

Abstract
Background and Purpose
Enhanced late Na+ current (late INa) in the myocardium is pro‐arrhythmic. Inhibition of this current is a promising strategy to stabilize ventricular repolarization and suppress arrhythmias. Here, we describe GS‐6615, a selective inhibitor of late INa, already in clinical development for the treatment of long QT syndrome 3 (LQT3).
Experimental Approach
The effects of GS‐6615 to inhibit late INa, versus other ion currents to shorten the ventricular action potential duration (APD), monophasic APD (MAPD) and QT interval, and decrease to the incidence of ventricular arrhythmias was determined in rabbit cardiac preparations. To mimic the electrical phenotype of LQT3, late INa was increased using the sea anemone toxin (ATX‐II).
Key Results
GS‐6615 inhibited ATX‐II enhanced late INa in ventricular myocytes (IC50 = 0.7 μM), shortened the ATX‐II induced prolongation of APD, MAPD, QT interval, and decreased spatiotemporal dispersion of repolarization and ventricular arrhythmias. Inhibition by GS‐6615 of ATX‐II enhanced late INa was strongly correlated with shortening of myocyte APD and isolated heart MAPD (R2 = 0.94 and 0.98 respectively). In contrast to flecainide, GS‐6615 had the minimal effects on peak INa. GS‐6615 did not decrease the maximal upstroke velocity of the action potential (Vmax) nor widen QRS intervals.
Conclusions and Implications
GS‐6615 was a selective inhibitor of late INa, stabilizes the ventricular repolarization and suppresses arrhythmias in a model of LQT3. The concentrations at which the electrophysiological effects of GS‐6615 were observed are comparable to plasma levels associated with QTc shortening in patients with LQT3, indicating that these effects are clinically relevant.
Abbreviations
- AP
action potential
- APD
AP duration
- INa
Na+ current
- J‐Tp
J‐Tpeak interval
- K‐H
Krebs–Henseleit
- LQT3
long QT syndrome type 3
- MAP
monophasic AP
- MAPD90
duration of the MAP at 90% repolarization
- Tp‐e
Tpeak‐Tend interval
- VT
ventricular tachycardia
- VF
ventricular fibrillation
- UDB
use‐dependent block
- TB
tonic block
- IKr
rapid component of the delayed rectifier potassium current
- Vmax
maximum rate of depolarization of ventricular AP
Tables of Links
| TARGETS |
|---|
| Voltage‐gated ion channels |
| NaV1.5 channels |
| LIGANDS |
|---|
| Eleclazine |
| Flecainide |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
In response to a threshold depolarization of the cardiomyocyte, Na+ channels open briefly to allow influx of Na+ ions generating a large inward Na+ current (peak INa), which elicits the upstroke of the action potential (AP) and then quickly inactivate. Following repolarization, Na+ channels return to a resting state. During the plateau phase of the cardiac AP, inward and outward currents are small and balanced, and repolarization occurs promptly as inward current gradually decays and outward current increases. The ‘late’ portion of inward INa (late INa) that persists during the plateau phase of the AP in a ventricular myocyte is normally small (≤0.1% of peak INa, about 30–50 pA) and may contribute to the AP duration (APD) by several tens of milliseconds (Gintant et al., 1984; Kiyosue and Arita, 1989). In the last 10 years, an increasing number of studies have indicated that the amplitude of late INa is greater than normal in cardiac myocytes isolated from the hearts of patients and of laboratory animals with various acquired or congenital conditions, toxins and drugs (for a comprehensive list and references see Table 1 in Belardinelli et al., 2015).
Table 1.
Inhibition by GS‐6615 or flecainide of peak INa in rabbit ventricular myocytes
| Holding Potential | GS‐6615 (10 μM) | Flecainide (10 μM) | ||
|---|---|---|---|---|
| Stimulating frequency | Stimulating frequency | |||
| 0.1 Hz | 3 Hz | 0.1 Hz | 3 Hz | |
| −120 mV | 8.8 ± 2.1% (n = 5) | 16.6 ± 3.2% (n = 5) | 7.0 ± 2.9% (n = 6) | 40 ± 2.6% (n = 6) |
| −90 mV | 12.0 ± 1.5% (n = 5) | 19.5 ± 1.7% (n = 8) | 21.6 ± 1.0% (n = 5) | 65.3 ± 2.4% (n = 4) |
An enhanced late INa in the myocardium has detrimental effects (Shryock et al., 2013; Belardinelli et al., 2015). Late INa‐induced Na+ influx increases intracellular Na+ and Ca2 + concentrations ([Na+]i and [Ca2 +]i) (Makielski and Farley, 2006; Noble and Noble, 2006), reduces net repolarizing current during the plateau phase of the AP and increases APD and the variability of APD both spatially and temporally in the ventricle (Antzelevitch and Belardinelli, 2006; Maltsev et al., 2007; Song et al., 2008). These late INa‐induced effects may cause the initiation of both early and delayed afterdepolarizations, which can act as triggers of arrhythmic activity (Maltsev et al., 1998; Song et al., 2008). On the other hand, late INa can increase the variability of APD (temporal dispersion) and repolarization times in adjacent cells and among regions of the ventricle (spatial dispersion), thus creating the substrate for propagation of ectopic activity and potentially lead to torsades de pointes ventricular tachycardia (VT) (Shimizu and Antzelevitch, 1997; Belardinelli et al., 2003; Weiss et al., 2010).
Pharmacological inhibition of late INa is a logical approach to enhance the net repolarizing current, reduce abnormal Na+ influx into myocytes and consequently to improve intracellular Ca2 + homeostasis and suppress arrhythmias. The amplitude of late INa in ventricular myocytes can be reduced by several anti‐arrhythmic agents, such as amiodarone, flecainide, lidocaine and mexiletine, as well as by the anti‐anginal drug ranolazine (Zaza et al., 2008; Shryock et al., 2013; Makielski, 2015). However, in addition to the Na+ channel, each of these drugs acts on many other targets, such as other ion channels and receptors, and these actions often cause side effects and/or safety concerns. In addition to extra‐cardiac effects, safety concerns may include prolongation of the QT interval, slowing of electrical impulse conduction in the ventricle (especially in patients with structural heart diseases in whom conduction is already altered) and a consequent increased risk of life‐threatening ventricular arrhythmias and sudden cardiac death.
A prototype inhibitor of late INa is ranolazine, an anti‐anginal drug (Chaitman, 2006), which also has anti‐arrhythmic activity in patients (Scirica et al., 2007). At clinically relevant doses (500–1000 mg, twice daily), ranolazine is estimated to inhibit late INa by 20–30%. In addition, ranolazine inhibits the rapid component of the delayed rectifier K+ current, IKr, and has minimal effects on peak INa in the ventricle, and these effects explain why this drug prolongs the QT interval, but it does not widen the QRS interval in humans (Antzelevitch et al., 2011). Inhibitors of late INa reported to be more potent than ranolazine have been described, such as GS‐458 967 and F15845 (Le Grand et al., 2008; Le Grand et al., 2009; Belardinelli et al., 2013), but none of these agents were developed for clinical use. A ‘second‐generation’ inhibitor of late INa with appropriate pharmaceutical properties is clearly needed. This drug should retain the positive characteristics of ranolazine, including its relative safety and effectiveness as an anti‐ischaemic and anti‐arrhythmic agent, but also have greater potency, efficacy and selectivity to inhibit late INa and to reduce [Na+]i‐dependent Ca2 +‐overload without inhibiting IKr and/or prolonging the QT interval. In addition, the drug should not reduce either peak INa or cardiac contractile function. In this report, we describe key electrophysiological properties of such compound, the selective inhibitor of late INa, GS‐6615. This compound is in clinical development ( as eleclazine) for cardiac diseases such as long QT syndrome type 3 (LQT3) and hypertrophic cardiomyopathy, in which late INa is increased.
Methods
Animals
All animal care and experimental procedures conformed to the ‘Guide for the Care and Use of Laboratory Animals’ (National Academy of Sciences, edition 2011) and were approved by the Institutional Animal Care and Use Committees of Gilead Sciences. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath & Lilley, 2015).
Rabbit hearts were used in four different series of experiments carried out in the following preparations: (1) single ventricular myocytes; (2) isolated papillary muscles; (3) perfused left ventricular wedges; and (4) perfused intact hearts. New Zealand White female rabbits (adult females, 2–4 kg; Western Oregon Rabbit Company, Philomath, OR) were used in this study. Each rabbit was sedated using an intramuscular administration of a mixture of 6 mg·kg−1 xylazine and 40 mg·kg−1 ketamine and then anaesthetised by i.v. administration of 15 mg·kg−1 ketamine and 4 mg·kg−1 xylazine in 1.5 mL saline via the marginal ear vein. After anaesthesia was complete, the thorax was quickly opened. The heart was excised and placed in a dissecting chamber filled with saline solution at room temperature or cooled to 4°C, as detailed below.
Single ventricular myocytes
Ventricular myocytes were isolated using standard enzymic techniques (Belardinelli et al., 2013). Membrane currents were recorded using whole‐cell patch clamp technique (Hamill et al., 1981; Belardinelli et al., 2013). pCLAMP 10.0 software (Molecular Devices, Sunnyvale, CA, USA) was used to generate voltage‐clamp protocols and acquire data, which were analysed using pCLAMP 10.0 and Microcal Origin (MicroCal, Northampton, MA, USA) software. The Axopatch‐700B patch clamp amplifier (Molecular Devices) was used to record membrane currents or transmembrane APs and cell capacitance. Series resistance compensation was performed to 70–80%, and membrane currents were recorded using square pulse protocols at 23 ± 1°C or 36.5 ± 1°C, and APs were recorded at 36.5 ± 1°C.
Isolated papillary muscle preparations
Papillary muscles 2–4 mm in length and 0.5–1 mm in width were excised from the right ventricle. Papillary muscles were placed in a 1.5 mL tissue chamber and superfused with Tyrode's solution at the average rate of 2–3 mL·min−1. The composition of the Tyrode's solution was (in mM): NaCl 139, KCl 4.0, CaCl2 1.8, MgSO4 0.5, NaH2PO4 0.9, NaHCO3 20, glucose 5.5. The pH of the solution was adjusted to 7.4 with a gas equilibration of 95% O2 + 5% CO2. The temperature of the superfusate was maintained at 36 ± 0.5°C. Preparations were stimulated through bipolar electrodes by an EP‐4 stimulator (St. Jude Medical, Austin, TX, USA) with rectangular pulses of 2 ms duration and at least twice the threshold intensity. Transmembrane APs were recorded using glass microelectrodes filled with 3 M KCl with tip resistances of 8–15 MΩ. AP signals were amplified through an Intracellular Micrometre IE‐210 amplifier (Warner Instrument Corporation, Hamden, CT, USA), digitized in real time using a Biopac MP 150 signal processor and displayed on a computer screen by Biopac system (MP150 and Acqknowledge software 4.1, Goleta, California, USA). The maximum rate of depolarization of phase 0 (upstroke) of the ventricular AP (Vmax) was calculated by differentiating the membrane potential recordings.
The papillary muscle was allowed to equilibrate in the chamber for at least 1 h in the Tyrode's solution at the stimulation rate of 60 beats·min−1 (bpm), prior to starting the experiments. In some experiments, trains of stimuli, 1, 3 and 3.5 Hz, (yielding rates of 60, 180 and 210 bpm respectively) were applied. Each train was composed of 20–50 pulses, depending on how rapidly the Vmax reached the steady‐state. Trains at 180 and 210 bpm were separated by a 60 bpm train to allow the recovery of the Vmax after higher frequency rates of stimulation. After the control recordings of Vmax at various frequencies of the trains, a single drug was applied for about 15 min to allow a steady‐state response. The drug effect on Vmax was tested at 60, 180 and 210 bpm. The Vmax of the AP was measured in the absence and presence of compounds during superfusion with extracellular K+ concentrations ([K+]O) of 4.0 or 6.0 mM. Vmax after drug application was normalized to the Vmax before the application of drugs at the same rates of stimulation.
Perfused intact hearts
Hearts were perfused by the method of Langendorff, as previously described (Wu et al., 2006; Belardinelli et al., 2013). The atrioventricular nodal area was thermally ablated to reduce ventricular rate. The hearts were paced at a rate of 1 Hz and perfused with a modified Krebs–Henseleit (K‐H) buffer: 118 NaCl, 2.8 KCl, 1.2 KH2PO4, 2.5 CaCl2, 0.5 MgSO4, 2.0 sodium pyruvate, 5.5 glucose, 0.57 Na2EDTA and 25 NaHCO3 at pH 7.4, gassed with 95% O2 and 5% CO2, 36.5°C using an isolated‐heart apparatus (Harvard Apparatus, Holliston, MA, USA). The total duration of the experimental protocol was limited to 2.5 h. Continuous recordings were made of the monophasic AP (MAP) from the left ventricular epicardium and the 12‐lead pseudo‐ECG signals. ECGs and coronary perfusion pressure signals were amplified, filtered and digitized in real time using a Biopac MP 150 signal processor and displayed on a computer screen. All signals were saved on a computer hard disc for subsequent analysis. Response to drug was measured after the effect had achieved steady‐state. The duration of the MAP at 90% repolarization (MAPD90) and the duration of the ECG QRS interval were determined. Occurrence of VT was defined as ≥3 sequential, non‐paced beats in the MAP recording.
Perfused left ventricular wedge preparation
Hearts were placed in a dissecting chamber filled with a cardioplegic solution consisting of cold (4°C) normal Tyrode's solution. The left circumflex branch of the coronary artery was cannulated and perfused with the cardioplegic solution (Wu et al., 2006). Unperfused areas of the left ventricle, which were easily identified by their reddish appearance due to the existence of unflushed erythrocytes, were removed. The preparation was then placed in a small tissue bath and arterially perfused with Tyrode's solution containing 4 mM K+ buffered with 95% O2 and 5% CO2 (temperature: 35.7 ± 0.1°C; mean perfusion pressure: 35–45 mmHg). The ventricular wedge was allowed to equilibrate in the tissue bath for 1 h prior to electrical recordings.
Assay for electrophysiological recordings in left ventricular wedge preparations
A transmural ECG signal was recorded using extracellular Ag/AgCl electrodes placed in the Tyrode's solution bathing the preparation 1.0–1.5 cm from the epicardial and endocardial surfaces, along the same vector as the transmembrane recordings. The QT interval was defined as the time from the onset of the Q wave to the point at which the final downslope of the T wave crossed the isoelectric line. Transmembrane APs were recorded from epicardium and endocardium, using floating glass microelectrodes. The J‐Tpeak (J‐Tp) and Tpeak‐Tend (Tp‐e) intervals, which closely approximates transmural dispersion of early and late repolarization, were defined as the time from the J‐point to peak of the T wave (J‐Tp) and from the peak of the T wave to the end of the T wave (Tp‐e). The QT, J‐Tp and Tp‐e intervals were measured manually from three consecutive beats within the last minute of the recording, and the values were then averaged. Spatial dispersion of repolarization was measured as the difference in APDs across the left ventricular wedge from the endocardial to the epicardial surface, for the QT interval, and J‐Tp and Tp‐e durations from pseudo‐ECG recordings (Wu et al., 2006).
Data and statistical analysis
These studies comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Results are expressed as mean ± SEM. Data were analysed using pCLAMP 10 and Microcal Origin (MicroCal) or Prism version 5 (Graph Pad Software, San Diego, CA, USA) and The significance of differences of measurements before and after interventions in the same heart was determined by repeated measures one‐way ANOVA followed by Student–Newman–Keul's test. A P value <0.05 was considered statistically significant.
Materials
Research grade eleclazine (GS‐6615; Figure 1) was synthesized at Gilead Sciences and prepared as 10 mM stock solutions in DMSO in glass vials. GS‐6615 (mol. wt. 415) is 4‐(pyrimidin‐2‐ylmethyl)‐7‐(4‐(trifluoromethoxy)phenyl)‐3,4‐dihydrobenzo[f][1,4]oxazepin‐5(2H)‐one. Flecainide and ATX‐II were purchased from Sigma‐Aldrich (St. Louis, MO, USA) and prepared as a 10 mM and 10 μM stock solutions in DMSO and distilled water, respectively. The stock solutions were aliquoted and stored at −20°C. Stock solutions were freshly diluted in the bath recording solution on the day of the experiment. The DMSO concentration was maintained at 0.1% in all external solutions.
Figure 1.
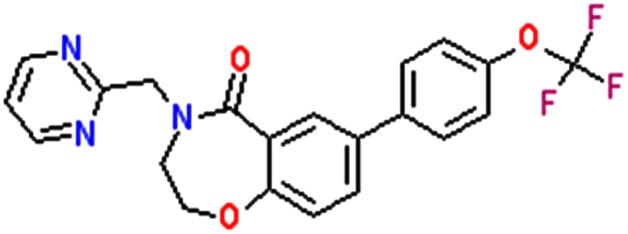
Chemical structure of GS‐6615, eleclazine.
Results
Effects of GS‐6615 on peak INa, late INa, IKr and other ion channel currents in rabbit ventricular myocytes
The late INa expressed by rabbit ventricular myocytes was enhanced by the sea anemone toxin (ATX‐II, 3 nM), as shown in Figure 2A. Addition of GS‐6615 (1 μM) reduced this enhancement of the late INa, with an IC50 value of 0.72 ± 0.06 μM at a holding potential of −120 mV (Figure 2B) and 0.26 ± 0.01 μM at a holding potential of −80 mV (n = 5–7). In contrast, at the same stimulating frequency (0.1 Hz) and holding potential (−120 mV), GS‐6615 at 10 μM reduced peak INa by only 8.8 ± 2.1% (n = 5, Figure 2B). The ratio of IC50 values for GS‐6615 to inhibit late, as distinct from peak, INa could not be determined, as the compound did not cause 50% inhibition of peak INa at the highest tested concentration (10 μM). GS‐6615 was a weak inhibitor of IKr with an IC50 value of approximately 14.2 μM (Figure 2B). Thus, the relative selectivity of GS‐6615 to inhibit late INa over IKr in rabbit ventricular myocytes was about 20‐fold.
Figure 2.
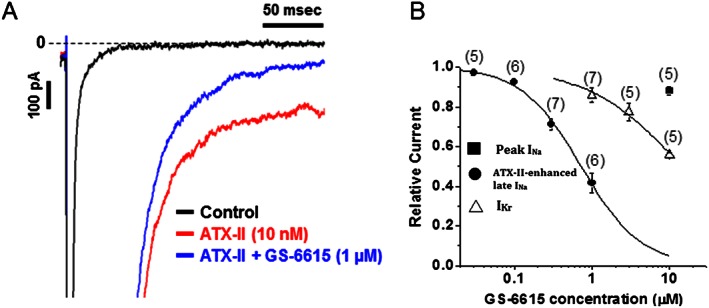
Panel A shows a typical recording of late INa measured in a rabbit ventricular myocyte under control conditions, following ATX‐II enhancement of late INa and 1 μM GS‐6615 in the continued presence of ATX‐II. Panel B shows the concentration–response curve for GS‐6615 to inhibit peak INa, ATX‐II‐enhanced late INa and IKr in rabbit ventricular myocytes. Data presented are mean ± SEM ; n = number of myocytes indicated in parenthesis.
The effects of Na+ channel‐blocking agents, such as flecainide, to inhibit peak INa are known to be enhanced by rapid stimulation and depolarization of myocytes (Anno and Hondeghem, 1990). Figure S1 shows representative experiments for the inhibition of rabbit ventricular myocyte peak INa by either GS‐6615 or flecainide. The magnitude of inhibition of peak INa by GS‐6615 was determined at stimulating frequencies of 0.1 Hz for tonic block (TB) and 3 Hz for use‐dependent block (UDB) and at holding potentials of −120 and −90 mV. GS‐6615, at a concentration of 10 μM, slightly inhibited TB (about 10%) and UDB (about 20%) at each holding potential (Table 1).
GS‐6615 at a concentration of 1 μM had no effect (≤10%) on L‐type and T‐type Ca2 + currents (ICaL and ICaT), pacemaker current (If), acetylcholine‐dependent K+ current (IKACh), ATP‐sensitive K+ current (IKATP), ultra‐rapid K+ current (IKur), transient outward current (Ito), delayed rectifier K+ current (IKs) and inward rectifier K+ current (INCX) of rabbit ventricular myocytes (Table S1) or on the heterologous expression of the pore‐forming subunit of the respective ion channel current (S2). Among human Na channel isoforms (Nav1.1–Nav1.8) expressed heterologously, the potencies of GS‐6615, measured at a depolarizing frequency of 10 Hz, were 5 μM for Nav1.7 and up to 96 μM for Nav1.1 (S3). In addition, GS‐6615 at a concentration of 10 μM had minimal (≤22%, except at adenosine A3 receptors) activity on a total of 67 different GPCRs, including β‐adrenoceptors and muscarinic receptors, or transporters and enzymes. GS‐6615 (10 μM) caused minimal inhibition of calmodulin kinase II (CaMKII) and did not inhibit signalling induced by β1, β2 or β3–adrenoceptor agonists (see section 4 in the Supporting Information).
Effects of GS‐6615 on APD of isolated myocytes and perfused hearts
The toxin ATX‐II (3 nM) also markedly prolonged the AP of rabbit ventricular myocytes (Figure 3B). GS‐6615 (0.03–0.3 μM), in a concentration‐dependent manner, shortened the APD (Figure 3C, D and F) and blunted the beat‐to‐beat variability of APD (Figure 3E). Consistent with the effects on single ventricular myocytes, in isolated perfused hearts, GS‐6615 significantly reduced the increase in the duration of the ventricular MAP (MAPD) caused by 3 nM ATX‐II (Figure 4A–D). The IC50 value for GS‐6615 to reduce the ATX‐II‐induced prolongation of MAPD was 0.74 ± 0.15 μM (n = 5; Figure 4E). ATX‐II induced VT was also suppressed by GS‐6615 with an IC50 value of 0.6 ± 0.1 μM (n = 5; Figure 4F).
Figure 3.

GS‐6615 (0.03 and 0.3 μM) attenuated the effects of ATX‐II (3 nM) to increase APD and beat‐to‐beat variability of APD in rabbit ventricular myocytes. Shown in panels A–D are representative sequential recordings of APs stimulated at 0.2 Hz and obtained in the absence of drug (control), in the presence of 3 nM ATX‐II and following additions of 0.03 and 0.1 μM GS‐6615 in the continued presence of ATX‐II. The durations of 10 consecutive APs recorded during each period are shown in panels E. Shown in panel F is the concentration‐dependent shortening by GS‐6615 of ATX‐II induced prolongation of APD in ventricular myocytes. Data shown here are means+ SEM; n=4‐7. * P <0.05; significantly different from ATX‐II alone; one‐way ANOVA with Student–Newman–Keul's test.
Figure 4.
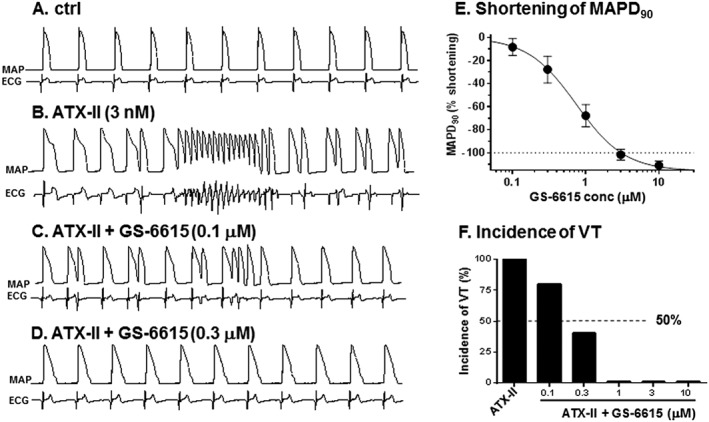
Effects of GS‐6615 to shorten the prolonged MAP and to suppress ventricular tachyarrhythmia induced by ATX‐II in rabbit isolated heart preparations. Shown in panels A–D are representative sequential recordings of epicardial MAP (top record in each panel) and pseudo‐ECG (bottom record in each panel) during exposure of a heart to perfusate alone (control, A), ATX‐II alone (B) and GS‐6615 (0.1 and 0.3 μM) in the continued presence of ATX‐II (C and D). Shown in panels E and F are concentration–response relationships for GS‐6615 to shorten the MAPD90 and to reduce the incidence of VT during exposure of hearts to ATX‐II. Data shown are means±SEM ; n=5. *P<0.05; repeated measures one‐way ANOVA, with Student–Newman–Keul's test.
In order to determine whether the changes in the magnitude of late INa and APD were correlated, their relationship during exposure of myocytes to ATX‐II and GS‐6615 was plotted. As shown in Figure 5A, the reductions by GS‐6615 of myocyte late INa and APD in the presence of 3 nM ATX‐II were strongly correlated (R2 = 0.94). Likewise, the percentage reductions of late INa in ventricular myocytes by GS‐6615 was also strongly correlated (R2 = 0.98) with the shortening of the MAPD of isolated hearts (Figure 5B).
Figure 5.
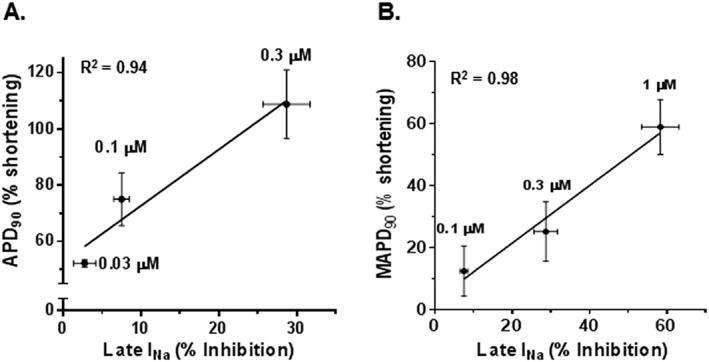
Relationships between inhibition of late INa and shortening of either APD90 (A) or MAPD90 (B) of rabbit isolated ventricular myocytes and perfused heart preparations respectively. Late INa, APD90 and MAPD90 were enhanced by ATX‐II (3 nM). Data for inhibition of late INa and shortening of APD90 or MAPD90 are from Figures 2, 3 and 4 respectively.
GS‐6615 reduces spatial dispersion of electrical activity in the isolated left ventricular wedge, a model of LQT3
Increased spatial dispersion of repolarization in the ventricle, including APD, QT, J‐Tp and Tp‐e intervals, are associated with an increased incidence of VT such as torsade de pointes (Antzelevitch, 2005). Therefore, the effect of GS‐6615 (0.3 μM) on spatial dispersion of repolarization in left ventricular wedge preparations exposed to ATX‐II was determined. ATX‐II (2 nM) significantly increased spatial dispersions of APD90 and increased QT, J‐Tp and Tp‐e intervals in preparations stimulated at basic cycle lengths of 500 and 1000 ms respectively (Figure 6). As shown in Figure 6, these effects of ATX‐II were significantly reversed by 0.3 μM GS‐6615 (n = 4; Figure 6, panels A–D).
Figure 6.
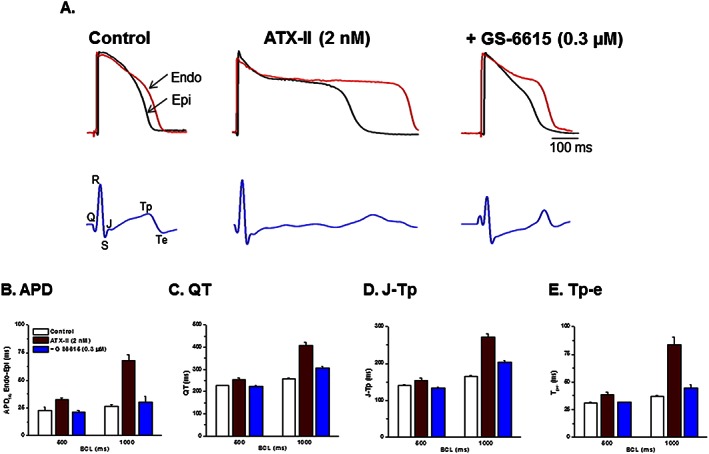
Effects of GS‐6615 to attenuate ATX‐II induced ventricular repolarization abnormalities associated with regional differences (epicardium, Epi vs. endocardium, Endo) in APD of rabbit left ventricular wedge preparations. Panel A shows representative transmembrane action potentials (upper panel) and pseudo‐electrocardiograms (lower panel) during exposure of a heart to perfusate alone (control), ATX‐II (2 nM) and GS‐6615 (1 μM) in the continued presence of ATX‐II. Panels B, C, D and E show summary of values of APD, QT, J‐Tp and Tp‐e during control, in the presence of ATX‐II and GS‐6615 (0.3 μM) in the continued presence of ATX‐II in left ventricular wedges paced at basic cycle lengths of 500 and 1000 ms respectively. Data shown are means ± SEM; n=4 per group.
Comparison of the electrophysiological effects of GS‐6615 and flecainide
Flecainide is a potent prototypical inhibitor of peak INa, a class IC anti‐arrhythmic agent, clinically used to treat patients with atrial fibrillation (Aliot et al., 2011). Flecainide is also known to shorten the QTc intervals of patients with LQT3, an effect due to inhibition of late INa (Benhorin et al., 2000). Notably, both preclinical and clinical data have shown that flecainide, as well as propafenone, is pro‐arrhythmic in ischaemic myocardium, because of their enhanced reduction of peak INa in depolarized myocytes and during rapid pacing rates (Anno and Hondeghem, 1990). Therefore, the effect of GS‐6615 on peak INa was compared with that of flecainide in ventricular myocytes stimulated at a frequency of 0.1 and 3 Hz and holding potentials of −120 and −90 mV (Figure S1). Note the rapid development of block from pulse 1–100 observed for GS‐6615 as compared with the slow binding kinetics of flecainide. In addition, the effects of GS‐6615 and flecainide on the Vmax of papillary muscle APs and QRS interval of isolated perfused hearts with K‐H buffer containing normal and elevated [K+]O and paced at various rates (to mimic depolarized conditions and tachycardia) were determined. The resting membrane potentials achieved during superfusion of papillary muscles with K‐H buffer containing 4, 6 and 8 mM [K+]O were −88 ± 0.3, −80 ± 0.6 and −71 ± 0.9 mV (n = 6 each, data not shown) respectively.
Effects on peak INa
The effect of GS‐6615 (10 μM) on peak INa has been described above and is also summarized in Table 1. In comparison, flecainide (10 μM) caused a much greater inhibition of peak INa (Table 1). Both the voltage‐dependent and UDB of peak INa by flecainide were greater than that of GS‐6615, in the range of 1.5‐fold to 2.3‐fold for voltage‐dependent and 1.9‐fold to 3.2‐fold for UDB (Table 1).
Effects on Vmax
The effects of vehicle (control), GS‐6615 and flecainide on the Vmax of papillary muscles stimulated at 60, 180 and 210 bpm superfused with K‐H buffer containing either 4 or 6 mM [K+]o, are shown in Figure 7. The control Vmax values were 118.9 ± 13.6, 111.0 ± 12.1 and 106.7 ± 11.5 V·s−1 at 4 mM [K+]o and at stimulation rates of 60, 180 and 210 bpm; while at 6 mM [K+]o and the same stimulation rates, the Vmax values were 125.0 ± 14.2, 109.4 ± 12.5 and 102.3 ± 11.9 V·s−1 (Figure 7) respectively. There were no differences in Vmax at 6 mM [K+]o compared with Vmax at 4 mM [K+]o (P > 0.5 at all three pacing intervals). The time matched control differences in Vmax were 0.52 ± 1.8, 0.2 ± 1.2 and 1.18 ± 1.6% at 4 mM [K+]o and stimulation rates of 60, 180 and 210 bpm; while they were 0.48 ± 1.01, 2.75 ± 0.31 and 3.37 ± 1.29% at 6 mM [K+]o at stimulation rates of 60, 180 and 210 bpm respectively (Figure 7).
Figure 7.
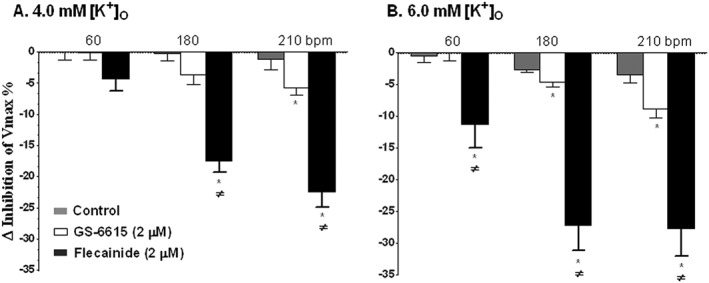
Effects of GS‐6615 and flecainide in the presence of 4.0 (panel A) and 6.0 mM (panel B) [K+]O on the Vmax of ventricular action potentials of rabbits papillary muscles stimulated at rates of 60, 180 and 210 bpm. Data are presented as the percentage change relative to control at the same pacing rate (means ± SEM; n=5 per group). *P < 0.05, significantly different from control at the same pacing frequency; ≠ P < 0.05, significantly different from effects of GS‐6615; repeated measures one‐way ANOVA, with Student–Newman–Keul's test.
GS‐6615 at 2 μM, a concentration that is about twice the mean estimated clinical therapeutic concentration (based on initial clinical studies), in the presence of 4.0 or 6 mM [K+]O caused less than 10% decrease in Vmax at stimulation rates of 60, 180 and 210 bpm respectively (n = 5 each; Figure 7). Flecainide, at a concentration of 2 μM, a concentration within its therapeutic range, caused much greater decreases in Vmax (n = 5 each, Figure 7) at all tested conditions, compared with the effects of GS‐6615.
Effects on QRS interval
The effects of GS‐6615 and flecainide on the QRS interval duration of isolated perfused hearts are shown in Figure 8. In hearts perfused with K‐H buffer containing 4 and 6 mM [K+]o and paced at 60, 180 and 210 bpm, GS‐6615 (3 μM) had no significant effect on the QRS interval (n = 6; Figure 8, panels A and B). Only with8 mM [K+]o, was there a significant but less than 10 ms prolongation of the QRS intervals (Figure 8C). In comparison, the magnitude of the effect of flecainide (2 μM) on QRS interval duration, at all [K+]O tested (4, 6 and 8 mM) irrespective of the pacing rate, was clearly greater than that caused by 3 μM GS‐6615 (Figure 8). For instance, at a pacing rate of 210 bpm and in the presence of 8 mM [K+]O, flecainide (2 μM) prolonged the QRS interval by about 60 ms (n = 5), whereas GS‐6615 (3 μM) under the same experimental conditions widened the QRS interval by just under 10ms (n = 6).
Figure 8.
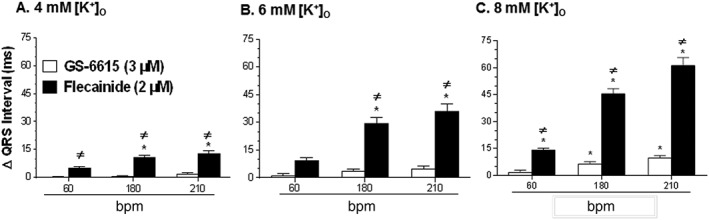
Effects of GS‐6615 (3 μM) and flecainide (2 μM) in the presence of 4 (panel A), 6 (panel B), and 8 mM (panel C) [K+]O on the duration of the QRS interval at a pacing rate of 60, 180 and 210 bpm. Data (means ± SEM; n=6) are presented as the difference (Δ) in the duration (ms) of QRS intervals in the absence and presence of drugs. *P < 0.05, significantly different from baseline at the same pacing rate; ≠ P < 0.05, significantly different from effects of GS‐6615; repeated measures one‐way ANOVA, with Student–Newman–Keul's test.
Discussion
The results of the present study indicate that GS‐6615, which is now being tested in clinical trials, is a highly selective inhibitor of late INa in cardiac tissues. GS‐6615 inhibited late INa of ventricular myocytes with a potency of 0.7 μM and attenuated the effect of ATX‐II to prolong APs of ventricular myocytes and isolated perfused hearts. At a concentration of 0.3 μM, GS‐6615 significantly reduced both the incidence of VT in isolated hearts (Figure 4) and the spatial dispersion of repolarization and the duration of the Tp‐e in LV wedge preparations (Figures 5 and 6). An important finding was that relationships between the magnitude of inhibition of late INa and the resulting shortening of APD and MAPD of single myocytes and perfused hearts, respectively, were linear and strongly correlated (R2 = 0.94 and 0.98 respectively). Taken together, these findings are consistent with the conclusion that all effects of GS‐6615 reported in this study are the result of a single action, namely inhibition of late INa, which increases APD stability and repolarization reserve (Belardinelli et al., 2015).
Drugs that inhibit Na+ channel current may reduce peak INa, late INa or both, and these effects are voltage‐dependent and rate (use)‐dependent. Among inhibitors of the Na+ channels in ventricular myocytes, ranolazine appears to be the most selective inhibitor of late, as distinct from peak, INa that is currently used in clinical practice (Antzelevitch et al., 2011). Consistent with this observation, ranolazine does not reduce the conduction velocity of electrical impulses (or increase the QRS interval) in the ventricle. In contrast, Class IC Na+ channel inhibitors such as flecainide are potent inhibitors of both peak and late INa (Belardinelli et al., 2013). The selectivity of GS‐6615 to reduce late, but not peak, INa is likely to be more than 40‐fold but could not be estimated in this study, because the minimal effect of the drug on peak INa was observed at 10 μM, the solubility limit for the drug in the assay. As expected, the effect of GS‐6615 to reduce peak INa was increased at depolarized resting potentials. Voltage‐dependent Na+ channel inhibition is the characteristic of all compounds that bind to the ‘local anaesthetic’ site of the channel (Catterall and Swanson, 2015). Although the site of action of GS‐6615 on the Na+ channel has not been identified, we speculate that like other local anaesthetic drugs, it binds to the inside of the channel pore in a state‐dependent manner.
Safety concerns have limited the use of Na+ channel inhibitors (Class I anti‐arrhythmic agents) in clinical practice. Among these, conduction slowing in the ventricle due to inhibition of peak INa, especially in hearts with structural disease, has been well described (Lu et al., 2010). The finding that GS‐6615, at concentrations about two‐fold greater than the estimated therapeutic concentration, caused a relatively small reduction of Vmax and a minimal increase in QRS intervals of rabbit cardiac preparations during conditions of depolarized membrane potential (−80 or −71 mV, at [K+]O of 6 and 8 mM respectively) and tachycardia (at a pacing rate of 210 bpm) suggests that the compound would be safe in the ischaemic myocardium. We have shown that GS‐6615 (2.7 μM) in anaesthetised rabbits undergoing acute myocardial ischaemia, caused by ligation of the coronary artery for 20 min followed by reperfusion for 40 min, suppressed VT/ventricular fibrillation (VF) (Zablocki et al.), while flecainide, propafenone and mexiletine increased the incidence of both VT and VF, as well as mortality (Belardinelli et al., 2013). Another safety concern with anti‐arrhythmic agents is their potential to induce torsades de pointes due to non‐selective inhibition of repolarizing currents such as IKr. GS‐6615 was found to reduce IKr with an IC50 value of 14.2 μM and to be devoid of significant effects on other cardiac ion channel currents up to 10 μM (Table 1; Tables S1 and S2 respectively). At concentrations of 0.3 to 1 μM required to inhibit late INa by 29–58% and to reduce arrhythmic activity in a model of LQT3, GS‐6615 is not expected to alter cardiac conduction (Figures 7 and 8).
Importantly, the electrophysiological effects of GS‐6615 described in present study, using a pharmacological model of LQT3, are consistent with the observations of the initial clinical studies that GS‐6615 at a wide range of plasma concentrations (Cmax values between 0.28 and 1.5 μM), shortened the QTc interval of patients with LQT3 syndrome without affecting heart rate, PR and QRS interval and left ventricular function (Zareba et al., 2014).
In conclusion, GS‐6615 is a novel, and selective inhibitor of late INa, that shortens the APD (and QTc) and reduces spatiotemporal dispersion of repolarization and ventricular arrhythmic activity in various cardiac preparations from rabbits. Further studies are needed to identify the site of action of GS‐6615 in the Na+ channel, to characterize its effects in additional models of atrial and ventricular arrhythmias, myocardial ischaemia and heart failure and to continue to evaluate its safety in disease models.
Author contributions
G.L., N.E.‐B., D.G., C.L., X.‐L.C., K.K. and N.M. performed the experiments. S.R., K.K. and L.B. designed the study and experiments and wrote the manuscript. E.E. and J.Z. synthesized GS‐6615 (eleclazine).
Conflict of interest
All authors are employees of Gilead Sciences.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Potencies of GS‐6615 to inhibit ion channel currents in rabbit isolated ventricular myocytes.
Table S2 Potencies of GS‐6615 to inhibit ion channel currents expressed in a heterologous system.
Table S3 Extracellular saline solutions used to record currents from human cardiac ion channels.
Table S4 Intracellular saline solutions used to record currents from human cardiac ion channels.
Table S5 Potencies of GS‐6615 to inhibit NaCh isoforms expressed in a heterologous system.
Table S6 Inhibition of various receptors and transporter by 10 μM GS‐6615.
Figure S1 Effects of GS‐6615 and Flecainide on rabbit ventricular myocyte peak INa This Figure shows representative recordings of peak INa, in rabbit ventricular myocytes. Panel A is peak INa measured at a stimulating frequency of 0.1 Hz in control (left) and following superfusion of either 10μM GS‐6615 (top, right) or 10μM Flecainide (bottom top, right). Panel B is peak INa measured at a stimulating frequency of 3 Hz in control (left) and following superfusion of either 10μM GS‐6615 (top, right) or 10μM Flecainide (bottom top, right). Note the rapid development of block from pulse 1 to 100 observed for GS‐6615 as compared to the slow binding kinetics of the Class IC agent flecainide. Average data from all experiments are reported in Table 1.
Supporting info item
Acknowledgements
The authors thank Dr Richard Verrier for a critical review of the manuscript.
Rajamani, S. , Liu, G. , El‐Bizri, N. , Guo, D. , Li, C. , Chen, X. ‐L. , Kahlig, K. M. , Mollova, N. , Elzein, E. , Zablocki, J. , and Belardinelli, L. (2016) The novel late Na+ current inhibitor, GS‐6615 (eleclazine) and its anti‐arrhythmic effects in rabbit isolated heart preparations. British Journal of Pharmacology, 173: 3088–3098. doi: 10.1111/bph.13563.
References
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliot E, Capucci A, Crijns HJ, Goette A, Tamargo J (2011). Twenty‐five years in the making: flecainide is safe and effective for the management of atrial fibrillation. Europace 13: 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anno T, Hondeghem LM (1990). Interactions of flecainide with guinea pig cardiac sodium channels. Importance of activation unblocking to the voltage dependence of recovery. Circ Res 66: 789–803. [DOI] [PubMed] [Google Scholar]
- Antzelevitch C (2005). Role of transmural dispersion of repolarization in the genesis of drug‐induced torsades de pointes. Heart Rhythm 2 (2 Suppl): S9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Belardinelli L (2006). The role of sodium channel current in modulating transmural dispersion of repolarization and arrhythmogenesis. J Cardiovasc Electrophysiol 17 (Suppl 1): S79–S85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Burashnikov A, Sicouri S, Belardinelli L (2011). Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm 8: 1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardinelli L, Antzelevitch C, Vos MA (2003). Assessing predictors of drug‐induced torsade de pointes. Trends Pharmacol Sci 24: 619–625. [DOI] [PubMed] [Google Scholar]
- Belardinelli L, Giles WR, Rajamani S, Karagueuzian HS, Shryock JC (2015). Cardiac late Na(+) current: proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress. Heart Rhythm 12: 440–448. [DOI] [PubMed] [Google Scholar]
- Belardinelli L, Liu G, Smith‐Maxwell C, Wang WQ, El‐Bizri N, Hirakawa R et al. (2013). A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. J Pharmacol Exp Ther 344: 23–32. [DOI] [PubMed] [Google Scholar]
- Benhorin J, Taub R, Goldmit M, Kerem B, Kass RS, Windman I et al. (2000). Effects of flecainide in patients with new SCN5A mutation: mutation‐specific therapy for long‐QT syndrome? Circulation 101: 1698–1706. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Swanson TM (2015). Structural basis for pharmacology of voltage‐gated sodium and calcium channels. Mol Pharmacol 88: 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaitman BR (2006). Ranolazine for the treatment of chronic angina and potential use in other cardiovascular conditions. Circulation 113: 2462–2472. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gintant GA, Datyner NB, Cohen IS (1984). Slow inactivation of a tetrodotoxin‐sensitive current in canine cardiac Purkinje fibers. Biophys J 45: 509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981). Improved patch‐clamp techniques for high‐resolution current recording from cells and cell‐free membrane patches. Pflugers Archiv: Eur J Physiol 391: 85–100. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyosue T, Arita M (1989). Late sodium current and its contribution to action potential configuration in guinea pig ventricular myocytes. Circ Res 64: 389–397. [DOI] [PubMed] [Google Scholar]
- Le Grand B, Pignier C, Letienne R, Colpaert F, Cuisiat F, Rolland F et al. (2009). Na + currents in cardioprotection: better to be late. J Med Chem 52: 4149–4160. [DOI] [PubMed] [Google Scholar]
- Le Grand B, Pignier C, Letienne R, Cuisiat F, Rolland F, Mas A et al. (2008). Sodium late current blockers in ischemia reperfusion: is the bullet magic? J Med Chem 51: 3856–3866. [DOI] [PubMed] [Google Scholar]
- Lu HR, Rohrbacher J, Vlaminckx E, Van Ammel K, Yan GX, Gallacher DJ (2010). Predicting drug‐induced slowing of conduction and pro‐arrhythmia: identifying the ‘bad’ sodium current blockers. Br J Pharmacol 160: 60–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makielski JC (2015). Late sodium current: A mechanism for angina, heart failure, and arrhythmia. Trends Cardiovasc Med pii: S1050‐1738 (15): 00151–00156. doi:10.1016/j.tcm.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makielski JC, Farley AL (2006). Na(+) current in human ventricle: implications for sodium loading and homeostasis. J Cardiovasc Electrophysiol 17 (Suppl 1): S15–S20. [DOI] [PubMed] [Google Scholar]
- Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M, Undrovinas AI (1998). Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 98: 2545–2552. [DOI] [PubMed] [Google Scholar]
- Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI (2007). Chronic heart failure slows late sodium current in human and canine ventricular myocytes: implications for repolarization variability. Eur J Heart Fail 9: 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble D, Noble PJ (2006). Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium‐calcium overload. Heart 92 (Suppl 4): iv1–iv5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scirica BM, Morrow DA, Hod H, Murphy SA, Belardinelli L, Hedgepeth CM et al. (2007). Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST‐segment elevation acute coronary syndrome: results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST‐Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN‐TIMI 36) randomized controlled trial. Circulation 116: 1647–1652. [DOI] [PubMed] [Google Scholar]
- Shimizu W, Antzelevitch C (1997). Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade des pointes in LQT2 and LQT3 models of the long‐QT syndrome. Circulation 96: 2038–2047. [DOI] [PubMed] [Google Scholar]
- Shryock JC, Song Y, Rajamani S, Antzelevitch C, Belardinelli L (2013). The arrhythmogenic consequences of increasing late INa in the cardiomyocyte. Cardiovasc Res 99: 600–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Shryock JC, Belardinelli L (2008). An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am J Physiol Heart Circ Physiol 294: H2031–H2039. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JN, Garfinkel A, Karagueuzian HS, Chen PS, Qu Z (2010). Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm 7: 1891–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Shryock JC, Song Y, Belardinelli L (2006). An increase in late sodium current potentiates the proarrhythmic activities of low‐risk QT‐prolonging drugs in female rabbit hearts. J Pharm Exp Ther 316: 718–726. [DOI] [PubMed] [Google Scholar]
- Zareba W, Rosero S, Zeng D, Moss A, Robinson J, Couderc JP et al. (2014). QTc shortening by GS‐6615, a new late sodium current blocker, in LQT3 patients [abstract]. Heart Rhythm 11: 35. [Google Scholar]
- Zaza A, Belardinelli L, Shryock JC (2008). Pathophysiology and pharmacology of the cardiac "late sodium current.". Pharmacol Ther 119: 326–339. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Potencies of GS‐6615 to inhibit ion channel currents in rabbit isolated ventricular myocytes.
Table S2 Potencies of GS‐6615 to inhibit ion channel currents expressed in a heterologous system.
Table S3 Extracellular saline solutions used to record currents from human cardiac ion channels.
Table S4 Intracellular saline solutions used to record currents from human cardiac ion channels.
Table S5 Potencies of GS‐6615 to inhibit NaCh isoforms expressed in a heterologous system.
Table S6 Inhibition of various receptors and transporter by 10 μM GS‐6615.
Figure S1 Effects of GS‐6615 and Flecainide on rabbit ventricular myocyte peak INa This Figure shows representative recordings of peak INa, in rabbit ventricular myocytes. Panel A is peak INa measured at a stimulating frequency of 0.1 Hz in control (left) and following superfusion of either 10μM GS‐6615 (top, right) or 10μM Flecainide (bottom top, right). Panel B is peak INa measured at a stimulating frequency of 3 Hz in control (left) and following superfusion of either 10μM GS‐6615 (top, right) or 10μM Flecainide (bottom top, right). Note the rapid development of block from pulse 1 to 100 observed for GS‐6615 as compared to the slow binding kinetics of the Class IC agent flecainide. Average data from all experiments are reported in Table 1.
Supporting info item