

Summary
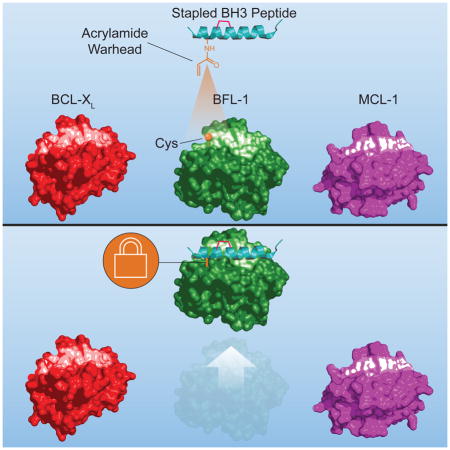
Anti-apoptotic BCL-2 family proteins block cell death by trapping the critical α-helical BH3 domains of pro-apoptotic members in a surface groove. Cancer cells hijack this survival mechanism by overexpressing a spectrum of anti-apoptotic members, mounting formidable apoptotic blockades that resist chemotherapeutic treatment. Drugging the BH3-binding pockets of anti-apoptotic proteins has become a highest priority goal, fueled by the clinical success of ABT-199, a selective BCL-2 inhibitor, in reactivating apoptosis in BCL-2 dependent cancers. BFL-1 is a BCL-2 homologue implicated in melanoma, lymphoma, and other cancers, and remains undrugged. A natural juxtaposition of two unique cysteines at the binding interface of the NOXA BH3 helix and BFL-1 pocket informed the development of stapled BH3 peptides bearing acrylamide warheads to irreversibly inhibit BFL-1 by covalent targeting. Given the frequent proximity of native cysteines to regulatory binding surfaces, covalent stapled peptide inhibitors provide a new therapeutic strategy for targeting pathologic protein interactions.
eTOC Blurb
Huhn et al. report the development of stapled peptides that covalently react with a discrete cysteine at the BH3-binding interface of anti-apoptotic BFL-1, representing a new strategy for selective covalent targeting of pathologic proteins in cancer and other diseases.
Introduction
Anti-apoptotic BCL-2 family members have emerged as ripe targets for therapeutic development, especially in human cancers that overexpress these proteins to enforce cellular immortality. The canonical mechanism for apoptotic suppression involves sequestration of the BH3 killer domain helices of pro-apoptotic members in a binding pocket composed of the BH1, BH2, and BH3 domains of the anti-apoptotic BCL-2, BCL-XL, BCL-w, MCL-1, BFL-1, and BCL-B proteins(Sattler et al., 1997). Thus, structural mimicry of pro-apoptotic BH3 helices has been pursued to pharmacologically “inhibit the inhibitors” of apoptosis. For example, small molecule BH3 mimetics, such as ABT-737(Oltersdorf et al., 2005) and ABT-263(Tse et al., 2008), were initially designed to target the BH3-binding pockets of both BCL-2 and BCL-XL, and the next-generation clinical agent, ABT-199(Souers et al., 2013), was refined for selective BCL-2 inhibition at least in part to avoid the adverse effect of BCL-XL inhibition on platelet survival(Mason et al., 2007; Souers et al., 2013). Given the diversity of anti-apoptotic BCL-2 family proteins at the cancer cell’s disposal, developing inhibitors for each of these oncogenic proteins, including compounds active against subsets or all of the targets, is a priority. To that end, the race is on for advancing the first, selective, small-molecule inhibitor of MCL-1 to the clinic(Leverson et al., 2015; Pelz et al., 2016), given the prominence of this protein as one of the top ten most expressed pathologic proteins across all subtypes of human cancers(Beroukhim et al., 2010).
We have taken an alternative approach to BCL-2 family targeting by transforming the spectrum of natural BH3 domain sequences into structurally-reinforced α-helices that resist proteolysis in vivo and, when appropriately designed, achieve intracellular access through macropinosomal import(Walensky and Bird, 2014; Walensky et al., 2004). Our earliest classes of all-hydrocarbon stapled peptides were modeled after the BH3 domains of BID(Walensky et al., 2004; Walensky et al., 2006) and BIM(Gavathiotis et al., 2010; Gavathiotis et al., 2008; LaBelle et al., 2012), two pro-apoptotic BCL-2 family members of the “BH3-only” subclass that can directly bind and activate the executioner proteins BAX and BAK, and also inhibit the entire spectrum of anti-apoptotic pockets. We have since identified the MCL-1 BH3 domain as the only exclusive inhibitor of MCL-1 across natural BH3 sequences and solved the structure of a stapled MCL-1 BH3 peptide in complex with MCL-1 to characterize the binding and selectivity determinants(Stewart et al., 2010). Here, we sought to apply our stapling technology to develop a selective inhibitor of anti-apoptotic BFL-1, a relatively understudied anti-apoptotic BCL-2 family protein that has been implicated in the development, maintenance, and chemoresistance of human cancers.
The pathologic expression of BFL-1 has been reported as an oncogenic driver of melanoma, lymphoma, and leukemia(Fan et al., 2010; Haq et al., 2013; Mahadevan et al., 2005; Placzek et al., 2010; Yecies et al., 2010). In melanoma, for example, BFL-1 overexpression correlates with chemoresistance and metastasis(Hind et al., 2015; Riker et al., 2008), and is directly regulated by the microphthalmia-associated transcription factor (MITF), which has proven essential to melanomagenesis(Haq et al., 2013). In lymphoma, upregulation of BFL-1 underlies the resistance to selective inhibition of BCL-2 and BCL-XL, underscoring the importance of developing BFL-1 inhibitors in the era of ABT-199(Yecies et al., 2010). Likewise, BFL-1 overexpression in the context of BRAF V600E mutation, which is found in ~80% of BRAF-mutant melanomas, blunts the pharmacologic benefit of small molecule BRAF inhibitors, whereas siRNA knockdown of BFL-1 sensitized the cells to apoptosis induction(Davies et al., 2002; Haq et al., 2013). Taken together, there is a compelling rationale for developing a targeted inhibitor of anti-apoptotic BFL-1 for cancer treatment.
In planning a strategy for selective BFL-1 inhibition, we noted the unique juxtaposition of cysteines at the binding interface of BFL-1 and the BH3 domain of pro-apoptotic NOXA. We reasoned that combining the high-affinity noncovalent interactions of a natural BH3 domain helix with the irreversible blockade afforded by covalent reaction could yield a high fidelity BFL-1 inhibitor, a strategy that could also be applied to a broad spectrum of helix-in-groove interactions containing native cysteines within or near the protein binding surface. Indeed, the development of small molecule covalent inhibitors of more focal binding sites on kinases has seen recent, remarkable success. For example, ibrutinib, which covalently targets C426 of Bruton’s tyrosine kinase (BTK), is FDA approved for the treatment of Waldenstrom’s macroglobulinemia, chronic lymphocytic leukemias, and mantle cell lymphoma(Burger et al., 2015; Dreyling et al., 2016; Treon et al., 2015). Afatinib is an irreversible covalent inhibitor that selectively targets the receptor tyrosine kinases EGFR and HER2, and has been approved by the FDA for treatment of metastatic, EGFR T790M-mutant non-small cell lung cancer(Li et al., 2008; Miller et al., 2012; Solca et al., 2012; Wu et al., 2014). Motivated by this resurgence of covalent inhibitor drugs and our identification of a uniquely-positioned cysteine residue in the BH3-binding pocket of BFL-1, we designed, characterized, and validated a new class of stapled peptide inhibitors with highly-selective covalent reactivity.
Results
Covalent Reaction Between Cysteines at the Binding Interface of NOXA BH3 and BFL-1
The BH3-only protein NOXA exhibits natural, dual selectivity for interaction with anti-apoptotic MCL-1 and BFL-1(Rooswinkel et al., 2012; Stewart et al., 2010), and therefore, its BH3 sequence was selected as a starting point for developing a BFL-1 inhibitor. In examining the crystal structure of human BFL-1ΔC in complex with NOXA BH3 (PDB ID 3MQP), we observed the proximity of NOXA C25 to BFL-1ΔC C55 at a distance of 3.9 Å, compatible with disulfide bond formation (Figure 1A). As no other anti-apoptotic BCL-2 family member contains a cysteine in its BH3-binding pocket, we reasoned that C55-targeting by a stapled BH3 peptide could yield a BFL-1 inhibitor with selective covalent reactivity. To test our hypothesis, we first generated stapled NOXA BH3 peptides and recombinant BFL-1ΔC constructs bearing their native cysteines (NOXA: C25, BFL-1: C4, C19, C55) and a series of serine mutants (NOXA: C25S, BFL-1: C4S/C19S, C4S/C19S/C55S) for binding studies. For the stabilized alpha-helices of BCL-2 domains (SAHBs) modeled after NOXA BH3 (aa 19–43), we positioned the i, i+4 all-hydrocarbon staple at our classic “A” position(Walensky et al., 2004) (substitution of R31 and K35) and derivatized the N-termini with PEG-biotin for biolayer interferometry analyses. We found that the peptide/protein pairs all demonstrated dissociation constants within a 46–165 nM range (Figures 1B, S1). Thus, serine mutagenesis, in and of itself, appeared to have no detrimental effect on binding affinity and, if anything, somewhat enhanced BFL-1 interaction by up to 3.5-fold.
Figure 1. Disulfide Bond Formation Between NOXA BH3 and BFL-1.

(A) Structure of the NOXA BH3/BFL-1ΔC complex (PDB ID 3MQP) highlighting the juxtaposition between NOXA C25 and BFL-1 C55.
(B) Dissociation constants for the binding interactions between BFL-1ΔC constructs and NOXA SAHBA peptides bearing the indicated native cysteines and cysteine-to-serine mutations. Binding experiments were performed in technical and biological duplicate.
(C) Exposure of BFL-1ΔC and FITC-NOXA SAHBA constructs to oxidizing conditions yielded a molecular weight shift only for peptide/protein pairs that retain native NOXA C25 and BFL-1 C55, as detected by Coomassie staining (top). Disulfide bond formation between BFL-1ΔC bearing C55 and wild-type NOXA SAHBA was confirmed by FITC scan (bottom).
(D) Incubation of FITC-NOXA SAHBA peptides with alternate anti-apoptotic BCL-2 family proteins, such as MCL-1ΔNΔC or BCL-XLΔC, under oxidizing conditions caused no molecular weight shift, as evaluated by Coomassie staining (top), or FITC-peptide labeling of protein, as assessed by FITC scan (bottom).
See also Figure S1.
We then sought to determine if disulfide bond formation between NOXA C25 and BFL-1ΔC C55 was biochemically feasible. Indeed, upon DTT (10 mM) reduction followed by GSSG oxidation (12 mM), we observed a shift in the molecular weight of wild-type BFL-1ΔC when incubated with NOXA SAHBA but not its C25S mutant, as assessed by gel electrophoresis under denaturing and nonreducing conditions and Coomassie staining (Figure 1C, top). Our use of FITC-NOXA SAHBA peptides provided confirmation that the BFL-1 protein was labeled by the wild-type but not C25S mutant peptide, as detected by FITC scan (Figure 1C, bottom). We likewise determined that NOXA C25 formed a disulfide bond with BFL-1ΔC C55, as demonstrated both by the molecular weight shift (Coomassie stain) and FITC-labeling of the BFL-1ΔC C4S/C19S construct (in which only C55 is present), but no adduct with the BFL-1ΔC C4S/C19S/C55S construct that lacks C55 (Figure 1C). As a measure of cysteine specificity, we repeated the experiment using MCL-1ΔNΔC and BCL-XLΔC, both of which contain cysteines (MCL-1 C286, BCL-XL C151), and observed no molecular weight shift or FITC labeling upon incubation with NOXA SAHBA under oxidizing conditions (Figure 1D). These data confirm that the juxtaposed cysteines at the NOXA BH3/BFL-1 interface can indeed form a disulfide bond and, what’s more, in a selective fashion.
Selective BFL-1 Reactivity of Stapled BH3 Peptides Bearing Electrophilic Warheads
The capacity of NOXA SAHBA and BFL-1ΔC to engage through disulfide bond formation suggested a novel opportunity to develop stapled peptides for covalent targeting of cysteines localized to key regulatory surfaces, such as the BH3-binding pocket of BFL-1. Because relying on intracellular disulfide bond formation as a basis for protein target inhibition is not a tractable pharmacologic strategy, we instead examined possible sites for insertion of non-natural amino acids bearing reactive acrylamide moieties, and identified NOXA L21 as having even closer proximity to BFL-1 C55 than NOXA C25 (3.3 vs. 3.9 Å, respectively) based on the crystal structure of the NOXA BH3/BFL-1ΔC complex (PDB ID 3MQP) (Figure 2A). In the case of the more promiscuous BIM BH3 sequence, W147 manifests optimal adjacency to BFL-1 C55 (3.6 Å) based on the crystal structure of the BIM BH3/BFL-1ΔC complex (PDB ID 2VM6) (Figure 2A). Thus, we capped NOXA SAHBA(Stewart et al., 2010) and BIM SAHBA(Walensky et al., 2006) at positions L21 and W147, respectively, with a series of non-natural amino acids bearing distinct acrylamide species (Figure 2B). In comparing the reactivity of the electrophilic “warhead”-bearing NOXA (aa 21–43) and BIM SAHBA (aa 147–166) panels, we observed efficient conversion of BFL-1ΔC to the heavier, conjugated adduct for SAHBs bearing warheads 1, 3, 5, and 8, as assessed by reducing and denaturing gel electrophoresis and Coomassie staining (Figure 2C). We advanced NOXA and BIM SAHBs bearing one of the most effective warheads, D-nipecotic acid (3), to specificity testing. First, we tested the selectivity of NOXA SAHBA-3 and BIM SAHBA-3 for BFL-1 C55. Upon incubation of SAHBA-3 compounds with BFL-1ΔC constructs bearing all native cysteines (BFL-1 WT), C55-only (BFL-1 C4S/C19S), C4 and C19-only (BFL-1 C55S), or no cysteines (BFL-1 C4S/C19S/C55S), we observed exclusive reactivity with the WT and BFL-1 C4S/C19S constructs, underscoring the cysteine-selectivity of NOXA SAHBA-3 and BIM SAHBA-3 for C55 of the BH3-binding pocket (Figure 2D). As a further measure of compound specificity, we repeated the experiment using MCL-1ΔNΔC and BCL-XLΔC and observed no nonspecific reactivity, despite the presence of cysteines in these anti-apoptotic BCL-2 family proteins (Figure 2E). Thus, we found that installing a cysteine-reactive warhead in stapled NOXA and BIM BH3 peptides results in efficient and selective covalent-targeting of the BFL-1 BH3-binding groove.
Figure 2. Incorporation of Electrophilic Warheads into Stapled NOXA and BIM BH3 Helices for Covalent Targeting of BFL-1 C55.

(A) The structures of the NOXA BH3/BFL-1ΔC (top, PDB ID 3MQP) and BIM BH3/BFL-1ΔC (bottom, PDB ID 2VM6) complexes demonstrate the proximity of discrete BH3 residues to C55 for replacement with electrophilic warheads.
(B) Chemical structures of the reactive acrylamide moieties installed at the N-termini of NOXA and BIM SAHB peptides.
(C) Reactivity of BIM and NOXA SAHBs bearing warheads 1–8 with BFL-1ΔC C4S/C19S, which only retains the native C55.
(D) BIM and NOXA SAHBA-3 peptides selectively reacted with BFL-1ΔC protein bearing C55.
(E) BIM and NOXA SAHBA-3 peptides did not react with MCL-1ΔNΔC or BCL-XLΔC, despite the presence of cysteines in these anti-apoptotic targets.
We next explored how conversion of NOXA and BIM SAHBs to BFL-1 C55-reactive agents influenced the balance between noncovalent and covalent SAHB interactions in the context of an anti-apoptotic protein mixture. First, we generated recombinant MCL-1ΔNΔC, BCL-XLΔC and BFL-1ΔC proteins with differential N-terminal tags (GST, tagless, and His, respectively) so that each could be readily identified upon gel electrophoresis and silver stain (Figure 3A–B). Upon incubation of the anti-apoptotic mixture with biotinylated NOXA SAHBA or NOXA SAHBA-3 (1:1:1:1 for each component), we only see a shift in the molecular weight of BFL-1ΔC, corresponding to the selective covalent reaction (Figure 3A, left). Streptavidin (SA) pull-down revealed prominent non-covalent capture of MCL-1ΔNΔC by NOXA SAHBA, but a notable shift in the interaction propensity of NOXA SAHBA-3, with relatively less MCL-1ΔNΔC and notably more BFL-1ΔC engagement as a result of covalent BFL-1ΔC conjugation (Figure 3A, right). Consistent with the broader anti-apoptotic binding spectrum of BIM BH3, the corresponding BIM SAHBs engaged BCL-XLΔC in addition to MCL-1ΔNΔC and BFL-1ΔC, but an increased BFL-1ΔC targeting propensity was again observed for BIM SAHBA-3 relative to BIM SAHBA as a consequence of covalent conjugation (Figure 3B). Thus, the capacity for selective covalent reaction with BFL-1ΔC can shift the competitive balance of SAHB interactions toward BFL-1.
Figure 3. Covalent Conjugation to the BH3-binding Pocket Enhances BFL-1 Targeting and Neutralizes Anti-Apoptotic Activity.

(A–B) Incorporation of an acrylamide moiety into NOXA and BIM SAHBs provided a competitive advantage for BFL-1 targeting, as demonstrated by SA pull-down of a 1:1:1:1 mixture (1 μM each) of biotinylated NOXA (A) or BIM (B) SAHBs with recombinant His-BFL-1ΔC, BCL-XLΔC (tagless), and GST-MCL-1ΔNΔC.
(C) Coomassie stain of recombinant BFL-1ΔC and its NOXA SAHBA-3 and BIM SAHBA-3 conjugates employed in liposomal release assays.
(D–E) BH3-only protein tBID directly activated BAX-mediated liposomal poration, as monitored by ANTS/DPX release. Whereas BFL-1ΔC completely blocked tBID-triggered BAX poration, covalent engagement of BFL-1ΔC by either NOXA SAHBA-3 (D) or BIM SAHBA-3 (E) effectively inhibited the functional activity of BFL-1ΔC. Liposomal experiments were performed in triplicate with exemplary release profiles shown.
Targeted Blockade of BFL-1 in Liposomes, Lysates and Cells
To determine the functional consequences of covalent targeting of the BFL-1 BH3-binding pocket, we performed liposomal release assays designed to monitor the influence of BFL-1 on direct BAX activation. We generated ANTS/DPX-encapsulated large unilamellar vesicles (LUV) and monitored liposomal release of fluorophore upon BAX-mediated membrane poration. Whereas BAX alone had no effect on the liposomes, the addition of direct activator BH3-only protein tBID, triggered time-responsive, BAX-mediated release, a process that was suppressed by BFL-1ΔC (Figure 3C–E). However, upon addition of either NOXA SAHBA-3 or BIM SAHBA-3 conjugated BFL-1ΔC (Figure 3C), the inhibitory function of BFL-1 was lost (Figures 3D–E). These data highlight that covalently “plugging” the BH3-binding pocket of BFL-1 with NOXA SAHBA-3 or BIM SAHBA-3 irreversibly neutralizes its anti-apoptotic function.
We next sought to test whether our covalent stapled peptide inhibitors could selectively react with BFL-1 in more complex protein mixtures. To specifically track C55 derivatization, we transiently expressed HA-BFL-1ΔC C4S/C19S in 293T cells and, after 24 hours, harvested cell lysates for crosslinking analyses with C-terminal Lys-biotin derivatized SAHB constructs that either did or did not contain the electrophilic warhead. Anti-HA western analyses revealed prominent molecular weight shifts only for warhead-bearing SAHBs, consistent with covalent incorporation of both NOXA SAHBA-3 and BIM SAHBA-3 into the BFL-1 protein at C55 (Figure 4A, top). To confirm that the observed molecular weight shifts reflected NOXA SAHBA-3 and BIM SAHBA-3 incorporation, we performed biotin western analyses. We found that the shifted HA-BFL-1ΔC bands were indeed biotin-immunoreactive and, importantly, there was little to no non-specific reactivity with other electrophoresed proteins from the 293T lysates (Figure 4A, bottom).
Figure 4. Covalent Targeting of BFL-1 C55 in Lysates and Cells.

(A) Biotinylated NOXA and BIM SAHBA-3 peptides crosslinked to HA-BFL-1ΔC C4S/C19S in lysates from transfected 293T lysates, as evidenced by the shift in molecular weight of BFL-1ΔC observed upon anti-HA western analysis. Anti-biotin blotting confirmed the selective incorporation of biotin into the HA-BFL-1ΔC band, with little to no crossreactivity with other proteins in the cellular lysate.
(B) Treatment of transfected 293T cells with biotinylated BIM SAHBA-3 followed by cellular lysis, HA immunoprecipitation, and biotin western analysis demonstrated the capacity of a warhead-bearing BIM SAHB to gain intracellular access and covalently target expressed HA-BFL-1ΔC C4S/C19S containing the native C55.
(C–D) BIM SAHBA-3, but not BIM SAHBA, effectively competed with tBID for HA-BFL-1ΔC C4S/C19S interaction in 293T lysates, achieving robust covalent conjugation (C), whereas in the context of exclusive non-covalent FLAG-MCL-1 interaction, the compounds were equally effective at competing with tBID (D), as measured by the indicated immunoprecipitation and western analyses.
(E) Covalent modification of HA-BFL-1ΔC C4S/C19S by cellular treatment with BIM SAHBA-3, but not the corresponding construct lacking the acrylamide-based warhead. Crosslinked BFL-1ΔC was observed by 2 hours and levels continued to increase in a time-dependent fashion throughout the 12 hour treatment period, as monitored by HA western analysis.
See also Figure S2.
To advance our strategy to cellular testing, we first evaluated the cellular uptake potential of our biotinylated NOXA SAHBA-3 and BIM SAHBA-3 constructs. We incubated 293T cells with the compounds at 20 μM dosing for 24 hours, trypsinized and washed the cells to remove any adherent peptide, and then generated lysates for anti-biotin western analyses. We found that cellular uptake of NOXA SAHBA-3 was relatively limited and therefore proceeded with BIM SAHBA-3 for cellular testing (Figure S2A). We further confirmed that the transfection conditions themselves did not independently influence the cellular uptake of BIM SAHBA-3 (Figure S2B). 293T cells transiently overexpressing HA-BFL-1ΔC C4S/C19S were treated with biotinylated BIM SAHBA (aa 148–166) or BIM SAHBA-3 (20 μM, 6 h) and then lysates, generated as above, were subjected to anti-HA immunoprecipitation. Biotin western analysis of the input revealed a single, prominent protein band only in the denatured and reduced electrophoresed lysate of cells treated with BIM SAHBA-3 (Figure 4B, left). Subjecting the immunoprecipitate to anti-HA western analysis revealed the BFL-1 doublet, and biotin western analysis confirmed that the upper band indeed corresponded to biotinylated HA-BFL-1ΔC (Figure 4B, right).
Having documented the feasibility of specific labeling of intracellular BFL-1 upon treating cells with biotinylated BIM SAHBA-3, we then examined the relative influence of covalent vs. non-covalent engagement on the capacity of BIM SAHBs to disrupt BFL-1 complexes. For this experiment, we added tBID to the lysates from 293T cells transiently transfected with HA-BFL-1ΔC C4S/C19S, incubated the mixture with biotinylated BIM SAHBA or BIM SAHBA-3, performed anti-HA immunoprecipitation and blotted for HA and tBID. Strikingly, BIM SAHBA was incapable of competing with tBID for HA-BFL-1 binding, whereas the warhead-bearing BIM SAHBA-3 construct covalently trapped HA-BFL-1, as exemplified by complete protein conversion to the higher molecular weight species and near total inhibition of tBID co-immunoprecipitation (Figure 4C). When the experiment was repeated using lysates from 293T cells transiently expressing FLAG-MCL-1, which bears no cysteine in its BH3-binding pocket, both BIM SAHBA peptides were equally effective as non-covalent disruptors of tBID/FLAG-MCL-1 co-immunoprecipitation (Figure 4D). Thus, by installing the warhead and enabling stapled peptide covalent reaction, we can selectively enhance the BFL-1 targeting efficacy of BIM SAHBA.
Finally, in anticipation of cancer cell treatment studies, we turned to the corresponding non-biotinylated BIM SAHBA constructs to probe the kinetics and efficiency of covalent targeting of BFL-1 in cells. Comparing BIM SAHBA- and BIM SAHBA-3-treated 293T cells transiently overexpressing HA-BFL-1ΔC C4S/C19S, we observed a discrete molecular weight shift in BFL-1 by anti-HA western analysis within 2 hours of BIM SAHBA-3 exposure, with a progressive increase in the crosslinked species over the 12 hour evaluation period (Figure 4E). Taken together, these data highlight the capacity of a stapled peptide bearing an electrophilic warhead to covalently target BFL-1 in treated lysates and cells.
Preferential Activation of Apoptosis by a Cysteine-Reactive BIM SAHBA in BFL-1-Expressing Melanoma
BFL-1 has recently been implicated as a lineage-specific driver of human melanoma, with gene amplification observed in ~30% of cases and BFL-1 overexpression mediated by the MITF transcription factor, a melanoma oncogene(Haq et al., 2013). Thus, to explore the functional impact of covalent BFL-1 targeting in cancer cells driven by BFL-1 expression, we tested the comparative effect of BIM SAHBA-3 with our lead noncovalent stapled peptide modulator of BCL-2 family proteins, BIM SAHBA1(LaBelle et al., 2012) in A375P melanoma cells(Haq et al., 2013). We first confirmed that BIM SAHBA1 and BIM SAHBA-3 have equivalent cellular uptake, as quantified by ImageXpress Micro (IXM) high content epifluorescence microscopy of treated A375P cells and mouse embryonic fibroblasts (MEFs), which we have used previously to benchmark the comparative cell penetrance of FITC-labeled stapled peptides(Bird et al., 2016) (Figure S3A–B). The mechanism of uptake for BIM SAHBs is consistent with macropinocytosis, as previously reported(Edwards et al., 2015; Walensky et al., 2004) and evidenced here by the epifluorescence microscopy pattern of treated A375P and MEF cells at 4 hours (Figure S3C–D).
Upon exposure of A375P cells to BIM SAHBs, we observed significant enhancement in cytotoxicity over time for the warhead-bearing BIM SAHBA-3 compared to BIM SAHBA1 (Figure 5A). We confirmed by LDH release assay that BIM SAHBs had no membranolytic effect on the cells (Figure 5B). The observed cytotoxicity was instead consistent with mitochondrial apoptosis induction, as reflected by time-responsive caspase 3/7 activation (Figure 5C) and mitochondrial cytochrome c release (Figure 5D). In accordance with its more pronounced impairment of cell viability, BIM SAHBA-3 treatment induced higher levels of caspase 3/7 activation and cytochrome c release compared to that observed for BIM SAHBA1 (Figure 5C–D). To mechanistically link the enhanced susceptibility of A375P cells to preferential BIM SAHBA-3 engagement of BFL-1, we incubated A375P lysates with the corresponding biotinylated BIM SAHBs, followed by SA pull-down and anti-BFL-1 and MCL-1 western analyses. Whereas BIM SAHBA and BIM SAHBA-3 demonstrated equivalent binding to anti-apoptotic MCL-1, as previously observed in the context of competitive interaction with recombinant anti-apoptotic proteins (Figure 3B), the warhead-bearing construct again showed markedly increased engagement of BFL-1 (Figure 6A). To verify that BIM SAHBA-3 can indeed label native mitochondrial BFL-1(Hind et al., 2015), we incubated mitochondria purified from A375P cells with biotinylated BIM SAHBs and observed BIM SAHBA-3-selective biotinylation of mitochondrial protein at the identical molecular weight as immunoreactive BFL-1 (Figure 6B). Live confocal microscopy imaging of A375P cells treated with FITC-BIM SAHBA-3 further revealed the stapled peptide’s striking intracellular localization at the mitochondria, the physiologic site of BFL-1 activity, in both morphologically normal A375P cells (Figure 6C) and those undergoing apoptosis induction, as reflected by cell shrinkage, nuclear condensation, and membrane blebbing (Figure 6D).
Figure 5. Enhanced Apoptotic Response of BFL1-driven Melanoma Cells upon Treatment with a Cysteine-reactive BIM SAHB.

(A) A375P cells were treated with BIM SAHBA1 or BIM SAHBA-3 (40 μM) and viability measure by CellTiter-Glo assay at the indicated time points. Data are mean ± s.d. for experiments performed in technical sextuplicate, and repeated twice using independent cell cultures with similar results.
(B) Quantitation of LDH release upon treatment of A375P cells BIM SAHBA1 or BIM SAHBA-3 (40 μM) for 30 min. Data are mean ± s.d. for experiments performed in technical triplicate.
(C) A375P cells were treated with BIM SAHBA1 or BIM SAHBA-3 (40 μM) and caspase 3/7 activation measured by CaspaseGlo assay at the indicated time points. Data are mean ± s.d. for experiments performed in technical sextuplicate, and repeated twice using independent cell cultures with similar results.
(D) Mitochondrial cytochrome c release in A375P cells treated with BIM SAHBA1 or BIM SAHBA-3 (40 μM), as detected by cytochrome c western analysis of cytosolic and mitochondrial fractions harvested at the indicated time points.
*, p < 0.001 by two-tailed Student’s t test.
See also Figures S3–S5.
Figure 6. Endogenous BFL-1 Targeting and Mitochondrial Localization of a Warhead-bearing BIM SAHB in A375P melanoma cells.

(A) Enhanced targeting of native BFL-1 by biotinylated BIM SAHBA-3, as compared to BIM SAHBA, in A375P lysates, as monitored by SA pull-down and BFL-1 western analysis (top). In contrast, both compounds are equally effective at engaging MCL-1, which bears no cysteine in its BH3-binding groove and thus provides no competitive advantage for BIM SAHBA-3 (bottom).
(B) BIM SAHBA-3, but not BIM SAHBA, biotinylates mitochondrial protein that migrates at the same molecular weight as immunoreactive BFL-1.
(C) Live confocal microscopy of A375P cells treated with FITC-BIM SAHBA-3 reveals its localization at the mitochondria, the intracellular site of native BFL-1. Blue, Hoechst; green, FITC-BIM SAHBA-3; red, MitoTracker; yellow, colocalization of FITC-BIM SAHBA-3 and MitoTracker. Bar, 10 μm.
(D) A FITC-BIM SAHBA-3-treated A375P cell is observed to undergo apoptosis induction, as manifested by cell shrinkage, nuclear condensation, and membrane blebbing. The colocalization FITC-BIM SAHBA-3 and MitoTracker is also evident, as described above. Bar, 10 μm.
Importantly, we observed comparative enhancement in cytotoxicity and caspase 3/7 activation for BIM SAHBA-3 in two additional BFL-1 expressing melanoma cell lines (SK-MEL-2, SK-MEL-28)(Haq et al., 2013; Hind et al., 2015) (Figure S4), but no evidence of this phenomenon in non-melanoma lines that either lack or maintain BFL-1 expression, but are driven by alternate oncogenic mechanisms (e.g. A549, MCF7, H929)(Acquaviva et al., 2012; Haq et al., 2013; Leverson et al., 2015) (Figure S5). Taken together, these data highlight the mechanistic advantage of the warhead-bearing BIM SAHBA-3 in the context of BFL-1-dependent cancer, as reflected by more effective engagement of native BFL-1 and greater efficacy in triggering apoptosis. Thus, in addition to harnessing a cysteine-reactive targeting strategy to selectively trap BFL-1, heightened susceptibility to covalent BFL-1 inhibitors such as BIM SAHBA-3 may provide a diagnostic approach for identifying BFL-1 dependency in human cancers.
Discussion
BFL-1, like its homologues BCL-2, BCL-XL, and MCL-1, has emerged as an oncogenic protein that drives discrete subtypes of human cancer, and promotes chemoresistance and metastasis(Fan et al., 2010; Haq et al., 2013; Hind et al., 2015; Mahadevan et al., 2005; Placzek et al., 2010; Riker et al., 2008; Yecies et al., 2010). Whereas selective small molecule targeting of BCL-2 with ABT-199 has shown remarkable success in BCL-2 dependent cancers, including relapsed chronic lymphocytic leukemia(Roberts et al., 2016), recapitulating this achievement for the diversity of anti-apoptotic targets remains a formidable challenge. The capacity to harness the natural selectivity of BH3 domain sequences for targeting individual, subsets, and all anti-apoptotic targets is an attractive feature of hydrocarbon-stapled BH3 peptides(Edwards et al., 2013; LaBelle et al., 2012; Stewart et al., 2010). Here, we identified the first example of a BH3-only and anti-apoptotic protein pair that juxtapose cysteines at their binding interface in a manner that could support disulfide bond formation. We translated this insight into the first proof-of-concept for generating covalent stapled peptide inhibitors that can selectively derivatize an intracellular protein target to obstruct its key regulatory binding surface.
The development of covalent inhibitors for intracellular protein targets has seen a recent resurgence, owing to the breakthrough success of such agents as ibrutinib and afitinib(Burger et al., 2015; Miller et al., 2012; Treon et al., 2015; Wu et al., 2014). An important hurdle for covalent drugs is balancing reactivity with selectivity, since nonspecific protein derivatization can lead to off-target activities and unwanted toxicities(Singh et al., 2011). Combining the natural selectivity and relatively large noncovalent binding interface of bioactive alpha-helices with embedded, focally-reactive electrophilic warheads could provide a new opportunity to engage otherwise intractable protein targets. Given the presence of native cysteines either within or immediately adjacent to the regulatory “helix-in-groove” binding surfaces of a host of therapeutic targets (Figure S6)(Allen and Taatjes, 2015; Kise et al., 2004; Kitagawa et al., 2006; Margarit et al., 2003; Scott et al., 2009), we envision that our covalent stapled peptide inhibitor approach could be broadly applied.
Tuning the specificity of alpha-helical domains by installing electrophilic warheads provides a new dimension to stapled peptide design. In the case of BFL-1, we demonstrate that a semi-selective NOXA BH3 peptide and an otherwise pan-interacting BIM BH3 sequence can be fashioned to react with C55 at the critical BH3-binding groove, yet not form analogous covalent bonds with alternative cysteines within BFL-1, its homologues MCL-1 or BCL-XL, or other cellular proteins. The enhanced BFL-1 targeting capacity of warhead-bearing stapled BH3 peptides, as compared to the corresponding constructs capable of noncovalent interaction alone, translated into enhanced apoptosis induction of BFL-1-dependent melanoma cells. This selective reactivity feature, coupled with the general proteolytic resistance of stapled peptides, their capacity for cellular uptake and recent advancement to Phase I testing (NCT02264613), suggests that incorporation of electrophilic warheads could yield a new class of chimeric molecules that combine the advantages of stapled peptides and covalent inhibitors for preclinical and clinical development.
Experimental Procedures
Stapled Peptide Synthesis
Hydrocarbon-stapled peptides corresponding to the BH3 domains of BCL-2 family proteins, and either N-terminally derivatized with acetyl, FITC-βAla, biotin-PEG, or electrophilic warheads, or C-terminally derivatized with Lys-biotin, were synthesized, purified, and quantitated using our previously reported methods(Bird et al., 2008; Bird et al., 2011). Acrylamide-bearing peptides were synthesized by either coupling acrylic acid or trans-crotonic acid to the peptide N-terminus, or by first coupling the Fmoc protected cyclic amino acids (Chem-Impex International) followed by Fmoc deprotection and acylation with acrylic acid, using standard Fmoc coupling and deprotection methods. FITC derivatization of acrylamide-bearing peptides is detailed in the Supplemental Information. Stapled peptide compositions, and their observed masses and use by figure, are listed in Table S1.
Recombinant Protein Expression and Purification
The recombinant anti-apoptotic proteins, BFL-1ΔC (aa 1–153), MCL-1ΔNΔC (aa 170–327) and BCL-XLΔC (aa 1–212) were cloned into the pET19b (Novagen; BFL-1ΔC) or pGEX-4T-1 (GE Healthcare; MCL-1ΔNΔC, BCL-XLΔC) expression vectors, expressed in Escherichia coli BL21(DE3), and purified by sequential affinity and size exclusion chromatography as described(Pitter et al., 2008) and detailed in the Supplemental Information.
In Vitro Covalent Conjugation Assay
BFL-1ΔC constructs (40 μM) were combined with NOXA SAHBA or NOXA SAHBA C25S (120 μM) and 10 mM DTT in 50 mM Tris pH 8.0, 100 mM NaCl (final volume, 5 μL), and then incubated in the dark for 1 h at room temperature (RT). After this incubation in a reducing environment, the mixture was diluted 5-fold into 50 mM Tris pH 8.0, 100 mM NaCl, 12 mM GSSG and incubated in the dark for an additional 30 min at RT. The samples were then boiled in 4x loading buffer lacking DTT and electrophoresed on 12% Bis-Tris gel. The gel was rinsed with water, subjected to FITC scan (Typhoon FLA 9500, GE Healthcare) and then Coomassie staining.
For warhead-bearing SAHBs, His-BFL-1ΔC C4S/C19S protein was pretreated with 10 mM DTT in 50 mM Tris pH 8.0, 100 mM NaCl for 30 min at RT (final volume, 9.5 μL), and then combined with a 10:1 molar ratio of NOXA SAHBA or BIM SAHBA peptides bearing warheads 1–8 (final volume, 10 μL) for an additional 2 h incubation at RT. Processing for gel electrophoresis and Coomassie staining was performed as above.
Streptavidin Pull-down
Recombinant His-BFL-1ΔC, BCL-XLΔC (tagless) and GST-MCL-1ΔNΔC (1 μM each) were combined and reduced with 3 mM DTT in PBS for 30 min at RT, and then incubated with 1 μM biotinylated SAHBA, SAHBA-3 or vehicle for 4h at RT. The mixtures were then combined with PBS-washed high-capacity SA agarose (Thermo Fisher Pierce) and incubated with rotation for 2 h at RT. The beads were centrifuged at 3000 rpm, washed twice with NP-40 lysis buffer (1% NP40, 50 mM Tris pH 8.0, 100 mM NaCl, 2.5 mM MgCl2), once with PBS, and then bound protein eluted by boiling in 10% SDS containing 10 mg/mL biotin. Inputs (10%) and eluates were electrophoresed on a 12% Bis-Tris gel and then subjected to silver stain and imaging.
Liposomal Release Assay
Liposomal release assays were performed as detailed in the Supplemental Information, with SAHBA-3/BFL-1ΔC conjugates prepared by treating BFL-1ΔC (10 μM) with DTT (20 mM) for 30 min at 4°C, followed by seq uential incubation with NOXA SAHBA-3 or BIM SAHBA-3 peptides at peptide:protein molar ratios of 1.2x, 0.75x, and 0.5x for 1h each at 4°C.
BFL-1 Targeting in Lysates and Cells
A series of NOXA and BIM SAHB constructs, with and without installed biotin handles and/or acrylamide warheads, were employed in comparative BFL-1 targeting assays in lysates containing or intact cells expressing HA-BFL-1ΔC C4S/C19S (transfected 293T cells) or native BFL-1 (A375P), performed as described in detail in the Supplemental Information.
Cell Viability, LDH Release, and Caspase-3/7 Activation Assays
Cancer cells were cultured using their standard culture media containing 10% FBS and penicillin/streptomycin (A375P: DMEM; SK-MEL-2, SK-MEL-28 and MCF-7: EMEM; A549, H929: RPMI). Cells were plated in 96-well plates (5 × 103 cells per well) and, after overnight incubation, treated with the indicated concentrations of BIM SAHBA1 or BIM SAHBA-3 in the corresponding media supplemented with 5% FBS for the indicated durations. Cell viability and caspase 3/7 activation was measured using CellTiter-Glo and Caspase-Glo 3/7 chemiluminescence reagents (Promega), respectively, and luminescence detected by a microplate reader (Spectramax M5, Molecular Devices). LDH release was quantified after 30 min peptide incubation by plate centrifugation at 1500 rpm for 5 min at 4°C, transfer of 100 μL cell culture media to a clear plate (Corning), incubation with 100 μL LDH reagent (Roche) for 30 min while shaking, and measurement of absorbance at 490 nm on a Spectramax M5 microplate reader.
Mitochondrial Cytochrome c Release and Biotinylation Assays
A375P cells were plated in 6-well Corning plates (3 × 105 cells/well) and cultured as above. After 24 h, the cells were treated with BIM SAHBA1 or BIM SAHBA-3 (40 μM) in DMEM containing 5% FBS for the indicated durations, and then trypsinized, washed with PBS, and cytosol (supernatant) and mitochondrial (pellet) fractions isolated as described(Dewson, 2015). Briefly, pelleted cells were resuspended at 1×107 cells/mL in permeabilization buffer (20 mM HEPES/KOH pH 7.5, 250 mM sucrose, 50 mM KCl, 2.5 mM MgCl2) supplemented with 0.025% digitonin and protease inhibitors, followed by incubation on ice for 10 min and centrifugation at 13,000g. The resultant supernatant and pellet fractions were boiled in LDS buffer and subjected to western analysis using a 1:1000 dilution of cytochrome c antibody (BD Pharmingen #556433). For biotinylation studies, A375P mitochondria were isolated as above, resuspended in permeabilization buffer, and then treated with biotinylated BIM SAHBA or BIM SAHBA-3 (50 μM) for 4 h at RT. Samples were then boiled in LDS buffer and subjected to western analysis using 1:1000 dilutions of BFL1 (Abcam #125259), biotin (Abcam, #53494), and VDAC1 (Abcam #14734) antibodies.
Confocal Microscopy
A375P cells were plated in chambered coverglass (1.5 × 104 cells/well) and cultured as above. After 24 h, cells were treated with FITC-BIM SAHBA1 or BIM SAHBA-3 (1 μM) for 4 h in phenol-free DMEM containing 5% FBS. Cells were washed, stained with MitoTracker Red (Thermo) and Hoechst 33342, and imaged live. Confocal images were collected with a Yokogawa CSU-X1 spinning disk confocal (Andor Technology) mounted on a Nikon Ti-E inverted microscope (Nikon Instruments). Images were acquired using a 100× 1.4 NA Plan Apo objective lens with an Orca ER CCD camera (Hamamatsu Photonics) and 488 nm laser. Acquisition parameters, shutters, filter positions and focus were controlled by Andor iQ software (Andor Technology).
Statistical Analysis
Datasets were analyzed by two-tailed Student’s t test, with p < 0.05 considered statistically significant.
Supplementary Material
Significance.
Selective targeting of intracellular proteins implicated in disease pathogenesis remains a high priority goal for the chemical biology and drug development fields. We combined the advantages of stapling peptide alpha-helices modeled after natural pro-apoptotic domains with the cysteine-targeting efficiency of electrophilic warheads to provide proof-of-concept for specific, covalent inhibition of anti-apoptotic BFL-1, a BCL-2 family protein linked to the development and chemoresistance of select human cancers. Covalent stapled peptide inhibitors modeled after the NOXA and BIM BH3 domains irreversibly “plugged” the critical BH3-binding pocket of BFL-1 by reacting with a unique cysteine residue embedded within its canonical BH3-binding pocket. Strikingly, no cross-reactivity to alternate cysteines in BFL-1, its close homologues MCL-1 or BCL-XL, or other cellular proteins was observed, highlighting the ideal partnership between the relatively expansive non-covalent binding interface of stapled alpha-helices and focally-reactive, acrylamide-bearing warheads. Whereas renewed interest in small molecule covalent inhibitors has successfully led to a series of new drugs to block the deep hydrophobic “holes” that typify kinase targets, covalent stapled peptide inhibitors have the potential to address a series of larger and previously undruggable protein interfaces with strikingly selective reactivity, as exemplified here for electrophilic warhead-bearing SAHBs and anti-apoptotic BFL-1.
Highlights.
A unique juxtaposition of cysteines occurs in the NOXA BH3/BFL-1 interaction.
Oxidizing conditions yields a disulfide bond between NOXA BH3 and BFL-1.
Stapled BH3 peptides bearing electrophilic warheads selectively react with BFL-1.
Covalent stapled peptide inhibitors represent a new class of protein modulators.
Acknowledgments
We thank E. Smith for graphics support and T. Oo for technical assistance with stapled peptide synthesis. This research was supported by NIH grant 1R35CA197583, a Melanoma Research Alliance Team Science Award, a Leukemia and Lymphoma Society (LLS) Marshall A. Lichtman Specialized Center of Research project grant, and an LLS Scholar Award to L.D.W. L.D.W. is a scientific advisory board member and consultant for Aileron Therapeutics.
Footnotes
Author Contributions
G.H.B and L.D.W. designed the study; G.H.B. generated stapled peptides and performed microscopy-based peptide uptake studies; A.J.H. executed the biochemical experiments including in vitro binding and crosslinking analyses, with assistance from E.P.H. in generating peptide-protein conjugates for the liposomal assays; R.M.G. conducted all of the cellular work including immunoprecipitation, in situ crosslinking, viability, caspase 3/7, cytochrome c release and cell imaging studies. A.J.H., R.M.G., G.H.B., and L.D.W. analyzed the data; and L.D.W. wrote the manuscript, which was reviewed by all co-authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acquaviva J, Smith DL, Sang J, Friedland JC, He S, Sequeira M, Zhang C, Wada Y, Proia DA. Targeting KRAS-mutant non-small cell lung cancer with the Hsp90 inhibitor ganetespib. Mol Cancer Ther. 2012;11:2633–2643. doi: 10.1158/1535-7163.MCT-12-0615. [DOI] [PubMed] [Google Scholar]
- Allen BL, Taatjes DJ. The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol. 2015;16:155–166. doi: 10.1038/nrm3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird GH, Bernal F, Pitter K, Walensky LD. Synthesis and biophysical characterization of stabilized alpha-helices of BCL-2 domains. Methods Enzymol. 2008;446:369–386. doi: 10.1016/S0076-6879(08)01622-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird GH, Crannell WC, Walensky LD. Chemical synthesis of hydrocarbon-stapled peptides for protein interaction research and therapeutic targeting. Curr Protoc Chem Biol. 2011;3:99–117. doi: 10.1002/9780470559277.ch110042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird GH, Mazzola E, Opoku-Nsiah K, Lammert MA, Godes M, Neuberg DS, Walensky LD. Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat Chem Biol. 2016 doi: 10.1038/nchembio.2153. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, Bairey O, Hillmen P, Bartlett NL, Li J, et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N Engl J Med. 2015;373:2425–2437. doi: 10.1056/NEJMoa1509388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Dewson G. Investigating Bax subcellular localization and membrane integration. Cold Spring Harb Protoc. 2015;2015:467–471. doi: 10.1101/pdb.prot086447. [DOI] [PubMed] [Google Scholar]
- Dreyling M, Jurczak W, Jerkeman M, Silva RS, Rusconi C, Trneny M, Offner F, Caballero D, Joao C, Witzens-Harig M, et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet. 2016;387:770–778. doi: 10.1016/S0140-6736(15)00667-4. [DOI] [PubMed] [Google Scholar]
- Edwards AL, Gavathiotis E, LaBelle JL, Braun CR, Opoku-Nsiah KA, Bird GH, Walensky LD. Multimodal interaction with BCL-2 family proteins underlies the proapoptotic activity of PUMA BH3. Chem Biol. 2013;20:888–902. doi: 10.1016/j.chembiol.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AL, Wachter F, Lammert M, Huhn AJ, Luccarelli J, Bird GH, Walensky LD. Cellular Uptake and Ultrastructural Localization Underlie the Pro-apoptotic Activity of a Hydrocarbon-stapled BIM BH3 Peptide. ACS Chem Biol. 2015;10:2149–2157. doi: 10.1021/acschembio.5b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan G, Simmons MJ, Ge S, Dutta-Simmons J, Kucharczak J, Ron Y, Weissmann D, Chen CC, Mukherjee C, White E, et al. Defective ubiquitin-mediated degradation of antiapoptotic Bfl-1 predisposes to lymphoma. Blood. 2010;115:3559–3569. doi: 10.1182/blood-2009-08-236760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis E, Reyna DE, Davis ML, Bird GH, Walensky LD. BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol Cell. 2010;40:481–492. doi: 10.1016/j.molcel.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavathiotis E, Suzuki M, Davis ML, Pitter K, Bird GH, Katz SG, Tu HC, Kim H, Cheng EH, Tjandra N, et al. BAX activation is initiated at a novel interaction site. Nature. 2008;455:1076–1081. doi: 10.1038/nature07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq R, Yokoyama S, Hawryluk EB, Jonsson GB, Frederick DT, McHenry K, Porter D, Tran TN, Love KT, Langer R, et al. BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc Natl Acad Sci U S A. 2013;110:4321–4326. doi: 10.1073/pnas.1205575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hind CK, Carter MJ, Harris CL, Chan HT, James S, Cragg MS. Role of the pro-survival molecule Bfl-1 in melanoma. Int J Biochem Cell Biol. 2015;59:94–102. doi: 10.1016/j.biocel.2014.11.015. [DOI] [PubMed] [Google Scholar]
- Kise Y, Lee SW, Park SG, Fukai S, Sengoku T, Ishii R, Yokoyama S, Kim S, Nureki O. A short peptide insertion crucial for angiostatic activity of human tryptophanyl-tRNA synthetase. Nat Struct Mol Biol. 2004;11:149–156. doi: 10.1038/nsmb722. [DOI] [PubMed] [Google Scholar]
- Kitagawa D, Kajiho H, Negishi T, Ura S, Watanabe T, Wada T, Ichijo H, Katada T, Nishina H. Release of RASSF1C from the nucleus by Daxx degradation links DNA damage and SAPK/JNK activation. EMBO J. 2006;25:3286–3297. doi: 10.1038/sj.emboj.7601212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C, Fisher JK, Godes M, Pitter K, Kung AL, et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. 2012;122:2018–2031. doi: 10.1172/JCI46231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, Nimmer P, Jin S, Smith M, Xiao Y, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax) Cell Death Dis. 2015;6:e1590. doi: 10.1038/cddis.2014.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, Padera RF, Shapiro GI, Baum A, Himmelsbach F, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan D, Spier C, Della Croce K, Miller S, George B, Riley C, Warner S, Grogan TM, Miller TP. Transcript profiling in peripheral T-cell lymphoma, not otherwise specified, and diffuse large B-cell lymphoma identifies distinct tumor profile signatures. Mol Cancer Ther. 2005;4:1867–1879. doi: 10.1158/1535-7163.MCT-05-0146. [DOI] [PubMed] [Google Scholar]
- Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi D, Kuriyan J. Structural evidence for feedback activation by Ras.GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112:685–695. doi: 10.1016/s0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–1186. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, Zhou C, Su WC, Wang M, Sun Y, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012;13:528–538. doi: 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Pelz NF, Bian Z, Zhao B, Shaw S, Tarr JC, Belmar J, Gregg C, Camper DV, Goodwin CM, Arnold AL, et al. Discovery of 2-Indole-acylsulfonamide Myeloid Cell Leukemia 1 (Mcl-1) Inhibitors Using Fragment-Based Methods. J Med Chem. 2016;59:2054–2066. doi: 10.1021/acs.jmedchem.5b01660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitter K, Bernal F, Labelle J, Walensky LD. Dissection of the BCL-2 family signaling network with stabilized alpha-helices of BCL-2 domains. Methods Enzymol. 2008;446:387–408. doi: 10.1016/S0076-6879(08)01623-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placzek WJ, Wei J, Kitada S, Zhai D, Reed JC, Pellecchia M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 2010;1:e40. doi: 10.1038/cddis.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riker AI, Enkemann SA, Fodstad O, Liu S, Ren S, Morris C, Xi Y, Howell P, Metge B, Samant RS, et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med Genomics. 2008;1:13. doi: 10.1186/1755-8794-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016;374:311–322. doi: 10.1056/NEJMoa1513257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooswinkel RW, van de Kooij B, Verheij M, Borst J. Bcl-2 is a better ABT-737 target than Bcl-xL or Bcl-w and only Noxa overcomes resistance mediated by Mcl-1, Bfl-1, or Bcl-B. Cell Death Dis. 2012;3:e366. doi: 10.1038/cddis.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- Scott FL, Stec B, Pop C, Dobaczewska MK, Lee JJ, Monosov E, Robinson H, Salvesen GS, Schwarzenbacher R, Riedl SJ. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature. 2009;457:1019–1022. doi: 10.1038/nature07606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, Kraemer O, Himmelsbach F, Haaksma E, Adolf GR. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343:342–350. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- Stewart ML, Fire E, Keating AE, Walensky LD. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat Chem Biol. 2010;6:595–601. doi: 10.1038/nchembio.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, Argyropoulos KV, Yang G, Cao Y, Xu L, et al. Ibrutinib in previously treated Waldenstrom’s macroglobulinemia. N Engl J Med. 2015;372:1430–1440. doi: 10.1056/NEJMoa1501548. [DOI] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- Walensky LD, Bird GH. Hydrocarbon-stapled peptides: principles, practice, and progress. J Med Chem. 2014;57:6275–6288. doi: 10.1021/jm4011675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. A stapled BID BH3 helix directly binds and activates BAX. Mol Cell. 2006;24:199–210. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]
- Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:213–222. doi: 10.1016/S1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.