay. The clonogenicity of cells depend on the integrity of DNA structure, which was severely compromised during IR. IR-induced DNA damages can be repaired over time through a cascade of DNA-repair mechanism. Therefore the 24-hour recovery group had higher survival fractions than the no-recovery group in all the cell lines. On the other hand, M-CPA significantly reduced survival fractions in Miapaca-2, L3.6pl, and HPSC cells after 24-hour recovery at IR doses of 2, 4, and 8 Gy (p < 0.05), indicating that the presence of M-CPA during the recovering period disrupted DNA repair (Fig. 4 and Fig. S8A&B). In contrast, Panc-1 cells had similar survival fractions regardless of the presence of M-CPA. Mean inactivation doses were calculated from the fitted survival curves (Table 1) 15a. M-CPA sensitized IR in Miapaca-2, L3.6pl, and HPSC cell lines by enhancement factors of 1.8 ± 0.2 (p < 0.05), 1.5 ± 0.2 (p < 0.05), and 1.5 ± 0.2 (p < 0.05), respectively. No IR sensitization was observed in Panc-1 cells, with an enhancement factor of 1.0 ± 0.1 (p > 0.05). In order to compare the radiosensitization effects of CPA and M-CPA, cells were irradiated at 4 Gy in the presence of 10 μM CPA or M-CPA, and then seeded for colony growth (Fig. 4D-F). Compared to CPA, fewer colonies were formed in the presence of M-CPA in Miapaca-2 and HPSC cells (p< 0.05); while neither CPA nor M-CPA reduced the number of colonies in Panc-1 cells (p> 0.05).
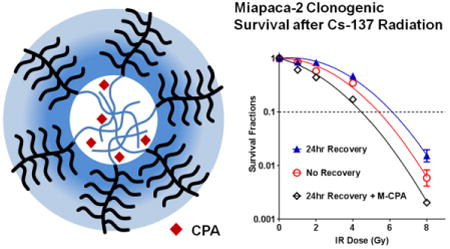
Figure 4.
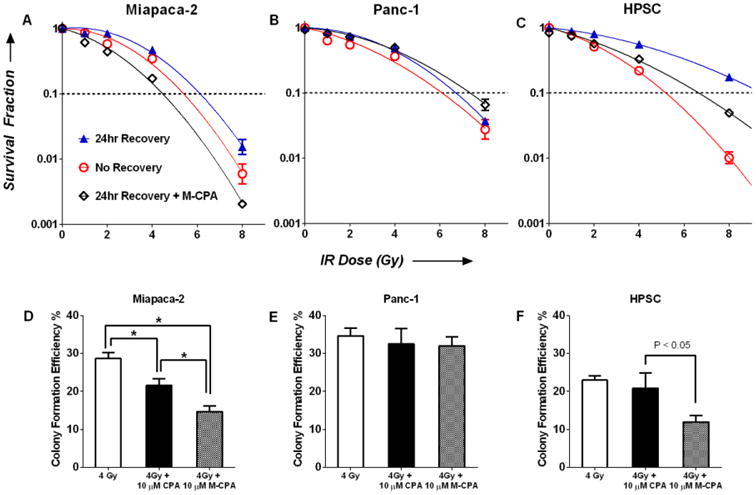
Clonogenic survival of Miapaca-2 (A), Panc-1 (B), and HPSC (C) cells as a function of IR dose. There were three treatment groups: no recovery (open red circle), 24-hr recovery (blue triangles) and 24-hr recovery with 10 μM M-CPA (open black diamond). Data are expressed as fraction of colonies formed relative to untreated cells (mean ± SEM, N = 4). Survival curves were fitted using Log10(survival fraction) = -α(Dose)-β(Dose)2. Mean inactivation dose (MID) was calculated as , see Ref 15a. (D-E) Efficiency of colony formation after 4Gy IR, 4Gy + 10 μM CPA, and 4Gy + 10 μM M-CPA.
Table 1. Effect of cell recovery and M-CPA on mean inactivation dose.
| Mean Inactivation Dose (Gy) | ||||
|---|---|---|---|---|
|
| ||||
| No Recovery | 24-hr Recovery | 24-hr Recovery + 10 μM M-CPA | Enhancement Factor of M-CPA | |
| Miapaca-2 | 5.4 ± 0.6 | 7.0 ± 0.7 | 3.8 ± 0.2 | 1.8 ± 0.2 |
| L3.6pl | 4.0 ± 0.4 | 5.1 ± 0.5 | 3.5 ± 0.2 | 1.5 ± 0.2 |
| HPSC | 4.3 ± 0.3 | 8.2 ± 0.6 | 5.4 ± 0.4 | 1.5 ± 0.2 |
| Panc-1 | 5.1 ± 0.6 | 6.7 ± 0.4 | 6.7 ± 0.5 | 1.0 ± 0.1 |
3.5. M-CPA Impaired Repair of IR-induced DNA Damage in Miapaca-2 and HPSC Cells
Cells were treated as described in Section 2.5. DNA single-strand breaks were analyzed by comet assay (Fig. 5 A, D and G). The percentage of damaged DNA in Miapaca-2 cells was 32.6 ± 1.1% without recovery and 12.9 ± 0.7% after 24-hour recovery. The percentage increased to 24.9 ± 1.1% when M-CPA was present during recovery (all groups significantly different from each other, p < 0.05, Fig. 5A). A similar trend was observed in HPSC cells. The percentages of damaged DNA were 38.1 ± 0.7%, 18.5 ± 0.5%, and 24.5 ± 0.5%, respectively (all group significantly different from each other, p < 0.05, Fig. 5G). M-CPA did not affect DNA repair in Panc-1 cells; the percentages of damaged DNA after 24-hour recovery were 13.8 ± 0.5% without M-CPA and 14.5 ± 0.5% with M-CPA (p > 0.05) (Fig. 5D).
Figure 5.
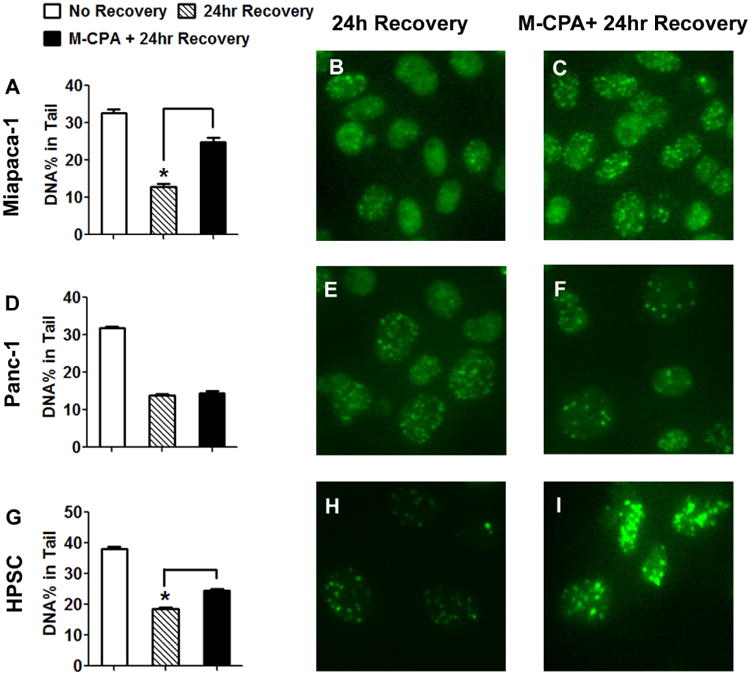
Effect of M-CPA on the repair of IR-induced DNA damage. (A, D, and G) Single-strand breaks were analyzed by comet assay. The percentages of damaged DNA were averaged from at least 100 nuclei (mean ± SEM). Double-strand breaks were visualized by 53BP1 staining: Miapaca-2 (B&C), Panc-1 (E&F), and HPSC (H&I). Treatment doses: radiation = 10 Gy, M-CPA = 10 μM. Significant differences are denoted by * (p < 0.05).
To further characterize the response in DNA double-strand breaks, expression of 53BP1, a marker of DNA double-strand breaks, was monitored using fluorescence microscopy 16. The results agreed with those of the comet assay. The presence of M-CPA retained 53BP1 staining 24 hrs after radiation in both Miapaca-2 (Fig.5 B&C) and HPSC (Fig.5 H&I). The 53BP1 intensity was not affected by M-CPA in Panc-1 cells (Fig.5 E&F).
4. Discussion
We hereby prepared a novel core-crosslinked polymeric micelle system to deliver CPA. The resultant M-CPA was a potent SMO inhibitor and more toxic than free CPA. M-CPA enhanced the radiation response of Miapaca-2, L3.6pl, and HPSC cells by factors of 1.8 ± 0.2, 1.5 ± 0.2, and 1.5 ± 0.2, respectively; and M-CPA radio-sensitization correlated with its ability to disrupt the repair of radiation-induced DNA damage. The efficacy of combined M-CPA-IR treatment in HPSC cells suggests that this tactic is also effective against tumor-associated stroma.
The first goal of this study was to develop a drug delivery system for CPA. Several nano-formulations have been explored to deliver CPA 11. In this study, CPA was encapsulated in core-crosslinked polymeric micelles, which had the following advantages. The micelles were surrounded by a dense layer of brush-like P(OEGMA) corona that should reduce the non-specific micelle uptake in healthy organs 17. The termini of the P(OEGMA) corona contained protected amine groups that can be de-protected and conjugated with targeting moieties, if needed. The micelle core was crosslinked with silica to increase micellar stability during circulation; the crosslinking was achieved spontaneously in aqueous solution. The degradability of oligo-caprolactone chains ensured CPA release and the eventual clearance of micelles from the body. M-CPA had the same mechanism of action as CPA with regard to its ability to down-regulate the expression of SHH pathway in CPA-sensitive cells (Fig. 2E). We also found that M-CPA exhibited higher cytotoxicity than free CPA in all four cell lines studied (Fig. 2 B-D and Fig. S2). M-CPA also had better radiosensitizing effects than CPA (Fig. 3&4). The improved cell killing activity may be attributed to the increased intra-cellular delivery of CPA through M-CPA.
The second goal of this study was to combine M-CPA with IR to treat PDAC. IR is an important option for inoperable PDAC, yet it has limited success against dormant tumor cells or cancer stem cells. In some cases, IR-induced tumor shrinkage was followed by tumor regrowth 18. Recent studies found that SHH activation could protect tumor cells against IR 19; while co-treatment with CPA, a potent SHH inhibitor, could enhance tumor response to IR 19b, 20. CPA-induced radio-sensitization was attributed to factors including cell cycle arrest, depletion of cancer stem cells and disruption of DNA damage repair 21. As for our study, in both Miapaca-2 and HPSC cells, the combined M-CPA/IR therapy reduced cell proliferation more than either mono-therapy did (Fig. 3 B&D). Since it is well known that IR causes DNA damage, we sought to determine whether M-CPA radio-sensitized through the disruption of DNA damage repair (Fig. 4 and 5). The decrease in clonogenicity of IR-treated cells was in accordance with the disruption of repair of DNA damage. In all three cell lines, the 24-hour recovery group had less DNA damage and formed more colonies than the no-recovery group. Moreover, the presence of M-CPA during the 24-hour recovery period disrupted DNA damage repair in Miapaca-2 and HPSC cells, and fewer colonies were formed than their counterparts without M-CPA. No radio-sensitization and no disruption in DNA damage repair were observed in Panc-1 cells treated with M-CPA, suggesting that the CPA-induced sensitization was associated with the DNA repair mechanism.
In order to test whether radiosensitization of M-CPA was related to sensitivity of cells to CPA treatment, we selected pancreatic cancer cell lines with variant sensitivities to CPA according to previous reports 13. CPA-resistant Panc-1 cells were used as negative control. Steg et al. 22 showed that the high expression of Gli-3 may contribute to its CPA resistance; where Gli-3 knockdown by siRNA treatment restored the CPA efficacy in Panc-1 cells. In another study, Lauth et al. 23 suggested that Panc-1 cells had mutations downstream of SMO, thus making the CPA treatment ineffective. As shown in Fig. 3, 4 and Table 1, M-CPA potentiated radioresponse in Miapaca-2 and HPSC cells but not in Panc-1 cells. Additionally, radio-sensitization was also observed in L3.6pl cell line with either delayed plating (Fig. S7A) or immediate plating (Fig. S7B). Miapaca-2 is known to be sensitive to CPA treatment, and Panc-1 cells are known to be resistant to CPA treatment 7b, 13. Both HPSC and L3.6pl cell lines were moderately sensitive to M-CPA (Fig. 2D and Fig. S2). Immunofluorescence staining found SMO expression in all tested cell lines including Panc-1 (Fig. S8). Taking together, our data suggest that SMO expression is not a prerequisite for radiosensitization effect of M-CPA. Redistribution to cell cycle to more radiosensitive phases is not responsible for M-CPA's radiosensitization activity. The radiosensitization effect of M-CPA appears to be linked to sensitivity of cells to CPA treatment and inhibition of DNA repair by M-CPA. Further studies are needed to clarify the role of SHH pathway in DNA repair and radiosensitization effect M-CPA.
The third goal of this study was to investigate the M-CPA-mediated radio-sensitization of pancreatic stellate cells. Stellate cells are a major source of tumor-associated stroma. Both stellate cells and stroma tissue contribute to the resilience of PDAC. Stellate cells may protect tumor cells against radiation and chemotherapy 24. They also enhance the stemness of cancer cells that may cause tumor regrowth 25. Stroma tissue impairs the efficiency of drug delivery, as well as creates a hypoxic environment that could aggravate pancreatic tumors growth 26. SHH is a key component regulating stroma deposition, since stellate cells can be stimulated by tumor-secreted SHH ligands to produce more stroma. Blockage of SHH pathway by CPA could permeate stroma and restore the delivery of chemotherapy drugs 9a. Similarly, we found in this study that M-CPA had significant cytotoxicity to HPSC cells and abrogated Gli-1 expression in HPSC cells (Fig. 2). M-CPA enhanced HPSC cell response to IR (Fig. 3D and 4C). Therefore, the combination therapy may be an effective tactic to damage tumor-associated stroma.
The limitations of current study is that the in vitro work was performed in normoxia. Hypoxia is an important player in the development of pancreatic cancer, because the oxygen supply is restricted by the poorly developed vasculatures as well as the dense tumor stroma 27. Hypoxia has been shown to contribute to the drug-resistance and epithelial-to-mesenchymal transition (EMT) in pancreatic cancer 28. It also decrease the efficacy of radiation since fewer reactive oxidizing species (ROS) could be generated by ionizing radiation under hypoxia than under normoxia 4b. The present study showed that M-CPA treatment could disrupt the stroma-producing stellate cells, suggesting that M-CPA may disrupt the tumor-associated stroma in vivo to alleviate the hypoxia within tumor microenvironment. Therefore, our future studies will be conducted in animal models or hypoxic cell culture systems to evaluate the effects of M-CPA on hypoxia.
In conclusion, the M-CPA delivery system improves CPA cytotoxicity compared to the free CPA. M-CPA sensitized radiation damage to both pancreatic cancer cells and stellate cells that are responsive to CPA. M-CPA-IR combination therapy may be an effective treatment for pancreatic cancer. We are aware that current manuscript is limited that only in vitro studies are presented. Future studies are needed to show that combined M-CPA and radiotherapy are also effective in treating pancreatic cancer in appropriate preclinical animal models of pancreatic cancer
Supplementary Material
Supplemental figures: S1 (1H-NMR spectra of the initiator and block copolymers); S2 (Effects of CPA and M-CPA on viability of L3.6pl Cell cells); S3 (Cytotoxicity of blank micelles and CPA-loaded micelles (M-CPA) against Miapaca-2 cells); S4 (Effects of CPA and M-CPA on cell apoptosis); S5 (Effect of CPA and M-CPA on cell cycles); S6 (Effect of combined M-CPA and IR treatments at different IR doses); S7 (Comparison of CPA and M-CPA in radiosensitization); S8 (Clonogenic survival of L3.6pl cell as a function of IR dose; 53BP1 staining and SHH immunoblotting of L3.6pl); S9 (Immunofluorescence staining against SMO).
Acknowledgments
We thank D. Chalaire for editing the manuscript. This work was supported in part by grants from the Viragh Family Foundation and the John S. Dunn Foundation. The University of Texas MD Anderson Cancer Center is supported in part by the National Institutes of Health through Cancer Center Support Grant P30CA016672 (This work used the Nuclear Magnetic Resonance Spectroscopy core and Flow Cytometry Core of Cancer Center Support Grant shared resources).
Abbreviations
- CPA
cyclopamine
- MID
mean inactivation dose
Footnotes
Supporting Information: Detailed procedures of polymer synthesis and characterization, flowcytometry, and calculation of mean inactivation dose are presented in supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Paulson AS, Tran Cao HS, Tempero MA, Lowy AM. Therapeutic advances in pancreatic cancer. Gastroenterology. 2013;144(6):1316–26. doi: 10.1053/j.gastro.2013.01.078. [DOI] [PubMed] [Google Scholar]
- 2.McRee AJ, Cowherd S, Wang AZ, Goldberg RM. Chemoradiation therapy in the management of gastrointestinal malignancies. Future oncology. 2011;7(3):409–26. doi: 10.2217/fon.11.7. [DOI] [PubMed] [Google Scholar]
- 3.Ogawa K, Ito Y, Hirokawa N, Shibuya K, Kokubo M, Ogo E, Shibuya H, Saito T, Onishi H, Karasawa K, Nemoto K, Nishimura Y Japanese Radiation Oncology Study Group Working Subgroup of Gastrointestinal, C. Concurrent radiotherapy and gemcitabine for unresectable pancreatic adenocarcinoma: impact of adjuvant chemotherapy on survival. International journal of radiation oncology, biology, physics. 2012;83(2):559–65. doi: 10.1016/j.ijrobp.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 4.(a) Kizaka-Kondoh S, Itasaka S, Zeng L, Tanaka S, Zhao T, Takahashi Y, Shibuya K, Hirota K, Semenza GL, Hiraoka M. Selective killing of hypoxia-inducible factor-1-active cells improves survival in a mouse model of invasive and metastatic pancreatic cancer. Clin Cancer Res. 2009;15(10):3433–41. doi: 10.1158/1078-0432.CCR-08-2267. [DOI] [PubMed] [Google Scholar]; (b) Schwartz DL, Bankson JA, Lemos R, Jr, Lai SY, Thittai AK, He Y, Hostetter G, Demeure MJ, Von Hoff DD, Powis G. Radiosensitization and stromal imaging response correlates for the HIF-1 inhibitor PX-478 given with or without chemotherapy in pancreatic cancer. Mol Cancer Ther. 2010;9(7):2057–67. doi: 10.1158/1535-7163.MCT-09-0768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waghray M, Yalamanchili M, di Magliano MP, Simeone DM. Deciphering the role of stroma in pancreatic cancer. Curr Opin Gastroen. 2013;29(5):537–43. doi: 10.1097/MOG.0b013e328363affe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.di Magliano MP, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer. 2003;3(12):903–911. doi: 10.1038/nrc1229. [DOI] [PubMed] [Google Scholar]
- 7.(a) Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, Marshall D, Fu L, Januario T, Kallop D, Nannini-Pepe M, Kotkow K, Marsters JC, Rubin LL, de Sauvage FJ. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455(7211):406–10. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]; (b) Hwang RF, Moore TT, Hattersley MM, Scarpitti M, Yang B, Devereaux E, Ramachandran V, Arumugam T, Ji B, Logsdon CD, Brown JL, Godin R. Inhibition of the hedgehog pathway targets the tumor-associated stroma in pancreatic cancer. Mol Cancer Res. 2012;10(9):1147–57. doi: 10.1158/1541-7786.MCR-12-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Castillo CFd, Yajnik V, Antoniu B, Mc Mahon M, Warshaw AL, Hebrok M. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425(6960):851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, Mc Intyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Ruckert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, Mc Govern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Feldmann G, Habbe N, Dhara S, Bisht S, Alvarez H, Fendrich V, Beaty R, Mullendore M, Karikari C, Bardeesy N, Ouellette MM, Yu W, Maitra A. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut. 2008;57(10):1420–1430. doi: 10.1136/gut.2007.148189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, Karikari C, Alvarez H, Iacobuzio-Donahue C, Jimeno A, Gabrielson KL, Matsui W, Maitra A. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007;67(5):2187–96. doi: 10.1158/0008-5472.CAN-06-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shafaee Z, Schmidt H, Du W, Posner M, Weichselbaum R. Cyclopamine increases the cytotoxic effects of paclitaxel and radiation but not cisplatin and gemcitabine in Hedgehog expressing pancreatic cancer cells. Cancer Chemother Pharmacol. 2006;58(6):765–70. doi: 10.1007/s00280-006-0227-4. [DOI] [PubMed] [Google Scholar]
- 11.(a) Zhou Y, Yang J, Kopecek J. Selective inhibitory effect of HPMA copolymer-cyclopamine conjugate on prostate cancer stem cells. Biomaterials. 2012;33(6):1863–72. doi: 10.1016/j.biomaterials.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cho H, Lai TC, Kwon GS. Poly(ethylene glycol)-block-poly(epsilon-caprolactone) micelles for combination drug delivery: evaluation of paclitaxel, cyclopamine and gossypol in intraperitoneal xenograft models of ovarian cancer. J Control Release. 2013;166(1):1–9. doi: 10.1016/j.jconrel.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chitkara D, Singh S, Kumar V, Danquah M, Behrman SW, Kumar N, Mahato RI. Micellar Delivery of Cyclopamine and Gefitinib for Treating Pancreatic Cancer. Mol Pharm. 2012;9(8):2350–2357. doi: 10.1021/mp3002792. [DOI] [PubMed] [Google Scholar]
- 12.Torchilin VP. Targeted polymeric micelles for delivery of poorly soluble drugs. Cell Mol Life Sci. 2004;61(19-20):2549–2559. doi: 10.1007/s00018-004-4153-5. [DOI] [PubMed] [Google Scholar]
- 13.Feldmann G, Dhara S, Fendrich V, Bedja D, Beaty R, Mullendore M, Karikari C, Alvarez H, Iacobuzio-Donahue C, Jimeno A, Gabrielson KL, Matsui W, Maitra A. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 2007;67(5):2187–96. doi: 10.1158/0008-5472.CAN-06-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) van Bree C, Franken NA, Rodermond HM, Stalpers LJ, Haveman J. Repair of potentially lethal damage does not depend on functional TP53 in human glioblastoma cells. Radiat Res. 2004;161(5):511–6. doi: 10.1667/3154. [DOI] [PubMed] [Google Scholar]; (b) Frankenberg-Schwager M, Frankenberg D, Harbich R. Potentially lethal damage repair is due to the difference of DNA double-strand break repair under immediate and delayed plating conditions. Radiat Res. 1987;111(2):192–200. [PubMed] [Google Scholar]
- 15.(a) Jacobson BS. Optimum inactivation dose and indices of radiation response based on the linear quadratic survival equation. Radiat Environ Biophys. 1993;32(4):311–7. doi: 10.1007/BF01225918. [DOI] [PubMed] [Google Scholar]; (b) Fertil B, Dertinger H, Courdi A, Malaise EP. Mean Inactivation Dose: A Useful Concept for Intercomparison of Human Cell Survival Curves. Radiat Res. 1984;99(1):73–84. [PubMed] [Google Scholar]
- 16.Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. P53 Binding Protein 1 (53bp1) Is an Early Participant in the Cellular Response to DNA Double-Strand Breaks. The Journal of Cell Biology. 2000;151(7):1381–1390. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Z, Zheng SY, Harrison WJ, Harder J, Wen XX, Gelovani JG, Qiao A, Li C. Long-circulating near-infrared fluorescence core-cross-linked polymeric micelles: Synthesis, characterization, and dual nuclear/optical imaging. Biomacromolecules. 2007;8(11):3422–3428. doi: 10.1021/bm7005399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fowler JF, Lindstrom MJ. Loss of local control with prolongation in radiotherapy. Int J Radiat Oncol Biol Phys. 1992;23(2):457–467. doi: 10.1016/0360-3016(92)90768-d. [DOI] [PubMed] [Google Scholar]
- 19.(a) Chen YJ, Lin CP, Hsu ML, Shieh HR, Chao NK, Chao KS. Sonic hedgehog signaling protects human hepatocellular carcinoma cells against ionizing radiation in an autocrine manner. Int J Radiat Oncol Biol Phys. 2011;80(3):851–9. doi: 10.1016/j.ijrobp.2011.01.003. [DOI] [PubMed] [Google Scholar]; (b) Ma J, Tian L, Cheng J, Chen Z, Xu B, Wang L, Li C, Huang Q. Sonic Hedgehog Signaling Pathway Supports Cancer Cell Growth during Cancer Radiotherapy. PLoS ONE. 2013;8(6):e65032. doi: 10.1371/journal.pone.0065032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Sims-Mourtada J, Izzo JG, Apisarnthanarax S, Wu TT, Malhotra U, Luthra R, Liao Z, Komaki R, van der Kogel A, Ajani J, Chao KS. Hedgehog: an attribute to tumor regrowth after chemoradiotherapy and a target to improve radiation response. Clin Cancer Res. 2006;12(21):6565–72. doi: 10.1158/1078-0432.CCR-06-0176. [DOI] [PubMed] [Google Scholar]; (b) Gonnissen A, Isebaert S, Haustermans K. Hedgehog signaling in prostate cancer and its therapeutic implication. Int J Mol Sci. 2013;14(7):13979–4007. doi: 10.3390/ijms140713979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Onishi H, Morifuji Y, Kai M, Suyama K, Iwasaki H, Katano M. Hedgehog inhibitor decreases chemosensitivity to 5-fluorouracil and gemcitabine under hypoxic conditions in pancreatic cancer. Cancer Sci. 2012;103(7):1272–9. doi: 10.1111/j.1349-7006.2012.02297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Che J, Zhang FZ, Zhao CQ, Hu XD, Fan SJ. Cyclopamine is a novel Hedgehog signaling inhibitor with significant anti-proliferative, anti-invasive and anti-estrogenic potency in human breast cancer cells. Oncol Lett. 2013;5(4):1417–1421. doi: 10.3892/ol.2013.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gu D, Liu H, Su GH, Zhang X, Chin-Sinex H, Hanenberg H, Mendonca MS, Shannon HE, Chiorean EG, Xie J. Combining hedgehog signaling inhibition with focal irradiation on reduction of pancreatic cancer metastasis. Mol Cancer Ther. 2013;12(6):1038–48. doi: 10.1158/1535-7163.MCT-12-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wu XY, Che J, Sun KK, Shen XJ, Yang D, Zhong N, Zhao H. Cyclopamine increases the radiosensitivity of human pancreatic cancer cells by regulating the DNA repair signal pathway through an epidermal growth factor receptordependent pathway. Mol Med Rep. 2013;8(4):979–83. doi: 10.3892/mmr.2013.1605. [DOI] [PubMed] [Google Scholar]
- 22.Steg A, Amm HM, Novak Z, Frost AR, Johnson MR. Gli3 mediates cell survival and sensitivity to cyclopamine in pancreatic cancer. Cancer Biol Ther. 2010;10(9):893–902. doi: 10.4161/cbt.10.9.13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lauth M, Bergstrom A, Toftgard R. Phorbol esters inhibit the Hedgehog signalling pathway downstream of Suppressor of Fused, but upstream of Gli. Oncogene. 2007;26(35):5163–8. doi: 10.1038/sj.onc.1210321. [DOI] [PubMed] [Google Scholar]
- 24.(a) Fujita H, Ohuchida K, Mizumoto K, Egami T, Miyoshi K, Moriyama T, Cui L, Yu J, Zhao M, Manabe T, Tanaka M. Tumor-stromal interactions with direct cell contacts enhance proliferation of human pancreatic carcinoma cells. Cancer Sci. 2009;100(12):2309–17. doi: 10.1111/j.1349-7006.2009.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chang Q, Foltz WD, Chaudary N, Hill RP, Hedley DW. Tumor-stroma interaction in orthotopic primary pancreatic cancer xenografts during hedgehog pathway inhibition. Int J Cancer. 2013;133(1):225–34. doi: 10.1002/ijc.28006. [DOI] [PubMed] [Google Scholar]
- 25.(a) Hamada S, Masamune A, Takikawa T, Suzuki N, Kikuta K, Hirota M, Hamada H, Kobune M, Satoh K, Shimosegawa T. Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem Biophys Res Commun. 2012;421(2):349–54. doi: 10.1016/j.bbrc.2012.04.014. [DOI] [PubMed] [Google Scholar]; (b) Kikuta K, Masamune A, Watanabe T, Ariga H, Itoh H, Hamada S, Satoh K, Egawa S, Unno M, Shimosegawa T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem Biophys Res Commun. 2010;403(3-4):380–4. doi: 10.1016/j.bbrc.2010.11.040. [DOI] [PubMed] [Google Scholar]
- 26.Spivak-Kroizman TR, Hostetter G, Posner R, Aziz M, Hu C, Demeure MJ, Von Hoff D, Hingorani SR, Palculict TB, Izzo J, Kiriakova GM, Abdelmelek M, Bartholomeusz G, James BP, Powis G. Hypoxia triggers hedgehog-mediated tumor-stromal interactions in pancreatic cancer. Cancer Res. 2013;73(11):3235–47. doi: 10.1158/0008-5472.CAN-11-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, Bastidas AJ, Vierra M. Pancreatic tumors show high levels of hypoxia. International Journal of Radiation Oncology*Biology*Physics. 2000;48(4):919–922. doi: 10.1016/s0360-3016(00)00803-8. [DOI] [PubMed] [Google Scholar]
- 28.Bao B, Azmi AS, Ali S, Ahmad A, Li Y, Banerjee S, Kong D, Sarkar FH. The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochim Biophys Acta. 2012;1826(2):272–96. doi: 10.1016/j.bbcan.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figures: S1 (1H-NMR spectra of the initiator and block copolymers); S2 (Effects of CPA and M-CPA on viability of L3.6pl Cell cells); S3 (Cytotoxicity of blank micelles and CPA-loaded micelles (M-CPA) against Miapaca-2 cells); S4 (Effects of CPA and M-CPA on cell apoptosis); S5 (Effect of CPA and M-CPA on cell cycles); S6 (Effect of combined M-CPA and IR treatments at different IR doses); S7 (Comparison of CPA and M-CPA in radiosensitization); S8 (Clonogenic survival of L3.6pl cell as a function of IR dose; 53BP1 staining and SHH immunoblotting of L3.6pl); S9 (Immunofluorescence staining against SMO).