

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease. Mutations in the Fused in Sarcoma/Translocated in Liposarcoma (FUS/TLS) gene cause a subset of familial ALS cases and are also implicated in sporadic ALS. FUS is typically localized to the nucleus. The ALS-related FUS mutations cause cytoplasmic mis-localization and the formation of stress granule-like structures. Abnormal cytoplasmic FUS localization was also found in a subset of frontotemporal dementia (FTLD) cases without FUS mutations. To better understand the function of FUS, we performed wild-type and mutant FUS pull-downs followed by proteomic identification of the interacting proteins. The FUS interacting partners we identified are involved in multiple pathways, including chromosomal organization, transcription, RNA splicing, RNA transport, localized translation, and stress response. FUS interacted with hnRNPA1 and Matrin-3, RNA binding proteins whose mutations were also reported to cause familial ALS, suggesting that hnRNPA1 and Matrin-3 may play common pathogenic roles with FUS. The FUS interactions displayed varied RNA dependence. Numerous FUS interacting partners that we identified are components of exosomes. We found that FUS itself was present in exosomes, suggesting that the secretion of FUS might contribute to the cell-to-cell spreading of FUS pathology. FUS interacting proteins were sequestered into the cytoplasmic mutant FUS inclusions that could lead to their mis-regulation or loss of function, contributing to ALS pathogenesis. Our results provide insights into the physiological functions of FUS as well as important pathways where mutant FUS can interfere with cellular processes and potentially contribute to the pathogenesis of ALS.
Keywords: ALS, FUS, proteomics, protein interaction network, neurodegeneration
1. Introduction
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a progressive and ultimately fatal neurodegenerative disease of the upper and lower motor neurons of the brain and spinal cord. At any given time, approximately 30,000 people in the United States are living with the disease [1]. There is currently no cure available for those afflicted [2]. General symptoms of ALS are muscle weakness and wasting triggered by the degeneration of motor neurons. In order to develop effective preventive measures or cures, we have to gain a better understanding of the molecular etiology of the disease.
Approximately 10% of the ALS cases are familial (fALS) and caused by heritable mutations in a number of different genes. A subset of fALS cases is caused by autosomal dominant mutations in the gene encoding Fused in Sarcoma/Translocated in Liposarcoma (FUS/TLS) [3, 4]. FUS mutations were also identified in a subset of the sporadic ALS (sALS) cases [5–7]. In most tissues, FUS is predominantly localized to the nucleus with a notable cytoplasmic presence in neurons [8]. FUS has several known functions in multiple cellular pathways. FUS binds RNA and shuttles between the nucleus and the cytoplasm, participating in nucleo-cytoplasmic RNA shuttling [9]. FUS also plays roles in DNA repair [10–12], transcription [13–23], RNA splicing [22, 24, 25], dendritic RNA transport [26–28] and miRNA biogenesis and function [29, 30]. However, the physiological functions of FUS are still not fully understood. The ALS-related FUS mutations cause varied degrees of cytoplasmic mis-localization of FUS and the formation of stress granule-like structures [31–34]. In a subset of frontotemporal dementia cases, pathological cytoplasmic mis-localization of FUS was found without FUS mutations [35, 36]. The depletion of FUS in the nucleus likely results in partial loss of its nuclear function(s). In addition, its cytoplasmic accumulation and the formation of stress granule-like structures and other ribonucleoprotein complexes might lead to a gained toxicity phenotype. These loss-of-function and gain-of-function/gain-of-toxicity mechanisms are both plausible and are not mutually exclusive [37, 38].
To gain insight into the normal functions of FUS and the pathogenesis caused by its mutations, we performed GST-FUS pull-downs from human cells expressing wild-type and R521G mutant FUS, followed by proteomic identification of the FUS interacting partners. In addition to previously reported interacting proteins, many novel interacting partners were identified in this study, including members of the spliceosome, IMP1-dependent ribonucleoprotein particles, transport RNA granules and stress granules. Many of the identified interacting partners are shared between two or more of these ribonucleoprotein particles. We selected a set of the identified FUS interacting partners that participate in a wide range of functions for further analysis. We validated the interaction of FUS with the selected partners and determined the RNA dependence of those interactions. We found that the interactions of FUS with some partners were enhanced, whereas with others were partly or completely abrogated by RNase digestion. Importantly, we found that FUS interacted with hnRNPA1 and Matrin-3, proteins whose mutations were also identified to cause familial ALS [39, 40]. Numerous identified FUS interacting partners are exosomal components. We found that FUS itself was present in exosomes, suggesting that the secretion of FUS might contribute to the cell-to-cell spreading of FUS pathology. We found that ALS mutants of FUS co-localized with Caprin-1, DDX3X, and DXH9 in cytoplasmic inclusions that could lead to the mis-regulation of their respective pathways, providing further clues to the mechanism of ALS pathogenesis.
2. Materials and Methods
2.1. Cell culture and transfection
The HEK293T (293T) and N2A cells were cultured in Dulbecco’s Modified Eagle’s Medium (Sigma-Aldrich, D5796) with 10% fetal bovine serum and penicillin-streptomycin at 37°C in 5% CO2/95% air. The SH-SY5Y cells were cultured in a 1:1 mixture of Dulbecco’s Modified Eagle’s Medium/F-12 Ham (Sigma-Aldrich, D8437) with 10% fetal bovine serum and penicillin-streptomycin at 37°C in 5% CO2/95% air. The 293T cells were transfected with Polyethylenimine “Max” (Polysciences, Inc.). The SH-SY5Y and N2A cells were transfected with Lipofectamine 2000 (Life Technologies, 11668).
2.2. Culture and transfection of primary cortical neurons
The mouse primary cortical neuron cultures were prepared as reported [41]. Briefly, neonatal mouse pups (strain C57BL/6, Jackson Laboratory) were sacrificed via decapitation within 24 hours of birth. The cells were maintained in Neurobasal Medium (Life Technologies, 21103049) with B27 supplement (Life Technologies, 17504044), L-glutamine and penicillin/streptomycin. Neurons were dissociated from glial cells using pipette lavage after incubation with trypsin. The cells were treated with 5-Fluoro-2′-deoxyuridine (Sigma-Aldrich, F0503). Fourteen days after 5-Fluoro-2′-deoxyuridine treatment, the primary neurons were transfected with EGFP-tagged WT or P525L mutant FUS expression constructs using Lipofectamine 2000 (Life Technologies, 11668). The primary neurons were fixed 48 hours post-transfection for immunofluorescence.
2.3. GST-FUS pull-downs
The GST-FUS pull-downs were performed similarly as reported before [32]. Briefly, the wild type, R521G or P525L mutant human FUS coding sequences were inserted into the pEBG glutathione S-transferase (GST) fusion vector [42] using the BamHI and KpnI sites. pEBG was a gift from David Baltimore (Addgene plasmid # 22227). 293T cells were transfected with the GST-FUS constructs. Two days post-transfection, cell lysates were prepared in 1x RIPA buffer (Millipore, 20–188) supplemented with protease inhibitor cocktail (Sigma, P-8340, 1:500) and 1mM sodium orthovanadate with syringe homogenization. The lysates were pre-cleared with Sepharose CL-4B (Sigma, CL4B200), then subjected to overnight pull-down with Glutathione Sepharose 4B (GE Healthcare, 17-0756-01). Where indicated, a cocktail of RNase A and RNase T1 (Life Technologies, AM2286) was added to the pre-cleared lysates before the pull- down at 1:100 dilution. The bound proteins were eluted by boiling with Laemmli sample buffer (Biorad, 161-0737).
2.4. Endogenous FUS immunoprecipitation
Cellular extracts of SH-SY5Y cells were prepared in 1x RIPA buffer (Millipore, 20-188) supplemented with protease inhibitor cocktail (Sigma, P-8340, 1:500) and 1mM sodium orthovanadate with syringe homogenization. The cellular lysates were first pre-cleared with Protein G UltraLink Resin (Thermo Scientific Pierce, 53126), then endogenous FUS was immunoprecipitated with mouse anti-FUS antibody (Santa Cruz, sc-47711) and Protein G UltraLink Resin. The negative control immunoprecipitation antibody was mouse anti-HA (Santa Cruz, sc-7392). The bound proteins were eluted by boiling with Laemmli sample buffer (Biorad, 161-0737).
2.5. Proteomic identification of FUS interaction partners
The proteins eluted from the Glutathione Sepharose 4B beads were resolved by denaturing polyacrylamide gel electrophoresis (SDS-PAGE) on 4–15% gradient gel, followed by staining with Sypro Ruby protein gel stain (Molecular Probes, S-12000). Each lane was cut into 20 slices of equal size and each slice was subjected to in-gel trypsin digestion [43]. The resulting tryptic peptides were extracted and subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) using an LTQ Orbitrap Velos mass spectrometer (Thermo Scientific). The LC-MS/MS results were processed with a local Mascot server (version 2.3, Matrix Science) for protein identification including methionine oxidation and cysteine carbamidomethylation as allowed side chain modifications. The LC-MS/MS data of the 20 slices were combined for protein identification using the MASCOT algorithm. False discovery rate of 1% was used in decoy search for the high-confidence peptides summarized in Supplemental Tables 1 and 2. Proteins with a score of at least 30 for single high-confidence peptides were considered positive identifications. MS/MS spectra of high-confidence peptides with scores lower than 50 were manually inspected and confirmed. UniProt protein names and identifier numbers [44] are used throughout this work.
2.6. Immunoblotting
The nitrocellulose membranes were blocked and antibodies were applied in 5% milk in TRIS-buffered saline/Tween-20 (TBST, 0.1M TRIS-HCl, 0.9% [w/v] NaCl, 0.1% [v/v] Tween-20, pH 7.5). The antibodies used were anti-NF45/ILF2 (Bethyl, A303-147A-M), anti-DHX9 (Bethyl, A300-855A-M), anti-Matrin-3 (Bethyl, A300-591A-M), anti-hnRNPA1 (Novus, NB100-672), anti-Caprin-1 (Proteintech, 15112-1-AP), anti-GST (Santa Cruz, sc-459), anti-DDX3X (Sigma, HPA001648), anti-actin (Santa Cruz, sc-1616), anti-FLAG (Sigma, A8592), anti-Alix (Santa Cruz, sc-49268), anti-TSG101 (GeneTex, GTX70255), anti-CD63 (Santa Cruz, sc-15363), and anti-Histone H3 (Cell Signaling Technology, 9715). The immunoblotting images were acquired with the Chemidoc MP Imaging System (Bio-Rad) and quantified with the Image Lab software (Bio-Rad).
2.7. Exosome isolation
The SH-SY5Y and N2A cells were transfected with 3×FLAG-tagged FUS constructs [32]. After transfection, the media were replaced with fresh medium every 24 hours. 72 hours post-transfection, the collected conditioned media were combined and sequentially centrifuged at 300 g for 10 min, 20,000 g for 30 min, and 120,000 g for 90 min. The 120,000 g exosome pellets were washed in 1x phosphate buffered saline (PBS) by centrifuging at 120,000 g for 90 min and the pellets were re-suspended in 1xRIPA buffer (Millipore, 20–188). The protein concentrations were measured and equal amounts of protein samples were subjected to SDS-PAGE followed by immunoblotting with the indicated antibodies.
2.8. Fluorescence microscopy
SH-SY5Y cells were seeded on gelatin-treated glass coverslips and transfected with EGFP-tagged FUS constructs [32]. Next day, the cells were fixed in 4% (w/v) paraformaldehyde in 1x PBS, permeabilized with 0.25% (v/v) Triton-X100 in 1x PBS and blocked with 10% (w/v) bovine serum albumin (BSA) in 1x PBS. The slides were incubated with primary antibodies diluted in 3% (w/v) BSA/1x PBS. The primary antibodies were the same as those used for immunoblotting. The secondary antibodies were Alexa Fluor 568 Donkey Anti-Rabbit IgG or Alexa Fluor 568 Donkey Anti-Mouse IgG (Life Technologies, A10042 and A10037, respectively). The nuclei were visualized with 4′,6-diamidino-2-phenylindole (DAPI). The coverslips were mounted on glass slides with Vectashield Mounting Medium (Vector Laboratories, H-1000-10). Confocal microscopic images were acquired using a Nikon A1 confocal microscope with a 60x objective.
3. Results
3.1. The identification of FUS interaction partners
We generated GST-FUS expression constructs using the pEBG vector that expresses the glutathione S-transferase (GST) gene of Schistosoma japonicum under control of the human EF-1α promoter [42]. We performed GST pull-downs from human HEK293T (293T) cells transfected with wild-type (WT) or R521G mutant GST-FUS or the GST vector. The GST-FUS pull-downs were performed both in the presence and absence of added RNase in the cellular lysates. SDS-PAGE revealed major banding pattern differences between the GST vector control and the GST-FUS lanes (Figure 1). Proteins that were identified in the vector lane (several human glutathione S-transferases and other proteins) and common contaminants such as keratin, heat shock proteins and abundant cytoskeletal components were excluded from the analysis of the GST-FUS lanes. It is noted that no endogenous FUS was identified in the GST vector control lane, suggesting that the endogenous FUS did not bind non-specifically to the Glutathione Sepharose 4B beads.
Figure 1. The identification of FUS interaction partners.
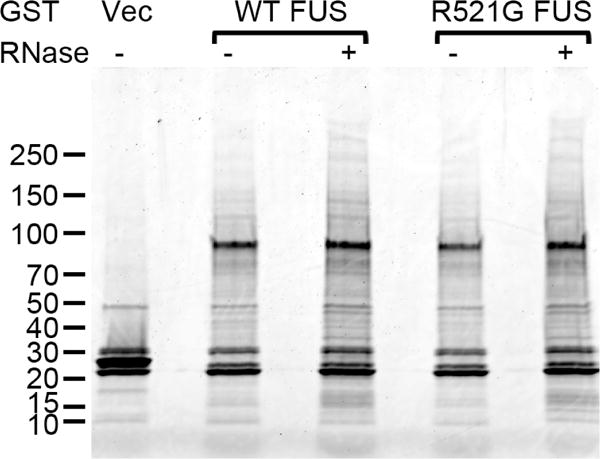
GST pull-downs were performed from cellular extracts of 293T cells transfected with the indicated GST expression constructs. RNase was included in the cellular lysates as indicated. The pull-down mixtures were subjected to SDS-PAGE on 4–15% gradient gel, followed by Sypro Ruby staining. Vec, vector. The molecular weights of marker bands are shown on the left (kDa).
We identified the FUS interacting proteins using the Mascot software (Matrix Science). Mascot uses probability-based scoring to judge whether identifications are significant. The peptide score shows the probability of whether the observed match is a random event. Scores are reported as −10 × log10(P), where P is the probability. For example, a Mascot score of 30 means that the probability of a peptide identification being a random event is 0.001. The protein score is calculated from the scores of the individual peptides. A total of 112 proteins were identified as FUS-interacting partners in this study. Among them, 70 were non-ribosomal proteins (Supplemental Table 1) and 42 were ribosomal proteins (Supplemental Table 2). The count of the peptides identified at high confidence level (false discovery rate < 1% in decoy search) is shown in the tables, and only proteins identified with a compounded score of 30 or higher are listed. The MS/MS spectra of high-confidence peptides with scores lower than 50 were manually inspected and confirmed. Proteins identified with these parameters are considered highly confident identifications.
3.2. Validation and characterization of selected FUS interactions
We demonstrated the co-precipitation of endogenous DHX9, Matrin-3, ILF2 and hnRNPA1 with endogenous FUS immunoprecipitated from SH-SY5Y cells (Figure 2). We also validated the interaction of FUS with endogenous DHX9, Matrin-3, DDX3X, Caprin-1, ILF2 and hnRNPA1 in 293T cells using GST-FUS pull-down followed by immunoblot with specific antibodies (Figure 3A–B, lanes 1 and 3).
Figure 2. Verification of selected FUS interacting partners with endogenous FUS immunoprecipitation.
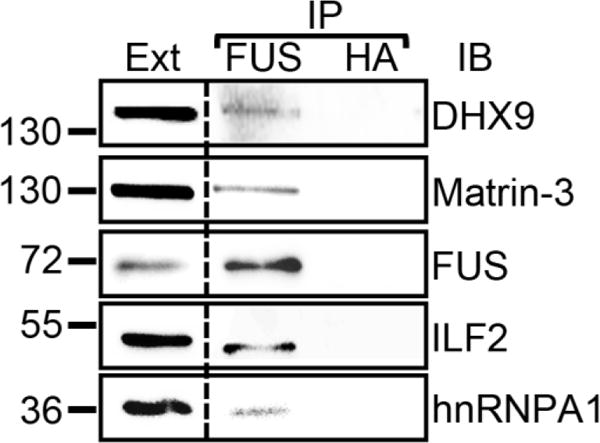
Immunoprecipitations were performed from cellular extracts of SH-SY5Y cells with a FUS-specific and a control (HA) antibody. The immunoprecipitates were subjected to SDS-PAGE followed by immunoblot with the indicated antibodies. IP, immunoprecipitation; Ext, extract; IB, immunoblot. The molecular weights of nearby marker bands are shown on the left (kDa).
Figure 3. Verification of selected FUS interacting partners with GST-FUS pulldown.
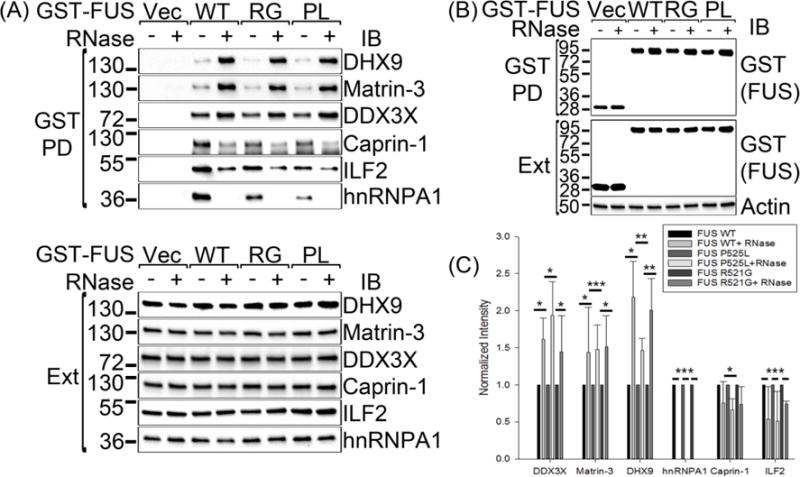
(A–B) GST pull-downs were performed from cellular extracts of 293T cells transfected with the indicated GST expression constructs. RNase was included in the cellular lysates as indicated. The pull-down mixtures were subjected to SDS-PAGE followed by immunoblot with the indicated antibodies. PD, pull-down; Ext, extract; Vec, vector; RG, R521G; PL, P525L; IB, immunoblot. The molecular weights of nearby marker bands are shown on the left (kDa). (C) Quantification of the RNA dependence of the FUS interactions shown in Figure 3A. The band intensities from three independent experiments were quantified and normalized to the respective band with no RNase added. The error bars stand for standard deviation. Statistical significance was determined with Student’s t-test. *: 0.01 < p < 0.05; **: 0.001 < p < 0.01; ***: p < 0.001.
We determined whether the interactions of FUS with the above proteins depended on RNA. Cells were transfected with GST vector or GST-tagged WT, R521G or P525L FUS expression constructs and the cell lysates were subjected to GST pull-down either with or without the addition of RNase (Figure 3). The precipitation of the baits was verified with anti-GST immunoblot (Figure 3B). The immunoblots of the co-precipitating proteins were quantified from three independent experiments (Figure 3C).The binding of FUS to its interaction partners was differentially affected by RNase treatment (Figure 3A and C). The amounts of the co-precipitated Matrin-3 and that of the RNA helicases DHX9 and DDX3X were significantly enhanced by RNase treatment. The interactions of FUS with Caprin-1 and ILF2 were weakened, but not fully abrogated, by the RNase treatment. The interaction of FUS with hnRNPA1 was entirely dependent on RNA, as the RNase treatment completely abolished its interaction with FUS. Thus, the interactions of FUS with DHX9, Matrin-3 and DDX3X are likely protein-protein interactions that do not require RNA, whereas the interactions with Caprin-1, ILF2, and hnRNPA1 are partly or fully RNA-dependent.
3.3. Co-localization of FUS interaction partners with cytoplasmic ALS mutant FUS inclusions
We tested whether the selected interaction partners co-localized with cytoplasmic inclusions of mutant FUS in the SH-SY5Y human neuroblastoma cell line transfected with EGFP-tagged WT, R521G or P525L FUS. We found that EGFP-tagged WT FUS localized to the nuclei of SH-SY5Y cells. The localization of R521G mutant FUS ranged widely from mostly nuclear to mostly cytoplasmic. The localization of P525L FUS was even further shifted towards the cytoplasm. Both the R521G and the P525L FUS mutants formed cytoplasmic inclusions (Figure 4).The DXH9 protein was primarily localized to the nuclei of SH-SY5Y cells sharing a similar distribution pattern with that of nuclear FUS (Figure 4A). The expression of both R521G and P525L FUS resulted in the accumulation of small amounts of DHX9 in cytoplasmic mutant FUS inclusions (Figure 4A).The DDX3X protein was mostly localized to the cytoplasm (Figure 4B). DDX3X was efficiently sequestered by the cytoplasmic inclusions of both R521G and P525L mutant FUS (Figure 4B). We had similar observations with the mostly cytoplasmic CAPR1/Caprin-1. The expression of R521G and P525L FUS induced the formation of cytoplasmic Caprin-1-positive inclusions, most of which co-localized with the mutant FUS inclusions (Figure 4C). We also demonstrated the co-localization of DHX9, DDX3X and Caprin-1 with cytoplasmic EGFP-P525L mutant FUS inclusions in primary cortical neurons (Figure 5).
Figure 4. The co-localization of DHX9, DDX3X and Caprin-1 with ALS mutant FUS inclusions in SH-SY5Y cells.
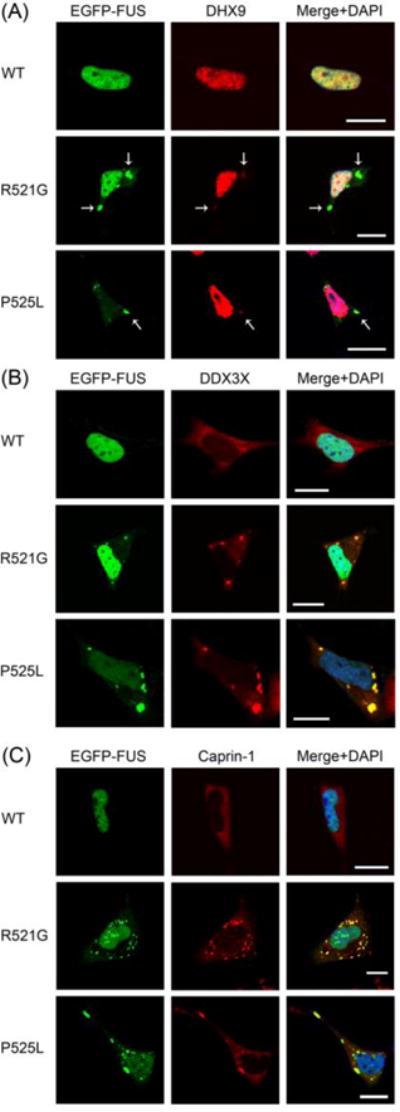
Confocal microscopic images of EGFP-tagged FUS (WT, R521G or P525L) fluorescence and DHX9 (A), DDX3X (B) and Caprin-1 (C) immunofluorescence in SH-SY5Y cells. The co-localization of mutant FUS and DHX9 in cytoplasmic inclusions is shown by arrows in (A). Scale bars, 10 μm.
Figure 5. The co-localization of DHX9, DDX3X and Caprin-1 with ALS mutant FUS inclusions in primary cortical neurons.
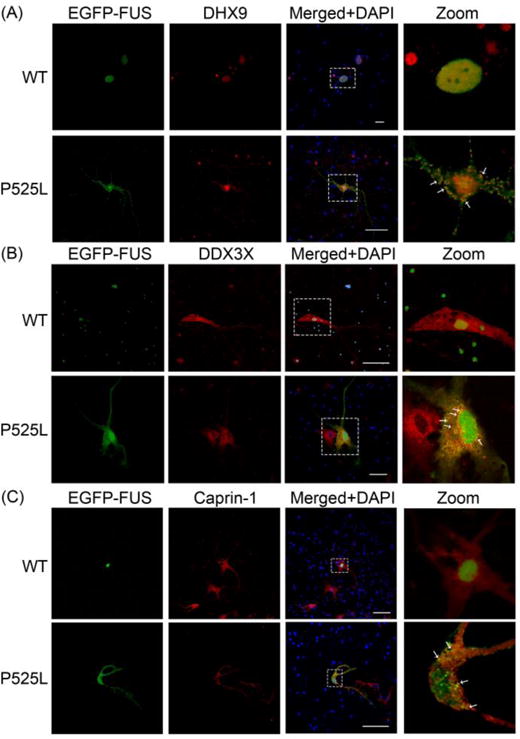
Confocal microscopic images of EGFP-tagged WT and P525L mutant FUS fluorescence and DHX9 (A), DDX3X (B) and Caprin-1 (C) immunofluorescence in cultured mouse primary cortical neurons. The boxed areas are magnified in the respective panels on the right. The co-localization of mutant FUS with the respective interaction partners in cytoplasmic inclusions is shown by arrows. Scale bars, 50μm.
The co-localization of DHX9, DDX3X and Caprin-1 with mutant FUS inclusions was quantified in SH-SY5Y cells transfected with EGFP-tagged P525L mutant FUS in four independent experiments, each representing over 400 transfected cells. On average, 24.5% of the transfected cells contained cytoplasmic P525L mutant FUS inclusions (StDev: 3.46%). DHX9 co-localized with 89.7% (StDev: 5.45%), DDX3X with 97.9% (StDev: 4.17%), and Caprin-1 with 88.7% (StDev: 6.27%) of the cytoplasmic P525L mutant FUS inclusions, respectively.
The FUS interaction partners Matrin-3 and hnRNPA1 were localized to the nuclei of SH-SY5Y cells and were not detected in cytoplasmic mutant FUS inclusions (Supplemental Figure 1A–B). The ILF2 protein was primarily in the nuclei and was not detected in cytoplasmic mutant FUS inclusions, either (Supplemental Figure 1C). These results suggest that the detected complexes of FUS with Matrin-3, hnRNPA1, and ILF2 were likely nuclear that will be explored in future studies.
3.4. Significantly enriched Gene Ontology (GO) terms among the identified FUS interacting partners
We searched for significantly enriched GO terms for the identified FUS interacting partners using the STRING server (http://www.string-db.org/, [45]). The enriched GO Biological Processes, Molecular Functions and Cellular Components of FUS interacting partners are summarized in Tables 1, 2 and 3, respectively. The p-values shown in the tables are Benjamini-Hochberg corrected p-values for enrichment of the terms in question, representing the estimated false discovery rate [46].
Table 1. Selected enriched GO Biological Processes among the identified FUS interacting partners.
The corresponding p-values and the numbers of FUS interaction partners are shown without/with the inclusion of ribosomal FUS interaction partners in the search.
| Term | p-value | Number of FUS interaction partners |
|---|---|---|
| RNA processing | 1.659e-32/3.050e-36 | 37/46 |
| RNA splicing | 4.750e-27/2.960e-22 | 27/27 |
| mRNA processing | 9.890e-27/1.499e-21 | 28/28 |
| mRNA metabolic process | 1.149e-25/6.829e-73 | 30/67 |
| mRNA splicing, via spliceosome | 1.149e-25/1.760e-21 | 23/23 |
| Gene expression | 1.220e-18/3.089e-33 | 49/80 |
| Posttranscriptional regulation of gene expression | 6.050e-13/1.650e-13 | 17/20 |
| mRNA stabilization | 2.940e-7/1.150e-6 | 6/6 |
| Regulation of translation | 8.299e-7/3.019e-8 | 11/14 |
| RNA localization | 1.980e-5/2.180e-4 | 8/8 |
| Transcription, DNA-templated | 6.779e-5/4.710e-2 | 25/26 |
| mRNA 3′-end processing | 3.570e-4/1.649e-3 | 5/5 |
| tRNA splicing, via endonucleolytic cleavage and ligation | 8.429e-4/1.699e-3 | 3/3 |
| ncRNA metabolic process | 1.000e-3/3.459e-10 | 9/18 |
| Termination of RNA polymerase II transcription | 2.360e-3/8.259e-3 | 4/4 |
| Transcription from RNA polymerase II promoter | 5.130e-3/1.499e-1 | 11/11 |
| Chromosome organization | 1.149e-2/2.939e-1 | 11/11 |
Table 2. Selected enriched GO Molecular Functions among the identified FUS interacting partners.
The corresponding p-values and the numbers of FUS interaction partners are shown without/with the inclusion of ribosomal FUS interaction partners in the search.
| Term | p-value | Number of FUS interaction partners |
|---|---|---|
| RNA binding | 5.000e-43/6.379e-74 | 53/87 |
| Single-stranded RNA binding | 2.070e-10/9.300e-9 | 9/9 |
| ATP-dependent RNA helicase activity | 2.220e-10/1.099e-8 | 9/9 |
| Double-stranded RNA binding | 8.889e-6/1.289e-4 | 6/6 |
| mRNA 3′-UTR binding | 4.350e-5/4.180e-4 | 5/5 |
| DNA binding | 3.330e-4/2.750e-2 | 22/25 |
| RNA polymerase II core binding | 1.690e-3/7.629e-3 | 3/3 |
| mRNA 5′-UTR binding | 1.359e-2/5.000e-4 | 2/3 |
Table 3.
Selected enriched GO Cellular Components among the identified FUS interacting partners.
| Term | p-value | Number of FUS interaction partners |
|---|---|---|
| Ribonucleoprotein complex | 2.369e-68 | 66 |
| Ribosome | 1.769e-46 | 38 |
| Spliceosomal complex | 1.080e-20 | 20 |
| Ribonucleoprotein granule | 3.200e-16 | 16 |
| Cytoplasmic stress granule | 3.430e-10 | 8 |
| Extracellular exosome | 1.430e-9 | 42 |
| tRNA-splicing ligase complex | 1.919e-6 | 4 |
| SMN-Sm protein complex | 5.140e-5 | 4 |
| Chromosome | 2.449e-3 | 13 |
Based on the GO Biological Processes, the majority of significantly enriched terms were related to various aspects of gene expression and RNA metabolism, such as chromosomal organization, transcription, RNA splicing, RNA processing, RNA transport, translation and RNA stability (Table 1). Based on the GO Molecular Functions, the majority of the identified FUS interaction partners are RNA and/or DNA binding proteins with a considerable enrichment for RNA helicases (Table 2). The FUS interacting partners CN166, CTR9 and ZN326 represent three of the 21 UniProt entries annotated with the significantly enriched GO Molecular Function “RNA polymerase II core binding” (Table 2). Among the significantly enriched GO Cellular Components were various ribonucleoprotein complexes including ribosomes, splicing complexes, the SMN-Sm protein complex and cytoplasmic stress granules (SGs). Also enriched were chromosomal proteins and components of extracellular exosomes (Table 3).
3.5. The identification of FUS in exosomes
Forty two of the FUS interacting partners identified in this work (18 non-ribosomal and 24 ribosomal proteins) were annotated as extracellular exosome components (Table 3), including the validated interaction partners DDX3X and hnRNPA1. This suggested that the FUS protein may be secreted via exosomes. Thus, we examined whether WT FUS or the ALS mutants R521G or R495X were detectable in exosomes isolated from the culture supernatant of SH-SY5Y (Figure 6) and N2A cells (Supplemental Figure 2). The isolated exosome fractions were very significantly enriched for the exosome markers Alix, TSG101 and CD63, but were devoid of the cytosolic marker actin and the nuclear marker histone H3 (Figure 6 and Supplemental Figure 2). We detected both WT and mutant FUS in the exosome fractions isolated from conditioned medium, and the severe ALS mutant R495X was present at a significantly higher level. Our results confirm that FUS was secreted from cells via exosomes (Figure 6 and Supplemental Figure 2).
Figure 6. The identification of WT and ALS mutant FUS in the exosome fraction of SH-SY5Y cells.
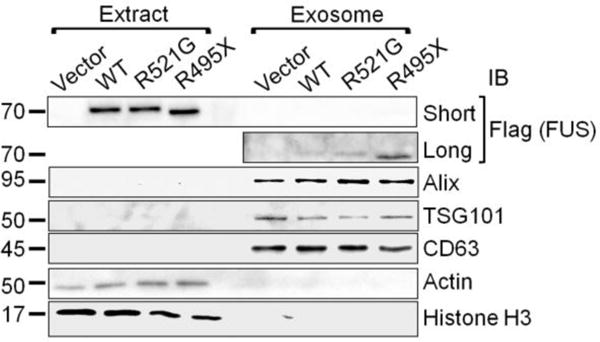
Exosomes were isolated as described in Section 2.7. in Materials and Methods. Equal amounts of protein samples were subjected to SDS-PAGE, followed by immunoblot with the indicated antibodies. The FLAG immunoblot of the exosome fractions is shown with a short and a long exposure time. The molecular weights of nearby marker bands are shown on the left (kDa).
3.6. FUS interacts with overlapping ribonucleoprotein complexes
Our analysis showed that the majority of the identified non-ribosomal FUS interaction partners (Supplemental Table 1) are known members of at least one of four types of ribonucleoprotein complexes: spliceosomes, IMP1-dependent ribonucleoprotein (RNP) granules, transport granules or stress granules (Figure 7). In Figure 7, only the FUS-interacting ribosomal proteins that were specifically identified as part of the above complexes are shown.
Figure 7. FUS interacts with overlapping ribonucleoprotein complexes.
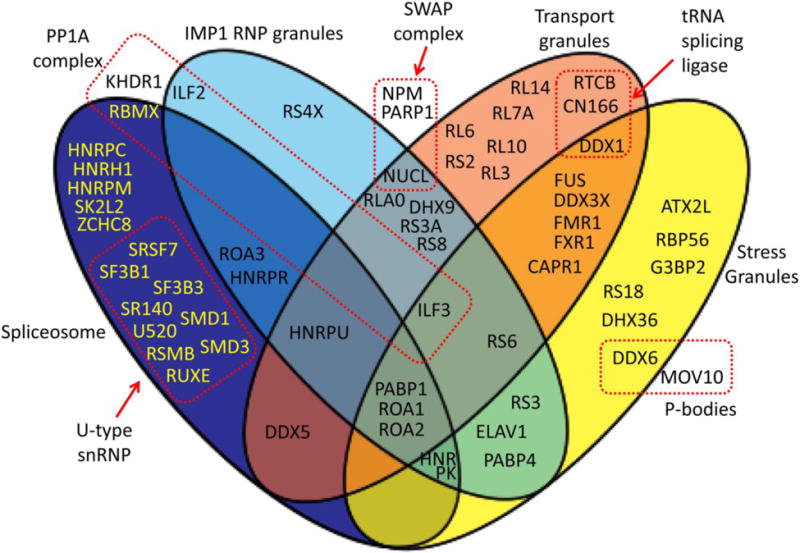
Venn diagram of a subset of the identified FUS interaction partners. Please see Section 3.6. of Results for details. The background diagram was obtained from Wikimedia Commons, https://commons.wikimedia.org/wiki/File:4-way-venn_vector.svg
This study identified the following FUS-interacting proteins that were previously identified as spliceosomal components: ROA1/hnRNPA1, ROA2/hnRNPA2B1, ROA3/hnRNPA3, HNRPC/hnRNPC, RBMX/hnRNPG, HNRH1/hnRNPH, HNRPK/hnRNPK, HNRPM/hnRNPM, HNRPR/hnRNPR, HNRPU/hnRNPU, PABP1, RSMB/SNRPB, SMD1, SMD3, RUXE/SNRPE, SF3B1, SF3B3, U520/SNRNP200, SK2L2, DDX5 and ZCHC8 [47], and SR140 and SRSF7 [48, 49]. In addition, FUS-interacting proteins are involved in a tRNA splicing ligase complex that is not part of the spliceosome: RTCB/HSPC117, CN166/CGI-99 and DDX1 [50].
Another group of FUS interacting proteins identified in this study belong to the IMP1-dependent mRNP granules. They include: DHX9, HNRPU/hnRNPU, NUCL/Nucleolin, PABP1, ILF2, ILF3, HNRPR/hnRNPR, PABP4, ROA1/hnRNPA1, ROA2/hnRNPA2B1, RLA0, RS3, RS3a, RS4X, RS6, RS8 [51], as well as HNRPK/hnRNPK, ROA3/hnRNPA3, and ELAV1/HuR [52].
The third group of our identified FUS-interacting proteins contains components of RNA transporting granules. DDX1, DDX3X, DDX5, RTCB/HSPC117, CN166/CGI-99, FMR1, FXR1, RL3, ROA1/hnRNPA1, HNRPU/hnRNPU, and NUCL are components of a FUS-positive kinesin-mediated RNA transporting dendritic granule complex [53]. Moreover, the DHX9, FMR1, HNRPU/hnRNPU, ILF3, NUCL, PABP1, RLA0, RL6, RL7A, RL10, RL14, RS2, RS3A, RS6, and RS8 proteins identified in this study were reported as components of Staufen-containing transport ribonucleoprotein complexes [54, 55]. In addition, ROA2/hnRNPA2 [56] and CAPR1/Caprin-1 [57] were also identified in RNA transport granules.
The fourth group among our identified FUS-interacting proteins belongs to stress granules. They include ATX2L, CAPR1/Caprin-1, DDX1, DDX3X, DDX6, DHX36/RHAU, ELAV1/HuR, FMR1, FXR1, G3BP2, ROA1/hnRNPA1, ROA2/hnRNPA2, HNRPK/hnRNPK, ILF3, PABP1, RS3, RS6, RS18, RBP56/TAF15 and PABP4 [58, 59]. Moreover, the two FUS-interacting proteins DDX6/RCK and MOV10 are components of processing bodies (P-bodies) [60, 61] that functionally interact with stress granules [62].
NPM, PARP1 and NUCL were identified as part of the SWAP complex, a B-cell-specific DNA recombination complex [63].
4. Discussion
4.1. Identification and validation of FUS interaction partners
We identified 70 non-ribosomal and 42 ribosomal proteins as FUS interaction partners in this study (Figure 1 and Supplemental Tables 1–2). No endogenous FUS was identified in the GST vector control pull-down. Proteins identified in the GST vector control experiment were excluded from the analysis of the GST-FUS pull-downs. Most FUS interaction partners we identified are known RNA and DNA binding proteins (Table 2) involved in pathways regulating various aspects of gene expression and RNA metabolism including transcription, splicing, RNA processing, RNA stability, RNA transport and translation (Table 1). Selected interactions were validated by immunopreciptation and GST pull-down followed by immunoblotting (Figures 2–3). Immunostaining followed by confocal microscopy showed that the FUS interaction partners DHX9, DDX3X and Caprin-1 localized to cytoplasmic FUS inclusions (Figures 4–5).
Our method to generate cellular lysates efficiently extracted not only cytosolic proteins but also predominantly nuclear proteins such as WT FUS, DHX9, Matrin-3 and hnRNPA1 (Figures 2–5, and Supplemental Figure 1). In the pull-down studies, we detected interactions between proteins mostly residing in different cellular compartments such as the interactions between the predominantly nuclear WT FUS and the almost exclusively cytoplasmic DDX3X or Caprin-1 (Figures 3–5). This observation demonstrates the significance of examining interacting proteins with imaging techniques in cells to determine whether the proteins interact with each other in live cells under physiological conditions.
Many FUS interacting partners participate in two or more of the ribonucleoprotein complexes shown in Figure 7. Ribonucleoprotein complexes are highly dynamic structures that can exchange components and transition from one kind of complex to another. It is conceivable that FUS cycles between different molecular complexes and ribonucleoprotein granules depending on a number of cellular influences including subcellular localization and stress conditions.
4.2. The RNA dependence of FUS interactions
The identified interactions were differentially impacted by the introduction of RNase into the pull-down mixtures. Whereas the interactions of FUS with DHX9, Matrin-3 and DDX3X are likely protein-protein interactions that were enhanced by RNase digestion, the interactions of FUS with CAPR1, ILF2, and hnRNPA1 were partly or fully RNA-dependent (Figure 3). It is likely that FUS molecules cycle between RNA-bound and RNA-unbound states as they interact with successive target RNA molecules. Our results showed that FUS displayed differential affinities to its protein interacting partners in the RNA-bound and RNA-unbound states, suggesting that the protein interacting partners of FUS likely also change during the RNA binding cycles. RNase treatment can free up molecular surfaces for protein-protein interactions otherwise occupied by RNA. Alternatively, the RNA-bound and unbound states of FUS might assume different conformations, regulating the preference of FUS to varied interaction partners.
4.3. The interaction of FUS with the ALS-related proteins Matrin-3 and hnRNPA1
We discovered that FUS interacted with MATR3/Matrin-3 and ROA1/hnRNPA1 (Figures 2–3 and Supplemental Table 1), proteins whose mutations were also identified to cause familial ALS [39, 40]. This suggests that FUS might be involved in common pathogenic pathways with Matrin-3 and hnRNPA1. There are intriguing differences between these complexes, though. The interaction between FUS and Matrin-3 was enhanced by RNase treatment, whereas the FUS-hnRNPA1 interaction was entirely dependent on RNA (Figure 3). It is relevant that hnRNPA1 was reported to locate to stress granules [39, 64] similarly to FUS [8, 31–34, 65]. The analysis of the role of the FUS-Matrin-3 and FUS-hnRNPA1 complexes in ALS pathogenesis is underway.
4.4. FUS is present in extracellular exosomes
Based on our results that 42 of the FUS interaction partners (18 non-ribosomal and 24 ribosomal proteins) were annotated as extracellular exosome components (Table 3), we hypothesized that FUS could be secreted via the exosome pathway. We tested and confirmed that FUS was indeed present in exosomes in both SH-SY5Y and N2A cells (Figure 6 and Supplemental Figure 2). It is also noted that the level of the R495X mutant in exosomes was significantly higher than that of WT or the R521G mutant. Exosomes have been reported to play a role in the intercellular propagation of SOD1 and TDP-43 pathology in ALS models [66–69]. It raises the possibility that the exosome-mediated secretion of FUS may contribute to the cell-to-cell propagation of FUS pathology. The role and relevance of the presence of FUS in exosomes is as yet unknown. It was recently reported in an HEK293 cell culture model that during the timeframe in which human WT SOD1 misfolding propagated to recipient cells, no mutant FUS pathology was transmitted to recipient cells when incubated with conditioned medium from mutant FUS-transfected cells [70]. We are currently investigating the role and functional consequences of the presence of FUS in exosomes.
4.5. FUS interacts with regulators of protein phosphorylation
We identified the FUS interaction partners KHDR1, ILF2, ILF3 and RBMX/hnRNPG that were previously reported to be in a complex with the serine/threonine-protein phosphatase PP1- α catalytic subunit PP1A (Figure 7) [71]. The ILF2 and ILF3 proteins were also reported to interact with and regulate the DNA-dependent protein kinase (DNA-PK) [72]. The phosphorylation of FUS by DNA-PK was recently reported [73]. Our results suggest that the KHDR1/ILF2/ILF3/RBMX complex might regulate FUS phosphorylation. FUS also interacted with CSK21, the catalytic alpha subunit of casein kinase II and the serine/threonine-protein kinase Nek10. The casein kinase II-mediated FUS phosphorylation and its functional relevance are currently being investigated in our laboratory.
4.6. Loss of nuclear FUS functions due to ALS mutations
We found that FUS interacted with numerous proteins that regulate nuclear processes. Twenty six of the FUS interacting partners we isolated were annotated as regulators of DNA-templated transcription (Table 1). Twenty three of the FUS interaction partners we identified are known spliceosomal components (Table 1, Figure 7). The splicing-related “Survival motor neuron” (SMN)-Sm protein complex was also among the significantly enriched GO Cellular Components (Table 3). Additionally, the FUS interacting partners RTCB, CN166 and DDX1 are components of a tRNA splicing ligase complex that is not part of the spliceosome (Figure 7) [50]. Although we did not identify FA98B/FAM98B, a fourth component of the tRNA splicing ligase complex as a FUS interacting partner, we did identify its closest human homolog, FA98A/FAM98A (Supplemental Table 1). The FUS-interacting RTCB/CN166/DDX1 complex was also reported to play a role in nucleo-cytoplasmic RNA shuttling [74], a function shared by FUS [9], suggesting potential functional cooperation. The familial ALS-related FUS mutations cause cytoplasmic mis-localization of FUS [3, 4, 31–34]. It is conceivable that the resulting lower nuclear FUS level leads to depletion of its nuclear complexes and loss of its nuclear functions.
4.7. Mis-regulation of cytoplasmic processes by ALS mutant FUS
We identified 34 FUS interacting partners that are known components of either IMP1-dependent RNP granules, RNA transport granules, or both (Figure 7). IMP1, also called insulin-like growth factor 2 mRNA-binding protein 1 is an RNA binding protein that recruits target mRNAs to cytoplasmic protein-RNA complexes. IMP1 stabilizes the recruited mRNAs allowing their storage and transport, and modulates the location at which the target mRNAs are translated [52, 75–77]. IMP1 plays a direct role in the transport and translation of transcripts required for axonal regeneration [78]. The IMP1-dependent RNP granules are distinct from RNA transport granules and stress granules [51], although they share components (Figure 7). The ALS-related FUS mutations cause the formation of FUS-positive cytoplasmic inclusions [31–34] that sequestered the IMP1-dependent RNP granule and/or transport granule components DHX9, DDX3X, and CAPR1 (Figures 4–5 and 7). The sequestration of these FUS interacting partners could lead to loss of their function in RNA transport and localized translation. Because of the extreme proportions of motor neurons, they rely on mRNA transport and localized translation more than other cell types. Accordingly, motor neurons could be especially sensitive to the sequestration of mRNA transport granule components.
We identified 21 reported stress granule (SG) components as FUS interacting partners (Figure 7). We found that the cytoplasmic ALS mutant FUS inclusions were immunopositive for the SG components DDX3X and CAPR1 (Figures 4–5 and 7). We previously reported that the mutant FUS inclusions were also immunopositive for another SG component, PABP1 [32]. Stress granules are stress-induced ribonucleoprotein complexes in eukaryotic cells that contain translationally silent pre-initiation complexes and a host of RNA-binding proteins [58, 79]. Two of the FUS interacting proteins we identified were processing body or P-body components (Figure 7). P-bodies are discrete cytoplasmic foci where mRNA degradation takes place [80]. P-bodies functionally interact with stress granules [62]. Among normal conditions, stress granules exist for up to a few hours. The normal dynamics of stress granules is critical for the survival of stressed neurons [81]. The perturbation of stress granule dynamics might be a pathological process of central importance in ALS [58, 82, 83]. Accordingly, triggering the formation of unusually stable stress granule-like structures by mutant FUS and the resulting perturbation of stress granule dynamics might cause a gained toxicity phenotype.
During the revision of this manuscript, a comparative interactomics study of varied ALS-associated proteins was published that identified a partially overlapping set of FUS interaction partners [84].
5. Conclusions
In summary, our results demonstrate that the FUS interacting partners participate in multiple pathways, providing insights into the physiological functions of FUS as well as to the potential mis-regulation of these pathways by ALS mutant FUS. Among the pathways, mRNA transport and stress granule dynamics are particularly important for the health of motor neurons. Mis-regulation of the dendritic and/or axonal mRNA transport and the generation of abnormally stable ectopic stress granule-like structures by ALS mutant FUS are likely contributors to ALS pathogenesis.
Supplementary Material
Highlights.
We performed proteomic analysis of FUS interacting proteins from human cells.
FUS interacts with numerous regulators of gene expression and RNA metabolism.
The interactions were differently impacted by RNase treatment.
FUS interacted with Matrin-3 and hnRNPA1, proteins also involved in ALS pathogenesis.
FUS interacted with numerous exosome components and was itself secreted by exosomes.
Acknowledgments
We thank Dr. Jose Abisambra (Sanders-Brown Center on Aging, University of Kentucky) for his help with the establishment of the primary cortical neuron cultures. This study was in part supported by the National Institutes of Neurological Disorder and Stroke grant R01NS077284, ALS Association grant 6SE340 and VA MERIT award I01 BX002149 (to HZ) as well as a Research Support Grant from the University of Kentucky Office of the Vice President for Research (to JG). MK is supported by the National Institute of Environmental Health Sciences training grant T32 ES007266.
Abbreviations
- ALS
amyotrophic lateral sclerosis
- fALS
familial ALS
- sALS
sporadic ALS
- 293T
human embryonic kidney (HEK293T) cells
- FUS/TLS
Fused in Sarcoma/Translocated in Liposarcoma
- GST
glutathione S-transferase
- RIPA
radioimmunoprecipitation assay buffer
- SDS-PAGE
denaturing polyacrylamide gel electrophoresis
- TBST
TRIS-buffered saline/Tween-20 buffer
- PBS
phosphate-buffered saline buffer
- EGFP
enhanced green fluorescent protein
- BSA
bovine serum albumin
- DAPI
4′,6-diamidino-2-phenylindole
- WT
wild type
- RNase
ribonuclease
- StDev
standard deviation
- GO
Gene Ontology
- SG
stress granule
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Marisa Kamelgarn, Email: meka229@g.uky.edu.
Jing Chen, Email: jchen4@email.uky.edu.
Lisha Kuang, Email: lisha.kuang@uky.edu.
Alexandra Arenas, Email: maar223@g.uky.edu.
Jianjun Zhai, Email: jianjunzhai@hotmail.com.
Haining Zhu, Email: haining@uky.edu.
Jozsef Gal, Email: jgal2@uky.edu.
References
- 1.Walling AD. Amyotrophic lateral sclerosis: Lou Gehrig’s disease. American family physician. 1999;59:1489–1496. [PubMed] [Google Scholar]
- 2.Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet journal of rare diseases. 2009;4:3. doi: 10.1186/1750-1172-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 4.Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeJesus-Hernandez M, Kocerha J, Finch N, Crook R, Baker M, Desaro P, Johnston A, Rutherford N, Wojtas A, Kennelly K, Wszolek ZK, Graff-Radford N, Boylan K, Rademakers R. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Human mutation. 2010;31:E1377–1389. doi: 10.1002/humu.21241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belzil VV, Valdmanis PN, Dion PA, Daoud H, Kabashi E, Noreau A, Gauthier J, S.D. team. Hince P, Desjarlais A, Bouchard JP, Lacomblez L, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology. 2009;73:1176–1179. doi: 10.1212/WNL.0b013e3181bbfeef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corrado L, Del Bo R, Castellotti B, Ratti A, Cereda C, Penco S, Soraru G, Carlomagno Y, Ghezzi S, Pensato V, Colombrita C, Gagliardi S, Cozzi L, Orsetti V, Mancuso M, Siciliano G, Mazzini L, Comi GP, Gellera C, Ceroni M, D’Alfonso S, Silani V. Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. Journal of medical genetics. 2010;47:190–194. doi: 10.1136/jmg.2009.071027. [DOI] [PubMed] [Google Scholar]
- 8.Andersson MK, Stahlberg A, Arvidsson Y, Olofsson A, Semb H, Stenman G, Nilsson O, Aman P. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC cell biology. 2008;9:37. doi: 10.1186/1471-2121-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zinszner H, Sok J, Immanuel D, Yin Y, Ron D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. Journal of cell science. 1997;110(Pt 15):1741–1750. doi: 10.1242/jcs.110.15.1741. [DOI] [PubMed] [Google Scholar]
- 10.Baechtold H, Kuroda M, Sok J, Ron D, Lopez BS, Akhmedov AT. Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. The Journal of biological chemistry. 1999;274:34337–34342. doi: 10.1074/jbc.274.48.34337. [DOI] [PubMed] [Google Scholar]
- 11.Mastrocola AS, Kim SH, Trinh AT, Rodenkirch LA, Tibbetts RS. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. The Journal of biological chemistry. 2013;288:24731–24741. doi: 10.1074/jbc.M113.497974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang WY, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IR, Huang EJ, Tsai LH. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nature neuroscience. 2013;16:1383–1391. doi: 10.1038/nn.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, Kurokawa R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature. 2008;454:126–130. doi: 10.1038/nature06992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan AY, Manley JL. TLS inhibits RNA polymerase III transcription. Molecular and cellular biology. 2010;30:186–196. doi: 10.1128/MCB.00884-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du K, Arai S, Kawamura T, Matsushita A, Kurokawa R. TLS and PRMT1 synergistically coactivate transcription at the survivin promoter through TLS arginine methylation. Biochemical and biophysical research communications. 2011;404:991–996. doi: 10.1016/j.bbrc.2010.12.097. [DOI] [PubMed] [Google Scholar]
- 16.Haile S, Lal A, Myung JK, Sadar MD. FUS/TLS is a co-activator of androgen receptor in prostate cancer cells. PloS one. 2011;6:e24197. doi: 10.1371/journal.pone.0024197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan AY, Riley TR, Coady T, Bussemaker HJ, Manley JL. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6030–6035. doi: 10.1073/pnas.1203028109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwartz JC, Ebmeier CC, Podell ER, Heimiller J, Taatjes DJ, Cech TR. FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes & development. 2012;26:2690–2695. doi: 10.1101/gad.204602.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujioka Y, Ishigaki S, Masuda A, Iguchi Y, Udagawa T, Watanabe H, Katsuno M, Ohno K, Sobue G. FUS-regulated region- and cell-type-specific transcriptome is associated with cell selectivity in ALS/FTLD. Scientific reports. 2013;3:2388. doi: 10.1038/srep02388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bronisz A, Carey HA, Godlewski J, Sif S, Ostrowski MC, Sharma SM. The multifunctional protein fused in sarcoma (FUS) is a coactivator of microphthalmia-associated transcription factor (MITF) The Journal of biological chemistry. 2014;289:326–334. doi: 10.1074/jbc.M113.493874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dhar SK, Zhang J, Gal J, Xu Y, Miao L, Lynn BC, Zhu H, Kasarskis EJ, St Clair DK. FUsed in sarcoma is a novel regulator of manganese superoxide dismutase gene transcription. Antioxidants & redox signaling. 2014;20:1550–1566. doi: 10.1089/ars.2012.4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang L, Gal J, Chen J, Zhu H. Self-assembled FUS binds active chromatin and regulates gene transcription. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:17809–17814. doi: 10.1073/pnas.1414004111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uranishi H, Tetsuka T, Yamashita M, Asamitsu K, Shimizu M, Itoh M, Okamoto T. Involvement of the pro-oncoprotein TLS (translocated in liposarcoma) in nuclear factor-kappa B p65-mediated transcription as a coactivator. The Journal of biological chemistry. 2001;276:13395–13401. doi: 10.1074/jbc.M011176200. [DOI] [PubMed] [Google Scholar]
- 24.Yang L, Embree LJ, Tsai S, Hickstein DD. Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. The Journal of biological chemistry. 1998;273:27761–27764. doi: 10.1074/jbc.273.43.27761. [DOI] [PubMed] [Google Scholar]
- 25.Dichmann DS, Harland RM. fus/TLS orchestrates splicing of developmental regulators during gastrulation. Genes & development. 2012;26:1351–1363. doi: 10.1101/gad.187278.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fujii R, Takumi T. TLS facilitates transport of mRNA encoding an actin-stabilizing protein to dendritic spines. Journal of cell science. 2005;118:5755–5765. doi: 10.1242/jcs.02692. [DOI] [PubMed] [Google Scholar]
- 27.Fujii R, Okabe S, Urushido T, Inoue K, Yoshimura A, Tachibana T, Nishikawa T, Hicks GG, Takumi T. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Current biology: CB. 2005;15:587–593. doi: 10.1016/j.cub.2005.01.058. [DOI] [PubMed] [Google Scholar]
- 28.Sephton CF, Tang AA, Kulkarni A, West J, Brooks M, Stubblefield JJ, Liu Y, Zhang MQ, Green CB, Huber KM, Huang EJ, Herz J, Yu G. Activity-dependent FUS dysregulation disrupts synaptic homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E4769–4778. doi: 10.1073/pnas.1406162111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morlando M, Dini Modigliani S, Torrelli G, Rosa A, Di Carlo V, Caffarelli E, Bozzoni I. FUS stimulates microRNA biogenesis by facilitating co-transcriptional Drosha recruitment. The EMBO journal. 2012;31:4502–4510. doi: 10.1038/emboj.2012.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dini Modigliani S, Morlando M, Errichelli L, Sabatelli M, Bozzoni I. An ALS-associated mutation in the FUS 3′-UTR disrupts a microRNA-FUS regulatory circuitry. Nature communications. 2014;5:4335. doi: 10.1038/ncomms5335. [DOI] [PubMed] [Google Scholar]
- 31.Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ, Jr, Sapp P, McKenna-Yasek D, Brown RH, Jr, Hayward LJ. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Human molecular genetics. 2010;19:4160–4175. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gal J, Zhang J, Kwinter DM, Zhai J, Jia H, Jia J, Zhu H. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiology of aging. 2011;32:2323 e2327–2340. doi: 10.1016/j.neurobiolaging.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ito D, Seki M, Tsunoda Y, Uchiyama H, Suzuki N. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Annals of neurology. 2011;69:152–162. doi: 10.1002/ana.22246. [DOI] [PubMed] [Google Scholar]
- 34.Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Schmid B, Neumann M, Haass C. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. The EMBO journal. 2010;29:2841–2857. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Urwin H, Josephs KA, Rohrer JD, Mackenzie IR, Neumann M, Authier A, Seelaar H, Van Swieten JC, Brown JM, Johannsen P, Nielsen JE, Holm IE, Consortium FR, Dickson DW, Rademakers R, Graff-Radford NR, Parisi JE, Petersen RC, Hatanpaa KJ, White CL, 3rd, Weiner MF, Geser F, Van Deerlin VM, Trojanowski JQ, Miller BL, Seeley WW, van der Zee J, Kumar-Singh S, Engelborghs S, De Deyn PP, Van Broeckhoven C, Bigio EH, Deng HX, Halliday GM, Kril JJ, Munoz DG, Mann DM, Pickering-Brown SM, Doodeman V, Adamson G, Ghazi-Noori S, Fisher EM, Holton JL, Revesz T, Rossor MN, Collinge J, Mead S, Isaacs AM. FUS pathology defines the majority of tau- and TDP-43-negative frontotemporal lobar degeneration. Acta neuropathologica. 2010;120:33–41. doi: 10.1007/s00401-010-0698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain: a journal of neurology. 2009;132:2922–2931. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun S, Ling SC, Qiu J, Albuquerque CP, Zhou Y, Tokunaga S, Li H, Qiu H, Bui A, Yeo GW, Huang EJ, Eggan K, Zhou H, Fu XD, Lagier-Tourenne C, Cleveland DW. ALS-causative mutations in FUS/TLS confer gain and loss of function by altered association with SMN and U1-snRNP. Nature communications. 2015;6:6171. doi: 10.1038/ncomms7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Armstrong GA, Drapeau P. Loss and gain of FUS function impair neuromuscular synaptic transmission in a genetic model of ALS. Human molecular genetics. 2013;22:4282–4292. doi: 10.1093/hmg/ddt278. [DOI] [PubMed] [Google Scholar]
- 39.Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, Kanagaraj AP, Carter R, Boylan KB, Wojtas AM, Rademakers R, Pinkus JL, Greenberg SA, Trojanowski JQ, Traynor BJ, Smith BN, Topp S, Gkazi AS, Miller J, Shaw CE, Kottlors M, Kirschner J, Pestronk A, Li YR, Ford AF, Gitler AD, Benatar M, King OD, Kimonis VE, Ross ED, Weihl CC, Shorter J, Taylor JP. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, Pliner HA, Abramzon Y, Marangi G, Winborn BJ, Gibbs JR, Nalls MA, Morgan S, Shoai M, Hardy J, Pittman A, Orrell RW, Malaspina A, Sidle KC, Fratta P, Harms MB, Baloh RH, Pestronk A, Weihl CC, Rogaeva E, Zinman L, Drory VE, Borghero G, Mora G, Calvo A, Rothstein JD, Drepper C, Sendtner M, Singleton AB, Taylor JP, Cookson MR, Restagno G, Sabatelli M, Bowser R, Chio A, Traynor BJ. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nature neuroscience. 2014;17:664–666. doi: 10.1038/nn.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abisambra J, Jinwal UK, Miyata Y, Rogers J, Blair L, Li X, Seguin SP, Wang L, Jin Y, Bacon J, Brady S, Cockman M, Guidi C, Zhang J, Koren J, Young ZT, Atkins CA, Zhang B, Lawson LY, Weeber EJ, Brodsky JL, Gestwicki JE, Dickey CA. Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biological psychiatry. 2013;74:367–374. doi: 10.1016/j.biopsych.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanaka M, Gupta R, Mayer BJ. Differential inhibition of signaling pathways by dominant-negative SH2/SH3 adapter proteins. Molecular and cellular biology. 1995;15:6829–6837. doi: 10.1128/mcb.15.12.6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhai J, Strom AL, Kilty R, Venkatakrishnan P, White J, Everson WV, Smart EJ, Zhu H. Proteomic characterization of lipid raft proteins in amyotrophic lateral sclerosis mouse spinal cord. The FEBS journal. 2009;276:3308–3323. doi: 10.1111/j.1742-4658.2009.07057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.C. UniProt. UniProt: a hub for protein information. Nucleic acids research. 2015;43:D204–212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic acids research. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic acids research. 2013;41:D808–815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jurica MS, Licklider LJ, Gygi SR, Grigorieff N, Moore MJ. Purification and characterization of native spliceosomes suitable for three-dimensional structural analysis. Rna. 2002;8:426–439. doi: 10.1017/s1355838202021088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Will CL, Urlaub H, Achsel T, Gentzel M, Wilm M, Luhrmann R. Characterization of novel SF3b and 17S U2 snRNP proteins, including a human Prp5p homologue and an SF3b DEAD-box protein. The EMBO journal. 2002;21:4978–4988. doi: 10.1093/emboj/cdf480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Will CL, Schneider C, Hossbach M, Urlaub H, Rauhut R, Elbashir S, Tuschl T, Luhrmann R. The human 18S U11/U12 snRNP contains a set of novel proteins not found in the U2-dependent spliceosome. Rna. 2004;10:929–941. doi: 10.1261/rna.7320604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Popow J, Englert M, Weitzer S, Schleiffer A, Mierzwa B, Mechtler K, Trowitzsch S, Will CL, Luhrmann R, Soll D, Martinez J. HSPC117 is the essential subunit of a human tRNA splicing ligase complex. Science. 2011;331:760–764. doi: 10.1126/science.1197847. [DOI] [PubMed] [Google Scholar]
- 51.Jonson L, Vikesaa J, Krogh A, Nielsen LK, Hansen T, Borup R, Johnsen AH, Christiansen J, Nielsen FC. Molecular composition of IMP1 ribonucleoprotein granules, Molecular & cellular proteomics. MCP. 2007;6:798–811. doi: 10.1074/mcp.M600346-MCP200. [DOI] [PubMed] [Google Scholar]
- 52.Weidensdorfer D, Stohr N, Baude A, Lederer M, Kohn M, Schierhorn A, Buchmeier S, Wahle E, Huttelmaier S. Control of c-myc mRNA stability by IGF2BP1-associated cytoplasmic RNPs. Rna. 2009;15:104–115. doi: 10.1261/rna.1175909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron. 2004;43:513–525. doi: 10.1016/j.neuron.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 54.Brendel C, Rehbein M, Kreienkamp HJ, Buck F, Richter D, Kindler S. Characterization of Staufen 1 ribonucleoprotein complexes. The Biochemical journal. 2004;384:239–246. doi: 10.1042/BJ20040812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Villace P, Marion RM, Ortin J. The composition of Staufen-containing RNA granules from human cells indicates their role in the regulated transport and translation of messenger RNAs. Nucleic acids research. 2004;32:2411–2420. doi: 10.1093/nar/gkh552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoek KS, Kidd GJ, Carson JH, Smith R. hnRNP A2 selectively binds the cytoplasmic transport sequence of myelin basic protein mRNA. Biochemistry. 1998;37:7021–7029. doi: 10.1021/bi9800247. [DOI] [PubMed] [Google Scholar]
- 57.Solomon S, Xu Y, Wang B, David MD, Schubert P, Kennedy D, Schrader JW. Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2alpha, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Molecular and cellular biology. 2007;27:2324–2342. doi: 10.1128/MCB.02300-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aulas A, Vande Velde C. Alterations in stress granule dynamics driven by TDP-43 and FUS: a link to pathological inclusions in ALS? Frontiers in cellular neuroscience. 2015;9:423. doi: 10.3389/fncel.2015.00423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burgess HM, Richardson WA, Anderson RC, Salaun C, Graham SV, Gray NK. Nuclear relocalisation of cytoplasmic poly(A)-binding proteins PABP1 and PABP4 in response to UV irradiation reveals mRNA-dependent export of metazoan PABPs. Journal of cell science. 2011;124:3344–3355. doi: 10.1242/jcs.087692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wilczynska A, Aigueperse C, Kress M, Dautry F, Weil D. The translational regulator CPEB1 provides a link between dcp1 bodies and stress granules. Journal of cell science. 2005;118:981–992. doi: 10.1242/jcs.01692. [DOI] [PubMed] [Google Scholar]
- 61.Meister G, Landthaler M, Peters L, Chen PY, Urlaub H, Luhrmann R, Tuschl T. Identification of novel argonaute-associated proteins, Current biology. CB. 2005;15:2149–2155. doi: 10.1016/j.cub.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 62.Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. The Journal of cell biology. 2005;169:871–884. doi: 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Borggrefe T, Wabl M, Akhmedov AT, Jessberger R. A B-cell-specific DNA recombination complex. The Journal of biological chemistry. 1998;273:17025–17035. doi: 10.1074/jbc.273.27.17025. [DOI] [PubMed] [Google Scholar]
- 64.Guil S, Long JC, Caceres JF. hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Molecular and cellular biology. 2006;26:5744–5758. doi: 10.1128/MCB.00224-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sama RR, Ward CL, Kaushansky LJ, Lemay N, Ishigaki S, Urano F, Bosco DA. FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. Journal of cellular physiology. 2013;228:2222–2231. doi: 10.1002/jcp.24395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gomes C, Keller S, Altevogt P, Costa J. Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neuroscience letters. 2007;428:43–46. doi: 10.1016/j.neulet.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 67.Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O’Neill MA, Yanai A, Silverman JM, Zeineddine R, Corcoran L, Kumita JR, Luheshi LM, Yousefi M, Coleman BM, Hill AF, Plotkin SS, Mackenzie IR, Cashman NR. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:3620–3625. doi: 10.1073/pnas.1312245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ding X, Ma M, Teng J, Teng RK, Zhou S, Yin J, Fonkem E, Huang JH, Wu E, Wang X. Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure. Oncotarget. 2015;6:24178–24191. doi: 10.18632/oncotarget.4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Feiler MS, Strobel B, Freischmidt A, Helferich AM, Kappel J, Brewer BM, Li D, Thal DR, Walther P, Ludolph AC, Danzer KM, Weishaupt JH. TDP-43 is intercellularly transmitted across axon terminals. The Journal of cell biology. 2015;211:897–911. doi: 10.1083/jcb.201504057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pokrishevsky E, Grad LI, Cashman NR. TDP-43 or FUS-induced misfolded human wild-type SOD1 can propagate intercellularly in a prion-like fashion. Scientific reports. 2016;6:22155. doi: 10.1038/srep22155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ulke-Lemee A, Trinkle-Mulcahy L, Chaulk S, Bernstein NK, Morrice N, Glover M, Lamond AI, Moorhead GB. The nuclear PP1 interacting protein ZAP3 (ZAP) is a putative nucleoside kinase that complexes with SAM68, CIA, NF110/45, and HNRNP-G. Biochimica et biophysica acta. 2007;1774:1339–1350. doi: 10.1016/j.bbapap.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 72.Ting NS, Kao PN, Chan DW, Lintott LG, Lees-Miller SP. DNA-dependent protein kinase interacts with antigen receptor response element binding proteins NF90 and NF45. The Journal of biological chemistry. 1998;273:2136–2145. doi: 10.1074/jbc.273.4.2136. [DOI] [PubMed] [Google Scholar]
- 73.Deng Q, Holler CJ, Taylor G, Hudson KF, Watkins W, Gearing M, Ito D, Murray ME, Dickson DW, Seyfried NT, Kukar T. FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:7802–7813. doi: 10.1523/JNEUROSCI.0172-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perez-Gonzalez A, Pazo A, Navajas R, Ciordia S, Rodriguez-Frandsen A, Nieto A. hCLE/C14orf166 associates with DDX1-HSPC117-FAM98B in a novel transcription-dependent shuttling RNA-transporting complex. PloS one. 2014;9:e90957. doi: 10.1371/journal.pone.0090957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nielsen J, Christiansen J, Lykke-Andersen J, Johnsen AH, Wewer UM, Nielsen FC. A family of insulin-like growth factor II mRNA-binding proteins represses translation in late development. Molecular and cellular biology. 1999;19:1262–1270. doi: 10.1128/mcb.19.2.1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Patel GP, Ma S, Bag J. The autoregulatory translational control element of poly(A)-binding protein mRNA forms a heteromeric ribonucleoprotein complex. Nucleic acids research. 2005;33:7074–7089. doi: 10.1093/nar/gki1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Prokipcak RD, Herrick DJ, Ross J. Purification and properties of a protein that binds to the C-terminal coding region of human c-myc mRNA. The Journal of biological chemistry. 1994;269:9261–9269. [PubMed] [Google Scholar]
- 78.Donnelly CJ, Willis DE, Xu M, Tep C, Jiang C, Yoo S, Schanen NC, Kirn-Safran CB, van Minnen J, English A, Yoon SO, Bassell GJ, Twiss JL. Limited availability of ZBP1 restricts axonal mRNA localization and nerve regeneration capacity. The EMBO journal. 2011;30:4665–4677. doi: 10.1038/emboj.2011.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kedersha N, Anderson P. Mammalian stress granules and processing bodies. Methods in enzymology. 2007;431:61–81. doi: 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- 80.Sheth U, Parker R. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science. 2003;300:805–808. doi: 10.1126/science.1082320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aulas A, Stabile S, Vande Velde C. Endogenous TDP-43, but not FUS, contributes to stress granule assembly via G3BP. Molecular neurodegeneration. 2012;7:54. doi: 10.1186/1750-1326-7-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. The Journal of cell biology. 2013;201:361–372. doi: 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bentmann E, Haass C, Dormann D. Stress granules in neurodegeneration–lessons learnt from TAR DNA binding protein of 43 kDa and fused in sarcoma. The FEBS journal. 2013;280:4348–4370. doi: 10.1111/febs.12287. [DOI] [PubMed] [Google Scholar]
- 84.Blokhuis AM, Koppers M, Groen EJ, van den Heuvel DM, Dini Modigliani S, Anink JJ, Fumoto K, van Diggelen F, Snelting A, Sodaar P, Verheijen BM, Demmers JA, Veldink JH, Aronica E, Bozzoni I, den Hertog J, van den Berg LH, Pasterkamp RJ. Comparative interactomics analysis of different ALS-associated proteins identifies converging molecular pathways. Acta neuropathologica. 2016 doi: 10.1007/s00401-016-1575-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.