

Abstract
Background and Purpose
Glycine receptors are important players in pain perception and movement disorders and therefore important therapeutic targets. Glycine receptors can be modulated by the intravenous anaesthetic propofol (2,6‐diisopropylphenol). However, the drug is more potent, by at least one order of magnitude, on GABAA receptors. It has been proposed that halogenation of the propofol molecule generates compounds with selective enhancement of glycinergic modulatory properties.
Experimental Approach
We synthesized 4‐bromopropofol, 4‐chloropropofol and 4‐fluoropropofol. The direct activating and modulatory effects of these drugs and propofol were compared on recombinant rat glycine and human GABAA receptors expressed in oocytes. Behavioural effects of the compounds were compared in the tadpole loss‐of‐righting assay.
Key Results
Concentration–response curves for potentiation of homomeric α1, α2 and α3 glycine receptors were shifted to lower drug concentrations, by 2–10‐fold, for the halogenated compounds. Direct activation by all compounds was minimal with all subtypes of the glycine receptor. The four compounds were essentially equally potent modulators of the α1β3γ2L GABAA receptor with EC50 between 4 and 7 μM. The EC50 for loss‐of‐righting in Xenopus tadpoles, a proxy for loss of consciousness and considered to be mediated by actions on GABAA receptors, ranged from 0.35 to 0.87 μM.
Conclusions and Implications
We confirm that halogenation of propofol more strongly affects modulation of homomeric glycine receptors than α1β3γ2L GABAA receptors. However, the effective concentrations of all tested halogenated compounds remained lower for GABAA receptors. We infer that 4‐bromopropofol, 4‐chloropropofol and 4‐fluoropropofol are not selective homomeric glycine receptor modulators.
Abbreviations
- 4‐bp
4‐bromopropofol
- 4‐cp
4‐chloropropofol
- 4‐fp
4‐fluoropropofol
Tables of Links
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Glycine receptors are members of the Cys‐loop receptor family (Lynch, 2004; Lynch, 2009). These Cl−‐permeable channels mediate inhibitory neurotransmission in the retina, brainstem and spinal cord. Functional receptors are formed as homopentamers of one of four α subunits or heteropentamers consisting of α and β subunits. Subunit expression is region‐specific and regulated developmentally. For example, prenatal glycinergic transmission is largely mediated by α2 homomers, whose expression is reduced sharply following birth (Malosio et al., 1991). Receptors containing the α1 subunit are dominant in adult retina and spinal cord. Interference with α1 expression, or introduction of mutations disrupting receptor function, can result in the hyperekplexia phenotype (Schaefer et al., 2013; Bode and Lynch, 2014). Glycine receptors containing the α3 subunit are expressed in the retina and in nociceptive sensory neurons in the spinal cord dorsal horn where they may mediate pain associated with chronic peripheral inflammation (Haverkamp et al., 2003; Harvey et al., 2004). Thus, glycine receptors can be important potential targets in a variety of physiological processes.
Recent studies have reported that halogenated analogues of propofol act as potent activators and modulators of the glycine receptor. In mouse ventral horn neurons, submicromolar concentrations of 4‐bromopropofol (4‐bp) elicit tonic current that can be blocked by strychnine, but not bicuculline (Eckle et al., 2014). 4‐Chloropropofol (4‐cp) reportedly potentiates currents from heterologously expressed α1 receptor with the concentration producing half‐maximal effect below 1 nM (de la Roche et al., 2012). These results led to the proposal that these, and other novel halogenated analogues of propofol, may be clinically useful in treating conditions mediated by functional deficiencies of glycine receptors, such as hyperekplexia, or employed as a starting point in the development of non‐sedative analgesics.
Propofol and its analogues can also activate and potentiate the GABAA receptor (Hales and Lambert, 1991; Krasowski et al., 2001). These actions likely underlie the loss‐of‐righting reflex (LRR) and other anaesthetic endpoints following application of propofol (Jurd et al., 2003). Previous work on halogenated analogues of propofol has demonstrated that bromine substitution at the ortho position reduces anaesthetic and GABAergic activity of the analogues, while compounds with halogen substituents para to the phenolic hydroxyl group remain strong GABAergic modulators (Krasowski et al., 2001; Trapani et al., 1998).
To provide a direct comparison of activation and modulation of glycine and GABAA receptors by this class of compounds and to test the potential for receptor selectivity, we compared direct activation and modulation of recombinant α1, α2 and α3 homomeric glycine receptors, and α1β3γ2L GABAA receptors by propofol, 4‐bp, 4‐CP and 4‐fluoropropofol (4‐FP). We show that these compounds are weak activators of the glycine receptor, with maximal observed responses at approximately 20% of the response to saturating glycine. All halogenated analogues were more potent than propofol at potentiating responses to a low concentration of glycine, with the EC50 for potentiation left‐shifted by up to an order of magnitude. 4‐BP and 4‐CP, but not 4‐FP, were also more potent than propofol at modulating the α1β3γ2L GABAA receptor. The EC50 for the LRR, a proxy for loss of consciousness in humans (Franks, 2008), were measured in Xenopus tadpoles and found to be 0.35–0.87 μM for propofol and its analogues. We infer that while halogenation of the propofol molecule does alter target selectivity, biasing it towards modulation of glycine receptors, 4‐halogenated analogues of propofol do not attain sufficient selectivity for use as selective glycinergic modulators.
Methods
Chemical syntheses
4‐Chloropropofol was synthesized according to the published procedure (Wheatley and Holdrege, 1958).
The synthesis of 4‐BP was adapted from the published procedure (Wheatley and Holdrege, 1958) as follows. 2,6‐Diisopropylphenol (1.85 mL, 1.78 g, 10 mmol) was added to a stirred, orange solution of tetrabutylammonium tribromide (4.82 g, 10 mmol) in chloroform (20 mL), at room temperature. After 30 min, the chloroform was evaporated from the almost colourless solution, and the residue was treated with water (30 mL) and diethyl ether (30 mL). The mixture was shaken, and the two layers were separated. The organic layer was washed with water (3×) and dried (MgSO4). Filtration and concentration gave an orange oil which was chromatographed on silica gel using hexane : ethyl acetate (19:1) as eluent to give the product as a pale yellow oil (1.59 g, 62%).
4‐Fluoropropofol was synthesized as follows.
4‐Nitro‐2,6‐diisopropylanisole (Meek et al., 1968; Chastrette et al., 1986): 2,6‐Diisopropylanisole (760 mg, 4 mmol) was dissolved in chloroform (5 mL) and treated with potassium nitrate (456 mg, 4.56 mmol) and trifluoroacetic anhydride (2 mL). The flask was loosely stoppered, and the mixture was stirred at room temperature for 5 days. The mixture was filtered; the solid washed with chloroform (1 × 5 mL) and the combined filtrate and washings were concentrated to a light brown oil (1.41 g). This was chromatographed on silica gel using hexane : ethyl acetate (19:1) as eluent to give the product as a yellow oil (910 mg, 96%).
4‐Amino‐2,6‐diisopropylanisole (Ponomarev and Burmistrov, 1964): The 4‐nitro derivative (910 mg, 3.84 mmol) was dissolved in ethanol (10 mL) and treated with tin dichloride dihydrate (2.25 g, 10 mmol). The mixture was stirred and heated at 90°C for 4 h. The reaction mixture was cooled to room temperature, and then ice/water (50 mL) was added followed by solid sodium hydroxide until the pH of the mixture was >13. Dichloromethane (50 mL) was added, and the mixture was vacuum filtered. The resultant two phases were separated, and the aqueous layer was extracted with dichloromethane (2 × 20 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated to an orange–brown oil (900 mg). This was chromatographed on silica gel using hexane : ethyl acetate (3:2) as eluent to give the product as a red oil (564 mg, 71%).
4‐Fluoro‐2,6‐diisopropylanisole (Kriuchkova and Zavgorodnii, 1960): The 4‐amino derivative (564 mg, 2.72 mmol) was dissolved in dry THF (3 mL), stirred, cooled in an ice/acetone bath and treated with boron trifluoride etherate (0.65 mL, 5.2 mmol) and then t‐butyl nitrite (0.36 mL, 313 mg, 3.05 mmol). The dark brown solution was allowed to warm to room temperature over 10 min then diluted with perfluorodecalin (3 mL). The resultant two‐phase mixture was subjected to rotary evaporation to remove most of the THF and t‐BuOH (failure to do this or substituting a hydrocarbon solvent for perfluorodecalin results in some 2,6‐diisopropylanisole which is very difficult to remove from its fluorinated analogue) and then stirred and heated to 115°C over about 20 min. At 60–65°C, some gas evolution occurred. Once 115°C had been attained, the flask was removed from the oil bath and cooled to room temperature. Acetonitrile (50 mL) was added. The lower, colourless, perfluorodecalin layer was removed, and the acetonitrile evaporated to give a very dark brown, viscous liquid (1.367 g). This was chromatographed on silica gel using hexane : ethyl acetate (19:1) as eluent to give the product as a yellow oil (255 mg, 45%).
4‐Fluoropropofol (Picard et al., 1996). The 4‐fluoroanisole (255 mg, 1.22 mmol) was dissolved in dichloromethane (15 mL) and cooled in an ice bath. The stirred solution was added to a solution of boron tribromide in dichloromethane (1 M, 5 mL, 5 mmole) and the resultant solution stirred at 0°C for 45 min and then at room temperature for 3 h. The reaction mixture was cooled in an ice bath, then water (15 mL) was added and the whole mixture was poured into diethyl ether (50 mL). The layers were shaken and separated, and the organic layer was washed with saturated, aqueous sodium bicarbonate (1×) and water (1×) and dried (MgSO4). The drying agent was filtered and the filtrate was concentrated to a light brown oil (250 mg). This was chromatographed on silica gel using hexane : ethyl acetate (19:1) as eluent to give the product as an off‐white solid, m.p. 41–43°C, (178 mg, 74%).
Animals
All animal care and experimental protocols complied with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health and were approved by the Animal Studies Committee of Washington University in St. Louis (Approval no. 20140 150 and no. 20150076). Frogs and tadpoles (from the African clawed frog, Xenopus laevis) were housed and cared for in a Washington University Animal Care Facility under the supervision of the Washington University Division of Comparative Medicine. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Frogs were purchased from Xenopus 1 (Dexter, MI) and oocytes were harvested from mature female frogs under tricaine anaesthesia, twice per frog. Frogs were killed under tricaine anaesthesia by rapid decapitation. Early prelimb‐bud stage tadpoles from Xenopus laevis were used in behavioural assays. Tadpoles (purchased from Nasco, Fort Atkinson, WI) were kept in continuously aerated tadpole Ringer's solution in aquaria and used within 6 days of receipt. The aquaria are inspected and approved by the Washington University Animal Care Committee. Behavioural assays using Xenopus laevis tadpoles were conducted in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health. A total of 370 tadpoles were used in the experiments described here. Tadpoles were killed after each experiment by addition of lethal concentrations of tricaine.
Frog oocytes were used for expression of glycine and GABAA receptors. Oocytes are a widely used and standard expression system for recombinant receptor channels. Their use permits studies of a defined population of receptors to provide specific and reliable pharmacological information. The use of tadpoles is justified because there are no cell lines or computer modelling systems that replace the use of animals for these behavioural studies.
Molecular biology and expression of receptors
The glycine and GABAA receptors were expressed in Xenopus oocytes. The cDNAs in the pcDNA3 (glycine α3, GABAA α1, β3, γ2L), pcDNA3.1 (glycine α2) or pGEMHE vector (glycine α1) were linearized by digestion with Xba I, NheI, Hind III or BglII (NEB Labs, Ipswich, MA). The cRNAs were produced using mMessage mMachine (Ambion, Austin, TX). The oocytes were injected with a total of 18–33 ng cRNA in a final volume of 20–60 nl and incubated in ND96 (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 2.5 mM Na pyruvate, 5 mM HEPES; pH 7.4) at 16°C. The ratio of GABAA receptor cRNAs used for injection was 5:1:5 (α1:β3:γ2L). Electrophysiological measurements were conducted within 1–3 days after injection.
Electrophysiology
Electrophysiological experiments were conducted using the standard two‐electrode voltage clamp approach. Voltage and current electrodes were regular patch‐clamp electrodes that when filled with 3 M KCl had resistances of less than 1 MΩ. The oocytes were clamped at −60 mV. The chamber (RC‐1Z, Warner Instruments, Hamden, CT) was perfused continuously at approximately 5 mL∙min−1. Bath solution (92.5 mM NaCl, 2.5 mM KCl, 1 mM∙MgCl2, 10 mM HEPES; pH 7.4) was perfused between all test applications. Solutions were gravity‐applied from 30 mL glass syringes with glass luer slips via Teflon tubing to reduce adsorption and were switched manually. A typical experiment consisted of recording of a 10 s baseline, followed by a 20–60 s drug application, and then a bath application (up to 10 min) until full recovery. Duration of drug application depended on the nature of drug and its concentration and was aimed at reaching a saturated peak response. The current responses were amplified with an Axoclamp 900 A (Molecular Devices, Sunnyvale, CA) or OC‐725C amplifier (Warner Instruments, Hamden, CT), filtered at 40 Hz, digitized with a Digidata 1320 or 1200 series digitizer (Molecular Devices) at a 100 Hz sampling rate and stored using pClamp (Molecular Devices). The traces were subsequently analysed with Clampfit (Molecular Devices) to determine the maximal amplitude of current response.
The maximal final DMSO concentration in drug solutions was 0.25%. Control experiments showed that 0.5% DMSO was without effect on holding current (<0.1% of the response to saturating transmitter). Co‐application of 0.5% DMSO with an EC50 concentration of a transmitter had no effect (glycine α1, GABAA α1β3γ2L) or a small effect (<10%; glycine α2, α3) on the peak response.
Behavioural assays
Predetermined concentrations of 4‐bp, 4‐CP, 4‐FP or propofol were added to beakers containing 100 mL of oxygenated tadpole ringer's solution (5.8 mM NaCl, 67 μM KCl, 34 μM Ca(NO3)2, 83 μM MgSO4, 419 μM Tris–HCl, 80 μM Tris‐Base; pH 7.5). Ten randomly chosen tadpoles were distributed into each beaker and allowed to equilibrate in the tadpole ringer's solution for 3 h. In the end of the equilibration period, the LRR was measured by turning the tadpole over using a hooked glass rod. LRR was defined as the inability of a tadpole to right itself within 5 s on its back in three consecutive trials. The operator was blinded to the concentration of compound in the beaker.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Activation concentration–response curves were fitted for data from each individual cell with the following equation:
| (1) |
where EC50 is the concentration of drug producing a half‐maximal effect, nH characterizes the slope of relationship and Ymax is the high‐concentration asymptote. Fitting was conducted using the NFIT software (The University of Texas Medical Branch at Galveston, Galveston, TX). Parameters of the fit are reported as mean ± SEM. The concentration–response data are presented as response relative to Ymax.
Potentiation of concentration–response relationships were determined by exposing cells to a low concentration of agonist (EC4–14 of glycine or GABA), in the absence and presence of several concentrations of a modulator (4‐bp, 4‐CP, 4‐FP or propofol). Control applications to saturating glycine or GABA were typically done after every 2–3 test applications, to verify overall stability of responses. There were no apparent changes in potentiation parameters for data obtained within this range of EC values. The concentration–response curves for potentiation are presented as response relative to the peak response in the presence of saturating agonist. The curves were fitted for data from each individual cell with the following equation:
| (2) |
where EC50 is the concentration of drug producing a half‐maximal effect, nH characterizes the slope of relationship and Ymin and Ymax are the low and high concentration asymptotes respectively.
Statistical analysis was carried out with one‐way anova and Bonferroni's correction (Stata/IC 12.1, StataCorp, College Station, TX). P <0.05 was chosen to indicate significant differences between group means.
Materials
Rat α1, α2 and α3 homomeric glycine receptors and human α1β3γ2L GABAA receptors were expressed in Xenopus oocytes. The glycine α1 clone was obtained from Dr J. Lynch (The University of Queensland), α2 from Dr H. Akagi (Gunma University) and α3 from Dr H. Betz (Max‐Planck‐Institute for Brain Research). The GABAA receptor subunit clones were provided by Drs G. White (Neurogen Corporation) and D. Weiss (University of Texas Health Science Center, San Antonio). Inorganic salts used in buffers, glycine and GABA were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Stock solutions of glycine and GABA were made in bath solution at 500 mM. The stock solutions were stored in aliquots at −20°C and diluted as needed on the day of experiment. Stock solutions of propofol (200 mM) or its analogues (100–200 mM) were made in DMSO. Dilutions to working concentrations were made on the day of experiment.
Results
Direct activation of glycine and GABAA receptors by propofol and its 4‐halogenated analogues
Oocytes expressing glycine or GABAA receptors responded with inward current to applications of propofol or its halogenated analogues. In general, the peak currents in response to applications of 4‐halogenated analogues were small, compared with responses to saturating concentrations of transmitter. Application of 100 μM 4‐bp (the highest concentration tested) resulted in peak currents that were 12 ± 3% (mean ± SEM; n = 5 cells), 6 ± 1% (n = 4) or 2 ± 0.1% (n = 4) of the response to saturating glycine in cells expressing α1, α2 or α3 homomeric glycine receptors respectively. In oocytes expressing α1β3γ2L GABAA receptors, 100 μM 4‐bp elicited a response that was 27 ± 2% (n = 4) of the response to saturating (1 mM) GABA.
To attempt to gain insight into whether the failure of these drugs to efficiently activate glycine receptors results from impaired binding (low‐affinity) or gating (low‐efficacy), we recorded current responses at lower drug concentrations. Relative to peak responses to saturating glycine, the mean responses to 0.2 μM and 10 μM 4‐bp were 0.03 ± 0.02% (n = 5) and 0.12 ± 0.04% (n = 5) in α1, 0.01 ± 0.004% (n = 4) and 0.23 ± 0.12% (n = 4) in α2 and 0.22 ± 0.15% (n = 5) and 0.32 ± 0.18% (n = 6) in α3 glycine receptors. The responses to 0.2 μM and 10 μM 4‐bp in α1β3γ2L GABAA receptors were 0.02 ± 0.01% (n = 4) and 6.2 ± 0.6% (n = 8) of the peak response to saturating GABA.
Application of 100 μM 4‐CP elicited peak responses that were 14 ± 3% (n = 5), 21 ± 6% (n = 5) or 6 ± 1% (n = 5) of the response to saturating glycine in oocytes expressing α1, α2 or α3 glycine receptors respectively. The α1β3γ2L GABAA receptors responded to application of 100 μM 4‐CP with a peak response that was 50 ± 8% (n = 5) of the response to saturating GABA.
The 4‐fluorinated analogue of propofol was the weakest activator. Exposure to 100 μM 4‐FP resulted in peak responses that were 1.6 ± 0.2% (n = 4), 0.4 ± 0.1% (n = 4) or 0.3 ± 0.2% (n = 5) of the response to saturating glycine in oocytes expressing α1, α2 or α3 glycine receptors respectively. In oocytes expressing α1β3γ2L GABAA receptors, application of 100 μM 4‐FP elicited a peak response that was 33 ± 3% (n = 5) of the response to 1 mM GABA. Sample current traces are shown in Figure 1A.
Figure 1.

Direct activation of glycine and GABAA receptors. (A) Sample currents from oocytes expressing α1, α2 or α3 glycine (GlyR) or α1β3γ2L GABAA receptors (GABAAR). The receptors were activated by 100 μM 4‐bp, 4‐CP or 4‐FP (left trace in each pair) or a saturating concentration of glycine (1 mM, 0.5 mM or 2 mM for α1, α2 and α3 respectively) or GABA (1 mM). In each pair, the traces for propofol analogue and glycine (or GABA) are from the same cell. (B) Summary of direct activation of glycine and GABAA receptors by saturating concentrations of propofol and its analogues. The responses (mean ± SEM) are normalized to responses to saturating concentrations of the transmitter from the same sets of cells.
We also tested the effect of the parent compound, propofol. In agreement with previous data (Feng and Macdonald, 2004), the α1β3γ2L GABAA receptors were efficaciously activated by 100 μM propofol, which produced a response that was 90 ± 3% (n = 7) of the response to saturating GABA. In contrast, in glycine receptors, propofol was an ineffective agonist. At 100 μM, propofol elicited responses that were 0.8 ± 0.3% (n = 4), 1 ± 0.3% (n = 4) or 0.2 ± 0.01% (n = 4) of the response to saturating glycine in α1, α2 or α3 receptors. A summary of direct activation data showing the ratios of responses to propofol and its analogues compared with saturating glycine or GABA is given in Figure 1B.
Potentiation of glycine receptors by propofol and its 4‐halogenated analogues
We first determined the concentration–response relationships for receptor activation by glycine. Cells expressing each of the α subunits were exposed to 7–8 concentrations of glycine. The raw current amplitudes were fitted with the Hill equation for each cell individually. The EC50 of the glycine activation curves were 79 ± 14 μM (n = 5), 64 ± 3 μM (n = 5) and 398 ± 52 μM (n = 5) in oocytes expressing α1, α2 and α3 receptors respectively. These values are similar to those already reported (Downie et al., 1996; Mascia et al., 1996; Pistis et al., 1997; Zhang et al., 2008).
We next investigated the effect of propofol or its 4‐halogenated analogues on responses elicited by glycine. Co‐application of 4‐BP, with a low concentration of glycine, potentiated current responses (Figure 2). Homomeric α1 glycine receptors were activated by 17.5–22 μM glycine (<EC15) and additionally exposed to 1–500 μM 4‐bp. The test responses were normalized to control responses to a saturating concentration (0.5 mM) of glycine that was applied after every 2–3 test responses and at the end of recordings from each cell, to confirm overall stability of responses. The data showed that 4‐bp potentiated glycine‐activated α1 receptors with an EC50 of 42 μM (n = 6). In an analogous experimental protocol, the EC50 for potentiation of α1 glycine receptors by 4‐CP was 32 μM (n = 5), by 4‐FP 78 μM (n = 5) and for potentiation by propofol 138 μM (n = 6). Thus, the 4‐halogenated compounds were more potent than propofol at potentiating α1 glycine receptors. The EC50, maximal current levels in the presence of potentiator, and the Hill coefficients are given in Table 1.
Figure 2.
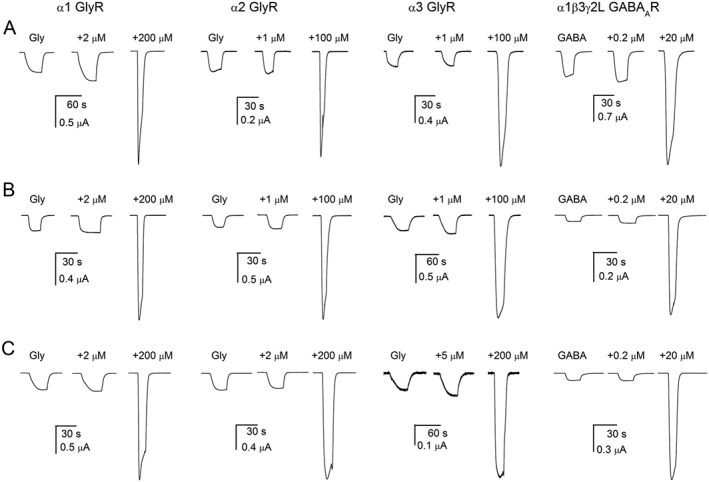
Potentiation of glycine and GABAA receptors. Sample currents from oocytes expressing α1, α2 or α3 glycine (GlyR) or α1β3γ2L GABAA receptors (GABAAR). The receptors were activated by a low concentration (<EC15) of transmitter alone or in the presence of two concentrations of 4‐bp (A), 4‐CP (B) or 4‐FP (C). The concentrations of potentiators were selected to show minimal and near‐maximal potentiating effects. Full concentration–response relationships are shown in Figure 3 and fitting results summarized in Table 1.
Table 1.
Summary of concentration–response data for potentiation of glycine and GABAA receptors
| Parameter | Gly α1 | Gly α2 | Gly α3 | GABA α1β3γ2L |
|---|---|---|---|---|
| 4‐bp : EC50 | 42 ± 5 μM * | 15 ± 2 μM ≠* | 11 ± 1 μM ≠* | 4 ± 1 μM ≠* |
| n H | 1.3 ± 0.2 | 1.3 ± 0.3 | 2.1 ± 0.3 | 2.5 ± 0.6 |
| I max | 0.63 ± 0.06 * | 0.62 ± 0.07 | 0.50 ± 0.07 | 0.68 ± 0.06 † |
| 4‐CP : EC50 | 32 ± 1 μM * | 23 ± 1 μM ≠* | 21 ± 1 μM * | 5 ± 1 μM ≠* |
| n H | 1.3 ± 0.2 | 1.5 ± 0.1 | 2.1 ± 0.3 | 1.3 ± 0.2 |
| I max | 0.81 ± 0.04 | 0.62 ± 0.03 | 0.56 ± 0.05 | 0.84 ± 0.05 #* |
| 4‐FP : EC50 | 78 ± 3 μM * | 62 ± 6 μM #†* | 42 ± 7 μM #* | 7 ± 1 μM #† |
| n H | 2.1 ± 0.3 | 1.5 ± 0.2 | 1.9 ± 0.2 | 3.6 ± 0.2 |
| I max | 0.69 ± 0.02 | 0.59 ± 0.05 | 0.70 ± 0.06 | 0.70 ± 0.02 |
| Propofol : EC50 | 138 ± 20 μM #†≠ | 95 ± 9 μM #†≠ | 112 ± 9 μM #†≠ | 7 ± 1 μM #† |
| n H | 1.2 ± 0.2 | 1.7 ± 0.2 | 1.6 ± 0.1 | 2.1 ± 0.3 |
| I max | 0.88 ± 0.04 # | 0.69 ± 0.08 | 0.63 ± 0.03 | 0.66 ± 0.05 † |
The Table summarizes the results from fitting the concentration‐response curves for potentiation for glycine and GABAA receptors expressed in Xenopus oocytes in the presence of 4‐BP, 4‐CP, 4‐FP or propofol. EC50 is the concentration producing a half‐maximal response, nH is the Hill coefficient and Imax is the fitted high‐concentration asymptote relative to the response to saturating concentration of transmitter. The data are presented as mean ± SEM for five to seven cells. EC50 and Imax values for a given receptor were compared between 4‐BP, 4‐CP, 4‐FP and propofol. The symbols next to the values denote significant differences (P < 0.05) from the corresponding values for 4‐BP (#), 4‐CP (†), 4‐FP (≠) or propofol (*); one‐way ANOVA with Bonferroni correction (Stata/IC 12.1, StataCorp, College Station, TX).
The α2 subunit‐containing glycine receptors were activated by 30–35 μM glycine (<EC15) in the presence of 1–200 μM 4‐bp, 1–200 μM 4‐CP, 5–500 μM 4‐FP or 5–1000 μM propofol. The test responses were normalized to control responses to a saturating concentration (0.5 mM) of glycine. The half‐maximal concentration for potentiation by 4‐BP was 15 μM (n = 5). 4‐CP potentiated α2 receptors with an EC50 of 23 μM (n = 5), and 4‐FP potentiated the receptors with an EC50 of 62 ± 6 μM (n = 5). Potentiation in the presence of propofol yielded an EC50 of 95 μM (n = 5). The data and the results of statistical analyses are provided in Table 1.
The α3 receptors were activated by 85–100 μM glycine (<EC15). Coapplication of 4‐bp with glycine enhanced current responses. The half‐maximal concentration of 4‐bp for potentiation was 11 μM (n = 5). 4‐CP and 4‐FP produced EC50s of 21 μM (n = 5) and 42 μM (n = 5) respectively. The EC50 for potentiation by propofol was 112 μM (n = 5). Sample traces are shown in Figure 2, and the data are summarized in Figure 3 and Table 1.
Figure 3.
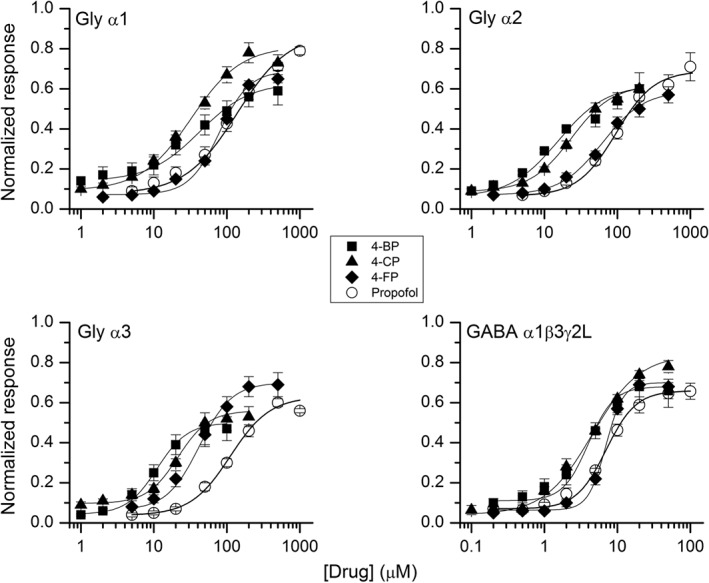
Concentration–response relationships for glycine and GABAA receptors. Oocytes expressing glycine α1, α2 or α3 subunit or GABAA α1β3γ2L subunits were activated by a low concentration of transmitter (<EC15) in the presence of 4‐bp, 4‐CP, 4‐FP or propofol. The data points (mean ± SEM from five to seven cells for each receptor‐drug combination) show responses normalized to peak current in the presence of saturating transmitter in the same cell. The curves show predicted concentration–response relationships based on data in Table 1.
Potentiation of α1β3γ2L GABAA receptors by propofol and its 4‐halogenated analogues
We tested potentiation of α1β3γ2L GABAA receptors by propofol and its halogenated analogues. The experiments were conducted using the experimental protocol described above for glycine receptors. A low concentration (1–3 μM) of GABA was applied in the presence of 0.2–50 μM 4‐bp, 0.1–50 μM 4‐CP, 0.2–50 μM 4‐FP or 0.2–100 μM propofol. The current amplitudes were normalized to the response to saturating (1 mM) GABA.
For propofol, recordings from five cells produced an EC50 of 7 μM. Potentiation data obtained in the presence of the halogenated analogues of propofol yielded EC50s of 4 μM (n = 7), 5 μM (n = 5) and 7 μM (n = 5) for 4‐bp, 4‐CP and 4‐FP respectively. The concentration–response data are summarized in Figure 3 and Table 1.
Propofol and its 4‐halogenated analogues elicit loss of the righting reflex (LRR) in Xenopus tadpoles
GABAA receptor‐mediated activity underlies the ability of propofol to induce or maintain sedation (see Franks, 2008). To gain insight into the sedative actions of the halogenated analogues, we examined the tadpole LRR. This anaesthetic endpoint correlates very well with anaesthesia in mammals, including humans (Franks and Lieb, 1993) and is considered as surrogate for loss of consciousness models (Franks, 2008).
The tadpoles were incubated in beakers containing 0.1–10 μM of 4‐bp, 0.06–3 μM 4‐CP, 0.03–10 μM 4‐FP or 0.03–10 μM propofol. The LRR was measured at the end of a 3 h exposure period. The number of tadpoles with LRR was plotted as a fraction of total and the EC50 estimated using the method described by Waud (1972).
All compounds were potent sedatives. The EC50 were 0.44 ± 0.06 μM, 0.35 ± 0.03 μM, 0.87 ± 0.10 μM and 0.52 ± 0.08 μM for 4‐bp, 4‐CP, 4‐FP and propofol respectively. The concentration–response data are shown in Figure 4.
Figure 4.
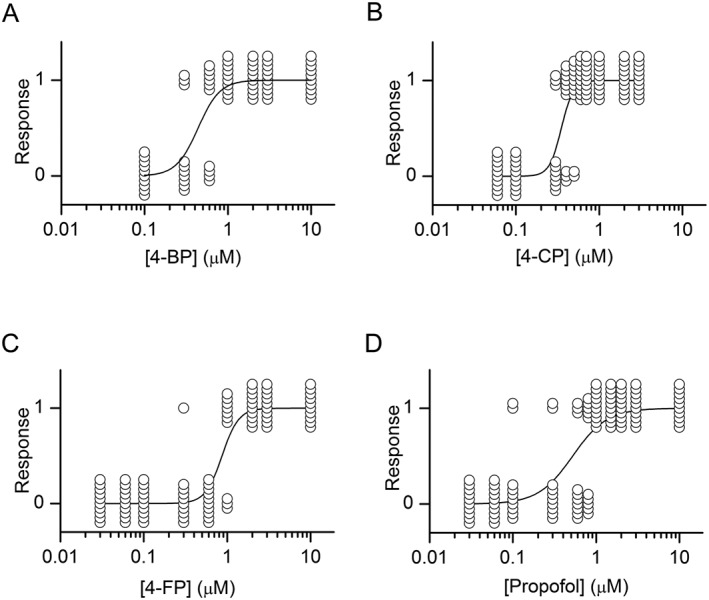
Tadpole behavioural data. Exposure to 4‐bp (A), 4‐CP (B), 4‐FP (C) or propofol (D) causes loss‐of‐righting in Xenopus tadpoles. The data are plotted as quantal dose–response relationships with each tadpole represented as one circle. Ten tadpoles were used at each concentration. The ordinate depicts the response as no effect (0) or LRR (1). The EC50 (and slopes) were 0.44 ± 0.06 μM (3.32 ± 0.93), 0.35 ± 0.03 μM (6.01 ± 1.69), 0.87 ± 0.10 μM (4.36 ± 1.30) and 0.52 ± 0.08 μM (2.10 ± 0.43) for 4‐bp, 4‐CP, 4‐FP and propofol respectively.
Discussion and conclusions
The intravenous anaesthetic propofol directly activates and potentiates currents from both glycine and GABAA receptors (Pistis et al., 1997). The sensitivities of the two receptors to the drug are, however, quite different, and it is considered that propofol's actions at clinical doses are mediated by GABAA, rather than glycine receptors (Rudolph and Antkowiak, 2004). Previous work has indicated that para‐halogenation (at position 4) of the propofol molecule may produce compounds with increased potency on glycine receptors. The 4‐chloro substituted analogue was reported to enhance currents elicited from heterologously expressed α1 homomeric glycine receptors at subnanomolar concentrations (de la Roche et al., 2012). Nanomolar concentrations of 4‐bp, but not propofol, elicited strychnine‐sensitive inward currents in spinal slices and reduced the discharge rate of ventral horn neurons (Eckle et al., 2014). In contrast, halogenation of propofol may have little effect on its ability to activate or modulate GABAA receptors. Compounds with chloro‐, bromo‐ or iodo‐ substituents at the para position of propofol have been shown to directly activate and potentiate the α1β1γ2 GABAA receptor with essentially the same half‐maximal concentrations and maximal effects as the parent compound, propofol (Trapani et al., 1998). In tadpoles, 4‐iodopropofol and propofol induce LRR with similar EC50 (Lingamaneni et al., 2001). Based on these findings, it has been proposed that halogenated propofol analogues may be considered selective glycinergic modulators.
Here, we have compared direct activation and modulation of recombinant glycine and GABAA receptors by propofol and its 4‐halogenated analogues 4‐bp, 4‐CP and 4‐FP. We studied these actions on α1, α2 and α3 homomeric glycine receptors. This choice was based on the previous study that implicated spinal cord receptors producing tonic, rather than synaptic, currents in the actions of 4‐bp (Eckle et al., 2014). Homomeric α subunit containing glycine receptors are likely to be located presynaptically where they regulate the release of transmitter or extrasynaptically where they mediate tonic glycinergic currents (Takahashi et al., 1992; Turecek and Trussell, 2002; Grudzinska et al., 2005) because synaptic localization requires a presence of the β subunit (Meyer et al., 1995). The α4 subunit, that is expressed in the chick brain and mouse retina, was not studied here. We also note that inclusion of the β subunit is not expected to change receptor sensitivity to propofol or its halogenated analogues (Haeseler et al., 2005). We also used the α1β3γ2L GABAA receptor. This subtype, along with one where β2 replaces β3, is the major target for propofol to produce various anaesthetic endpoints (Jurd et al., 2003; Reynolds et al., 2003).
The key finding is that halogenation of propofol at the para position leads to a 2–10‐fold reduction in the EC50 for potentiation of glycine receptors. The greatest effect was observed with α3 receptors for which addition of the bromide group to propofol resulted in a leftward shift from 112 to 11 μM in the midpoints of the concentration–response curves (Table 1). The changes in EC50 were not accompanied by a significant or systematic increase in relative current levels from maximally potentiated receptors. We infer that halogenation at the para position increases affinity of the compound, rather than its efficacy, on glycine receptors.
Propofol and its 4‐halogenated analogues were weak direct activators of glycine receptors. Comparison of currents obtained in the presence of 0.2, 10 and 100 μM 4‐bp suggests that the small responses are due to low affinity of the compound rather than low efficacy.
Halogenation of propofol weakly affected modulation of GABAA receptors. In oocytes expressing α1β3γ2L GABAA receptors, the potentiation curves for 4‐bp and 4‐CP were shifted to the left by less than two‐fold compared with propofol, whereas the concentration–response relationships for 4‐FP and propofol were indistinguishable. Halogenation of propofol also had small effect on the EC50 for LRR in Xenopus tadpoles. The EC50 for 4‐bp and 4‐CP were slightly lower while that for 4‐FP was higher than the EC50 for propofol. The LRR is considered to be mediated by actions on the GABAA receptor (Reith and Sillar, 1999; Belelli et al., 2003), and there is good correlation between LRR in tadpoles and inhibition of binding of t‐butylbicyclophosphorothionate or modulation of GABAA receptor function (Krasowski et al., 2001; Akk et al., 2007). Of note, the numerical values for EC50 of potentiation by all tested analogues were lower for GABAA than glycine receptors. Thus, we infer that 4‐BP, 4‐CP or 4‐FP cannot plausibly be used as selective glycine receptor modulators.
This inference is not in agreement with conclusions made in an earlier study that found significant effects on the discharge rate of mouse ventral horn neurons and strychnine‐sensitive tonic currents in organotypic spinal cultures by 50–200 nM 4‐BP (Eckle et al., 2014). In our experiments on recombinant homomeric glycine receptors, 200 nM 4‐BP was essentially inert in the absence of glycine (relative to saturating glycine mean responses ranged from 0.01–0.22%), while potentiation of glycine‐elicited currents was reliably observed at micromolar concentrations. As Eckle et al. (2014) did not provide a full concentration–response relationship for their observed effects, precise comparison of our results is difficult. The increase in tonic current in the presence of 200 nM 4‐bp may have represented the low‐concentration tail of the potentiation curve. However, using the data in Table 1, we calculate that potentiation by 200 nM 4‐bp is minimal, ranging from 0.2 to 4.1% for currents elicited by EC5 glycine.
Our data also contradict another study reporting a subnanomolar EC50 for potentiation of glycine α1 receptors by 4‐CP (de la Roche et al., 2012). We found an EC50 of approximately 30 μM for potentiation of α1 receptors by 4‐CP. The cause for discrepancy is unclear to us. We note that other phenol analogues with the 4‐chloro substituent, 3‐methyl‐4‐chlorophenol and 3,5‐dimethyl‐4‐chlorophenol also show potentiation EC50 in the micromolar range (Haeseler et al., 2005).
In summary, our findings indicate that halogenation of propofol at the para position generates compounds more capable of potentiating glycine receptors than the parent compound, propofol. The effect is manifested as a reduction in the EC50 of the concentration–response curves for potentiation. However, we emphasize that 4‐halogenated analogues of propofol remain more potent as potentiators of the α1β3γ2L GABAA, than of homomeric glycine, receptors. It may be argued that these compounds are less desirable as selective clinical agents because halogenation at the para position leads to reduction in selectivity for the GABAA receptors that characterizes the parent compound propofol.
Author contributions
N.P.F., A.S.E. and G.A. conceived the study and designed the experiments; C.J.E. and E.H.S. synthesized the halogenated analogues of propofol; A.G., D.S. and B.D.M. conducted the experiments; A.G., D.S., B.D.M., C.J.E. and G.A. analysed the data; and C.J.E., E.H.S., N.P.F., A.S.E. and G.A. wrote the manuscript. All authors approved the final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
We thank Amanda Taylor and Steve Mennerick for assistance with harvesting Xenopus oocytes and Joe Henry Steinbach for invaluable help with programming. This study was supported by grants from the National Institutes of Health, USA (GM108799 to A.S.E. and GM108580 to G.A.), by a grant from the Medical Research Council, UK (G0901892 to N.P.F.) and funds from the Taylor Family Institute for Innovative Psychiatric Research (to A.S.E. and G.A.).
Germann, A. L. , Shin, D. J. , Manion, B. D. , Edge, C. J. , Smith, E. H. , Franks, N. P. , Evers, A. S. , and Akk, G. (2016) Activation and modulation of recombinant glycine and GABAA receptors by 4‐halogenated analogues of propofol. British Journal of Pharmacology, 173: 3110–3120. doi: 10.1111/bph.13566.
References
- Akk G, Covey DF, Evers AS, Steinbach JH, Zorumski CF, Mennerick S (2007). Mechanisms of neurosteroid interactions with GABAA receptors. Pharmacol Ther 116: 35–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated Ion Channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Muntoni AL, Merrywest SD, Gentet LJ, Casula A, Callachan H et al. (2003). The in vitro and in vivo enantioselectivity of etomidate implicates the GABAA receptor in general anaesthesia. Neuropharmacology 45: 57–71. [DOI] [PubMed] [Google Scholar]
- Bode A, Lynch JW (2014). The impact of human hyperekplexia mutations on glycine receptor structure and function. Mol Brain 7: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chastrette M, Zakarya D, Elmouaffek A (1986). Structure‐odor relations of the nitrobenzene musks. Eur J Med Chem 21: 505–510. [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Roche J, Leuwer M, Krampfl K, Haeseler G, Dengler R, Buchholz V et al. (2012). 4‐Chloropropofol enhances chloride currents in human hyperekplexic and artificial mutated glycine receptors. BMC Neurol 12: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downie DL, Hall AC, Lieb WR, Franks NP (1996). Effects of inhalational general anaesthetics on native glycine receptors in rat medullary neurones and recombinant glycine receptors in Xenopus oocytes. Br J Pharmacol 118: 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckle VS, Grasshoff C, Mirakaj V, O'Neill PM, Berry NG, Leuwer M et al. (2014). 4‐bromopropofol decreases action potential generation in spinal neurons by inducing a glycine receptor‐mediated tonic conductance. Br J Pharmacol 171: 5790–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng HJ, Macdonald RL (2004). Multiple actions of propofol on αβγ and αβδ GABAA receptors. Mol Pharmacol 66: 1517–1524. [DOI] [PubMed] [Google Scholar]
- Franks NP (2008). General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci 9: 370–386. [DOI] [PubMed] [Google Scholar]
- Franks NP, Lieb WR (1993). Selective actions of volatile general anaesthetics at molecular and cellular levels. Br J Anaesth 71: 65–76. [DOI] [PubMed] [Google Scholar]
- Grudzinska J, Schemm R, Haeger S, Nicke A, Schmalzing G, Betz H et al. (2005). The β subunit determines the ligand binding properties of synaptic glycine receptors. Neuron 45: 727–739. [DOI] [PubMed] [Google Scholar]
- Haeseler G, Ahrens J, Krampfl K, Bufler J, Dengler R, Hecker H et al. (2005). Structural features of phenol derivatives determining potency for activation of chloride currents via α1 homomeric and α1β heteromeric glycine receptors. Br J Pharmacol 145: 916–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales TG, Lambert JJ (1991). The actions of propofol on inhibitory amino acid receptors of bovine adrenomedullary chromaffin cells and rodent central neurones. Br J Pharmacol 104: 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey RJ, Depner UB, Wassle H, Ahmadi S, Heindl C, Reinold H et al. (2004). GlyR α3: an essential target for spinal PGE2‐mediated inflammatory pain sensitization. Science 304: 884–887. [DOI] [PubMed] [Google Scholar]
- Haverkamp S, Muller U, Harvey K, Harvey RJ, Betz H, Wassle H (2003). Diversity of glycine receptors in the mouse retina: localization of the α3 subunit. J Comp Neurol 465: 524–539. [DOI] [PubMed] [Google Scholar]
- Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F et al. (2003). General anesthetic actions in vivo strongly attenuated by a point mutation in the GABAA receptor β3 subunit. FASEB J 17: 250–252. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL (2001). General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of γ‐aminobutyric acid (GABA) current at the GABAA receptor but not with lipid solubility. J Pharmacol Exp Ther 297: 338–351. [PubMed] [Google Scholar]
- Kriuchkova VG, Zavgorodnii SV (1960). Alkylation of 4‐fluoroanisole by olefins in the presence of BF3, BF3.H3PO4 and BF3.O(C2H5)2. Dokl Akad Nauk SSSR 130: 775–778. [Google Scholar]
- Lingamaneni R, Krasowski MD, Jenkins A, Truong T, Giunta AL, Blackbeer J et al. (2001). Anesthetic properties of 4‐iodopropofol: implications for mechanisms of anesthesia. Anesthesiology 94: 1050–1057. [DOI] [PubMed] [Google Scholar]
- Lynch JW (2004). Molecular structure and function of the glycine receptor chloride channel. Physiol Rev 84: 1051–1095. [DOI] [PubMed] [Google Scholar]
- Lynch JW (2009). Native glycine receptor subtypes and their physiological roles. Neuropharmacology 56: 303–309. [DOI] [PubMed] [Google Scholar]
- Malosio ML, Marqueze‐Pouey B, Kuhse J, Betz H (1991). Widespread expression of glycine receptor subunit mRNAs in the adult and developing rat brain. EMBO J 10: 2401–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascia MP, Mihic SJ, Valenzuela CF, Schofield PR, Harris RA (1996). A single amino acid determines differences in ethanol actions on strychnine‐sensitive glycine receptors. Mol Pharmacol 50: 402–406. [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek JS, Fowler JS, Monroe PA, Clark TJ (1968). Reaction of hindered phenols with diazomethane. J Org Chem 33: 223–226. [Google Scholar]
- Meyer G, Kirsch J, Betz H, Langosch D (1995). Identification of a gephyrin binding motif on the glycine receptor β subunit. Neuron 15: 563–572. [DOI] [PubMed] [Google Scholar]
- Picard JA, O'Brien PM, Sliskovic DR, Anderson MK, Bousley RF, Hamelehle KL et al. (1996). Inhibitors of acyl‐CoA: cholesterol O‐acyltransferase. 17. Structure–activity relationships of several series of compounds derived from N‐chlorosulfonyl isocyanate. J Med Chem 39: 1243–1252. [DOI] [PubMed] [Google Scholar]
- Pistis M, Belelli D, Peters JA, Lambert JJ (1997). The interaction of general anaesthetics with recombinant GABAA and glycine receptors expressed in Xenopus laevis oocytes: a comparative study. Br J Pharmacol 122: 1707–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev NV, Burmistrov SI (1964). Alkylation of amines. VII. Dialkylation of p‐alkoxyanilines. Zhurnal Obshchei Khimii 34: 3828–3831. [Google Scholar]
- Reith CA, Sillar KT (1999). Development and role of GABAA receptor‐mediated synaptic potentials during swimming in postembryonic Xenopus laevis tadpoles. J Neurophysiol 82: 3175–3187. [DOI] [PubMed] [Google Scholar]
- Reynolds DS, Rosahl TW, Cirone J, O'Meara GF, Haythornthwaite A, Newman RJ et al. (2003). Sedation and anesthesia mediated by distinct GABAA receptor isoforms. J Neurosci 23: 8608–8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Antkowiak B (2004). Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci 5: 709–720. [DOI] [PubMed] [Google Scholar]
- Schaefer N, Langlhofer G, Kluck CJ, Villmann C (2013). Glycine receptor mouse mutants: model systems for human hyperekplexia. Br J Pharmacol 170: 933–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (Database Issue): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Momiyama A, Hirai K, Hishinuma F, Akagi H (1992). Functional correlation of fetal and adult forms of glycine receptors with developmental changes in inhibitory synaptic receptor channels. Neuron 9: 1155–1161. [DOI] [PubMed] [Google Scholar]
- Trapani G, Latrofa A, Franco M, Altomare C, Sanna E, Usala M et al. (1998). Propofol analogues. Synthesis, relationships between structure and affinity at GABAA receptor in rat brain, and differential electrophysiological profile at recombinant human GABAA receptors. J Med Chem 41: 1846–1854. [DOI] [PubMed] [Google Scholar]
- Turecek R, Trussell LO (2002). Reciprocal developmental regulation of presynaptic ionotropic receptors. Proc Natl Acad Sci U S A 99: 13884–13889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waud DR (1972). On biological assays involving quantal responses. J Pharmacol Exp Ther 183: 577–607. [PubMed] [Google Scholar]
- Wheatley WB, Holdrege CT (1958). Dialkylaminoalkyl ethers of some 2,6‐dialkylphenols. J Org Chem 23: 568–571. [Google Scholar]
- Zhang XB, Sun GC, Liu LY, Yu F, Xu TL (2008). α2 subunit specificity of cyclothiazide inhibition on glycine receptors. Mol Pharmacol 73: 1195–1202. [DOI] [PubMed] [Google Scholar]