

Abstract
The serine/threonine protein kinase, mammalian target of rapamycin (mTOR) is regulated by insulin and nutrient availability and has been proposed to play a central role as a nutrient sensor in skeletal muscle. mTOR associates with its binding partners, raptor and rictor, to form two structurally and functionally distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) respectively. We have investigated the assembly of mTORC1/2 and the activation of their downstream substrates (i.e. Akt, S6K1) in response to known effectors of mTOR, excess lipid availability and AMP-activated protein kinase (AMPK) activation/exercise training in rat skeletal muscle. The in vivo formation of mTORC1 and 2 and the activation of their respective downstream substrates were increased in response to chronic (8 weeks) consumption of a high-fat diet. Diet-induced mTORC activation and skeletal muscle insulin resistance were reversed by 4 weeks of exercise training, which was associated with enhanced muscle AMPK activation. In order to determine whether AMPK activation reverses lipid-induced mTOR activation, L6 myotubes were exposed to 0·4 mM palmitate to activate mTORC1/2 in the absence or presence of 5′-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR). Palmitate exposure (4 h) increased insulin-stimulated S6K1 Thr389 phosphorylation by 60%, indicating activation of mTORC1. AMPK activation with 1 mM AICAR abolished lipid-induced mTOR activation in vitro. Our data implicates reductions in mTOR complex activation with the reversal of lipid-induced skeletal muscle insulin resistance in response to exercise training or AICAR and identifies mTOR as a potential target for the treatment of insulin resistance.
Introduction
The insulin signal transduction cascade regulates many processes in skeletal muscle including glucose and lipid metabolism, cellular growth and differentiation and protein synthesis (Avruch 1998, Kimball et al. 2002, Taniguchi et al. 2006). Accordingly, impaired insulin signalling has been implicated in several disease states including type 2 diabetes (Zamboni et al. 2005, Taniguchi et al. 2006). Insulin action in muscle is a tightly regulated process with inputs from several diverse stimuli such as nutrient availability, hormonal milieu and exercise/muscle contraction (Hawley et al. 2006, Taniguchi et al. 2006). Specifically, chronic consumption of a high-fat (HF) diet increases the circulation of non-esterified fatty acids (NEFA) resulting in marked skeletal muscle insulin resistance in rodents and humans (Barnard & Youngren 1992, Boden 2002, Lessard et al. 2007). In contrast, one of the most potent means to improve insulin action in muscle is exercise training (Barnard & Youngren 1992, Hawley 2004, Hawley & Lessard 2008). We and others have previously demonstrated that exercise reverses the effects of an HF diet on skeletal muscle insulin resistance (Barnard & Youngren 1992, Lessard et al. 2007). However, the precise mechanism(s) by which lipids induce insulin resistance and exercise training reverses these defects in skeletal muscle is currently unknown.
The serine/threonine protein kinase, mammalian target of rapamycin (mTOR), is a member of the phosphatidylinositol 3 kinase (PI3K) family of enzymes. This kinase is regulated by insulin and nutrient availability and plays a central role as a nutrient sensor in skeletal muscle (Patti & Kahn 2004, Marshall 2006). mTOR associates with its binding partners, raptor and rictor, to form two structurally and functionally distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) respectively (Kim et al. 2002, Sarbassov et al. 2004, Bhaskar & Hay 2007). Activation of mTORC1 through a negative feedback loop inhibits insulin signalling via the activation of its downstream substrate ribosomal protein S6 kinase 1 (S6K1) and subsequent inhibitory serine phosphorylation of insulin receptor substrate 1 (IRS1; Tremblay & Marette 2001). In contrast, mTORC2 is a positive regulator of insulin signal transduction through its phosphorylation of protein kinase B/Akt on its serine 473 activation site (Hresko & Mueckler 2005, Sarbassov et al. 2005). Its ability to differentially modulate both proximal and distal components of the insulin signalling cascade raises the intriguing possibility that mTOR may be a critical regulator of insulin action in skeletal muscle.
Recent evidence demonstrates that in obese rodents or in rodents consuming an HF diet, overactivation of mTOR may be associated with impairments to skeletal muscle insulin action (Khamzina et al. 2005, Tremblay et al. 2007). In contrast, S6K1 null mice are protected from HF diet-induced insulin resistance (Um et al. 2004). Taken collectively, these results (Um et al. 2004, Khamzina et al. 2005, Tremblay et al. 2007) implicate mTORC1 activation as a possible mechanism for impaired insulin action in response to elevated lipid availability. However, it is unclear what role, if any, mTORC2 activation plays in lipid-induced insulin resistance. Activation of Akt by mTORC2 has several potential consequences for myocellular metabolism, as the Akt pathway is responsible for mediating most of the metabolic actions of insulin (Taniguchi et al. 2006). At present, it is unknown whether in skeletal muscle mTORC1 and 2 are differentially regulated in response to increased lipid availability.
We have previously demonstrated that endurance training increases the activation of 5′-AMP-activated protein kinase (AMPK) in skeletal muscle of HF-fed rodents (Lessard et al. 2007). AMPK, because of its role in suppressing energy-consuming processes, is a known physiological inhibitor of the energy consuming mTOR signalling pathway (Kimball 2006, Deshmukh et al. 2008). At present, the effect of endurance training on mTOR activation in insulin resistant muscle has not been investigated.
mTOR complexes can positively or negatively affect insulin action and are regulated by nutrients and AMPK. Therefore, we hypothesised that improvements in muscle insulin action following exercise training in HF-fed animals would be associated with altered activation and formation of mTOR complexes 1/2. Accordingly, we determined the effects of HF feeding and exercise training on the regulation of mTORC1 and 2 and the activation of their downstream substrates (i.e. Akt, S6K1/IRS1) in skeletal muscle. In addition, we determined whether acute palmitate exposure and the activation of AMPK by 5′-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) regulate the formation and activation of mTOR complexes 1/2.
Materials and Methods
Animals
Sprague–Dawley rats (~4-week-old) were given ad libitum access to either control chow diet (n=8; D12328, Research Diets Inc., New Bruns wick, NJ, USA, 73·1% carbohydrates, 10·5% fat and 16·4% protein) or an HF diet (n=16; D12330, Research Diets Inc., 25·5% carbohydrates, 58% fat and 16·4% protein). After a 4 week dietary period to induce insulin resistance, HF-fed animals were randomly assigned to either exercise training (HF EXT, n=8) or sedentary control (HF CON, n=8) groups. Exercise training consisted of treadmill running for 1 h/day, 5 days/week on a 15% incline at a speed that was progressively increased to 32 m/min during the first 5 days of training and maintained for subsequent 4 weeks. The third group of animals (CF CON) remained on the control chow diet for the duration of the study (8 wk). Following the experimental period animals were fasted for 8–12 h before undergoing hind-limb perfusion (described subsequently). Exercise trained animals undertook their last exercise bout 36–48 h prior to hind-limb perfusion. All experimental procedures were approved by the Institutional Animal Care and Use Committee at California State University, Northridge.
Hind-limb perfusion and 3-O-methylglucose transport
Rats were anaesthetised with an i.p. injection of sodium pentobarbital (6·5 mg/100 g body mass) and surgically prepared for hind-limb perfusion as previously described (Lessard et al. 2007). Briefly, cannulae were inserted into the abdominal aorta and vena cava, and the animals were sacrificed via an intracardiac injection of pentobarbital as the hind limbs were washed out with 30 ml Krebs–Henseleit buffer (KHB, pH 7·55). Immediately, the cannulae were placed in line with a non-recirculating perfusion system, and the hind limbs were allowed to stabilise during a 5 min washout period which consisted of a basic perfusate medium that contained 30% washed time-expired human erythrocytes (Ogden Medical Center, Ogden, UT, USA) KHB, 4% dialysed BSA (Fisher Scientific, Fair Lawn, NJ, USA), 0·2 mM pyruvate and was continuously gassed with a mixture of 95% O2–5% CO2 and warmed to 37 °C. Perfusions were performed at a flow rate of 7·5 ml/min and glucose transport was measured over an 8 min period using 8 mM of the non-metabolised glucose analogue 3-O-methylglucose (3-MG; 32 μCi 3-[3H] mg/mM, Perkin-Elmer Life Sciences, USA) in the presence (INSULIN) or absence (BASAL) of 500 μU/ml insulin. As an extracellular space marker 2 mM mannitol (60 μCi-[1-14C] mannitol/mM, PerkinElmer Life Sciences) was used. Immediately following the perfusion, portions of the red gastrocnemius (RG), a muscle comprising 90–95% oxidative fibres (Wilson et al. 1998), were excised from both hind limbs, blotted on gauze dampened with cold KHB, freeze clamped in liquid N2 and stored at −80 °C for later analysis. Rates of basal and insulin-stimulated skeletal muscle 3-MG transport were calculated as previously described (Lessard et al. 2007).
Muscle homogenisation
Portions of muscle were cut from basal and insulin-stimulated RG, weighed frozen and homogenised in an ice-cold homogenisation buffer (1:8 wt/vol) containing 50 mM Tris–HCl (pH 7·5), 5 mM Na-pyrophosphate, 50 mM NaF, 1 mM EDTA, 1 mM EGTA, 10% glycerol (v/v), 1% Triton-X, 1 mM dithiothreitol, 1 mM benzamidine, 1 mM phenyl-methylsulphonyl fluoride, 10 μg/ml trypsin inhibitor and 2 μg/ml aprotinin. Following centrifugation (21 000 g, 4 °C) for 25 min the supernatant was collected and assayed for protein content.
Cell culture
Stock L6 myoblasts (American Type Culture Collection, Manassas, VA, USA) were maintained at 37 °C (95% O2–5% 2 collagen-coated flasks in α-modified CO2) on 75 cm Eagle’s medium (Invitrogen) containing 10% foetal bovine serum (Sigma–Aldrich) culture medium, 1% penicillin–streptomycin (Sigma–Aldrich) and 5·5 mM glucose as previously described (Lessard et al. 2006). Differentiation was induced by switching to medium containing 2% horse serum (Sigma–Aldrich) when the myoblasts were ~90% confluent. Experimental treatments were started after 5 days, by which time nearly all of the myoblasts had fused to form myotubes. For experimental procedures the cells were maintained in 75 cm2 flasks (co-immunoprecipitation experiments) or trypsinised and seeded in 6-well plates. All subsequent experiments were done after 4 h serum starvation. To determine the effect of acute palmitate treatment, cells were incubated with 0·4 mM palmitate in ethanol vehicle for 0, 2 or 4 h in 2% fatty-acid free BSA. The effect of AMPK activation was subsequently determined by incubating cells for 1 or 4 h with or without 1 mM AICAR (Sigma–Aldrich). A control group was maintained for each experiment by incubating in the presence of 2% fatty-acid free BSA (Sigma–Aldrich) and the appropriate vehicle (ethanol and/or PBS). For the co-immunoprecipitation experiments, cells were incubated with 0·4 mM palmitate for 4 h with or without 1 mM AICAR. For insulin-stimulated conditions 100 nM insulin was added to appropriate wells during the last 30 min of incubations. All experiments were run in triplicate.
Western blotting
Insulin-stimulated RG muscle lysates (60 μg) were solubilised in Laemmli buffer, separated by SDS-PAGE, and transferred to PVDF membranes. The membranes were then blocked (5% non-fat dry milk (NFDM)), and incubated overnight at 4 °C with primary antibodies specific for either mTOR (mTab1), phospho-Akt1 Ser473, phospho-Akt1 Thr308, (1:1000; Upstate (Millipore) Biotechnology, Billerica, MA, USA), AMPKα, Akt1, Akt2, S6K1, Raptor, Rictor (mAb), TSC2, phospho-mTOR Ser2448, phospho-S6K1 Thr389, phospho-TSC2 Thr1462, phospho-IRS1 Ser636/639, phospho-Akt substrate (1:1000; Cell Signaling, Danvers, MA, USA) or phospho-AMPKα Thr172 which was raised against AMPKα peptide (KDGEFLRpTSCGAPNY) as described previously (Lessard et al. 2006). The immunoreactive proteins were detected with enhanced chemiluminescence (Amersham (GE Healthcare) Biosciences) and quantified by densitometry.
Akt2 immunoprecipitation
The Catch and Release v2.0 Reversible Immunoprecipitation System (Upstate (Millipore) Biotechnology) was used for detection of phospho-specific Akt2 sites as per manufacturer’s instructions. Briefly, insulin-stimulated RG muscle lysates (600 μg) and Akt2 (rabbit) antibody (6 μg, Upstate (Millipore) Biotechnology) were rotated overnight at 4 °C, immunocomplexes were eluted and subjected to SDS-PAGE electrophoresis. Antibodies specific for phospho-Akt Ser 473 (mouse) or phospho-Akt Thr308 (mouse; Cell Signaling) were used to detect the immunoreactive proteins as described above.
Akt1 kinase activity assay
Akt1 Immunoprecipitation Kinase Assay Kit (Upstate (Millipore) Biotechnology) was used to determine Akt1 activation. Briefly, Akt1 antibody (4 μg/sample, Upstate (Millipore) Biotechnology) and insulin-stimulated RG muscle lystates (400 μg) were rotated overnight at 4 °C. Protein G (50 μl, Amersham (GE Healthcare) Biosciences) slurry was then added to the antibody–protein complex and rotated 120 min at 4 °C. The kinase activity assay was then performed as per manufacturer’s instructions.
mTOR complex co-immunoprecipitation
The mTOR complexes have been previously been shown to be detergent sensitive (Sarbassov et al. 2004). Therefore, 0·3% CHAPS was substituted for 1% Triton-X in the homo-genisation buffer for the co-immunoprecipitation of the mTOR complexes. Basal and insulin-stimulated RG homogenates (5 mg) or L6 cell lysates (500 μg) and mTOR (mTab1) antibody (8 μl/sample, Upstate (Millipore) Bio-technology) were rotated overnight at 4 °C. Protein G (100 μl, Amersham (GE Healthcare) Biotechnology) slurry was then added to the antibody–protein complex and rotated 120 min at 4 °C. The beads were then washed five times with homogenisation buffer and aliquoted for western blotting. Aliquots of the bead–antibody–protein complex were resuspended in Laemmli buffer, subjected to SDS-PAGE and transferred to PVDF membranes. The membranes were then blocked (5% NFDM) and incubated overnight at 4 °C with primary antibodies specific for raptor, rictor (1:1000; Cell Signaling) and mTOR. Results are normalised to total mTOR.
Statistical analyses
All results are presented as mean±S.E.M. Differences between treatment groups were determined using a one-way ANOVA with a Student–Newman–Keuls post hoc test.
Results
Insulin-stimulated 3-MG transport in RG skeletal muscle
Basal rates of 3-MG transport were similar among groups (Fig. 1). The rates of insulin-stimulated glucose transport were decreased by the HF-diet (P<0·05, versus CF CON; Fig. 1) but were normalised to control levels by exercise training (P<0·05, versus HF CON; Fig. 1).
Figure 1.
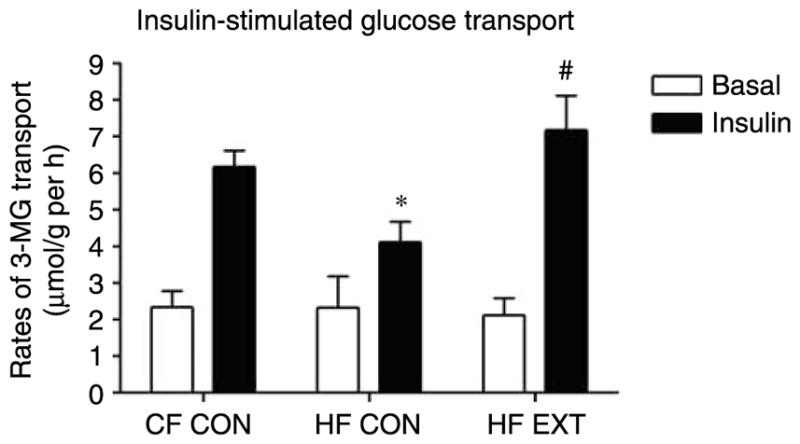
Muscle glucose transport was measured in the red gastrocnemius muscle after an 8 min hind-limb perfusion with 3H-3-O-methylglucose in the absence (basal) or presence (insulin) of insulin (500 μU/ml). Data expressed as rates μmol/g per hour (mean±S.E.M., n=6–8). Significant differences between groups *P<0·05 versus CF CON, #P<0·05 versus HF CON.
Chronically increased lipid availability enhances mTOR complex formation
The basal and insulin-stimulated formations of the mTORC1 and 2 complexes in response to increased chronic lipid availability and exercise were determined by immunoprecipitation of mTOR then probing for its binding partners (raptor and rictor, Fig. 2A). In the insulin-stimulated muscle, there was a 70% increase in co-immunoprecipitation of raptor and mTOR in HF CON (P<0·01, versus CF CON; Fig. 2C), indicating higher mTORC1 complex formation in response to HF feeding. Exercise training completely abolished the HF diet-induced increase in mTORC1 formation such that values were restored to those observed in CF CON (P<0·01, HF EXT versus HF CON; Fig. 2C). The co-immunoprecipitation of rictor and mTOR (i.e. mTORC2 complex formation), in the insulin-stimulated muscle, mirrored mTORC1 complex formation. HF feeding increased mTORC2 formation by 64% (P<0·05, versus CF CON; Fig. 2B), while exercise training decreased mTORC2 formation by 79% (P<0·01, HF CON versus HF EXT; Fig. 2B). These results demonstrate that in response to insulin both mTORC1 and mTORC2 formation are stimulated by chronically increased lipid availability, but these effects are reversed by exercise training. There were no significant differences in the formation of mTORC1 (Fig. 2C) and mTORC2 (Fig. 2B) in the basal muscle. Changes of mTOR complex formation occurred in the absence of changes in the total protein content of mTOR, raptor and rictor (Fig. 2D).
Figure 2.

The basal or insulin-stimulated formation of the mTOR complexes in response to high-fat feeding or after exercise training was determined by immunoprecipitation of mTOR followed by western blotting to detect its binding partners, (A) rictor and raptor, (B) rictor bound to mTOR, (C) raptor bound to mTOR are expressed as arbitrary units (mean±S.E.M., n=6–8). Relative protein levels of total mTOR, total rictor and total raptor (D) were quantified by densitometry. Representative blots are shown. Significant differences between groups *P<0·05 versus CF CON, #P<0·05 versus HF CON.
HF diet and exercise training differentially affects mTORC1 activity
In order to determine how mTORC1 complex formation translates to mTOR activation, the phosphorylation of its downstream substrates was measured under insulin-stimulated conditions. Increases in mTORC1 formation following an HF diet resulted in concomitant increases in mTOR activity, as demonstrated by phosphorylation of its downstream substrate S6K1 (Fig. 3A). In response to HF feeding phosphorylation of S6K1 was increased by 20% at Thr389 (P<0·001, CF CON versus HF CON; Fig. 3A). Exercise training normalised S6K1 phosphorylation on Thr389 (P<0·001, HF CON versus HF EXT; Fig. 3A). Increased S6K1 activation by HF feeding resulted in an 18% increase in inhibitory phosphorylation of IRS1 at Ser636/639 (P<0·05, CF CON versus HF CON; Fig. 3B), which was reversed in HF EXT (P<0·05, HF CON versus HF EXT; Fig. 3B). These results indicate that an HF diet increased mTORC1 activity, which induced a negative feedback loop on insulin signal transduction at the level of IRS1. Importantly, these diet-induced effects were reversed by exercise training. There were no changes in the protein concentration in total S6K1 or total IRS1 (Fig. 3A and B respectively).
Figure 3.

The insulin-stimulated phosphorylation of S6K1 on Thr389 (A), IRS1 on Ser636/639 (B) were quantified using western blot analysis and densitometry. Representative immunoblots are shown on each figure. Values are expressed as arbitrary units (mean±S.E.M., n=8). Significant differences between groups *P<0·05 versus CF CON, #P<0·05 versus HF CON.
Akt1 phosphorylation and activity mirrors the increase in mTORC2 complex formation
To assess mTORC2 activation in response to insulin stimulation, the isoform-specific phosphorylation of Akt1 and Akt2 at Ser473 was measured. In agreement with changes in mTORC2 formation, the phosphorylation of Akt1 was increased by 26% on the Ser473 site in HF CON (P<0·05 versus CF CON) but was decreased by 30% in HF EXT (P<0·05, versus HF CON; Fig. 4B). Changes in Akt1 Ser473 phosphorylation were accompanied by similar alterations in Akt1 kinase activity, which tended to increase in HF CON (12%, P<0·10 versus CF CON, Fig. 4C) and was decreased in HF EXT (P<0·05, HF EXT versus HF CON; Fig. 4C). Decreases in Akt1 Ser473 phosphorylation and activity following exercise training occurred despite an increase in total Akt1 protein (P<0·001 CF CON versus HF EXT and P<0·001 HF CON versus HF EXT; Fig. 4A). In contrast, following HF feeding or exercise training there were no changes in Ser473 phosphorylation or total protein content of the Akt2 isoform, or the phosphorylation of its downstream target the Akt substrate of 160 kDa (AS160; Fig. 4D). These results indicate that mTORC2 may preferentially phosphorylate and activate the Akt1 isoform. In addition, the phosphorylation of Akt1 and Akt2 on Thr308 were similar in all groups (Fig. 4B and D respectively), demonstrating that the phosphorylation of Thr308 is independent of mTORC2 formation.
Figure 4.

Total Akt1 (A) and its insulin-stimulated phosphorylation on Ser473 and Thr308 (B) were quantified using western blot analysis and densitometry. Insulin-stimulated Akt1 kinase activity was measured using the immunoprecipitation of 400 μg of protein (C). Site-specific Akt2 phosphorylation was measured by immunoprecipitation of 600 μg of protein and western blot analysis using antibodies specific for Akt Ser473 and Akt Thr308 phosphorylation sites (D). Total Akt2 and the phosphorylation of AS160 (D) were quantified using western blot analysis and densitometry. Representative immunoblots are shown. Values are expressed as arbitrary units (mean±S.E.M., n=7–8). Significant differences between groups *P<0·05 versus CF CON, #P<0·05 versus HF CON.
Exercise training regulates the phosphorylation of mTOR and its physiological inhibitors AMPK and TSC2
AMPK and tuberous sclerosis complex 2 (TSC2) are known inhibitors of mTOR activation. Thus, their phosphorylation in response to HF feeding and exercise training was determined in response to insulin. The phosphorylation of AMPK on its Thr172 activation site was increased by 19% in HF EXT (P<0·05, versus HF CON; Fig. 5B). No changes were noted in the total protein concentration of the AMPKα catalytic subunit (Fig. 5B). In contrast, exercise training decreased the phosphorylation of TSC2 on Thr1462 by 30% (P<0·01, versus HF CON; Fig. 5C). As TSC2 is a downstream target of Akt, our observed decreases in TSC2 phosphorylation likely reflect reduced Akt1 Ser473 phosphorylation and activation following exercise training (Fig. 4A). There was no difference in the total protein concentration of TSC2 between the groups (Fig. 5C). Despite the increases in mTORC1 activation by HF feeding, the phosphorylation of mTOR on its Ser2448 site was increased in HF EXT by 37% (P<0·05, versus HF CON) and 47% (P<0·01, versus CF CON; Fig. 5A).
Figure 5.

Insulin-stimulated phosphorylation of mTOR and its inhibitors AMPK and TSC2. The phosphorylation of mTOR on Ser2448 (A) AMPKα on Thr172 (B) and TSC2 on Thr1462 (C) was quantified using western blot analysis and densitometry. Representative immunoblots are shown on each figure. Values are expressed as arbitrary units (mean±S.E.M., n=7–8). Significant differences between groups *P<0·05 versus CF CON, #P<0·05 versus HF CON.
Acute palmitate and AICAR activation of mTOR complexes in L6 cell culture
It has been previously demonstrated that within 3–5 h circulating NEFA induce insulin resistance in skeletal muscle (Boden 2002). Treatment with the AMPK activator AICAR has been shown to enhance glucose transport and inhibit mTOR activation (Bolster et al. 2002, Ju et al. 2007). Therefore, we determined whether 4 h palmitate incubation could activate mTOR signalling in vitro and whether the stimulation of AMPK by AICAR could inhibit the formation of mTOR complexes 1/2 and the phosphorylation of their downstream substrates. We observed an incremental increase in palmitate-induced S6K1 and Akt1 phosphorylation after 2 and 4 h of palmitate exposure in L6 myotubes (Fig. 6A). AICAR decreased the phosphorylation of S6K1 after 1 and 4 h of treatment in both the absence (CON) and presence of palmitate (Fig. 6B). Similar to the results of the chronic in vivo experiments, 4 h palmitate treatment increased the phosphorylation of S6K1 Thr389 and Akt1 Ser473 by 60 and 30% respectively (P<0·05 versus CON; Fig. 6C). AICAR significantly increased the phosphorylation of AMPK on Thr172 (P<0·05 versus CON, Fig. 6C). AICAR-induced AMPK activation decreased palmitate-induced S6K1 Thr389 phosphorylation by 63% (P<0·05 versus Palm, Fig. 6C). There were no significant changes in the acute complex formation of either mTORC1 or mTORC2 in response to palmitate or AICAR (Fig. 6E and F).
Figure 6.

The basal and insulin-stimulated activation of mTORC1/2 substrates in response to palmitate and/or AICAR treatment was determined in L6 myotubes. Phosphorylation of S6K1 on Thr389 and Akt1 on Ser 473 was measured in response to treatment with 0·4 mM palmitate for 0 (control), 2 (2 h Palm) or 4 (4 h Palm) hour (A). The effect of 1 mM AICAR on the phosphorylation of S6K1 (Thr389), Akt1 (Ser473) and AMPK (Thr172) was measured in cells treated with (Palm) or without (Con) 0·4 mM palmitate during the entire 4 h incubation (4 h AICAR) or during the last 1 h (1 h AICAR) (B). In order to associate changes in mTORC activation with complex formation, 75 mm2 flasks of cells were incubated in 0·4 mM palmitate and 1 mM AICAR for 4 h to determine the phosphorylation of S6K1 on Thr389, Akt1 on Ser473 and AMPK on Thr172 (C). The formation of mTORC1/2 (D) was measured by immunoprecipitation of mTOR followed by western blot of mTOR binding partners’ raptor (E) and rictor (F) which were quantified by densitometry. Insulin (100 nM) was added to the appropriate groups during the last 30 min of incubation. Representative blots are shown. Actin was run as a loading control for each sample. Significant differences between groups *P<0·05 versus CF CON, #P<0·05 versus HF CON; n=3/treatment.
Discussion
The aim of the present study was to determine the effect of increased lipid oversupply on the regulation of mTOR complexes, mTORC1 and mTORC2. Furthermore, we sought to establish whether mTOR complex formation is linked to the activation of downstream substrates involved in insulin signal transduction in skeletal muscle. We had previously demonstrated that exercise training can reverse impairments to skeletal muscle insulin action in response to chronic HF feeding (Lessard et al. 2007). Given the potential for mTOR complexes to both positively and negatively regulate insulin action in skeletal muscle, it is plausible that HF feeding and exercise training divergently regulate mTOR complex formation. Accordingly, we hypothesised that a potential mechanism by which exercise and HF feeding exert divergent effects on insulin action may involve the differential regulation of mTOR complex formation. For the first time, we demonstrate in vivo that both mTORC1 and mTORC2 formation are up-regulated by chronic lipid availability (HF diet) and that these changes are completely reversed by exercise training. Additionally, increased mTORC1 and mTORC2 formation in response to an HF diet was associated with activation of their respective downstream substrates and altered insulin signal transduction.
The hyperactivation of mTORC1 by nutrient excess (amino acids, lipids, obesity) has been implicated in decreased insulin signal transduction at the level of IRS1 (Patti et al. 1998, Tremblay & Marette 2001, Um et al. 2004, Khamzina et al. 2005, Krebs et al. 2007, Tremblay et al. 2007). It has been proposed that an S6K1-associated negative feedback loop results in the inhibitory serine phosphorylation of IRS1 in response to mTOR activation (Patti & Kahn 2004, Um et al. 2006). We have previously observed decreases in IRS1-associated PI3K activity in skeletal muscle after HF feeding (Lessard et al. 2007). However, the regulation of mTORC1 activation by acute and chronic lipid availability is not well described. Recent in vitro work in skeletal muscle cell culture has directly implicated palmitate with the inhibition of insulin stimulated glucose uptake (Pimenta et al. 2008) likely through the activation of mTORC1 and its inhibition of insulin signalling (Mordier & Iynedjian 2007). In support of a role for mTOR activation in insulin resistance, Miller et al. (2008) observed increased mTOR activation in the skeletal muscle of ob/ob mice (Miller et al. 2008). However, the authors did not observe improvements in whole-body insulin or glucose tolerance after acute treatment with rapamycin, a potent inhibitor of mTOR activation (Miller et al. 2008). In contrast, in the current study we observed an exercise-induced inhibition of mTOR signalling that corresponded with an increase in skeletal muscle specific insulin sensitivity (Fig. 1).
A novel finding of the present study was that an HF diet increased the activation of mTORC1 in skeletal muscle, as evidenced by an increase in Thr389 S6K1 phosphorylation (Figs 3A and 6C), which led to a concomitant increase in the serine phosphorylation of IRS1 (Fig. 3B). Our results support the contention that one potential mechanism for the inhibition of proximal insulin signalling following acute and chronic lipid oversupply may be increased mTORC1 activation leading to IRS1 serine phosphorylation. In contrast, we show that exercise training reverses the effects of chronic lipid oversupply on mTORC1 activation (Fig. 3A), as evidenced by normalisation of S6K1 phosphorylation and decreased serine phosphorylation of IRS1 (Fig. 3B). We and others have previously shown that exercise training increases AMPK activation (Atherton et al. 2005, Lessard et al. 2007, Hawley & Lessard 2008), suggesting that exercise training may reverse the effect of mTOR activation through increased AMPK activity. In agreement with this hypothesis, Ju et al. (2007) have shown in cell culture that AMPK regulates insulin action through the inhibition of IRS1 serine phosphorylation by mTOR/S6K1 (Ju et al. 2007). Here, we demonstrate that treating palmitate incubated cells with the AMPK activator AICAR inhibits phosphorylation of S6K1 at its Thr389 site (Fig. 6C).
Analogous to the effects of increased lipid availability on mTORC1, we demonstrate that an HF-diet increases the formation of mTORC2, as indicated by increased mTOR/rictor association (Fig. 1B). Again, exercise training reversed the effects of HF feeding on mTORC2 formation (Fig. 1B). The simultaneous activation/deactivation of mTORC1 and mTORC2 in skeletal muscle seems paradoxical, as these two complexes have opposing effects on insulin signal transduction (Bhaskar & Hay 2007). While mTORC1 activation inhibits IRS1 activation, it has recently been reported that mTORC2 is an upstream kinase of Akt at Ser473, which is required for its full activation in response to insulin (Hresko & Mueckler 2005, Sarbassov et al. 2005, Kumar et al. 2008). Our results indicate that in skeletal muscle, mTORC2 formation was coupled with Ser473 phosphorylation of the Akt1 isoform, which has primarily been studied with respect to its role in cell growth and differentiation (Cho et al. 2001, Wilson & Rotwein 2007). Similar to our in vivo findings we observed a significant increase in Ser473 Akt1 phosphorylation in L6 myotubes after 4 h palmitate incubation (Fig. 6C). In contrast, we observed no changes in Ser473 phosphorylation of the Akt2 isoform, which is an important regulator of glucose uptake in muscle (Fig. 3D). In addition, we could detect no mTORC2-associated changes in the phosphorylation of AS160, a downstream substrate of Akt, with a putative role in insulin-stimulated glucose uptake (Fig. 3D). The selective activation of Akt1 and the absence of Akt2 regulation by mTORC2 with HF feeding may explain how an HF diet impairs insulin stimulated glucose transport despite activation of mTORC2 in muscle. The converse argument would apply with respect to the ability of exercise training to enhance insulin-stimulated glucose uptake despite inhibiting mTORC2 formation.
The results of previous investigations that have examined the effects of endurance exercise on skeletal muscle mTOR activation are equivocal, with some studies demonstrating decreased (Williamson et al. 2006, Glynn et al. 2008, Miranda et al. 2008), no change (Atherton et al. 2005, Coffey et al. 2006, Mascher et al. 2007) or increased (Fujita et al. 2007) activation of mTOR following endurance exercise. In the present study, we observed an unexpected increase in the exercise-induced phosphorylation of Ser2448 on mTOR despite decreased Akt activation (Fig. 4B and C). Our results are in agreement with recent studies that have demonstrated that the phosphorylation of mTOR on Ser2448 is independent of Akt activation (Parkington et al. 2003, Mothe-Satney et al. 2004, Chiang & Abraham 2005, Allemand et al. 2009). The results of the present investigation are also consistent with those of Miranda et al. (2008), who demonstrated that insulin-stimulated S6K1 activation was decreased following endurance training (Miranda et al. 2008). Thus, it appears that unlike resistance training, which is a potent activator of mTOR (Atherton et al. 2005, Coffey & Hawley 2007), endurance training may inhibit mTOR activation in skeletal muscle. Opposing roles for these two training modes on mTOR activation are not surprising given the highly contrasting nature of muscle adaptations resulting from endurance and resistance exercise (Atherton et al. 2005, Coffey & Hawley 2007).
One mechanism by which exercise training enhances insulin action in skeletal muscle is via the chronic activation of AMPK (Hawley & Lessard 2008). AMPK is a known physiological inhibitor of the energy consuming mTOR signalling pathway (Kimball 2006). Indeed, interventions that reduce intracellular ATP levels, or administration of the AMPK activator, AICAR, decreases mTOR activation as demonstrated by decreased phosphorylation of S6K1 (Kimball 2006, Deshmukh et al. 2008). In the present study, we observed an exercise-induced increase in AMPK Thr172 phosphorylation (Fig. 5B), suggesting that AMPK activation may be one possible mechanism to explain the inhibition of mTOR activation in response to exercise training (Fig. 5B). Indeed, when treating palmitate-incubated cells with the AMPK activator AICAR, we observed significant decreases in the phosphorylation of the mTORC1 substrate Thr389 S6K1 (Fig. 6C). Given that such opposing stimuli and cellular responses are associated with the AMPK and mTOR pathways it seems logical that AMPK activation results in the inhibition of mTOR.
It is known that the activation of mTORC1 and mTORC2 is sensitive to increased nutrient availability (Kim et al. 2002, Jacinto et al. 2004, Marshall 2006). Investigations using rapamycin, an mTOR inhibitor, have previously demonstrated that the formations of mTOR complexes are necessary for nutrient-induced activation of S6K1/IRS1 and Akt (Tremblay & Marette 2001, Sarbassov et al. 2006, Sipula et al. 2006, Krebs et al. 2007). Our results provide novel evidence that the in vivo formation of both mTOR complexes (mTORC1 and mTORC2) is responsive to both the chronic interventions of exercise training and an HF diet. Furthermore, we demonstrate that changes in mTOR complex formation are associated with the activation of their respective downstream substrates. In agreement with the results from the current study, Schieke et al. (2006) have shown that increases in mTOR-raptor association after immunoprecipitation correlated with the phosphorylation and activation of S6K1 (Schieke et al. 2006). In contrast, acute in vitro palmitate application in cell culture had no effect on the formation of the mTOR complexes (Fig. 6E and F) despite increased mTORC1/2 activation (Fig. 6C).
In conclusion, we demonstrate for the first time that an HF diet and exercise training have divergent in vivo effects on the formation and activation of mTORC1 and mTORC2. Our data also suggest that changes in the activation of the mTOR complexes may be one mechanism to explain the altered insulin signal transduction in response to lipid availability. The reversal of mTORC 1/2 activation by exercise training was associated with the activation of AMPK in skeletal muscle. This observation is supported by our in vitro work demonstrating that AICAR abolishes the lipid-induced activation of S6K1. As impaired insulin action in skeletal muscle is pivotal at the onset of type 2 diabetes, these findings have implications for both the understanding of insulin signal transduction in skeletal muscle and the discovery of therapeutic targets for the treatment of its dysregulation.
Acknowledgments
Funding
This work was supported by an RMIT University, School of Medical Sciences and Emerging Researcher Grants (S J Lessard), an Australian Research Council Grant (J A Hawley, DP0663862) and National Institutes of Health Grants (B B Yaspelkis III, GM-48680, GM-08395 and DK-57625).
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that would prejudice the impartiality of this scientific work.
References
- Allemand MC, Irving BA, Asmann YW, Klaus KA, Tatpati L, Coddington CC, Nair KS. Effect of testosterone on insulin stimulated IRS1 Ser phosphorylation in primary rat myotubes – a potential model for PCOS-related insulin resistance. PLoS ONE. 2009;4:e4274. doi: 10.1371/journal.pone.0004274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton PJ, Babraj J, Smith K, Singh J, Rennie MJ, Wackerhage H. Selective activation of AMPK-PGC-1α or PKB-TSC2-mTOR signaling can explain specific adaptive responses to endurance or resistance training-like electrical muscle stimulation. FASEB Journal. 2005;19:786–788. doi: 10.1096/fj.04-2179fje. [DOI] [PubMed] [Google Scholar]
- Avruch J. Insulin signal transduction through protein kinase cascades. Molecular and Cellular Biochemistry. 1998;182:31–48. [PubMed] [Google Scholar]
- Barnard RJ, Youngren JF. Regulation of glucose transport in skeletal muscle. FASEB Journal. 1992;6:3238–3244. doi: 10.1096/fasebj.6.14.1426762. [DOI] [PubMed] [Google Scholar]
- Bhaskar PT, Hay N. The two TORCs and Akt. Developmental Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Boden G. Interaction between free fatty acids and glucose metabolism. Current Opinion in Clinical Nutrition and Metabolic Care. 2002;5:545–549. doi: 10.1097/00075197-200209000-00014. [DOI] [PubMed] [Google Scholar]
- Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. Journal of Biological Chemistry. 2002;277:23977–23980. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- Chiang GG, Abraham RT. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. Journal of Biological Chemistry. 2005;280:25485–25490. doi: 10.1074/jbc.M501707200. [DOI] [PubMed] [Google Scholar]
- Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBα is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. Journal of Biological Chemistry. 2001;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- Coffey VG, Hawley JA. The molecular bases of training adaptation. Sports Medicine. 2007;37:737–763. doi: 10.2165/00007256-200737090-00001. [DOI] [PubMed] [Google Scholar]
- Coffey VG, Zhong Z, Shield A, Canny BJ, Chibalin AV, Zierath JR, Hawley JA. Early signaling responses to divergent exercise stimuli in skeletal muscle from well-trained humans. FASEB Journal. 2006;20:190–192. doi: 10.1096/fj.05-4809fje. [DOI] [PubMed] [Google Scholar]
- Deshmukh AS, Treebak JT, Long YC, Viollet B, Wojtaszewski JF, Zierath JR. Role of adenosine 5′-monophosphate-activated protein kinase subunits in skeletal muscle mammalian target of rapamycin signaling. Molecular Endocrinology. 2008;22:1105–1112. doi: 10.1210/me.2007-0448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita S, Rasmussen BB, Cadenas JG, Drummond MJ, Glynn EL, Sattler FR, Volpi E. Aerobic exercise overcomes the age-related insulin resistance of muscle protein metabolism by improving endothelial function and Akt/mammalian target of rapamycin signaling. Diabetes. 2007;56:1615–1622. doi: 10.2337/db06-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn EL, Lujan HL, Kramer VJ, Drummond MJ, DiCarlo SE, Rasmussen BB. A chronic increase in physical activity inhibits fed-state mTOR/S6K1 signaling and reduces IRS-1 serine phosphorylation in rat skeletal muscle. Applied Physiology, Nutrition, and Metabolism. 2008;33:93–101. doi: 10.1139/h07-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley JA. Exercise as a therapeutic intervention for the prevention and treatment of insulin resistance. Diabetes/Metabolism Research and Reviews. 2004;20:383–393. doi: 10.1002/dmrr.505. [DOI] [PubMed] [Google Scholar]
- Hawley JA, Lessard SJ. Exercise training-induced improvements in insulin action. Acta Physiologica. 2008;192:127–135. doi: 10.1111/j.1748-1716.2007.01783.x. [DOI] [PubMed] [Google Scholar]
- Hawley JA, Hargreaves M, Zierath JR. Signalling mechanisms in skeletal muscle: role in substrate selection and muscle adaptation. Essays in Biochemistry. 2006;42:1–12. doi: 10.1042/bse0420001. [DOI] [PubMed] [Google Scholar]
- Hresko RC, Mueckler M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. Journal of Biological Chemistry. 2005;280:40406–40416. doi: 10.1074/jbc.M508361200. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR, complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature Cell Biology. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- Ju JS, Gitcho MA, Casmaer CA, Patil PB, Han D-G, Spencer SA, Fisher JS. Potentiation of insulin-stimulated glucose transport by the AMP-activated protein kinase. American Journal of Physiology. Cell Physiology. 2007;292:C564–C572. doi: 10.1152/ajpcell.00269.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. 2005;146:1473–1481. doi: 10.1210/en.2004-0921. [DOI] [PubMed] [Google Scholar]
- Kim D-H, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with Raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Kimball SR. Interaction between the AMP-activated protein kinase and mTOR signaling pathways. Medicine and Science in Sports and Exercise. 2006;11:1958–1964. doi: 10.1249/01.mss.0000233796.16411.13. [DOI] [PubMed] [Google Scholar]
- Kimball SR, Farrell PA, Jefferson LS. Exercise effects on muscle insulin signaling and action: invited review: role of insulin in translational control of protein synthesis in skeletal muscle by amino acids or exercise. Journal of Applied Physiology. 2002;93:1168–1180. doi: 10.1152/japplphysiol.00221.2002. [DOI] [PubMed] [Google Scholar]
- Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, Roth E, Furnsinn C, Promintzer M, Anderwald C, et al. The mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes. 2007;56:1600–1607. doi: 10.2337/db06-1016. [DOI] [PubMed] [Google Scholar]
- Kumar A, Harris TE, Keller SR, Choi KM, Magnuson MA, Lawrence JC., Jr Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances basal glycogen synthase activity. Molecular and Cellular Biology. 2008;28:61–70. doi: 10.1128/MCB.01405-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard SJ, Chen Z-P, Watt MJ, Hashem M, Reid JJ, Febbraio MA, Kemp BE, Hawley JA. Chronic rosiglitazone treatment restores AMPKα2 activity in insulin-resistant rat skeletal muscle. American Journal of Physiology. Endocrinology and Metabolism. 2006;290:E251–E257. doi: 10.1152/ajpendo.00096.2005. [DOI] [PubMed] [Google Scholar]
- Lessard SJ, Rivas DA, Chen Z-P, Bonen A, Febbraio MA, Reeder DW, Kemp BE, Yaspelkis BB, III, Hawley JA. Tissue-specific effects of rosiglitazone and exercise in the treatment of lipid-induced insulin resistance. Diabetes. 2007;56:1856–1864. doi: 10.2337/db06-1065. [DOI] [PubMed] [Google Scholar]
- Marshall S. Role of insulin, adipocyte hormones, and nutrient-sensing pathways in regulating fuel metabolism and energy homeostasis: a nutritional perspective of diabetes, obesity, and cancer. Science’s STKE. 2006;2006:re7. doi: 10.1126/stke.3462006re7. [DOI] [PubMed] [Google Scholar]
- Mascher H, Andersson H, Nilsson P-A, Ekblom B, Blomstrand E. Changes in signalling pathways regulating protein synthesis in human muscle in the recovery period after endurance exercise. Acta Physiologica. 2007;191:67–75. doi: 10.1111/j.1748-1716.2007.01712.x. [DOI] [PubMed] [Google Scholar]
- Miller AM, Brestoff JR, Phelps CB, Berk EZ, Reynolds TH., IV Rapamycin does not improve insulin sensitivity despite elevated mammalian target of rapamycin complex 1 activity in muscles of ob/ob mice. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2008;295:R1431–R1438. doi: 10.1152/ajpregu.90428.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda L, Horman S, De Potter I, Hue L, Jensen J, Rider MH. Effects of contraction and insulin on protein synthesis, AMP-activated protein kinase and phosphorylation state of translation factors in rat skeletal muscle. Pflügers Archiv. 2008;455:1129–1140. doi: 10.1007/s00424-007-0368-2. [DOI] [PubMed] [Google Scholar]
- Mordier S, Iynedjian PB. Activation of mammalian target of rapamycin complex 1 and insulin resistance induced by palmitate in hepatocytes. Biochemical and Biophysical Research Communications. 2007;362:206–211. doi: 10.1016/j.bbrc.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Mothe-Satney I, Gautier N, Hinault C, Lawrence JC, Jr, Van Obberghen E. In rat hepatocytes glucagon increases mammalian target of rapamycin phosphorylation on serine 2448 but antagonizes the phosphorylation of its downstream targets induced by insulin and amino acids. Journal of Biological Chemistry. 2004;279:42628–42637. doi: 10.1074/jbc.M405173200. [DOI] [PubMed] [Google Scholar]
- Parkington JD, Siebert AP, LeBrasseur NK, Fielding RA. Differential activation of mTOR signaling by contractile activity in skeletal muscle. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2003;285:R1086–R1090. doi: 10.1152/ajpregu.00324.2003. [DOI] [PubMed] [Google Scholar]
- Patti ME, Kahn BB. Nutrient sensor links obesity with diabetes risk. Nature Medicine. 2004;10:1049–1050. doi: 10.1038/nm1004-1049. [DOI] [PubMed] [Google Scholar]
- Patti M-E, Brambilla E, Luzi L, Landaker EJ, Ronald Kahn C. Bidirectional modulation of insulin action by amino acids. Journal of Clinical Investigation. 1998;101:1519–1529. doi: 10.1172/JCI1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimenta AS, Gaidhu MP, Habib S, So M, Fediuc S, Mirpourian M, Musheev M, Curi R, Ceddia RB. Prolonged exposure to palmitate impairs fatty acid oxidation despite activation of AMP-activated protein kinase in skeletal muscle cells. Journal of Cellular Physiology. 2008;217:478–485. doi: 10.1002/jcp.21520. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim D-H, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Current Biology. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor–mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen J-H, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Molecular Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Schieke SM, Phillips D, McCoy JP, Jr, Aponte AM, Shen R-F, Balaban RS, Finkel T. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. Journal of Biological Chemistry. 2006;281:27643–27652. doi: 10.1074/jbc.M603536200. [DOI] [PubMed] [Google Scholar]
- Sipula IJ, Brown NF, Perdomo G. Rapamycin-mediated inhibition of mammalian target of rapamycin in skeletal muscle cells reduces glucose utilization and increases fatty acid oxidation. Metabolism. 2006;55:1637–1644. doi: 10.1016/j.metabol.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nature Reviews Molecular Cell Biology. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- Tremblay F, Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. Journal of Biological Chemistry. 2001;276:38052–38060. doi: 10.1074/jbc.M106703200. [DOI] [PubMed] [Google Scholar]
- Tremblay F, Brûlé S, Um SH, Li Y, Masuda K, Roden M, Sun XJ, Krebs M, Polakiewicz RD, George T, et al. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. PNAS. 2007;104:14056–14061. doi: 10.1073/pnas.0706517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- Um SH, D’Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metabolism. 2006;3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Williamson DL, Kubica N, Kimball SR, Jefferson LS. Exercise-induced alterations in extracellular signal-regulated kinase 1/2 and mammalian target of rapamycin (mTOR) signalling to regulatory mechanisms of mRNA translation in mouse muscle. Journal of Physiology. 2006;573:497–510. doi: 10.1113/jphysiol.2005.103481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EM, Rotwein P. Selective control of skeletal muscle differentiation by Akt1. Journal of Biological Chemistry. 2007;282:5106–5110. doi: 10.1074/jbc.C600315200. [DOI] [PubMed] [Google Scholar]
- Wilson MC, Jackson VN, Heddle C, Price NT, Pilegaard H, Juel C, Bonen A, Montgomery I, Hutter OF, Halestrap AP. Lactic acid efflux from white skeletal muscle Is catalyzed by the monocarboxylate transporter isoform MCT3. Journal of Biological Chemistry. 1998;273:15920–15926. doi: 10.1074/jbc.273.26.15920. [DOI] [PubMed] [Google Scholar]
- Zamboni M, Mazzali G, Zoico E, Harris TB, Meigs JB, Di Francesco V, Fantin F, Bissoli L, Bosello O. Health consequences of obesity in the elderly: a review of four unresolved questions. International Journal of Obesity. 2005;29:1011–1029. doi: 10.1038/sj.ijo.0803005. [DOI] [PubMed] [Google Scholar]