

INTRODUCTION
The well-stirred hepatic clearance model (WSHM) has been expanded to include drug transporters (i.e. extended clearance model [ECM]). But, the consequences of this expansion in understanding when transporters vs. metabolic enzymes will affect the PK and PD of drugs remains opaque. Identifying the rate-determining step(s) in systemic or tissue drug PK/PD will allow accurate predictions of drug PK/PD and drug-drug interactions (DDIs). Here we clarify the implications of the ECM on PK/PD of drugs.
COMMENTARY
Models describing hepatic clearance of drugs have provided significant insight into hepatic drug disposition including when intrinsic metabolic clearance or hepatic blood flow play a significant role in determining the hepatic drug clearance (henceforth called the rate-determining step). However, a major limitation of these models (e.g. WSHM, Eq. 1) is the assumption that the unbound drug concentrations in the blood and the liver are in instantaneous equilibrium. This assumption is correct for drugs that are lipophilic and are not transported across the sinusoidal membrane. But, this assumption is not correct for drugs that are transported by the sinusoidal transporters or have poor permeability across the sinusoidal membrane. With the discovery of transporters present on the sinusoidal and canalicular membranes that are important in the disposition of many drugs, these models need to be modified. While we (1) and others (2, 3) have described the ECM (Eq. 2, Fig. 1A), where the WSHM has been modified to include transporters, the consequences of this modification in understanding the PK/PD of drugs remains opaque. In this commentary we clarify the implications of the ECM on drug PK/PD through theoretical simulations followed by in vivo examples of drugs that exhibit paradoxical PK/PD behavior in the clinic that cannot be explained by the WSHM. While the focus is on the liver, the principles enunciated here apply to any organ where transporters are expressed (e.g. kidney, blood-brain barrier).
| (1) |
| (2) |
Figure 1.
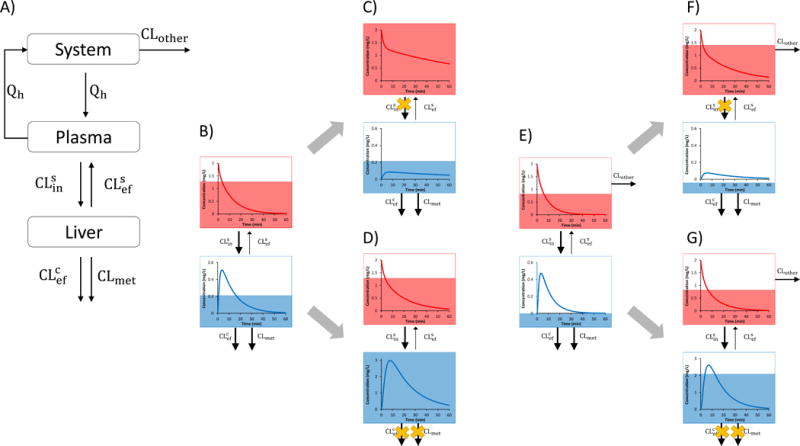
A) The extended clearance model (ECM) for hepatic disposition of a drug is described by sinusoidal influx (CLsin) and efflux clearances (CLsef), canalicular efflux (biliary) clearance (CLcef), metabolic clearance (CLmet), hepatic blood flow, Qh, and fraction unbound of the drug in the blood/plasma (fu). Transport at the sinusoidal membrane is represented by both active transport and passive diffusion while only active transport is assumed at the canalicular membrane. System represents the systemic circulation, while plasma refers to the hepatic plasma. CLother represents extra-hepatic CL and is assumed to be negligible for Figs 1B–D but significant for Figs 1E–G. B–G) The red and blue lines represent the systemic and hepatic drug concentration-time profile, respectively. The blue and red shaded areas represent the systemic and hepatic drug AUC, respectively. B) When CLsef is much smaller than CLmet plus CLcef, the liver effectively acts as a sink, which causes hepatic CL (as determined by systemic plasma/blood concentrations) to be rate-determined by CLsin (Scenario 1). C) Therefore, inhibition of CLsin will increase the systemic AUC of the drug but its hepatic AUC will not change. However, the shape of the hepatic drug concentration-time profile (e.g. Cmax and Tmax) will be altered. D) In contrast, when CLmet (or CLcef) is inhibited, the systemic AUC of the drug will not increase but the hepatic AUC will. The following values were used for simulation for parts B–D: CLsin = 1 L/min, CLsef = 0.1 L/min, CLmet + CLcef = 1.2 L/min, CLother= 0 L/min and Qh was set arbitrarily at 1 L/min. 90% inhibition of CLsin or CLmet + CLcef was simulated. E) Presence of significant extra-hepatic CL (CLother >> 0) of the drug depicted in B will decrease the fraction of drug eliminated via the liver. F) As a result, compared with the drug in B, inhibition of CLsin will result in a smaller increase in systemic AUC of the drug and now a decrease in hepatic AUC of the drug. G) Furthermore, when CLmet or CLcef is inhibited, the magnitude of change in hepatic drug AUC will be smaller compared to drug in part B. The simulations were conducted as in B except that CLother (0.2 L/min) was added.
Scenario 1: When does sinusoidal uptake clearance determine hepatic clearance of drugs?
Atorvastatin (ATV) (logD = 1.53) is extensively cleared by CYP3A4 (fm > 0.85) and has negligible extra-hepatic clearance. It is also a substrate of sinusoidal uptake transporters, mainly OATPs (4). According to the WSHM one would predict that inhibition of CYP3A4 would result in a significant increase in ATV plasma concentrations. But this is not the case. IV administration of itraconazole (a CYP3A4 inhibitor) does not affect plasma ATV AUC, while a single oral dose of rifampin (an OATP inhibitor but a weak CYP3A4 inhibitor) increases ATV plasma AUC by ~12-fold (5).
While the WSHM cannot explain the above paradoxical findings, the ECM can. When a drug enters the liver and is metabolized or excreted into the bile faster than the drug can exit the liver through the sinusoidal membrane, the loss of the drug from the systemic circulation will be determined only by sinusoidal uptake. That is, when metabolic (CLmet) plus canalicular efflux clearance (CLcef) are much greater than sinusoidal efflux clearance (CLsef), the liver acts as a sink, and thus sinusoidal influx clearance (CLsin) becomes the rate-determining step in the hepatic clearance of the drug. Under this scenario, the ECM model (Eq. 2) simplifies such that CLsin (as well as fup and Qh) determine hepatic clearance (Eq. 3 and Supplementary derivation).
| (3) |
This scenario is most likely to occur for a drug with permeability restrictions that is transported by sinusoidal membrane transporters. Since ATV is transported into the liver by OATPs and is extensively and rapidly metabolized by CYP3A enzymes, its hepatic clearance is rate-determined by CLsin by OATPs and not by CLmet. Therefore, inhibition of OATPs results in a 12-fold increase in systemic ATV AUC while inhibition of CYP3A does not affect systemic ATV AUC. In essence, there is a disconnect between the hepatic and systemic drug concentrations. As described below, the consequences of this disconnect with respect to impact of transport/metabolic DDI’s and SNPs on systemic PK or PD (in the liver) are profound.
At first sight, one would predict that rifampin, due to its inhibition of OATPs, would decrease the hepatic exposure to ATV. However, the ECM predicts this will not be the case because ATV is cleared solely by the liver. Therefore, although its hepatic clearance is reduced by rifampin, ATV is eventually entirely cleared by the liver and thus the hepatic exposure to the drug (i.e. hepatic AUC) is not affected (Fig. 1B&C). In addition, the increased systemic concentration works against a change in hepatic drug concentration. For example, if CLsin is decreased by 10-fold then systemic concentrations must increase by 10-fold which makes the amount of drug per time (or flux) entering the liver unchanged. This is an example of a disconnect between hepatic PK (and therefore hepatic PD) and systemic PK. If the cholesterol effect of ATV is dependent on only its hepatic AUC (and not Cmax), then its PD effect will not be altered even when OATPs are inhibited or have reduced function (e.g. SNPs). Indeed, patients with the OATP1B1 polymorphism c.521T>C had no change in their LDL cholesterol lowering effect of ATV even though there was a 1.6–2.5-fold increase in plasma ATV AUC (6). But, while ATV PD effect is not affected, higher systemic concentrations of ATV may lead to off-target toxicity, such as muscle myopathy. While inhibition of CLsin only impacts systemic AUC and not hepatic AUC, the hepatic Cmax and Tmax (Fig. 1B&C) will change as shown by our rosuvastatin PET imaging study (7). It is important to recognize that inhibition of CLsin not affecting hepatic drug AUC holds true for drugs predominately eliminated by the liver since presence of a significant secondary route (e.g. renal elimination) will change the fraction of drug available to the liver, and thus change hepatic exposure (Fig. 1C&F). Of note, inhibition of CLmet and CLcef will increase hepatic AUC independent of the routes of elimination (Fig. 1D&G).
Contrary to OATP inhibition, inhibition of CYP3A metabolism is not expected to alter systemic ATV concentrations but it should increase hepatic exposure and thus its PD effect. Indeed, patients that have homozygous CYP3A5*3 allele (low or undetectable CYP3A5 expression) have significantly higher serum total cholesterol reduction (8). The improved PD response is due to higher hepatic ATV concentrations, even if the systemic ATV concentrations remain unchanged (Fig. 1B&D). Again, there is a disconnect since impact of SNPs on enzyme function does not manifest in systemic drug concentrations but it does reflect changes in hepatic PD. Interestingly, when ATV is given with an oral dose of itraconazole there is a 1.5-fold increase in systemic ATV concentrations (4). This somewhat unexpected result is not due to a decrease in the liver’s ability to eliminate systemic drug but rather it is a decrease in the gut extraction which increases drug bioavailability (Fig. S1B).
Scenario 2: When does metabolic clearance determine hepatic clearance of drugs?
The common observation where metabolic enzymes (vs. transporters) determine the hepatic clearance of a drug occurs when the drug has high permeability across the sinusoidal membrane. When drugs can readily diffuse across the plasma membrane, the sinusoidal membrane effectively becomes “transparent” (i.e. not a barrier). Now systemic concentrations will reflect the loss of drug from the liver via metabolism. In other words, CLmet will be the rate-determining step in hepatic clearance when CLsin and CLsef are equal and much larger compared to CLmet (Fig. S2 and Supplementary derivation). For example, midazolam (logD = 3.48) is extensively metabolized by CYP3A enzymes and is not a substrate of sinusoidal or canalicular transporters. Therefore, the observed CYP3A DDI with protease inhibitors ritonavir and nelfinavir can be fully explained via the WSHM (Eq. 1).
Scenario 3: When do canalicular plus metabolic clearances determine hepatic clearance of drugs?
As described above, when the drug permeability is high such as the sinusoidal membrane becomes “transparent”, both CLmet and CLcef will determine systemic concentrations (Eq. 5, Fig. S2, and Supplementary derivation). For example, docetaxel (logD = 3.03) is a substrate of CYP3A enzymes, glutathione S-transferase, P-gp, MRP2, and OATP1B3 (4). In Mdr1a/1b−/−, Cyp3a−/−, and dual Cyp3a−/−/Mdr1a/1b−/− mice, docetaxel systemic AUC is increased 2-fold, 4.9-fold, and 17-fold, respectively (4). This example illustrates how inhibition of either CLcef or CLmet results in an increase in systemic concentrations. Notice how not accounting for canalicular transport can cause an overestimation of the impact of metabolism on drug disposition for inhibitors that are dual transporter/enzyme inhibitors. For a more detailed discussion on transport – enzyme interplay, see Endres et al (1).
| (5) |
Scenario 4: When do all hepatobiliary clearances determine hepatic clearance of drugs?
While scenarios 1–3 describe the extremes, many drugs will have characteristics (moderate or low passive diffusion or relatively low metabolic/biliary clearance) where their systemic clearance will be determined by both transport (sinusoidal/canalicular) and metabolism (Fig. S3 and Supplementary derivation). Therefore, the full ECM (Eq. 2) will be needed to predict the in vivo clearance of these drugs or the pact of DDI or SNPs on this clearance. For example, repaglinide (logD = 2.6) is a substrate of CYP2C8 (fm=0.7), CYP3A4 (fm=0.3), and OATP1B1 (4). Patients with OATP1B1 polymorphism c.521T>C had a 2.9-fold increase in systemic AUC (4). Co-administration of trimethoprim (a selective CYP2C8 inhibitor, but not a CYP3A or OATP inhibitor) or itraconazole increased repaglinide systemic AUC 1.6-fold and 1.4 fold, respectively (4). These studies demonstrate that both CLsin and CLmet are important determinants of repaglinide systemic clearance. Therefore, repaglinide DDI with gemfibrozil (CYP2C8 and OATP1B1 inhibitor) and cyclosporine (OATP1B1 and CYP3A4 inhibitor) which lead to an 8.0-fold and 2.4-fold increase in systemic repaglinide AUC, respectively, is a reflection of dual transporter and enzyme DDI (Ref 4 and see Fig. S3 for further details). Due to space constraints, consequences of inhibition of only CLsef are not presented here but are described in Table 1 and Fig. S3.
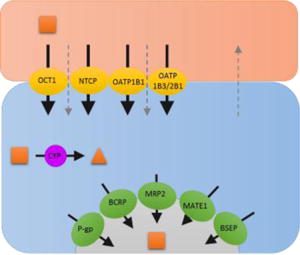
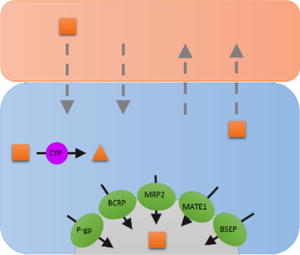
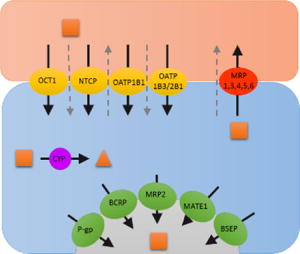
Table 1.
Scenarios to identify which clearance pathways determine hepatic CL of drugs and their impact on systemic and hepatic AUC and therefore hepatic PK/PD. In the drawing above, solid black arrows represent drug transport and metabolism while the gray dashed arrows represent passive diffusion. The direction of the arrows within the table represent the expected change in systemic and hepatic AUC, and the hepatic PD effect when each respective CL pathway is inhibited. The double arrows indicate higher impact to systemic or hepatic AUC when a particular CL pathway is inhibited in the presence and absence of significant extra-hepatic CL (CLother >> 0). − * − Assumes that hepatic efficacy and toxicity is related to hepatic drug AUC and not hepatic drug Cmax. Note that significant inhibition of a CL pathway can push a drug’s PK profile to change scenarios, that is its rate-determining step(s) in hepatic CL (for example, significant inhibition of CLsef can change the dynamic between CLsef and CLmet such as a drug switches from having CLmet [scenario 2] to CLsin [scenario 1] as its rate-determining step in hepatic CL). The simulations above were conducted to avoid such switching.
|
|
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Scerio 1 | Scenario 2/3 | Scenario 4 | |||||||
| Rate-determining step(s) in hepatic CL of drugs | Sinusoidal influx CL | Canalicular efflux and metabolic CL(s) | All hepatobiliary CL(s) | ||||||
|
| |||||||||
| Condition | Canalicular efflux plus metabolic CL(s) is much greater than sinusoidal efflux CL. The liver acts as a sink. | Sinusoidal influx and efflux CL(s) are equal (e.g. no active sinusoidal transport) and larger than canalicular and metabolic CL(s). The sinusoidal membrane is not a barrier, it becomes “transparent.” | Canalicular efflux plus metabolic CL(s) is NOT much greater than sinusoidal efflux CL. The liver does not act as a sink. | ||||||
|
| |||||||||
| Governing equation |
|
|
|
||||||
|
| |||||||||
| Inhibition of CLsin | CLother=0 | CLother >> 0 | CLother=0 | CLother >> 0 | CLother=0 | CLother >> 0 | |||
| Systemic AUC | ↑↑ | ↑ | ↔ | ↔ | ↑↑ | ↑ | |||
| Systemic toxicity/efficacy | ↑↑ | ↑ | ↔ | ↔ | ↑↑ | ↑ | |||
| Hepatic AUC | ↔ | ↓ | ↔ | ↓ | ↔ | ↓ | |||
| Hepatic efficacy/toxicity* | ↔ | ↓ | ↔ | ↓ | ↔ | ↓ | |||
|
| |||||||||
| Inhibition of CLsef | CLother=0 | CLother >> 0 | CLother=0 | CLother >> 0 | CLother=0 | CLother >> 0 | |||
| Systemic AUC | ↔ | ↔ | ↔ | ↔ | ↓ | ↓ | |||
| Systemic toxicity/efficacy | ↔ | ↔ | ↔ | ↔ | ↓ | ↓ | |||
| Hepatic AUC | ↔ | ↔ | ↔ | ↑ | ↔ | ↑ | |||
| Hepatic efficacy/toxicity* | ↔ | ↔ | ↔ | ↑ | ↔ | ↑ | |||
|
| |||||||||
| Inhibition of CLmet | CLother=0 | CLother >> 0 | CLother=0 | CLother >> 0 | CLother=0 | CLother >> 0 | |||
| Systemic AUC | ↔ | ↔ | ↑↑ | ↑ | ↑↑ | ↑ | |||
| Systemic toxicity/efficacy | ↔ | ↔ | ↑↑ | ↑ | ↑↑ | ↑ | |||
| Hepatic AUC | ↑↑ | ↑ | ↑↑ | ↑ | ↑↑ | ↑ | |||
| Hepatic efficacy/toxicity* | ↑↑ | ↑ | ↑↑ | ↑ | ↑↑ | ↑ | |||
|
| |||||||||
| Inhibition of CLcef | CLother=0 | CLother >> 0 | CLother=0 | CLother >> 0 | CLother=0 | CLother >> 0 | |||
| Systemic AUC | ↔ | ↔ | ↑↑ | ↑ | ↑↑ | ↑ | |||
| Systemic toxicity/efficacy | ↔ | ↔ | ↑↑ | ↑ | ↑↑ | ↑ | |||
| Hepatic AUC | ↑↑ | ↑ | ↑↑ | ↑ | ↑↑ | ↑ | |||
| Hepatic efficacy/toxicity* | ↑↑ | ↑ | ↑↑ | ↑ | ↑↑ | ↑ | |||
How does one measure sinusoidal, canalicular, and metabolic clearances for incorporation into the ECM?
As discussed above, quantifying various clearances pathways is necessary to predict the rate-determining step(s) in hepatic clearance of drugs. CLmet can be quantified using human liver microsomes (HLM’s). CLsin, CLsef, and CLcef can be quantified using sandwich-cultured human hepatocytes (SCHH) but the use of SCHH can be cumbersome with low or no CLcef depending on SCHH quality. Therefore, emphasis should be placed in developing alternative quantification methods. We and others have proposed such an alternative method, namely a bottom-up proteomics approach using activity data in transfected cells lines which can be scaled up to that in vivo using transporter protein expression in both human tissues and the transfected cells lines (9). Others have proposed in silico methods utilizing drug dependent parameters (for example the Extended Clearance Classification System) to classify drugs into categories of rate-determining step(s) (10). Collectively, these methods will help advance the predictions of PK and tissue concentration of drugs and the impact of DDIs and SNPs on these predictions.
SUMMARY
The ECM can help predict whether transporters or metabolic enzymes or both will be important in PK and tissue concentration of drugs and the impact of DDIs and SNPs on these predictions (Table 1, S4–6 animations). The concepts discussed here can be extended to other tissues important in drug disposition, (e.g. kidneys and brain).
Supplementary Material
Acknowledgments
Supported in part by NCATS TL1 TR000422 (GP) and NIH P01DA032507.
References
- 1.Endres CJ, Endres MG, Unadkat JD. Interplay of drug metabolism and transport: a real phenomenon or an artifact of the site of measurement? Mol Pharm. 2009;6:1756–65. doi: 10.1021/mp9002392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sirianni GL, Pang KS. Organ clearance concepts: new perspectives on old principles. J Pharmacokinet Biopharm. 1997;25:449–70. doi: 10.1023/a:1025792925854. [DOI] [PubMed] [Google Scholar]
- 3.Shitara Y, Horie T, Sugiyama Y. Transporters as a determinant of drug clearance and tissue distribution. Eur J Pharm Sci. 2006;27:425–46. doi: 10.1016/j.ejps.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 4.University of Washington Metabolism and Transport Drug Interaction Database. doi: 10.1186/1479-7364-5-1-61. < https://www.druginteractioninfo.org/>. [DOI] [PMC free article] [PubMed]
- 5.Maeda K, et al. Identification of the rate-determining process in the hepatic clearance of atorvastatin in a clinical cassette microdosing study. Clin Pharmacol Ther. 2011;90:575–81. doi: 10.1038/clpt.2011.142. [DOI] [PubMed] [Google Scholar]
- 6.Maeda K. Organic anion transporting polypeptide (OATP)1B1 and OATP1B3 as important regulators of the pharmacokinetics of substrate drugs. Biol Pharm Bull. 2015;38:155–68. doi: 10.1248/bpb.b14-00767. [DOI] [PubMed] [Google Scholar]
- 7.He J, et al. PET imaging of Oatp-mediated hepatobiliary transport of [(11)C] rosuvastatin in the rat. Mol Pharm. 2014;11:2745–54. doi: 10.1021/mp500027c. [DOI] [PubMed] [Google Scholar]
- 8.Kivistö KT, et al. Lipid-lowering response to statins is affected by CYP3A5 polymorphism. Pharmacogenetics. 2004;14:523–5. doi: 10.1097/01.fpc.0000114762.78957.a5. [DOI] [PubMed] [Google Scholar]
- 9.Prasad B, Unadkat JD. Optimized approaches for quantification of drug transporters in tissues and cells by MRM proteomics. AAPS J. 2014;16:634–48. doi: 10.1208/s12248-014-9602-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Varma MV, Steyn SJ, Allerton C, El-Kattan AF. Predicting Clearance Mechanism in Drug Discovery: Extended Clearance Classification System (ECCS) Pharm Res. 2015 doi: 10.1007/s11095-015-1749-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.