

Abstract
HD-GYPs make up a subclass of the metal-dependent HD phosphohydrolase superfamily and catalyze conversion of cyclic di(3′,5′)-guanosine monophosphate (c-di-GMP) to 5′-phosphoguanylyl-(3′→5′)-guanosine (pGpG) and GMP. Until now, the only reported crystal structure of an HD-GYP that also exhibits c-di-GMP phosphodiesterase activity contains a His/carboxylate ligated triiron active site. However, other structural and phylogenetic correlations indicate that some HD-GYPs contain dimetal active sites. Here we provide evidence that an HD-GYP c-di-GMP phosphodiesterase, TM0186, from Thermotoga maritima can accommodate both di- and trimetal active sites. We show that an as-isolated iron-containing TM0186 has an oxo/carboxylato-bridged diferric site, and that the reduced (diferrous) form is necessary and sufficient to catalyze conversion of c-di-GMP to pGpG, but that conversion of pGpG to GMP requires more than two metals per active site. Similar c-di-GMP phosphodiesterase activities were obtained with divalent iron or manganese. On the basis of activity correlations with several putative metal ligand residue variants and molecular dynamics simulations, we propose that TM0186 can accommodate both di- and trimetal active sites. Our results also suggest that a Glu residue conserved in a subset of HD-GYPs is required for formation of the trimetal site and can also serve as a labile ligand to the dimetal site. Given the anaerobic growth requirement of T. maritima, we suggest that this HD-GYP can function in vivo with either divalent iron or manganese occupying di- and trimetal sites.
Graphical abstract
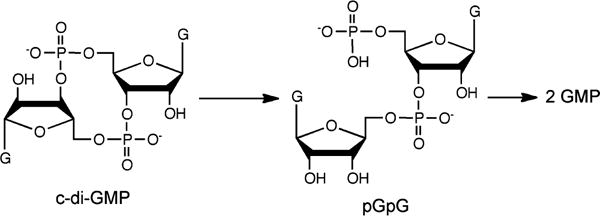
Cyclic di(3′,5′)-guanosine monophosphate (c-di-GMP) has been established as a “universal” bacterial second messenger regulating numerous cellular processes, including biofilm formation and virulence.1–6 Intracellular levels of c-di-GMP are controlled by its synthesis and degradation via diguanylate cyclases and c-di-GMP phosphodiesterases (c-di-GMP PDEs), respectively. The EAL family of c-di-GMP PDEs catalyzes conversion of c-di-GMP to pGpG, which is then further degraded, apparently, by other nonspecific PDE enzymes to GMP, as shown in Scheme 1. The other known family of c-di-GMP PDEs, termed HD-GYP because of its combination of HD and GYP amino acid sequence motifs, catalyzes conversion of c-di-GMP to GMP via pGpG,7,8 although in some cases pGpG has been reported as the only detected product.9,10 A set of HD-GYPs from Vibrio cholera has recently been reported to additionally use the mixed adenine/guanine cyclic dinucleotide, 3′,3′-cyclic GMP-AMP, as a PDE substrate in vivo.11
Scheme 1.
c-Di-GMP PDE Activity
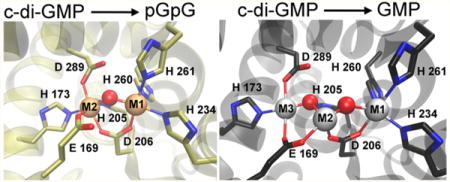
HD-GYPs make up a subclass of the metal-dependent HD phosphohydrolase superfamily.12 They typically contain N-terminal regulatory domains and C-terminal HD-GYP domains. X-ray crystal structures of three different proteins classified as HD-GYPs have been reported,8,13,14 but only one of them, PmGH from Persephonella marina, has been reported to show appreciable c-di-GMP PDE activity.8 When discussing the HD-GYP domain structures, we use the labels M1, M2, or M3 to discriminate the metal positions and to identify analogous positions in the three structures. Figure S1 shows an amino acid sequence alignment of these HD-GYP domains with the metal ligands highlighted. The PmGH crystal structure contained a non-heme triiron site with His, Asp, and Glu ligands, as shown in Figure 1A. This triiron structure also contained metal ligands from exogenous molecules, succinate and imidazole, present in the crystallization solutions (Figure S2). The crystal structure of an apparently inactive HD-GYP homologue from Bdellovibrio bacteriovorus (Bd1718) showed a non-heme His,Asp,Glu-ligated diiron site with the metals occupying the M1 and M2 positions (Figure 1B). The crystal structure of a third HD-GYP, PA4781 (Figure 1C), showed occupancy by two nickel ions, one at the M1 position and a second at approximately the same position as M3 in the PmGH structure, denoted as M3′ in Figure 1C.14 PA4781 was reported to bind various divalent metals with similar affinities and was proposed to be a pGpG binding protein. There is no structural or functional evidence of direct interactions of the GYP residues with the metals in any HD-GYP. When the Escherichia coli expression medium for PmGH was supplemented with a divalent manganese salt, a crystal structure identical to that in Figure 1A was obtained in which the M2 site retained iron, but the M1 and M3 sites were occupied by manganese.8 We have previously reported that an iron-containing HD-GYP from V. cholerae has c-di-GMP PDE activity and a UV–vis absorption spectrum characteristic of a diiron active site structure similar to that of Bd1718 (Figure 1B).10 The D of the HD motif provides a carboxylate bridge between two metal ions in all three crystal structures. When a solvent bridge is also present, the D bridges M1 and M2. The M1 and M3 sites are too far apart to accommodate a solvent bridge between them. Substitution of the D of the HD motif with A in other HD-GYPs invariably leads to loss of c-di-GMP PDE activity.7,8,10
Figure 1.
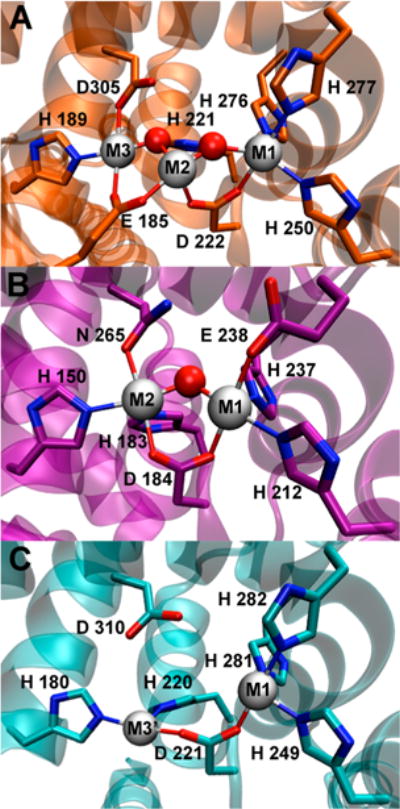
HD-GYP crystal structures containing a (A) triiron site in Persephonella marina PmGH (PDB entry 4MCW), (B) diiron site in B. bacteriovorus Bd1817 (PDB entry 3TMB), or (C) dinickel site in Pseudomonas aeruginosa PA4781 (PDB entry 4R8Z). Gray spheres represent metal positions, and red spheres represent metal-bridging solvent ligands. For the sake of clarity, exogenous succinate and imidazole ligands (shown in Figure S2) are omitted from the depiction of the triiron (4MCW) structure in panel A, a bridging phosphate ligand is omitted from the depiction of the Bd1817 (3TMB) structure in panel B, and solvent ligands (all nonbridging) are omitted from the depiction of the dinickel (4R8Z) structure in panel C.
How the nature of the metal, occupancy of the metal sites, and active site structure affect the successive PDE reactions shown in Scheme 1 is, thus, not clear. We report here our investigations of these issues using an HD-GYP from T. maritima (TM0186). The recombinant TM0186 was previously reported to contain one iron per protein monomer and to show c-di-GMP PDE activity with pGpG as the product.9 All the ligand residues in the PmGH triiron crystal structure (Figure 1A) are conserved in TM0186 (Figure S1). However, except for PmGH E185, which provides an M2,M3 bridging carboxylate (Figure 1A), all the metal ligand residues in the triiron PmGH structure are homologous to the M1,M2 ligand residues of the diiron site in the Bd1817 structure (Figure 1B). On the basis of a phylogenetic analysis, Bellini et al.8 proposed that HD-GYPs capable of forming a trimetal M1,M2,M3 active site contain a Glu residue homologous to E185 in PmGH, whereas HD-GYPs lacking this Glu more likely contain a M1,M2 dimetal site analogous to that in Bd1718. On this basis, TM0186, which has the E169 residue homologous to PmGH E185, was classified as a trimetal HD-GYP. To correlate activity with residues that could potentially provide ligands to an M3 metal in TM0186, we examined three variants, E169A, H173A, and D289A (starred in Figure S1), corresponding to E185, H189, and D305, respectively, in the M3 coordination sphere of the PmGH triiron site (Figure 1A). In this report, we show that TM0186 is capable of forming a solvent/carboxylate-bridged dimetal site and also provide evidence of binding of a third metal at the M3 position. We also correlate c-di-GMP PDE activities and products with metal site occupancy and the nature of the metal.
MATERIALS AND METHODS
Reagents
Reagents and buffers were of the highest grade commercially available. All reagents, protein, and medium solutions were prepared using water purified with a Millipore ultrapurification system to a resistivity of 18 MΩ to minimize trace metal ion contamination. c-Di-GMP and pGpG were purchased from Axxora, LLC (Farmingdale, NY), and GMP was from Acros Organics.
Protein Expression
Genes encoding the amino acid sequence of TM0186 and its E169A, H173A, and D289A variants with codons optimized for expression in E. coli were synthesized and inserted by Genscript (Piscataway, NJ) into a previously reported expression plasmid encoding an N-terminal six-His-MBP tag.10 These plasmids were transformed into E. coli strain BL21 pLysE DE3 (Invitrogen). Transformed cells were used to inoculate 50 mL of Luria-Bertani broth containing 100 μg/mL amp and 35 μg/mL Cm (LB/amp/Cm). This 50 mL culture was incubated while being shaken for approximately 12 h at 37 °C. Each of three flasks containing 1 L of M9 medium/amp/Cm was inoculated with approximately 15 mL of the 50 mL culture. The inoculated 1 L cultures were incubated while being shaken at 37 °C until the OD600 reached ~0.8. The temperature of the cultures was then lowered to 25 °C; 100 mg/L ferrous ammonium sulfate or 100 mg/L manganese chloride and 125 mg/L isopropyl β-D-thiogalactoside were added, and the cultures were incubated while being shaken for a further 18 h at 25 °C. Cells were then harvested by centrifugation at 8000g and frozen at −80 °C. This same protein expression protocol was used for the E169A, H173A, and D289A variants.
Protein Isolation and Purification
The frozen cell pellets from the protein expression cultures were thawed and suspended in 50 mM MOPS containing 250 mM NaCl (pH 7.3) (buffer A) at 5 mL/g of cell pellet, typically to a total volume of ~150 mL. The suspended cells were lysed by sonication; the cell debris was removed by centrifugation at 20000g, and the supernatant was diluted to 500 mL with buffer A. A few grains of ferricyanide were added to the diluted supernatants from the ferrous ammonium sulfate-spiked cultures. The diluted supernatant was passed over a 10 mL column of fast flow amylose resin (New England Biolabs) equilibrated with buffer A at 5 mL/min. The resin was then washed with buffer A until the A280 of the flow-through was less than 0.1 AU. Protein was eluted from the resin using 10 mM maltose in buffer A. Fractions with A280 values above baseline were evaluated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Fractions containing a predominant band at ~90000 Da (the approximate size of the six-His-MBP-TM0186) were pooled and concentrated using a 15 mL centrifugal concentrator with a 30 kDa MWCO (Millipore). The protein solution was diluted and reconcentrated multiple times with 50 mM Tris, 50 mM NaCl, and 10 mM MgCl2 (pH 7.6). The same protocol was used for the purification of the E169A and H173A variants.
Protein and Metal Quantifications
Protein concentration was determined by a bicinchoninic acid assay (Pierce) using bovine serum albumin as a standard, and iron content was assessed using a standard ferrozine assay. Further metal analysis was conducted using inductively coupled plasma-optical emission spectrometry at the Center for Applied Isotope Studies at the University of Georgia (Athens, GA).
UV–Vis Absorption Spectra
UV–vis absorption spectra were recorded using an Ocean Optics QE65 Pro or an Ocean Optics USB 2000 spectrophotometer.
Redox Cycling
The buffer for all solutions consisted of 50 mM Tris, 50 mM NaCl, and 10 mM MgCl2 (pH 7.6), and all samples were manipulated at room temperature unless explicitly stated otherwise; ~50 μL volumes of either Fe- or Mn-TM0186 at ~300 μM protein (monomer basis) were allowed to equilibrate with an anaerobic N2 atmosphere in a glovebox (Vacuum Atmospheres Co.) for 1 h, after which the solution vials were closed and left in the glovebox overnight at room temperature. The protein was then diluted to 50 μM with anaerobic buffer in a rubber-septum screw top 1 cm path length quartz cuvette, and its UV–vis absorption spectrum was recorded in the glovebox. The Fe-TM0186 was then treated with 20 molar equiv of sodium dithionite from a freshly prepared anaerobic stock solution. After 30 min, the dithionite was removed by several dilution/reconcentration cycles using a Millipore Amicon centrifugal filter with a 30 kDa MWCO, and a UV–vis absorption spectrum of the anaerobic dialyzed solution was recorded. The solution was then removed from the glovebox and exposed to air for at least 3 h, after which another spectrum was obtained. For some experiments, 1 equiv of Fe2+ was added to 100 μM reduced protein after removal of dithionite, and the solution was allowed to incubate for 30 min at 37 °C anaerobically prior to removal from the glovebox, re-exposure to air, dilution to 50 μM, and recording of a spectrum.
c-Di-GMP PDE Activities
All solutions were manipulated under a N2 atmosphere in the glovebox. Dithionite-treated Fe-TM0186 samples were prepared as described above. Activities were measured at 37 °C with 2 μM protein (monomer basis) in 50 mM Tris (pH 7.6), 50 mM NaCl, and 10 mM MgCl2 after removal of dithionite. The reaction was initiated by addition of c-di-GMP to a concentration of 100 μM in a final volume of 250 μL. The effects of excess divalent iron or manganese were examined by adding either ferrous ammonium sulfate or manganous chloride from a concentrated stock solution to achieve a concentration of 80 μM or 10 mM, respectively, and incubating the resulting assay solution for 30 min at 37 °C prior to addition of protein and nucleotide substrate. At various times following addition of c-di-GMP, 75 μL aliquots were transferred from the assay solution to Eppendorf tubes, where the reaction was quenched by addition of 2% TFA. After a 5 min centrifugation at maximal speed in an Eppendorf 5418 centrifuge, the supernatant was filtered through a 13 mm, 0.22 μm syringe filter (Fisher Scientific, Millex GV), and 20 μL of the filtrate was injected onto a Dionex Ultimate 3000 HPLC system with a Dionex Acclaim 120 reverse phase C18 HPLC column fitted with an Acclaim 120 C18 guard cartridge. The column was pre-equilibrated with 92% 20 mM potassium phosphate (pH 6.0) and 8% methanol. A mobile phase of 92% 20 mM potassium phosphate (pH 6.0) and 8% methanol in an isocratic flow was used for nucleotide elution, which was monitored by absorbance at 254 nm. Control experiments without protein were conducted analogously and run in parallel. Elution times were calibrated with pure commercial samples of the nucleotides. The “0 min” label in the figures indicates chromatograms obtained on samples withdrawn and TFA quenched within ~30 s of addition of c-di-GMP.
Structural Models
Molecular dynamics simulations were used to generate structural models of TM0186 containing a trimetal site analogous to that in PmGH (Figure 1A) and a dimetal site analogous to that in Bd1817 (Figure 1B). The programs visual molecular dynamics (VMD)15 and nanoscale molecular dynamics (NAMD)16 were used to prepare files and conduct simulations. The input file was a TM0186 structure homology model generated from its amino acid sequence using the SWISS-MODEL web server (http://swissmodel.expasy.org/) and the PmGH trimetal protein crystal structure (PDB entry 4MCW) as a template. A generic divalent closed shell metal ion was inserted into the M1 and M3 positions of this TM0186 homology model based on a structural alignment with the PmGH trimetal protein. To generate a TM0186 dimetal HD-GYP domain model, the M2 metal was replaced with a water molecule, and M1 and M3 ligand bonds and bridging solvent were constrained to those analogous to the dimetal site of Bd1817. A more detailed description is provided in the Supporting Information.
RESULTS AND DISCUSSION
Protein Isolation and Metal Contents
Recombinant TM0186, and engineered E169A, H173A, and D289A variants, were overexpressed as N-terminal six-His-MBP-tagged proteins and isolated from E. coli cultures. To avoid the possibility of metal contamination from the His tag affinity column, we purified these proteins using only amylose affinity resin, which utilizes the binding of MBP to amylose. We found that protease cleavage of this six-His-MBP tag following purification sometimes resulted in protein precipitation. For this reason, we used the six-His-MBP-tagged proteins for the experiments reported here.
Iron and manganese contents of the as-isolated TM0186 and variants are listed in Table 1. All of the as-isolated proteins examined here contained <0.1 per protein monomer of Zn, Co, or Ni. Incorporation of predominantly iron or manganese into the as-isolated proteins was achieved by addition of either ferrous ammonium sulfate or manganous chloride, respectively, to the culture medium at the time of induction of protein expression. Potassium ferricyanide was added to the cell extracts from the iron-spiked cultures to convert protein-bound iron to the less labile ferric oxidation state, which minimized iron loss during purification. Protein expressed and isolated in this fashion contained 2.2 irons/protein monomer and <0.1 Mn/protein monomer. This protein is hereafter termed as-isolated Fe-TM0186. Omitting ferricyanide treatment of the cell extract yielded protein containing approximately 1 iron per protein monomer, as reported previously.9 The TM0186 isolated from the Mn-spiked cultures contained 2.4 Mn atoms/protein monomer and <0.1 Fe/protein monomer. Protein expressed and purified in this fashion is termed Mn-TM0186. The metal content of E169A TM0186 isolated from iron-spiked cultures was indistinguishable from that of TM0186 (Table 1). E169A TM0186 from the manganese-spiked cultures contained 1.6 Mn atoms and ~0.1 Fe per protein monomer. The H173A TM0186 isolated from the iron- or manganese-spiked cultures contained 0.7–0.8 Fe or Mn, respectively, per protein monomer. Spiking cultures with zinc sulfate in place of iron or manganese salts did not result in significant incorporation of zinc into TM0186. We have previously reported10 that the six-His-MBP with no other protein domains, when expressed and isolated as described for the tagged TM0186, contained no detectable iron or UV–vis absorption spectral features other than the protein absorption at 280 nm. We have also verified that the analogously expressed six-His-MBP contains no detectable manganese upon being isolated from Mn-spiked cultures.
Table 1.
Iron and Manganese Content per Protein Monomer for TM0186 and Its Variants
| Fe | Mn | |
|---|---|---|
| Fe- | 2.2 | <0.1 |
| Mn- | 0.1 | 2.4 |
| E169A Fe- | 2.2 | <0.1 |
| E169A Mn- | 0.1 | 1.6 |
| H173A Fe- | 0.7 | <0.1 |
| H173A Mn- | <0.1 | 0.8 |
| D289A Fe- | 1.1 | <0.1 |
| D289A Mn- | <0.1 | 1.2 |
UV–Vis Absorption Spectra
The UV–vis absorption spectra of as-isolated Fe-TM0186 (Figure 2A) contained two features between 300 and 400 nm (ε350 ~ 7100 M−1 cm−1 per 2 Fe) and a weak shoulder between 460 and 500 nm. This spectrum constitutes a well-established signature of non-heme oxo/carboxylato-bridged diferric sites with terminal histidine and Asp/Glu-carboxylate ligands having a structure such as that shown in Figure 1B for the Bd1817 HD-GYP (PDB entry 3TMB).10,17,18 These characteristic spectral features were previously reported for the same TM0186 examined here but containing ~1 Fe/protein monomer rather than the 2.2 Fe atoms in our preparations.9 These similar spectral features are consistent with “all-or-nothing” formation of the diiron sites. The weak, featureless UV–vis absorption of the dithionite-treated Fe-TM0186 obtained on an anaerobic solution after removal of dithionite is consistent with reduction to the diferrous form. Re-exposure of this solution to air restored the diferric spectral features with nearly all the original intensity (Figure 2A), which supports reversible diferric/diferrous interconversion. The dithionite-treated/removed protein is hereafter termed “reduced Fe-TM0186”. The UV–vis absorption of Mn-TM0186 (Figure 2A) is relatively weak and featureless, as expected for Mn2+ coordinated by His and carboxylate ligands. These spectra serve as benchmarks for the activity and spectral correlations described below.
Figure 2.
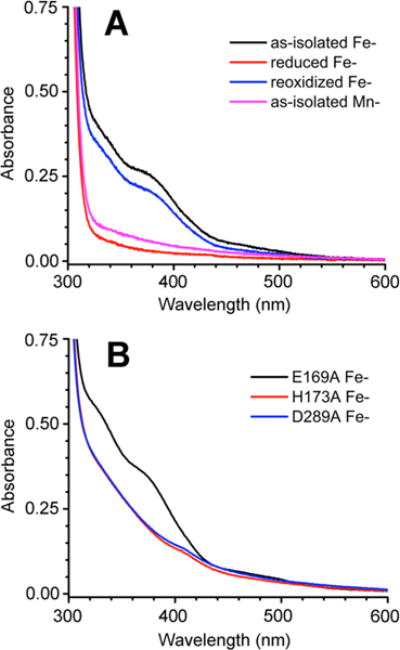
UV–vis absorption spectra of (A) Fe- and Mn-TM0186 and (B) E169A and H173A Fe-TM0186. Proteins were at a concentration of 50 μM in 50 mM Tris, 50 mM NaCl, and 10 mM MgCl2 (pH 7.6). The weak absorption feature near 410 nm in the H173A and D289A Fe-TM0186 spectra is due to a very small amount of contaminating heme.
Addition of 10 mM Mn2+ to the as-isolated Fe-TM0186 resulted in spectral changes (Figure S3A) indicating disruption of the oxo/carboxylato-bridged diferric site, which is consistent with some Mn2+ displacement of Fe3+. Addition of 1 equiv of Fe2+ to an anaerobic solution of reduced Fe-TM0186 and subsequent air oxidation yielded a UV–vis absorption spectrum that contained features of the oxo/carboxylato-bridged diferric site superimposed on a sloping less-featured absorption spectrum (Figure S3B), characteristic of a mononuclear His/carboxylate-ligated ferric iron site.
For reference, TM0186 residues E169, H173, and D289 are homologous to M3 ligand residues E185 (bridging to M2), H189, and D305, respectively, in the PmGH crystal structure (Figure 1A). TM0186 residues H173 and D289 are homologous to H180 and N265 ligand residues of M2 in the Bd1817 crystal structure (Figure 1B) (Bd1817 contains no E169 homologue). The UV–vis absorption spectrum of as-isolated E169A Fe-TM0186 (Figure 2B) contained the two spectral features between 300 and 400 nm indicating conservation of the oxo/carboxylato-bridged diferric site. Comparison of the E169A Fe-TM0186 spectrum with the Fe-TM1086 spectrum (Figure 2A) cannot distinguish whether E169 is a ligand to the diferric site. The UV–vis absorption spectra of as-isolated H173A Fe-TM0186 and D289A Fe-TM0186 (Figure 2B) contained less distinct features between 300 and 400 nm, which are consistent with His/carboxylate-ligated ferric centers but inconsistent with oxo/carboxylato-bridged diferric sites. These less featured spectra are consistent with disruption of the diferric site upon substitution of H173 or D289 with the nonligating Ala side chain, and also with the ~0.7 and ~1 iron per protein monomer in H173A and D289A Fe-TM0186, respectively.
c-Di-GMP PDE Activities
Conditions
A qualitative summary of activities determined in this study is compiled in Table 2. To test the metal dependence of c-di-GMP activities, we either omitted or added separately the 10 mM MnCl2 often present in c-di-GMP PDE assay solutions.7,8 For the same reason, we also omitted another comment ingredient, the metal-chelating EDTA. Under these conditions, no c-di-GMP consumption was observed in the presence of excess Fe2+ or Mn2+ in the absence of protein (Figure S4). Given the air sensitivity of the diferrous site in reduced Fe-TM0186, all PDE activities reported here were measured under a N2 atmosphere to compare activities under analogous conditions. No significant difference was observed for c-di-GMP PDE activities of Mn-TM0186 under aerobic versus anaerobic conditions. We also compared the effects of excess divalent Fe2+ or Mn2+ added prior to addition of c-di-GMP. Using >80 μM Fe2+ in the assay mixture sometimes resulted in a slow precipitation of the iron in the sample after exposure to air, which interfered with HPLC; 80 μM Fe2+ was chosen to ensure a significant excess over the 2 μM protein. We, therefore, compared activities at 80 μM Fe2+ with those at 10 mM Mn2+. Finally, we quenched our samples using TFA, as previously reported for TM0186,9 rather than the more commonly used heat quenching.7,8 TFA quenching avoids the possibility that TM0186, which is from a hyperthermophilic bacteria, retains some activity during heating.
Table 2.
Qualitative c-Di-GMP PDE Activities of TM0186 and Its Variants
| TM0186 or varianta | form | excess metal addedb | c-di-GMP PDE productsc
|
|
|---|---|---|---|---|
| pGpG | GMP | |||
| Fe- | as-isolated | none | − | − |
| Mn | + | − | ||
| reduced | none | + | − | |
| Mn or Fe | + | + | ||
| Mn- | as-isolated | none | + | + |
| Mn or Fe | + | + | ||
| E169A Fe- | as-isolated | none | − | − |
| Mn | + | − | ||
| reduced | none | + | − | |
| Mn or Fe | + | − | ||
| E169A Mn- | as-isolated | none | + | − |
| Mn or Fe | + | − | ||
| H173A Fe- | as-isolated | none | − | − |
| Mn | + | − | ||
| reduced | none | − | − | |
| Fe | <+ | − | ||
| Mn | + | − | ||
| H173A Mn- | as-isolated | none | − | − |
| Fe | <+ | − | ||
| Mn | + | − | ||
| D289A Fe- | as-isolated | none | − | − |
| Mn | − | − | ||
| reduced | none | − | − | |
| Fe | − | − | ||
| Mn | − | − | ||
| D289A Mn- | as-isolated | none | − | − |
| Fe | − | − | ||
| Mn | − | − | ||
At 2 μM (monomer basis).
At 80 μM Fe2+ or 10 mM Mn2+.
Products observed 120 min after addition of c-di-GMP: −, no product; +, product; <+, low yield of product.
Fe-TM0186
HPLC traces monitoring activities of Fe-TM086 are shown in Figure 3. The as-isolated (predominantly diferric) Fe-TM0186 showed very little consumption of c-di-GMP over 120 min (Figure 3A). A 30 min preincubation of as-isolated Fe-TM0186 with 10 mM MnCl2 prior to addition of c-di-GMP resulted in complete conversion of the c-di-GMP to pGpG within 30 min, but no production of GMP was observed over a period of 120 min (Figure 3B). The reduced Fe-TM0186 converted all the c-di-GMP to pGpG within 30 min, but no significant GMP was produced over 120 min (Figure 3C). Upon preincubation of the reduced Fe-TM0186 with 80 μM ferrous ammonium sulfate, all the starting c-di-GMP (100 μM) was converted to pGpG within 30 min, and ~70% of this enzymatically generated pGpG was converted to GMP over 120 min (Figure 3D). Essentially complete conversion of 500 μM c-di-GMP to GMP was observed within 30 min when 100 μM reduced Fe-TM0186 was preincubated with 1 equiv of Fe2+ followed by addition of the c-di-GMP (Figure S3C). A UV–vis absorption spectrum was obtained on a portion of this same solution (Figure S3B, green trace), and the spectrum indicated that the oxo/carboxylato-bridged diferric site was preserved along with an additional ferric site after c-di-GMP turnover and re-exposure to air. Preincubation of 2 μM reduced Fe-TM0186 with 10 mM MnCl2 stimulated conversion of ~30% of the enzymatically generated pGpG to GMP over 120 min (Figure 3E).
Figure 3.
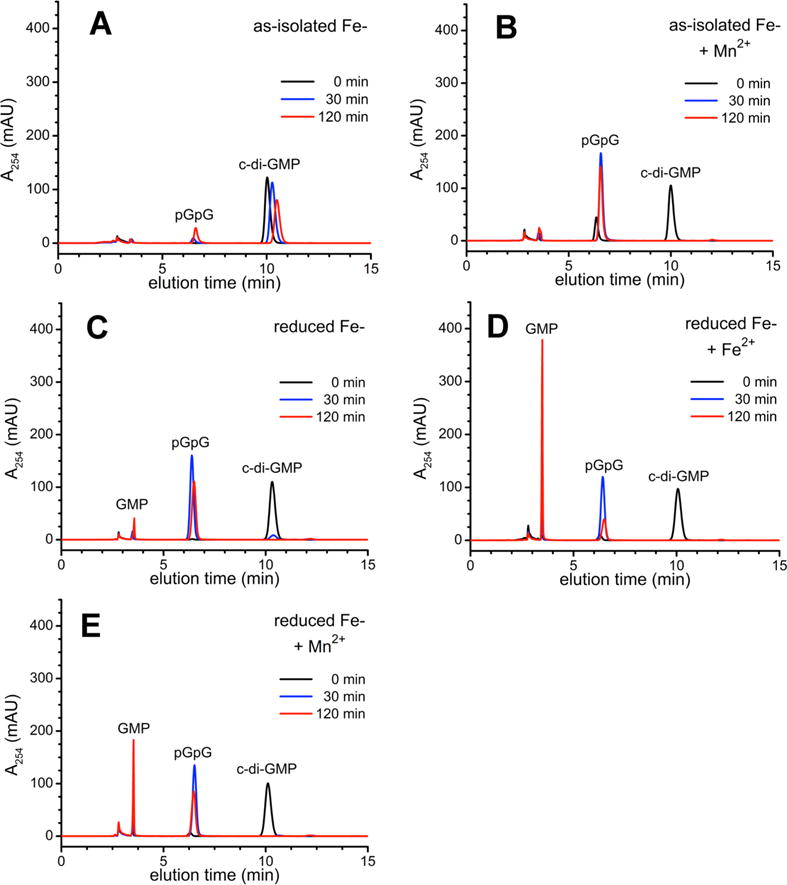
HPLC chromatograms for c-di-GMP PDE activities of Fe-TM0186: (A) as-isolated, (B) as-isolated and preincubated with 10 mM manganous chloride (+Mn2+), (C) reduced, (D), reduced and preincubated with 80 μM ferrous ammonium sulfate (+Fe2+), or (E) reduced and preincubated with 10 mM manganous chloride (+Mn2+), and sampled at the indicated times following addition of c-di-GMP.
Mn-TM086
Figure 4 shows HPLC traces monitoring activity of Mn-TM0186. As-isolated 2 μM Mn-TM0186 converted 100 μM c-di-GMP completely to pGpG within 30 min, and ~20% of the pGpG was converted to GMP within 120 min (Figure 4A). Preincubation of Mn-TM0186 with either 80 μM ferrous ammonium sulfate or 10 mM MnCl2 stimulated production of GMP via pGpG to ~45% (Figure 4B) and ~35% (Figure 4C), respectively.
Figure 4.
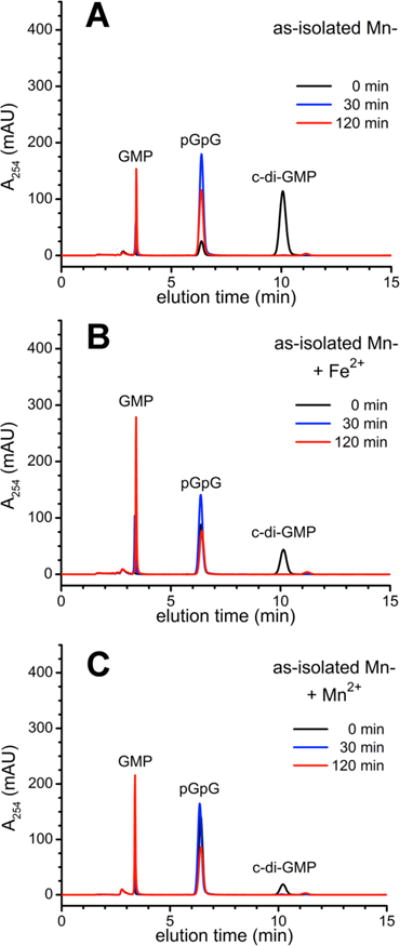
HPLC chromatograms for c-di-GMP PDE activities of Mn-TM0186: (A) as-isolated, (B) as-isolated and preincubated with 80 μM ferrous ammonium sulfate (+Fe2+), or (C) as-isolated and preincubated with 10 mM manganous chloride (+Mn2+).
E169A Variant
The PDE activities of the as-isolated and reduced E169A Fe-TM0186 (Figure 5) paralleled those of the corresponding redox states of Fe-TM0186; i.e., the as-isolated E169A variant showed no activity without added 10 mM Mn2+, and the reduced E169A variant completely converted c-di-GMP to pGpG. However, unlike the behavior of Fe-TM0186, preincubation of reduced E169A Fe-TM0186 with either 80 μM ferrous ammonium sulfate or 10 mM MnCl2 did not stimulate conversion of enzyme-generated pGpG to GMP. As-isolated E169A Mn-TM0186 (2 μM) completely converted 100 μM c-di-GMP to pGpG within 30 min (Figure 6), but here again, preincubation of E169A Mn-TM0186 with either 80 μM Fe2+ or 10 mM Mn2+ failed to stimulate GMP production.
Figure 5.
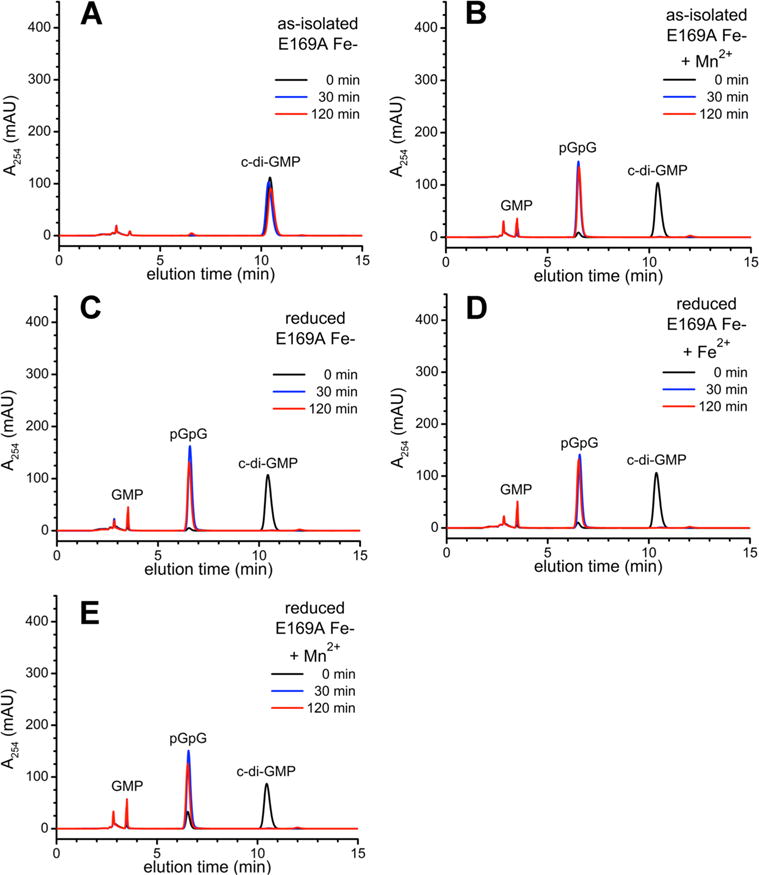
HPLC chromatograms for c-di-GMP PDE activities of E169A Fe-TM0186: (A) as-isolated, (B) as-isolated and preincubated with 10 mM manganous chloride (+Mn2+), (C) reduced, (D), reduced and preincubated with 80 μM ferrous ammonium sulfate (+Fe2+), or (E) reduced and preincubated with 10 mM manganous chloride (+Mn2+), and sampled at the indicated times following addition of c-di-GMP.
Figure 6.
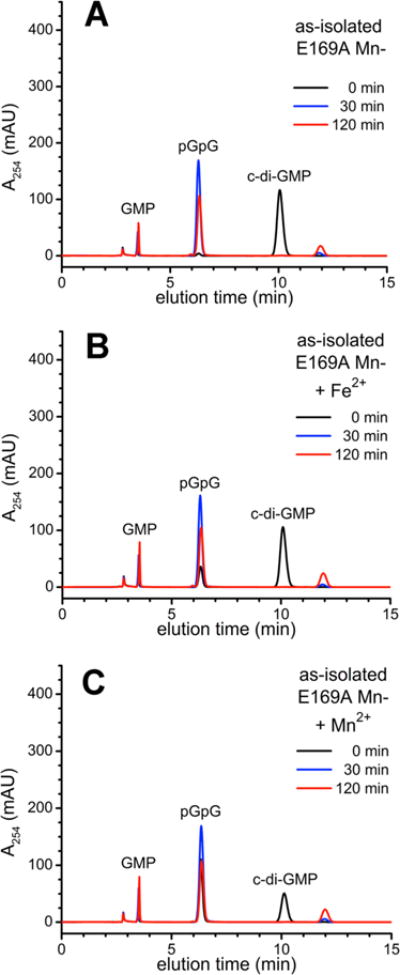
HPLC chromatograms for c-di-GMP PDE activities of E169A Mn-TM0186: (A) as-isolated, (B) as-isolated and preincubated with 80 μM ferrous ammonium sulfate (+Fe2+), or (C) as-isolated and preincubated with 10 mM manganous chloride (+Mn2+).
H173A Variant
Neither as-isolated nor reduced H173A Fe-TM0186 showed any c-di-GMP PDE activity over at least 120 min (Figure S5). Preincubation of H173A Fe-TM0186 with either 80 μM Fe2+ or 10 mM Mn2+ stimulated relatively slow conversion to pGpG but no production of GMP. Similar behavior was observed for H173A Mn-TM0186 (Figure S6). This residual activity presumably arises from the 0.7–0.8 metal/monomer in this variant.
D289A Variant
Neither as-isolated nor reduced D289A Fe-or Mn-TM0186 showed any c-di-GMP PDE activity over at least 120 min (Figures S7 and S8, respectively). Preincubation of reduced D289A Fe- or Mn-TM0186 with either 80 μM Fe2+ or 10 mM Mn2+ did not stimulate c-di-GMP PDE activity (Figure S8). This lack of activity was observed despite the 1.1–1.2 irons or manganeses/protein monomer observed in this as-isolated variant.
Correlations of c-Di-GMP PDE Activities with Di- and Trimetal Active Sites
The UV–vis absorption spectra of Fe-TM0186 establish the presence of a solvent- and carboxylate-bridged diiron site resembling that in Bd1817 (Figure 1B). The reduced Fe-TM0186 can catalyze conversion c-di-GMP to pGpG, but conversion to GMP required excess Fe2+ or Mn2+ and occurred only after complete conversion of c-di-GMP to pGpG. Similar activities were observed with Mn-TM0186 containing ~2.4 Mn2+ atoms/protein monomer.
On the basis of these activity correlations, we propose that in TM0186 an M1,M2 diiron or dimanganese site resembling that in Bd1817 is necessary and sufficient to catalyze conversion of c-di-GMP to pGpG, but conversion of pGpG to GMP requires an M1,M2,M3 occupied trimetal site resembling that in PmGH (Figure 1A). We used molecular dynamics simulations to test the possibility that a TM0186 HD-GYP domain structure resembling that of the triiron PmGH could accommodate both tri- and dimetal sites. A homology model of the TM0186 HD-GYP domain was first generated using the PmGH triiron structure (PDB entry 4MCW) as a template. This homology model then had divalent metal ions and water molecules inserted at the M1, M2, and M3 iron atom and bridging solvent positions, based on a root-mean-square deviation structural alignment with the PmGH structure. Figure 7A shows this trimetal site model for TM0186, and Figure S9 shows its overlay on the PmGH trimetal site. (Nonbridging water molecules were also added to the TM0186 trimetal model in place of the exogenous succinate and imidazole ligand atoms in the PmGH structure but are omitted from Figure 7 for the sake of clarity.) A water molecule was then substituted at the M2 position of this TM0186 model, and distance and angle constraints were imposed between the remaining M1 and M3 metals, the protein ligands, and the substituted water to mimic the corresponding interactions in the M1,M2 dimetal site of the Bd1817 structure. Because the Bd1817 structure does not contain a residue homologous to E169, no constraints were imposed on this residue. All atoms in the molecular dynamics simulation were then allowed to move. More details of the simulations are described in the Supporting Information. The resulting TM0186 dimetal site in the model is shown in Figure 7B, and an overlay of the TM0186 di- and trimetal site models is shown in Figure 7C. The dimetal site structure model is homologous to that in Bd1718, except that the E169 is a ligand to M2. A superimposition of the corresponding di- and trimetal protein backbones is shown in Figure S10. An analogous molecular dynamics simulation was performed on the HD-GYP domain of the PmGH 4MCW structure, which is a dimer and has a triiron site in both subunits. Dimetal site structures resembling that in Bd1817 were again obtained. The dimetal PmGH model and its backbone superposition on the triiron PmGH crystal structure are shown in Figure S11. These simulations indicate that, unlike the Bd1817 structure, the substrate entry and binding channel to the active site could remain open in the M1,M2 dimetal forms of TM0186 and PmGH.
Figure 7.
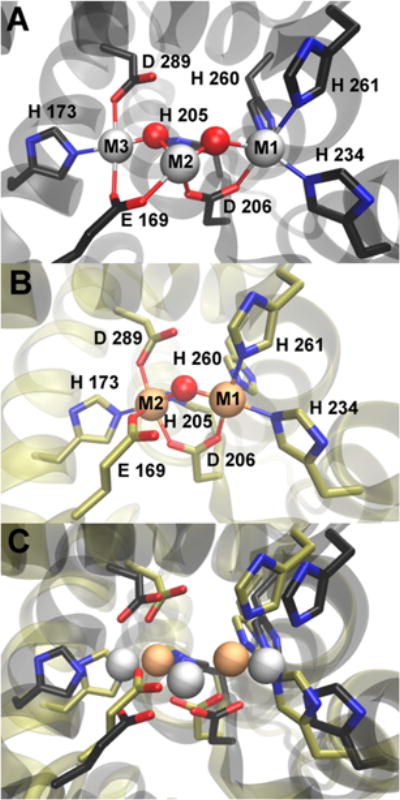
(A) Trimetal site in the TM0186 HD-GYP domain structural model. (B) Dimetal TM0186 site structure obtained from the molecular dynamics simulation. Lines represent M1-O/N or M2-O/N interatomic distances within the range 2.0–2.2 Å. (C) Overlay of the trimetal and dimetal TM0186 structures from panels A and B. In all panels, red and blue colors represent oxygen and nitrogen atoms, respectively. Red spheres represent metal-bridging solvent oxygens. Gray or tan spheres represent metal atoms in the tri- or dimetal sites, respectively.
The modeling thus indicates that the M3 ligand residues, E169, H174, and D289, in TM0186 can move to positions within ligating distance of M2 in a dimetal site and that this movement can be accomplished with only localized movements of the protein backbone. The TM0186 dimetal model showed the homologous E169 as a ligand to M2 even though no constraints were applied to induce this interaction. The M2 is, thus, six-coordinate in this model, which would seem to preclude substrate binding. However, the UV–vis absorption spectra and activity assays show that E169 in TM0186 is not required for oxo/carboxylato-bridged diiron site formation or for c-di-GMP-to-pGpG activity. Interestingly, the M2,M3 bridging E185 residue (analogous to E169 in TM0186) in the triiron PmGH crystal structure was found to be a ligand to M2 in one subunit of the PmGH dimetal model but was not a metal ligand in the other subunit (Figure S11). This observation and our spectral/activity correlations of the E169A variant could imply that E169 is either not coordinated or relatively weakly coordinated to M2, dissociates upon substrate binding, and remains dissociated during turnover to pGpG. E169A TM0186 cannot catalyze the second phosphodiester cleavage step, pGpG to GMP, even with excess metal ion. This observation supports our proposal that an M1,M2,M3 trimetal site, in which the E169 carboxylate bridges the M2,M3 pair, is required for pGpG phosphodiester cleavage. We found that TM0186 residues H173 and D289 were required for full dimetal occupancy and for any appreciable c-di-GMP PDE activity even with excess divalent metal. These observations are consistent with H173 and D289 supplying ligands to M2 in the dimetal site and to M3 in the trimetal site, as shown in Figure 7. A possible pathway for conversion of the M1,M2 dimetal site to a trimetal M1,M2,M3 site is attack of the incoming metal on the solvent bridge between M1 and M2 leading to insertion of this metal between M1 and M2 with concomitant movement of M2 to the M3 position. Alternatively, the incoming metal may initially bind to the E169 carboxylate, leading to entry of this metal directly into the M3 position.
Scheme 2 summarizes a proposed c-di-GMP phosphodiester cleavage pathway consistent with our results. The quantitative accumulation of a large excess of pGpG over enzyme and its subsequent conversion to GMP indicate that the consecutive c-di-GMP/pGpG phosphodiester cleavage pathway in TM0186 is distributive rather than processive. As suggested by others,19–21 c-di-GMP phosphodiester cleavage could be initiated by nucleophilic attack of a bridging hydroxo on a dimetal-bridging phosphodiester. We propose that this initial phosphodiester cleavage occurs at the M1,M2 dimetal form of the active site in TM0186. Subsequent pGpG phosphodiester cleavage could occur at either the M1,M2 or M2,M3 pair following insertion of the third metal. The additional possibility of a pGpG 5′-phosphate bridge across the M1,M2 or M2,M3 pairs is indicated by the dashed lines in Scheme 2. This possibility is consistent with the structure of PmGH obtained from crystals soaked in GMP (PDB entry 4ME4), which shows the phosphate of GMP (the product of pGpG phosphodiester cleavage) bridging the M2,M3 pair in the triiron site.8 Scheme 2 implies that the active site cycles between dimetal and trimetal forms during turnover, although our results do not rule out the possibility that c-di-GMP cleavage could additionally occur at the M1,M2 pair in the trimetal site.
Scheme 2.
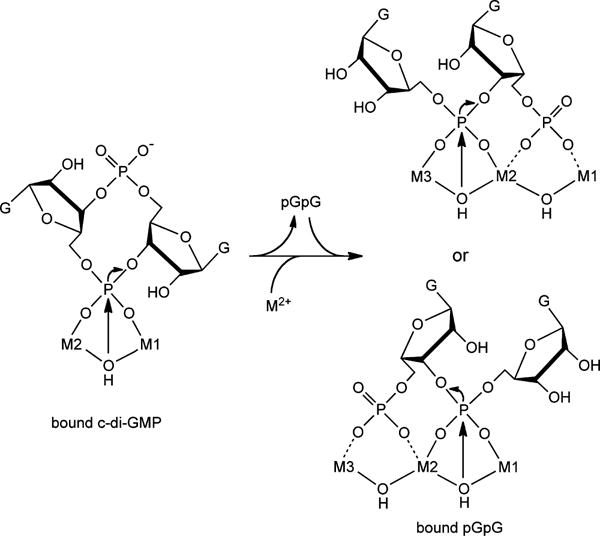
Proposed c-Di-GMP and pGpG Phosphodiester Cleavages in TM0186
Implications for HD-GYP Cyclic Dinucleotide PDE Activities in Vivo
This work represents the first clear demonstration of solely iron-based and solely manganese-based c-di-GMP PDE activity in the same HD-GYP and demonstrates that the activities with these two divalent metals are similar to each other. Manganese-containing PDEs are often reported to be more active than their iron-containing counterparts, but this observation may be due to the lower activity of the air stable trivalent (ferric) than air-sensitive divalent (ferrous) iron.22 We have in fact previously demonstrated this iron oxidation state/c-di-GMP PDE activity dependence for a dimetal HD-GYP from V. cholerae.10 The bacterial intracellular concentration of “free” ferrous iron under aerobic growth conditions is estimated to be ~10 μM,23 which is on the same order of magnitude as the 80 μM Fe2+ that we found can stimulate anaerobic conversion of c-di-GMP to GMP by Fe- or Mn-TM0186. It has recently been recognized that nonredox bacterial enzymes and some metalloregulatory proteins can function in vivo with either divalent iron or manganese depending on the availability of the metal, and intracellular Fe2+ availability tends to be relatively higher in reducing environments.24,25 T. maritima is classified as an anaerobe, although it can grow under an atmosphere containing low levels of O2.26,27 Given this reducing growth environment and our results showing comparable c-di-GMP PDE activity with divalent iron and manganese, TM0186 could function with di- and trimetal active sites occupied by either divalent iron or manganese in vivo and could switch between dimetal and trimetal forms depending on intracellular metal availability. TM0186 contains an N-terminal REC regulatory domain, which has been suggested to regulate substrate accessibility to the active site in other HD-GYPs.14 Activity may also be modulated by interactions with partner regulatory proteins or cofactors.28 The T. maritima genome contains nine other annotated but so far uncharacterized HD-GYP domain proteins.29 Only one of these nine contains a Glu residue homologous to E169 in TM0186. T. maritima may, therefore, contain both di- and trimetal HD-GYPs. These redox and metal occupancy/activity correlations add yet another layer onto the complex regulation of bacterial cyclic dinucleotide levels.
Supplementary Material
Acknowledgments
Funding
This work was supported by National Institutes of Health Grant R01 GM040388 to D.M.K.
ABBREVIATIONS
- amp
ampicillin
- Cm
chloramphenicol
- MOPS
3-(N-morpholino)propanesulfonic acid
- c-di-GMP
cyclic di-(3′,5′)-guanosine monophosphate
- pGpG
5′-phosphoguanylyl-(3′→ 5′)-guanosine
- MBP
maltose binding protein
- MWCO
molecular weight cutoff
- PDB
Protein Data Bank
- PDE
phosphodiesterase
- Tris
tris(hydroxymethyl)aminomethane
- TFA
trifluoroacetic acid
- diferric
two Fe(III) atoms per site
- diferrous
two Fe(II) atoms per site
- HPLC
high-performance liquid chromatography
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b01227.
Description of the molecular dynamics simulation methodology, amino acid sequence alignment of HD-GYPs, UV–vis absorption spectra and HPLC activity chromatograms for TM0186 metal ligand residue variants, control HPLC chromatograms in the absence of protein, view of the triiron PmGH crystal structure showing exogenous ligands, and views of PmGH and TM0186 dimetal site HD-GYP domain backbone models (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Hengge R. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol. 2009;7:263–273. doi: 10.1038/nrmicro2109. [DOI] [PubMed] [Google Scholar]
- 2.Römling U, Galperin MY, Gomelsky M. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev. 2013;77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caly DL, Bellini D, Walsh MA, Dow JM, Ryan RP. Targeting cyclic di-GMP signalling: a strategy to control biofilm formation? Curr Pharm Des. 2014;21:12–24. doi: 10.2174/1381612820666140905124701. [DOI] [PubMed] [Google Scholar]
- 4.Lori C, Ozaki S, Steiner S, Bohm R, Abel S, Dubey BN, Schirmer T, Hiller S, Jenal U. Cyclic di-GMP acts as a cell cycle oscillator to drive chromosome replication. Nature. 2015;523:236–239. doi: 10.1038/nature14473. [DOI] [PubMed] [Google Scholar]
- 5.Li W, Cui T, Hu L, Wang Z, Li Z, He ZG. Cyclic diguanylate monophosphate directly binds to human side-rocalin and inhibits its antibacterial activity. Nat Commun. 2015;6:8330. doi: 10.1038/ncomms9330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trampari E, Stevenson CE, Little RH, Wilhelm T, Lawson DM, Malone JG. Bacterial rotary export ATPases are allosterically regulated by the nucleotide second messenger cyclic-di-GMP. J Biol Chem. 2015;290:24470–24483. doi: 10.1074/jbc.M115.661439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ryan RP, Fouhy Y, Lucey JF, Crossman LC, Spiro S, He YW, Zhang LH, Heeb S, Camara M, Williams P, Dow JM. Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc Natl Acad Sci U S A. 2006;103:6712–6717. doi: 10.1073/pnas.0600345103. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Bellini D, Caly DL, McCarthy Y, Bumann M, An SQ, Dow JM, Ryan RP, Walsh MA. Crystal structure of an HD-GYP domain cyclic-di-GMP phosphodiesterase reveals an enzyme with a novel trinuclear catalytic iron centre. Mol Microbiol. 2014;91:26–38. doi: 10.1111/mmi.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plate L, Marletta MA. Nitric oxide modulates bacterial biofilm formation through a multicomponent cyclic-di-GMP signaling network. Mol Cell. 2012;46:449–460. doi: 10.1016/j.molcel.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miner KD, Klose KE, Kurtz DM., Jr An HD-GYP cyclic di-guanosine monophosphate phosphodiesterase with a non-heme diiron-carboxylate active site. Biochemistry. 2013;52:5329–5331. doi: 10.1021/bi4009215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao J, Tao J, Liang W, Zhao M, Du X, Cui S, Duan H, Kan B, Su X, Jiang Z. Identification and characterization of phosphodiesterases that specifically degrade 3′3′-cyclic GMP-AMP. Cell Res. 2015;25:539–550. doi: 10.1038/cr.2015.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galperin MY, Koonin EV. Divergence and convergence in enzyme evolution. J Biol Chem. 2012;287:21–28. doi: 10.1074/jbc.R111.241976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lovering AL, Capeness MJ, Lambert C, Hobley L, Sockett RE. The structure of an unconventional HD-GYP protein from Bdellovibrio reveals the roles of conserved residues in this class of cyclic-di-GMP phosphodiesterases. mBio. 2011;2:e00163. doi: 10.1128/mBio.00163-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rinaldo S, Paiardini A, Stelitano V, Brunotti P, Cervoni L, Fernicola S, Protano C, Vitali M, Cutruzzola F, Giardina G. Structural basis of functional diversification of the HD-GYP domain revealed by the Pseudomonas aeruginosa PA4781 protein, which displays an unselective bimetallic binding site. J Bacteriol. 2015;197:1525–1535. doi: 10.1128/JB.02606-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graphics. 1996;14:27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 16.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurtz DM., Jr Oxo- and hydroxo-bridged diiron complexes: A chemical perspective on a biological unit. Chem Rev. 1990;90:585–606. [Google Scholar]
- 18.Okamoto Y, Onoda A, Sugimoto H, Takano Y, Hirota S, Kurtz DM, Jr, Shiro Y, Hayashi T. Crystal structure, exogenous ligand binding, and redox properties of an engineered diiron active site in a bacterial hemerythrin. Inorg Chem. 2013;52:13014–13020. doi: 10.1021/ic401632x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wigren E, Liang ZX, Romling U. Finally! The structural secrets of a HD-GYP phosphodiesterase revealed. Mol Microbiol. 2014;91:1–5. doi: 10.1111/mmi.12463. [DOI] [PubMed] [Google Scholar]
- 20.Huynh TN, Luo S, Pensinger D, Sauer JD, Tong L, Woodward JJ. An HD-domain phosphodiesterase mediates cooperative hydrolysis of c-di-AMP to affect bacterial growth and virulence. Proc Natl Acad Sci U S A. 2015;112:E747–756. doi: 10.1073/pnas.1416485112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tchigvintsev A, Xu X, Singer A, Chang C, Brown G, Proudfoot M, Cui H, Flick R, Anderson WF, Joachimiak A, Galperin MY, Savchenko A, Yakunin AF. Structural insight into the mechanism of c-di-GMP hydrolysis by EAL domain phosphodiesterases. J Mol Biol. 2010;402:524–538. doi: 10.1016/j.jmb.2010.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mashhadi Z, Xu HM, White RH. An Fe2+- dependent cyclic phosphodiesterase catalyzes the hydrolysis of 7,8-dihydro-D-neopterin 2′,3′-cyclic phosphate in methanopterin biosyn-thesis. Biochemistry. 2009;48:9384–9392. doi: 10.1021/bi9010336. [DOI] [PubMed] [Google Scholar]
- 23.Andrews SC, Robinson AK, Rodriguez-Quinones F. Bacterial iron homeostasis. FEMS Microbiol Rev. 2003;27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- 24.Imlay JA. The mismetallation of enzymes during oxidative stress. J Biol Chem. 2014;289:28121–28128. doi: 10.1074/jbc.R114.588814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Latour JM. Manganese, the stress reliever. Metallomics. 2015;7:25–28. doi: 10.1039/c4mt00180j. [DOI] [PubMed] [Google Scholar]
- 26.Le Fourn C, Fardeau ML, Ollivier B, Lojou E, Dolla A. The hyperthermophilic anaerobe Thermotoga maritima is able to cope with limited amount of oxygen: insights into its defence strategies. Environ Microbiol. 2008;10:1877–1887. doi: 10.1111/j.1462-2920.2008.01610.x. [DOI] [PubMed] [Google Scholar]
- 27.Le Fourn C, Brasseur G, Brochier-Armanet C, Pieulle L, Brioukhanov A, Ollivier B, Dolla A. An oxygen reduction chain in the hyperthermophilic anaerobe Thermotoga maritima highlights horizontal gene transfer between Thermococcales and Thermotogales. Environ Microbiol. 2011;13:2132–2145. doi: 10.1111/j.1462-2920.2011.02439.x. [DOI] [PubMed] [Google Scholar]
- 28.Ryan RP, McCarthy Y, Andrade M, Farah CS, Armitage JP, Dow JM. Cell-cell signal-dependent dynamic interactions between HD-GYP and GGDEF domain proteins mediate virulence in Xanthomonas campestris. Proc Natl Acad Sci U S A. 2010;107:5989–5994. doi: 10.1073/pnas.0912839107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kyrpides NC, Ouzounis CA, Iliopoulos I, Vonstein V, Overbeek R. Analysis of the Thermotoga maritima genome combining a variety of sequence similarity and genome context tools. Nucleic Acids Res. 2000;28:4573–4576. doi: 10.1093/nar/28.22.4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.