

Abstract
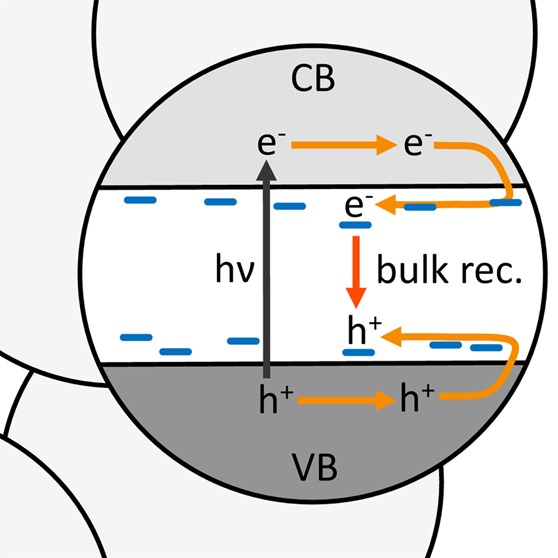
In nanostructured thin films, photogenerated charge carriers can access the surface more easily than in dense films and thus react more readily. However, the high surface area of these films has also been associated with enhanced recombination losses via surface states. We herein use transient absorption spectroscopy to compare the ultrafast charge carrier kinetics in dense and nanostructured TiO2 films for its two most widely used polymorphs: anatase and rutile. We find that nanostructuring does not enhance recombination rates on ultrafast time scales, indicating that surface state mediated recombination is not a key loss pathway for either TiO2 polymorph. Rutile shows faster, and less intensity-dependent recombination than anatase, which we assign to its higher doping density. For both polymorphs, we conclude that bulk rather than surface recombination is the primary determinant of charge carrier lifetime.
The conversion of solar energy into electricity or fuel critically relies on the stable separation of photogenerated charge carriers. Recombination of these charge carriers can compete with both charge separation and interfacial charge transfer, thus representing one of the major limiting factors in solar-to-energy conversion devices.1,2 Nanostructuring can considerably reduce the distance between the point of charge generation and the surface of the material, which facilitates interfacial charge separation and access to catalytically active surface sites. However, nanostructuring may also result in increased recombination losses due to the presence of surface recombination sites. For example, high surface areas in metal chalcogenides such as cadmium selenides and tellurides, in particular in quantum dot systems, are known to promote recombination via nonradiative decay pathways.3,4 These decay pathways involve surface-induced energy states within the band gap and lead to an overall increase in recombination.5 Because metal oxides are closely related to metal chalcogenides, recombination via surface states is often thought to limit their photovoltaic and photocatalytic function in a similar way, although the correlation between surface area and recombination rate has not directly been investigated for these systems. To elucidate the relevance of surface-enhanced recombination losses in metal oxides, we herein directly compare the ultrafast recombination kinetics of photogenerated charge carriers in nanocrystalline and dense TiO2 films.
TiO2 is a widely used transition metal oxide and has been subject to a wealth of studies, often serving as a model system. For instance, nanostructured TiO2 films have been used as electron transport layer and sensitizer scaffold in dye-sensitized solar cells for decades.6,7 Recently, the material has been used for both of those functions in perovskite solar cells8 and has also become a common photocatalyst for sunlight-driven water splitting and CO2 reduction.9,10 TiO2 occurs in the three polymorphs anatase, rutile, and brookite,11 among which anatase and rutile are typically used in photocatalytic applications. While the electronic structure of anatase and rutile is similar, anatase exhibits a band gap of 3.2 eV which is slightly larger than the 3.0 eV band gap found for rutile.12 Anatase is generally considered the most photocatalytically active polymorph.13 However, the performance of rutile was found to be superior for certain slow oxidation reactions, such as water oxidation.14 The extensive research on TiO2 has led to a profound understanding of its material properties. Nevertheless, fundamental issues such as its charge recombination behavior have received relatively little attention to date despite being fundamental to its photochemistry.15 Elucidating the origins of charge carrier recombination is therefore a crucial step on the way to improved efficiencies of metal oxide-based devices.
Charge carrier recombination may be enhanced where the chemical bonding of the periodic crystal lattice is altered, which predominantly is the case at crystal defects or at the surface of the material. The weaker bonding at such sites reduces splitting between bonding and antibonding orbitals compared to valence and conduction band states and thus gives rise to electronic states within the semiconductor band gap.16 Such intraband gap states can influence charge carrier dynamics because they offer pathways for the energetic relaxation of photogenerated charge carriers in the valence or conduction band. These states are therefore generally referred to as “trap states”, or as “surface states” if they originate from surface sites. Surface states are particularly relevant for photocatalytic applications because they are due to species that are situated directly at the catalytic interface. However, their exact role in photocatalysis has so far remained controversial. On one hand, surface states have been claimed to facilitate charge separation17 and help to localize charges close to the semiconductor interface, thereby potentially enhancing reactivity. On the other hand, they have been deemed to enhance recombination losses,18−21 thus being detrimental to device performance.
In this study, we aim to address this issue by investigating the correlation between surface area and charge recombination in TiO2. In the experiments shown herein, we use transient absorption spectroscopy on ultrafast time scales to gain insight into the kinetics of photogenerated carriers by monitoring the evolution of their optical absorption following pulsed photoexcitation. We compare carrier kinetics in nanocrystalline TiO2 films to those in dense ones in order to investigate the impact of morphology. The principal difference between these two morphologies is the larger surface-area-to-volume ratio of nanostructured films, which facilitates the access of photogenerated charge carriers to the TiO2/electrolyte interface.16 However, it is unclear whether this improved access to the surface also promotes their recombination and thus leads to an increased loss of photogenerated charges before reactions can take place. The possibility of modulating the ratio of surface charges to bulk charges in TiO2 by tuning the film morphology makes this material an ideal model system to study the role of surface states in the process of charge recombination. To elucidate this correlation, we compare charge carrier dynamics in both dense and nanostructured thin films for the two most commonly used TiO2 crystal phases, i.e., anatase and rutile.
Detailed methods of how samples were prepared are described in the Supporting Information. The crystal structures of the anatase and rutile TiO2 samples investigated in this study were verified by X-ray diffraction and showed no evidence of mixed phases, reflecting the phase-purity of our anatase and rutile films (Figure S2). Figure 1 shows scanning electron microscopy (SEM) images of the dense and mesoporous anatase films. The dense anatase films are compact layers composed of irregularly shaped domains with sizes ranging from ca. 50 to 200 nm. In contrast, the mesoporous anatase films consist of an extended interpenetrating network of spherical particles with ca. 20 nm diameter. Analogous data for dense and mesoporous rutile films are shown in Figure S3. These SEM images are consistent with our previous studies, which show a 100-fold increase in specific surface area for mesoporous versus dense films.22 In these previous studies, a range of assays of photocatalytic activity demonstrated a 30-fold enhancement of interfacial charge transfer and photocatalytic activity for the mesoporous anatase films compared to the dense films. As such we expect that for both the anatase and rutile films studied herein, charge carriers photogenerated in mesoporous films readily access the TiO2 surface. In contrast, charge carriers generated in the dense films do not easily reach the surface but rather remain in the TiO2 bulk. Because of this difference in surface accessibility of photogenerated charges, our films are particularly suitable to investigate the impact of surface states on charge recombination.
Figure 1.

Scanning electron micrographs of dense (left) and mesoporous (right) TiO2 anatase thin films on quartz glass substrates.
In this study, we use ultrafast transient absorption spectroscopy (TAS) to investigate the impact of charge access to the metal oxide surface on the recombination behavior of photogenerated charges. This technique allows us to monitor the population of photogenerated charges and track their kinetics on the relevant time scales. TAS has been extensively employed to investigate TiO2 materials.21−29 Previous studies have shown that photogenerated electrons in TiO2 give rise to transient absorption signals in the near-infrared to infrared region, whereas photogenerated holes can be monitored in the visible region (around 500 nm).23 Because signals from electrons and holes overlap more strongly in the visible region, herein we use the electron signal in the near-infrared region to track the recombination process. Our transient studies are conducted at an excitation wavelength of 355 nm near the TiO2 band gap under argon atmosphere. Upon excitation we observe a broad photoinduced absorption feature in the near-infrared region for all samples studied (Figure S4).
To gain insight into the electron decay kinetics, we monitor the change in absorption at 1200 nm as a function of excitation intensity, as shown in Figure 2a for the dense anatase film. At all excitation intensities studied, we find a fast decay within the first 0.6 ps after excitation. This early feature is intensity-independent and has previously been observed for TiO2 anatase and rutile following excitation at wavelengths close to the band gap.30 Under these excitation conditions, electron–hole pairs (excitons) are photogenerated with little excess energy relative to the band edges and thus lack additional driving force to aid their separation. Therefore, we tentatively assign this intensity-independent decay to geminate recombination of electron–hole pairs. Following this initial fast decay, an intensity-dependent component evolves (e.g., from ca. 10 ps for the highest intensity used). We find that the decay kinetics of this component decelerate with decreasing light intensity. Such behavior is characteristic of the bimolecular recombination of separated charges,31 which is characterized by power law decay kinetics and a decay half time inversely proportional to the excitation density (Figure S5). Figure 2b shows the decay kinetics at 1200 nm for the mesoporous anatase film. Strikingly, we observe decay kinetics very similar to that of dense anatase, although charge carriers in the mesoporous films readily access the TiO2 surface. This similarity suggests that surface state mediated recombination may not be a dominant loss pathway for these films.
Figure 2.

Normalized transient absorption decay kinetics probed at 1200 nm following 355 nm excitation under argon as a function of excitation intensity for (a) dense TiO2 anatase and (b) mesoporous TiO2 anatase, normalized at 1 ps.
The decay kinetics for dense and mesoporous rutile films are shown in Figure 3. As expected for the smaller rutile band gap, we do not observe geminate recombination below 1 ps upon excitation with 355 nm light because the higher excess energy facilitates the dissociation of electron–hole pairs. Dense rutile (Figure 3a) exhibits an exponential decay between ca. 3 and 100 ps with little intensity dependence. The mesoporous rutile film (Figure 3b) exhibits decay kinetics remarkably similar to the dense one. As for anatase, this similarity suggests that surface state recombination is not a key cause of charge carrier recombination. More detailed inspection shows that the kinetics of electron decay in mesoporous rutile exhibit a weak dependence on light intensity and are actually slower than dense rutile. This slower decay further emphasizes that access to surface states does not accelerate charge carrier recombination but may, if anything, retard it. We note that for both polymorphs, we observe small residual, long-lived TAS signals, assigned to photogenerated charge carriers trapped in intraband defect states.1,21,28
Figure 3.

Normalized transient absorption decay kinetics probed at 1200 nm following 355 nm excitation under argon as a function of excitation intensity for (a) dense TiO2 rutile and (b) mesoporous TiO2 rutile, normalized at 1 ps.
Our TA measurements demonstrate that, under the same illumination conditions, the decay kinetics of photogenerated carriers in dense and mesoporous TiO2 follow a qualitatively similar recombination behavior without any evidence for enhanced recombination due to different surface-area-to-volume ratios. These findings reveal that ultrafast recombination is not enhanced when more photogenerated charge carriers are able to access surface sites. Consequently, our data suggest that the benefits of nanostructuring in photocatalytic and photovoltaic devices are not attained at the expense of surface-enhanced recombination losses. Interestingly, our transient data also suggest that differences in crystal phase play a more dominant role in controlling recombination kinetics than differences in film morphology.
As shown in Figures 2 and 3, we observe faster, first-order recombination kinetics in rutile in contrast to slower, near-bimolecular recombination in anatase. These differences in decay kinetics may be indicative of different concentrations of intrinsic charge carriers in the material. In films with low intrinsic charge carrier densities, photogenerated electrons mostly recombine with photogenerated holes following a bimolecular recombination process, as observed for anatase herein. However, when one type of intrinsic charge carrier is present in higher densities (e.g., n-type doping due to oxygen vacancies), photogenerated carriers can readily recombine with these intrinsic carriers, leading to intensity-independent pseudo-first order kinetics, as shown for rutile herein. The faster decay kinetics observed in rutile compared to anatase TiO2 is also consistent with the doping density in rutile being higher than in anatase. This observation is in good agreement with doping densities reported in the literature for anatase (ranging from 1016 cm–3 to 1019 cm–3) anatase and rutile (from 1019 cm–3 to 1020 cm–3).25 For the number of charge carriers generated upon excitation in this study (from 1013 cm–3 to 1015 cm–3 for the different intensities), the decay kinetics are expected to be more strongly dominated by the intrinsic carriers in rutile than they are in anatase.
In conclusion, we have compared the charge carrier dynamics in TiO2 films as a function of surface area in order to elucidate the impact of morphology on the recombination dynamics of photogenerated charges. This comparison was done separately for the two most commonly used TiO2 crystal phases, i.e., anatase and rutile. We find that an increase in surface area as a result of nanostructuring does not lead to enhanced recombination rates of photogenerated charges. Instead, our results suggest that recombination is more dependent on the crystal phase, with faster recombination found in rutile than in anatase over the time scales examined. We tentatively assign this faster recombination in rutile to its higher doping level. Our results highlight that enhanced catalytic performance through the high surface-area-to-volume ratio of nanostructured TiO2 films can be achieved without detriment to the rates of charge carrier recombination in the device, even without the presence of surface passivation layers.
Given the surface-enhanced recombination rates observed for certain metal chalcogenides such as cadmium selenides or tellurides, it is striking that TiO2 does not exhibit such behavior. It appears likely that this absence of surface state mediated charge recombination may be a key factor behind the remarkably high photocatalytic efficiencies reported for TiO2 and potentially may be relevant to the promising photoelectrochemical performance reported for TiO2 and other metal oxide photoelectrodes.
Acknowledgments
J.R.D. and M.S. acknowledge support from ERC AdG Intersolar (291482) and the Imperial College Ph.D. Scholarship scheme. E.P. acknowledges the Engineering and Physical Sciences Research Council (EPSRC) for funding. A.K. thanks the Ramsay Memorial Fellowships Trust for funding.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpclett.6b01501.
Experimental details, XRD data, rutile SEM images, UV–NIR transmittance spectra, TAS spectra, and t50% TAS decay times as a function of excitation intensity (PDF)
Author Present Address
∥ E.P.: Physical Biosciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720.
The authors declare no competing financial interest.
Supplementary Material
References
- Cowan A. J.; Tang J.; Leng W.; Durrant J. R.; Klug D. R. Water Splitting by Nanocrystalline TiO2 in a Complete Photoelectrochemical Cell Exhibits Efficiencies Limited by Charge Recombination. J. Phys. Chem. C 2010, 114, 4208–4214. 10.1021/jp909993w. [DOI] [Google Scholar]
- Cowan A. J.; Durrant J. R. Long-Lived Charge Separated States in Nanostructured Semiconductor Photoelectrodes for the Production of Solar Fuels. Chem. Soc. Rev. 2013, 42, 2281–2293. 10.1039/C2CS35305A. [DOI] [PubMed] [Google Scholar]
- Ding T. X.; Olshansky J. H.; Leone S. R.; Alivisatos A. P. Efficiency of Hole Transfer from Photoexcited Quantum Dots to Covalently Linked Molecular Species. J. Am. Chem. Soc. 2015, 137, 2021–2029. 10.1021/ja512278a. [DOI] [PubMed] [Google Scholar]
- Olshansky J. H.; Ding T. X.; Lee Y. V.; Leone S. R.; Alivisatos A. P. Hole Transfer from Photoexcited Quantum Dots: The Relationship between Driving Force and Rate. J. Am. Chem. Soc. 2015, 137, 15567–15575. 10.1021/jacs.5b10856. [DOI] [PubMed] [Google Scholar]
- Smith A. M.; Nie S. Semiconductor Nanocrystals: Structure, Properties, and Band Gap Engineering. Acc. Chem. Res. 2010, 43, 190–200. 10.1021/ar9001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Regan B.; Grätzel M. A Low-Cost, High-Efficiency Solar Cell Based on Dye-Sensitized Colloidal TiO2 Films. Nature 1991, 353, 737–740. 10.1038/353737a0. [DOI] [Google Scholar]
- Hagfeldt A.; Boschloo G.; Sun L.; Kloo L.; Pettersson H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110, 6595–6663. 10.1021/cr900356p. [DOI] [PubMed] [Google Scholar]
- Snaith H. J. Perovskites: The Emergence of a New Era for Low-Cost, High-Efficiency Solar Cells. J. Phys. Chem. Lett. 2013, 4, 3623–3630. 10.1021/jz4020162. [DOI] [Google Scholar]
- Ma Y.; Wang X. L.; Jia Y. S.; Chen X. B.; Han H. X.; Li C. Titanium Dioxide-Based Nanomaterials for Photocatalytic Fuel Generations. Chem. Rev. 2014, 114, 9987–10043. 10.1021/cr500008u. [DOI] [PubMed] [Google Scholar]
- Habisreutinger S. N.; Schmidt-Mende L.; Stolarczyk J. K. Photocatalytic Reduction of CO2 on TiO2 and Other Semiconductors. Angew. Chem., Int. Ed. 2013, 52, 7372–7408. 10.1002/anie.201207199. [DOI] [PubMed] [Google Scholar]
- Kapilashrami M.; Zhang Y.; Liu Y.-S.; Hagfeldt A.; Guo J. Probing the Optical Property and Electronic Structure of TiO2 Nanomaterials for Renewable Energy Applications. Chem. Rev. 2014, 114, 9662–9707. 10.1021/cr5000893. [DOI] [PubMed] [Google Scholar]
- Scanlon D. O.; Dunnill C. W.; Buckeridge J.; Shevlin S. A.; Logsdail A. J.; Woodley S. M.; Catlow C. R. A.; Powell M. J.; Palgrave R. G.; Parkin I. P.; et al. Band Alignment of Rutile and Anatase TiO2. Nat. Mater. 2013, 12, 798–801. 10.1038/nmat3697. [DOI] [PubMed] [Google Scholar]
- Luttrell T.; Halpegamage S.; Tao J.; Kramer A.; Sutter E.; Batzill M. Why Is Anatase a Better Photocatalyst than Rutile? - Model Studies on Epitaxial TiO2 Films. Sci. Rep. 2014, 4, 4043. 10.1038/srep04043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakata A.; Vequizo J. J. M.; Matsunaga H. Distinctive Behavior of Photogenerated Electrons and Holes in Anatase and Rutile TiO2 Powders. J. Phys. Chem. C 2015, 119, 24538–24545. 10.1021/acs.jpcc.5b09236. [DOI] [Google Scholar]
- Ohtani B. Titania Photocatalysis beyond Recombination: A Critical Review. Catalysts 2013, 3, 942–953. 10.3390/catal3040942. [DOI] [Google Scholar]
- Heller A. Conversion of Sunlight into Electrical Power and Photoassisted Electrolysis of Water in Photoelectrochemical Cells. Acc. Chem. Res. 1981, 14, 154–162. 10.1021/ar00065a004. [DOI] [Google Scholar]
- Zawadzki P.; Laursen A. B.; Jacobsen K. W.; Dahl S.; Rossmeisl J. Oxidative Trends of TiO2—hole Trapping at Anatase and Rutile Surfaces. Energy Environ. Sci. 2012, 5, 9866–9869. 10.1039/c2ee22721e. [DOI] [Google Scholar]
- Linsebigler A. L.; Lu G.; Yates J. T. Photocatalysis on TiO2 Surfaces: Principles, Mechanisms, and Selected Results. Chem. Rev. 1995, 95, 735–758. 10.1021/cr00035a013. [DOI] [Google Scholar]
- Sudhagar P.; Devadoss A.; Nakata K.; Terashima C.; Fujishima A. Enhanced Photoelectrocatalytic Water Splitting at Hierarchical Gd3+:TiO2 Nanostructures through Amplifying Light Reception and Surface States Passivation. J. Electrochem. Soc. 2015, 162, H108–H114. 10.1149/2.0161503jes. [DOI] [Google Scholar]
- Peter L. M. Dynamic Aspects of Semiconductor Photoelectrochemistry. Chem. Rev. 1990, 90, 753–769. 10.1021/cr00103a005. [DOI] [Google Scholar]
- Colombo D. P.; Bowman R. M. Femtosecond Diffuse Reflectance Spectroscopy of TiO2 Powders. J. Phys. Chem. 1995, 99, 11752–11756. 10.1021/j100030a020. [DOI] [Google Scholar]
- Wang X.; Kafizas A.; Li X.; Moniz S. J. A.; Reardon P. J. T.; Tang J.; Parkin I. P.; Durrant J. R. Transient Absorption Spectroscopy of Anatase and Rutile: The Impact of Morphology and Phase on Photocatalytic Activity. J. Phys. Chem. C 2015, 119, 10439–10447. 10.1021/acs.jpcc.5b01858. [DOI] [Google Scholar]
- Tamaki Y.; Furube A.; Katoh R.; Murai M.; Hara K.; Arakawa H.; Tachiya M. Trapping Dynamics of Electrons and Holes in a Nanocrystalline TiO2 Film Revealed by Femtosecond Visible/near-Infrared Transient Absorption Spectroscopy. C. R. Chim. 2006, 9, 268–274. 10.1016/j.crci.2005.05.018. [DOI] [Google Scholar]
- Tamaki Y.; Furube A.; Murai M.; Hara K.; Katoh R.; Tachiya M. Dynamics of Efficient Electron–hole Separation in TiO2 Nanoparticles Revealed by Femtosecond Transient Absorption Spectroscopy under the Weak-Excitation Condition. Phys. Chem. Chem. Phys. 2007, 9, 1453–1460. 10.1039/B617552J. [DOI] [PubMed] [Google Scholar]
- Kafizas A.; Wang X.; Pendlebury S. R.; Barnes P.; Ling M.; Sotelo-Vazquez C.; Quesada-Cabrera R.; Li C.; Parkin I. P.; Durrant J. R. Where Do Photogenerated Holes Go in Anatase:Rutile TiO2? A Transient Absorption Spectroscopy Study of Charge Transfer and Lifetime. J. Phys. Chem. A 2016, 120, 715–723. 10.1021/acs.jpca.5b11567. [DOI] [PubMed] [Google Scholar]
- Antila L. J.; Santomauro F. G.; Hammarström L.; Fernandes D. L. a.; Sá J. Hunting for the Elusive Shallow Traps in TiO2 Anatase. Chem. Commun. 2015, 51, 10914–10916. 10.1039/C5CC02876K. [DOI] [PubMed] [Google Scholar]
- Cowan A. J.; Leng W.; Barnes P. R. F.; Klug D. R.; Durrant J. R. Charge Carrier Separation in Nanostructured TiO2 Photoelectrodes for Water Splitting. Phys. Chem. Chem. Phys. 2013, 15, 8772. 10.1039/c3cp50318f. [DOI] [PubMed] [Google Scholar]
- Philip Colombo D.; Roussel K. A.; Saeh J.; Skinner D. E.; Cavaleri J. J.; Bowman R. M. Femtosecond Study of the Intensity Dependence of Electron-Hole Dynamics in TiO2 Nanoclusters. Chem. Phys. Lett. 1995, 232, 207–214. 10.1016/0009-2614(94)01343-T. [DOI] [Google Scholar]
- Colombo D. P.; Bowman R. M. Does Interfacial Charge Transfer Compete with Charge Carrier Recombination? A Femtosecond Diffuse Reflectance Investigation of TiO2 Nanoparticles. J. Phys. Chem. 1996, 100, 18445–18449. 10.1021/jp9610628. [DOI] [Google Scholar]
- Noguchi H.; Ohtani B.; Uosaki K. Effect of Excitation Wavelength on Ultrafast Electron–Hole Recombination in Titanium(IV) Oxide Powders Irradiated by Femtosecond Laser Pulses. Chem. Lett. 2005, 34, 694–695. 10.1246/cl.2005.694. [DOI] [Google Scholar]
- Clarke T. M.; Durrant J. R. Charge Photogeneration in Organic Solar Cells. Chem. Rev. 2010, 110, 6736–6767. 10.1021/cr900271s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.