

Abstract
Hemophilia is the most well-known hereditary bleeding disorder, with an incidence of one in every 5000 to 30,000 males worldwide. The disease is treated by infusion of protein products on demand and as prophylaxis. Although these therapies have been very successful, some challenging and unresolved tasks remain, such as reducing bleeding rates, presence of target joints and/or established joint damage, eliminating the development of inhibitors, and increasing the success rate of immune-tolerance induction (ITI). Many preclinical trials are carried out on animal models for hemophilia generated by the hemophilia research community, which in turn enable prospective clinical trials aiming to tackle these challenges. Suitable animal models are needed for greater advances in treating hemophilia, such as the development of better models for evaluation of the efficacy and safety of long-acting products, more powerful gene therapy vectors than are currently available, and successful ITI strategies. Mice, dogs, and pigs are the most commonly used animal models for hemophilia. With the advent of the nuclease method for genome editing, namely the CRISPR/Cas9 system, it is now possible to create animal models for hemophilia other than mice in a short period of time. This review presents currently available animal models for hemophilia, and discusses the importance of animal models for the development of better treatment options for hemophilia.
Keywords: Hemophilia, Animal models, Genetically-engineered, CRISPR/Cas9
Background
Hemophilia, caused by deficiency or dysfunction of coagulation factor VIII (FVIII) or factor IX (FIX), is the most well-known hereditary bleeding disorder, with an incidence of one in every 5000 to 30,000 males worldwide. Current standard care involves intravenous infusions of protein products on demand, and prophylaxis. Although very successful, protein replacement therapy runs the risk of inducing neutralizing antibodies or “inhibitors”. Patients with inhibitors can no longer use FVIII/FIX concentrates and require more expensive bypass products [1] or elimination of the inhibitors by immune-tolerance induction (ITI) [2]. ITI may not be performed due to economical accessibility (affordability) of FVIII/FIX products and, in addition, ITI of hemophilia-B is performed less often because inhibitors are less well-developed and have lower success rates. Multiple factors are involved in the success of ITI of hemophilia A including the historical peak and pre-ITI level of inhibitors, age of patients, time period from inhibitor diagnosis to ITI, interruptions of ITI and peak inhibitors during ITI. Products and regimen may also play a role in ITI outcome, although current clinical data are contradictory or inconclusive with regard to this point.
Considerable effort has been exerted to improve hemophilia treatment through the development of long-acting products, powerful gene therapy vectors, and successful ITI strategies. Clinical trials aiming to tackle these challenges have been undertaken, and many preclinical trials have been carried out on animal models for hemophilia generated by the research community. Preclinical and clinical trials all require a suitable animal model of hemophilia. The most commonly used animal models for hemophilia include but are not limited to mice, dogs, and pigs. Animal models are important for basic biological and preclinical studies, such as studies aimed at understanding the pathophysiology of inhibitor formation, improving ITI protocols, estimating drug doses, and evaluating therapeutic efficacy before human trials. With the advent of the nuclease method for genome editing, named the CRISPR/Cas9 system, it is now possible to create animal models of hemophilia other than mice in a short period of time. This review presents an overview of currently available animal models for hemophilia, and discusses the importance of animal models for the development of better treatment options for hemophilia.
Review
Hemophilia animals with spontaneous mutations
Hemophilia A dogs
Dogs with naturally occurring hemophilia A were first documented following observations of abnormally prolonged bleeding that could be treated or prevented by infusion of canine FVIII. Currently, two colonies of hemophilia A dogs are most commonly used in studies: an Irish Setter colony maintained at the University of North Carolina in Chapel Hill [3], and a miniature schnauzer colony maintained at Queens University in Toronto, Canada [4]. Both colonies are deficient in circulating FVIII and show an aberrant inversion mutation between ~0.5 Mb upstream of the FVIII gene and its intron 22 [5, 6]. In addition to intron 22 inversion of FVIII gene, dogs with hemophilia A have also been described to have other spontaneous mutations in the FVIII gene [7, 8].
Hemophilia A dogs have been used extensively in preclinical trials of human FVIII protein products as well as in studies on the safety and efficacy of adeno-associated viral (AAV) vector-conducted gene therapy [9], in testing the gene therapy of platelet-specific expression of human FVIII [10], and have provided promising data for bypass therapy [11]. A subset of these dogs have a propensity to develop inhibitors [12] after infusion of canine FVIII [13, 14], with the Queens colony being more prone to inhibitor development than the Chapel Hill dogs. Dogs with inhibitors can facilitate the studies of inhibitor pathogenesis and ITI strategies, e.g., eradication of pre-existing neutralizing antibodies by liver gene therapy [15]. The development of inhibitors to FVIII is the most significant complication of protein replacement therapy. Because of the high homology of the canine and human immune system, assessments of FVIII immunogenicity using hemophilia A dogs may provide meaningful insights into the human immune response.
Hemophilia A sheep
Phenotypes consistent with human hemophilia A, including spontaneous bleeding and plasma FVIII activity of less than 1 %, have been found in sheep [16, 17]. Sheep with naturally occurring hemophilia A were re-established through reproductive technologies in 2009. Sequence mapping revealed a single nucleotide insertion-induced frame shift of the FVIII gene creating a premature stop codon at the base position 3112–4 in exon 14 and five additional stop codons within the next 183 bp [18]. Hemophilia sheep have been used in studies relevant to gene and cell therapies for hemophilia, including investigations of pre-existing immunity to AAV vectors [19] and the use of mesenchymal stem cells as cellular delivery vehicles for the FVIII gene [20]. However, the restricted availability of the recombinant ovine FVIII for treatment may limit the practical use of hemophiliac sheep.
Hemophilia A rats
Researchers identified an inbred WAG/RijY rat strain, designated WAG/RijYcb, which tended to show abnormal hemorrhaging, a prolonged activated partial thromboplastin time, and a mutated FVIII gene (proline was substituted for leucine at amino acid 176 in the A1 domain). In rats, the FVIII gene is located on chromosome 18. Thus, hemophilia in rats is autosomal recessive, which contrasts with humans and other animal models where it is an X-linked hereditary disease [21, 22]. The mutated FVIII region of WAG/RijYcb rats generally lacks immunodominant epitopes for inhibitor formation thus this model may not be suitable for studying the immune response to FVIII treatment or developing ITI strategies.
Hemophilia B dogs
Similar to human hemophilia B, canine hemophilia B has a sex-linked inheritance pattern, and no detectable circulating FIX exists in the plasma. Researchers have identified at least three colonies of hemophilia B dogs, each of which has a unique molecular FIX defect. Hemophilia B cairn terriers were identified in Toronto [23] and have been maintained in Chapel Hill since 1966 [24]. These animals have a point mutation that results in substitution of glutamic acid for glycine at AA 377 in the catalytic domain [24]. Lhasa Apso dogs with hemophilia B, established in Auburn, Alabama [25], carry a 5-bp deletion at nucleotides 772–776 and a C-to-T transition in the FIX gene, resulting in a premature stop codon at AA 146 [25]. A Labrador retriever colony at Cornell University, New York [26], carries complete deletion of the canine FIX gene. Chapel Hill and Auburn dogs have been used extensively for testing FIX products and gene therapy strategies. The Auburn dogs are prone to developing inhibitors to infused canine FIX [27, 28]. Translational data produced from hemophilia B dogs have supported the development of long-acting FIX [29, 30] with accompanying recent human clinical trials [12, 31–34]. In contrast to the higher success ITI rate in hemophilia A, the ITI outcome in hemophilia B patients with FIX inhibitors is poor and research in this area remains in its infancy [35]. Recently, an ITI strategy using AAV liver expression of a FIX variant, FIX-Padua was successfully applied to hemophilia B dogs prone to inhibitors. This strategy eradicated FIX inhibitor in dogs with pre-existing inhibitors [34], suggesting its value in the development of ITI.
Genetically engineered animal models of hemophilia
Hemophilia A mice
As small mammals that are easy to breed and relatively inexpensive to maintain in large numbers, mice are a popular animal for medical research and preclinical testing worldwide. Although spontaneous bleeding does not naturally occur in mice, genome-editing technologies have contributed to development of various hemophiliac mouse models. Hemophilia mice are the best initial model to use when attempting to test new therapeutics because they only require small amounts of drugs.
In 1995, the first two hemophilia A mouse models, with a mixed genetic background of 129SV and C57BL/6, were established. These two strains of FVIII knockout mice were generated through gene targeting and embryonic stem cell manipulation [36]. A neo cassette was inserted into the 3′ end of exon 16 of the mouse FVIII gene in one strain (E16 mice), whereas exon 17 was disrupted in the other strain (E17 mice). Mice had no detectable circulating FVIII, and their plasma FVIII activity was less than 1 %. Unlike human hemophilia A, little spontaneous bleeding is observed in mouse models of hemophilia A, whereas tail clipping or other invasive procedures could lead to death. Mouse models of hemophilia A are widely used for the evaluation of FVIII treatment efficacy [37, 38], investigation of mechanisms of inhibitor formation, and development of ITI protocols for FVIII [39].
To mimic patients with severe hemophilia A who express no endogenous FVIII (i.e., cross-reacting material) and are prone to form inhibitors after protein replacement therapy, researchers generated a total FVIII gene knockout hemophilia A mouse model in a pure C57BL/6 background. The entire coding sequence was deleted by a Cre recombinase-LoxP site-mediated deletion. Plasma FVIII activity and anti-FVIII inhibitor titer induced after FVIII treatment were comparable to those of E16 mice [40].
Hemophilia A pig
Pigs with hemophilia A were generated by nuclear transfer and cloning from porcine fetal fibroblasts carrying disruption of exon 16 of the porcine FVIII gene by the neomycin-resistant gene [41]. Species differences between humans and mice, such as size, general physiology, anatomy, and lifespan, limit the value of mouse models in preclinical trials. In contrast, the coagulation systems of pigs and humans are highly homologous. Moreover, hemophilia A pigs may develop arthropathy similar to humans because of repeated joint bleeding [41]. Thus, hemophilia A pigs provide another option for evaluating novel therapeutics for hemophilia A patients [42].
Hemophilia B mice
Although mice with naturally occurring deficiency of FIX have not been identified, a series of FIX knockout mice were engineered by homologous recombination in embryonic stem cells. Initially, the mouse FIX gene was disrupted by insertion of a neo cassette into exon 3 [43] or the coding region for the catalytic domain of FIX [44, 45], resulting in a mouse model with no detectable mRNA or plasma protein expression of FIX. In addition to mice with complete deletion of FIX, researchers have created knock-in mice expressing human FIX carrying a missense mutation (R333Q-hFIX) under the control of the mouse FIX promoter. The R333Q mutation is located in the catalytic domain of human FIX; the same mutation has been identified in several patients with severe hemophilia B. In R333Q hFIX mice, mutant human FIX transcript and circulating human FIX protein were detectable throughout development, but the FIX protein activity was <1 % [46]. Similar to hemophilia A mice, hemophilia B mice do not show spontaneous bleeding, but will bleed and die after tail clipping unless the wound is cauterized. Hemophilia B mice have been used to test the efficacy of FIX and FIX variants, including those FIX variants with very high clotting activities [47–52], as well as to evaluate the immunity and safety of gene therapy [53–55].
Researchers have also established several knock-in mice carrying a FIX variant coding for K5A in the Gla domain of FIX [56], a full-length wild-type (WT) human FIX coding sequence, and a FIX variant expressing FIX-Triple containing 3 amino acid modifications [47]. The K5A mutation impairs FIX binding to collagen type IV, and these mice show a relatively mild bleeding phenotype [56]. The knock-in mice with complete human WT and FIX-Triple FIX coding sequence exhibited clotting activity of, respectively, nearly 5 and 50 % that of mouse FIX, which was unexpected and was perhaps due to species specificity of FVIII [47].
Humanized hemophilia A mice for FVIII immunity
To understand the regulation of antibody responses against FVIII in hemophilia A, researchers modified the hemophilia A mouse model to be “humanized” for HLA class II antigen. Researchers crossed E17 mice with mice expressing chimeric human-mouse HLA-DRB1*1501, which is associated with an increased risk of inhibitor development in humans [57]. Use of this humanized hemophilia A mouse model allowed identification of immunodominant FVIII peptides that trigger inhibitor formation, as well as the characterization of interactions of T-cell receptors with disease-associated FVIII peptides and MHC class II molecules [58].
Novel hemophilia NSG mouse models established by CRISPR/Cas9 technology
A recent advance in genetic engineering technology provides a powerful tool to modify the genome in any living species [59]. CRISPR (clustered regularly interspaced short palindromic repeat)-associated RNA-guided endonuclease Cas9, which was identified from the microbial adaptive immune system [60], can be used to alter the mammalian genome with high efficiency and precision. Recently, a reliable animal model for elucidating the humoral and cellular immune responses of patients to FVIII/FIX treatment was developed. The CRISPR/Cas9 system and immunodeficient NSG mice (Nod/Scid-Il2γ−/−) were combined to mutate the FVIII and FIX genes, generating hemophilia A/B mice with the NSG background (HemoA/B-NSG mice). Oligonucleotides of 20 residues serving as specific guiding RNAs (gRNAs) were developed to target exon 1 of mouse FVIII and FIX. The gRNA and CRISPR/Cas9 RNA were microinjected into the NSG mouse zygotes to generate founders. Four male founder mice were obtained, each carrying a 1, 2 or 5-bp deletion in exon 1 of the FVIII gene, resulting in a premature stop codon (Fig. 1a). Hepatic FVIII mRNA level of these mice was 50 % lower than that of NSG mice (unpublished data), whereas their plasma FVIII activity was dramatically decreased to be comparable to that of hemophilia A mice with the 129SV/C57BL/6J mixed genetic background [36]. Two male founder mice were identified that had the same mutation, i.e., carrying an 8-nucleotide deletion in exon 1 of the FIX gene, which created a premature stop codon (Fig. 1b). NSG mice are more acceptable than NOD/SCID mice for transplanting with human hematopoietic cells and can, therefore, be used for direct assessment of the human immune response to FVIII/FIX treatment.
Fig. 1.
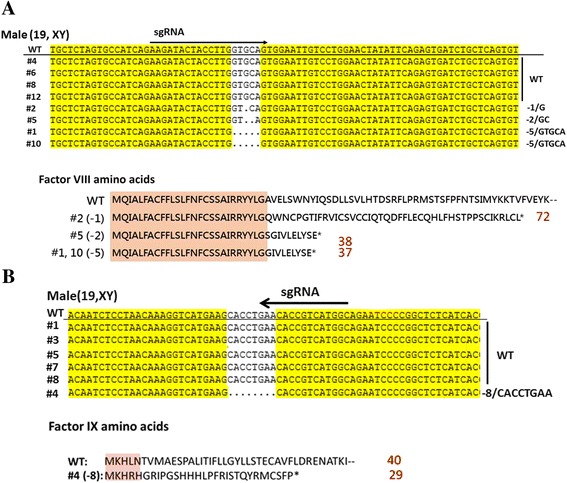
Illustration of NSG male mouse with missense mutation in exon 1 of the mouse FVIII or FIX gene. Sequences of tail DNA derived from male founders of hemophilia NSG mice are shown. a Four male founders carrying mutations in the FVIII gene were generated. Deletion of 1 bp (−1/G), 2 bp (−2/GC), and 5 bp (−5/GTGCA) in exon 1 created a premature stop codon at the 72, 38 and 37th AA residue of mouse FVIII, respectively. b One male founder carried an −8/CACCTGAA deletion in the FIX gene, which resulted in a premature stop codon at the 30th AA residue of mouse FIX
Conclusions
Translational research from hemophilia animal models gives valuable information about safety and efficacy, and guides design of human clinical trials. Despite their availability, hemophilia animal models have many potential disadvantages and limitations, including a short half-life of human FVIII (in mice), differences in tissue tropism of viral vectors compared to humans, and immune reactions to human FVIII/FIX. Reducing inhibitor incidence and elimination of inhibitors in hemophilia patients is a major task that needs to be resolved. It is considered that the use of different products and treatment protocols may be involved in inhibitor production [61], but the possible mechanism(s) is not fully understood. An appropriate humanized animal model for the evaluation of different products, regimens, and ITI strategies is still lacking. CRISPR/Cas9 technology is a very efficient method to generate hemophilia in rare and difficult-to-breed mice. We believe that our hemophilia NSG mice will be a very useful model for studying the human immune response to therapeutics.
Acknowledgements
The authors thank the Transgenic Mouse Model Core Facility (supported by the National Core Facility Program for Biotechnology, Ministry of Science and Technology, Taiwan) and the Gene Knockout Mouse Core Laboratory (supported by the National Taiwan University Center of Genomic Medicine of the National Taiwan University, Taiwan) for technical support. This work was supported by grants from the Ministry of Science and Technology, R.O.C (Grant No. 104-2325-B-002-023-, 105-2325-B-002-015-).
Declaration
Publication fees for this article have been funded by APSTH 2016.
This article has been published as part of Thrombosis Journal Volume 14 Supplement 1, 2016. The full contents of the supplement are available at https://thrombosisjournal.biomedcentral.com/articles/supplements/volume-14-supplement-1.
Availability of data and material
The genetically-engineered materials described in this article are available upon request.
Authors’ contributions
TZY and SWL contributed in the design and writing of the article. MNF and ISY contributed in executing experiments. YLY and SCC contributed to writing and discussion. All authors have read the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All animal experiments were approved by Institutional Animal Care and Use Committee (IACUC) of National Taiwan University College of Medicine and College of Public Health. No. 20140335, 20140383, and 20140428.
Abbreviations
- FVIII
Factor VIII
- FIX
Factor IX
- CRISPR/Cas9
Clustered regularly interspaced short palindromic repeat-associated RNA-guided endonuclease Cas9
- AA
Amino acid
- ITI
Immune tolerance induction
References
- 1.Makris M, Hay CR, Gringeri A, D’Oiron R. How I treat inhibitors in haemophilia. Haemophilia. 2012;18(Suppl 4):48–53. doi: 10.1111/j.1365-2516.2012.02829.x. [DOI] [PubMed] [Google Scholar]
- 2.DiMichele DM. Immune tolerance in haemophilia: the long journey to the fork in the road. Br J Haematol. 2012;159:123–34. doi: 10.1111/bjh.12028. [DOI] [PubMed] [Google Scholar]
- 3.Graham JB, Buckwalter JA, Hartley LJ, Brinkhous KM. Canine hemophilia; observations on the course, the clotting anomaly, and the effect of blood transfusions. J Exp Med. 1949;90:97–111. doi: 10.1084/jem.90.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giles AR, Tinlin S, Greenwood R. A canine model of hemophilic (factor VIII:C deficiency) bleeding. Blood. 1982;60:727–30. [PubMed] [Google Scholar]
- 5.Lozier JN, Dutra A, Pak E, Zhou N, Zheng Z, Nichols TC, et al. The Chapel Hill hemophilia A dog colony exhibits a factor VIII gene inversion. Proc Natl Acad Sci U S A. 2002;99:12991–6. doi: 10.1073/pnas.192219599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hough C, Kamisue S, Cameron C, Notley C, Tinlin S, Giles A, et al. Aberrant splicing and premature termination of transcription of the FVIII gene as a cause of severe canine hemophilia A: similarities with the intron 22 inversion mutation in human hemophilia. Thromb Haemost. 2002;87:659–65. [PubMed] [Google Scholar]
- 7.Joseph SA, Brooks MB, Coccari PJ, Riback SC. Hemophilia A in a German shorthaired pointer: clinical presentations and diagnosis. J Am Anim Hosp Assoc. 1996;32:25–8. doi: 10.5326/15473317-32-1-25. [DOI] [PubMed] [Google Scholar]
- 8.Lozier JN, Nichols TC. Animal models of hemophilia and related bleeding disorders. Semin Hematol. 2013;50:175–84. doi: 10.1053/j.seminhematol.2013.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nichols TC, Hough C, Agerso H, Ezban M, Lillicrap D. Canine models of inherited bleeding disorders in the development of coagulation assays, novel protein replacement and gene therapies. J Thromb Haemost. 2016;14:894–905. doi: 10.1111/jth.13301. [DOI] [PubMed] [Google Scholar]
- 10.Du LM, Nurden P, Nurden AT, Nichols TC, Bellinger DA, Jensen ES, et al. Platelet-targeted gene therapy with human factor VIII establishes haemostasis in dogs with haemophilia A. Nat Commun. 2013;4:2773. doi: 10.1038/ncomms3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Margaritis P, Roy E, Aljamali MN, Downey HD, Giger U, Zhou S, et al. Successful treatment of canine hemophilia by continuous expression of canine FVIIa. Blood. 2009;113:3682–9. doi: 10.1182/blood-2008-07-168377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–65. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giles AR, Tinlin S, Hoogendoorn H, Greenwood P, Greenwood R. Development of factor VIII:C antibodies in dogs with hemophilia A (factor VIII:C deficiency) Blood. 1984;63:451–6. [PubMed] [Google Scholar]
- 14.Nichols TC, Dillow AM, Franck HW, Merricks EP, Raymer RA, Bellinger DA, et al. Protein replacement therapy and gene transfer in canine models of hemophilia A, hemophilia B, von willebrand disease, and factor VII deficiency. ILAR J. 2009;50:144–67. doi: 10.1093/ilar.50.2.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finn JD, Ozelo MC, Sabatino DE, Franck HW, Merricks EP, Crudele JM, et al. Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood. 2010;116:5842–8. doi: 10.1182/blood-2010-06-288001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neuenschwander S, Kissling-Albrecht L, Heiniger J, Backfisch W, Stranzinger G, Pliska V. Inherited defect of blood clotting factor VIII (haemophilia A) in sheep. Thromb Haemost. 1992;68:618–20. [PubMed] [Google Scholar]
- 17.Neuenschwander S, Pliska V. Factor VIII in blood plasma of haemophilic sheep: analysis of clotting time-plasma dilution curves. Haemostasis. 1994;24:27–35. doi: 10.1159/000217077. [DOI] [PubMed] [Google Scholar]
- 18.Porada CD, Sanada C, Long CR, Wood JA, Desai J, Frederick N, et al. Clinical and molecular characterization of a re-established line of sheep exhibiting hemophilia A. J Thromb Haemost. 2010;8:276–85. doi: 10.1111/j.1538-7836.2009.03697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tellez J, Van Vliet K, Tseng YS, Finn JD, Tschernia N, Almeida-Porada G, et al. Characterization of naturally-occurring humoral immunity to AAV in sheep. PLoS One. 2013;8:e75142. doi: 10.1371/journal.pone.0075142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porada CD, Sanada C, Kuo CJ, Colletti E, Mandeville W, Hasenau J, et al. Phenotypic correction of hemophilia A in sheep by postnatal intraperitoneal transplantation of FVIII-expressing MSC. Exp Hematol. 2011;39:1124–35. doi: 10.1016/j.exphem.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Booth CJ, Brooks MB, Rockwell S. Spontaneous coagulopathy in inbred WAG/RijYcb rats. Comp Med. 2010;60:25–30. [PMC free article] [PubMed] [Google Scholar]
- 22.Booth CJ, Brooks MB, Rockwell S, Murphy JW, Rinder HM, Zelterman D, et al. WAG-F8(m1Ycb) rats harboring a factor VIII gene mutation provide a new animal model for hemophilia A. J Thromb Haemost. 2010;8:2472–7. doi: 10.1111/j.1538-7836.2010.03978.x. [DOI] [PubMed] [Google Scholar]
- 23.Mustard JF, Rowsell HC, Robinson GA, Hoeksema TD, Downie HG. Canine haemophilia B (Christmas disease) Br J Haematol. 1960;6:259–66. doi: 10.1111/j.1365-2141.1960.tb06241.x. [DOI] [PubMed] [Google Scholar]
- 24.Evans JP, Brinkhous KM, Brayer GD, Reisner HM, High KA. Canine hemophilia B resulting from a point mutation with unusual consequences. Proc Natl Acad Sci U S A. 1989;86:10095–9. doi: 10.1073/pnas.86.24.10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mauser AE, Whitlark J, Whitney KM, Lothrop CD., Jr A deletion mutation causes hemophilia B in Lhasa Apso dogs. Blood. 1996;88:3451–5. [PubMed] [Google Scholar]
- 26.Brooks MB, Gu W, Ray K. Complete deletion of factor IX gene and inhibition of factor IX activity in a labrador retriever with hemophilia B. J Am Vet Med Assoc. 1997;211:1418–21. [PubMed] [Google Scholar]
- 27.Herzog RW, Mount JD, Arruda VR, High KA, Lothrop CD., Jr Muscle-directed gene transfer and transient immune suppression result in sustained partial correction of canine hemophilia B caused by a null mutation. Mol Ther. 2001;4:192–200. doi: 10.1006/mthe.2001.0442. [DOI] [PubMed] [Google Scholar]
- 28.Brinkhous KM, Sigman JL, Read MS, Stewart PF, McCarthy KP, Timony GA, et al. Recombinant human factor IX: replacement therapy, prophylaxis, and pharmacokinetics in canine hemophilia B. Blood. 1996;88:2603–10. [PubMed] [Google Scholar]
- 29.Taupin P. Chimeric proteins comprising the constant region of immunoglobulins for treating hemophilia B (WO2005001025) Expert Opin Ther Pat. 2011;21:967–70. doi: 10.1517/13543776.2011.577070. [DOI] [PubMed] [Google Scholar]
- 30.Ostergaard H, Bjelke JR, Hansen L, Petersen LC, Pedersen AA, Elm T, et al. Prolonged half-life and preserved enzymatic properties of factor IX selectively PEGylated on native N-glycans in the activation peptide. Blood. 2011;118:2333–41. doi: 10.1182/blood-2011-02-336172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manno CS, Chew AJ, Hutchison S, Larson PJ, Herzog RW, Arruda VR, et al. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood. 2003;101:2963–72. doi: 10.1182/blood-2002-10-3296. [DOI] [PubMed] [Google Scholar]
- 32.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–7. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 33.Finn JD, Nichols TC, Svoronos N, Merricks EP, Bellenger DA, Zhou S, et al. The efficacy and the risk of immunogenicity of FIX Padua (R338L) in hemophilia B dogs treated by AAV muscle gene therapy. Blood. 2012;120:4521–3. doi: 10.1182/blood-2012-06-440123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crudele JM, Finn JD, Siner JI, Martin NB, Niemeyer GP, Zhou S, et al. AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood. 2015;125:1553–61. doi: 10.1182/blood-2014-07-588194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Batorova A, Morongova A, Tagariello G, Jankovicova D, Prigancova T, Horakova J. Challenges in the management of hemophilia B with inhibitor. Semin Thromb Hemost. 2013;39:767–71. doi: 10.1055/s-0033-1356574. [DOI] [PubMed] [Google Scholar]
- 36.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH., Jr Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10:119–21. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 37.Di Minno G, Cerbone AM, Coppola A, Cimino E, Di Capua M, Pamparana F, et al. Longer-acting factor VIII to overcome limitations in haemophilia management: the PEGylated liposomes formulation issue. Haemophilia. 2010;16(Suppl 1):2–6. doi: 10.1111/j.1365-2516.2009.02155.x. [DOI] [PubMed] [Google Scholar]
- 38.Siner JI, Iacobelli NP, Sabatino DE, Ivanciu L, Zhou S, Poncz M, et al. Minimal modification in the factor VIII B-domain sequence ameliorates the murine hemophilia A phenotype. Blood. 2013;121:4396–403. doi: 10.1182/blood-2012-10-464164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scott DW, Pratt KP, Miao CH. Progress toward inducing immunologic tolerance to factor VIII. Blood. 2013;121:4449–56. doi: 10.1182/blood-2013-01-478669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chao BN, Baldwin WH, Healey JF, Parker ET, Shafer-Weaver K, Cox C, et al. Characterization of a genetically engineered mouse model of hemophilia A with complete deletion of the F8 gene. J Thromb Haemost. 2016;14:346–55. doi: 10.1111/jth.13202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kashiwakura Y, Mimuro J, Onishi A, Iwamoto M, Madoiwa S, Fuchimoto D, et al. Porcine model of hemophilia A. PLoS One. 2012;7:e49450. doi: 10.1371/journal.pone.0049450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohmori T, Mizukami H, Ozawa K, Sakata Y, Nishimura S. New approaches to gene and cell therapy for hemophilia. J Thrombo Haemost. 2015;13(Suppl. 1):S133–42. doi: 10.1111/jth.12926. [DOI] [PubMed] [Google Scholar]
- 43.Lin HF, Maeda N, Smithies O, Straight DL, Stafford DW. A coagulation factor IX-deficient mouse model for human hemophilia B. Blood. 1997;90:3962–6. [PubMed] [Google Scholar]
- 44.Wang L, Zoppe M, Hackeng TM, Griffin JH, Lee KF, Verma IM. A factor IX-deficient mouse model for hemophilia B gene therapy. Proc Natl Acad Sci U S A. 1997;94:11563–6. doi: 10.1073/pnas.94.21.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kundu RK, Sangiorgi F, Wu LY, Kurachi K, Anderson WF, Maxson R, et al. Targeted inactivation of the coagulation factor IX gene causes hemophilia B in mice. Blood. 1998;92:168–74. [PubMed] [Google Scholar]
- 46.Jin DY, Zhang TP, Gui T, Stafford DW, Monahan PE. Creation of a mouse expressing defective human factor IX. Blood. 2004;104:1733–9. doi: 10.1182/blood-2004-01-0138. [DOI] [PubMed] [Google Scholar]
- 47.Kao CY, Lin CN, Yu IS, Tao MH, Wu HL, Shi GY, et al. FIX-Triple, a gain-of-function factor IX variant, improves haemostasis in mouse models without increased risk of thrombosis. Thromb Haemost. 2010;104:355–65. doi: 10.1160/TH09-11-0792. [DOI] [PubMed] [Google Scholar]
- 48.Lin CN, Lin CN, Kao CY, Miao CH, Hamaguchi N, Wu HL, et al. Generation of a novel factor IX with augmented clotting activities in vitro and in vivo. J Thromb Haemost. 2010;8:1773–8. doi: 10.1111/j.1538-7836.2010.03913.x. [DOI] [PubMed] [Google Scholar]
- 49.Wu YM, Kao CY, Huang YJ, Yu IS, Lee HS, Lai HS, et al. Genetic modification of donor hepatocyte improves therapeutic efficacy for hemophilia B in mice. Cell Transplant. 2010;19:1169–80. doi: 10.3727/096368910X503398. [DOI] [PubMed] [Google Scholar]
- 50.Kao CY, Yang SJ, Tao MH, Jeng YM, Yu IS, Lin SW. Incorporation of the factor IX Padua mutation into FIX-Triple improves clotting activity in vitro and in vivo. Thromb Haemost. 2013;110:244–56. doi: 10.1160/TH13-02-0154. [DOI] [PubMed] [Google Scholar]
- 51.Dietrich B, Schiviz A, Hoellriegl W, Horling F, Benamara K, Rottensteiner H, et al. Preclinical safety and efficacy of a new recombinant FIX drug product for treatment of hemophilia B. Int J Hematol. 2013;98:525–32. doi: 10.1007/s12185-013-1448-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cooley B, Funkhouser W, Monroe D, Ezzell A, Mann DM, Lin FC, et al. Prophylactic efficacy of BeneFIX vs Alprolix in hemophilia B mice. Blood. 2016;128:286–92. doi: 10.1182/blood-2016-01-696104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davidoff AM, Nathwani AC. Genetic targeting of the albumin locus to treat hemophilia. N Engl J Med. 2016;374:1288–90. doi: 10.1056/NEJMcibr1600347. [DOI] [PubMed] [Google Scholar]
- 54.Zhang R, Wang Q, Zhang L, Chen S. Optimized human factor IX expression cassettes for hepatic-directed gene therapy of hemophilia B. Front Med. 2015;9:90–9. doi: 10.1007/s11684-015-0390-2. [DOI] [PubMed] [Google Scholar]
- 55.Monahan PE, Sun J, Gui T, Hu G, Hannah WB, Wichlan DG, et al. Employing a gain-of-function factor IX variant R338L to advance the efficacy and safety of hemophilia B human gene therapy: preclinical evaluation supporting an ongoing adeno-associated virus clinical trial. Hum Gene Ther. 2015;26:69–81. doi: 10.1089/hum.2014.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gui T, Reheman A, Ni H, Gross PL, Yin F, Monroe D, et al. Abnormal hemostasis in a knock-in mouse carrying a variant of factor IX with impaired binding to collagen type IV. J Thromb Haemost. 2009;7:1843–51. doi: 10.1111/j.1538-7836.2009.03545.x. [DOI] [PubMed] [Google Scholar]
- 57.Reipert BM, Steinitz KN, van Helden PM, Unterthurner S, Schuster M, Ahmad RU, et al. Opportunities and limitations of mouse models humanized for HLA class II antigens. J Thromb Haemost. 2009;7(Suppl 1):92–7. doi: 10.1111/j.1538-7836.2009.03403.x. [DOI] [PubMed] [Google Scholar]
- 58.Steinitz KN, van Helden PM, Binder B, Wraith DC, Unterthurner S, Hermann C, et al. CD4+ T-cell epitopes associated with antibody responses after intravenously and subcutaneously applied human FVIII in humanized hemophilic E17 HLA-DRB1*1501 mice. Blood. 2012;119:4073–82. doi: 10.1182/blood-2011-08-374645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–78. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Calvez T, Chambost H, Claeyssens-Donadel S, d’Oiron R, Goulet V, Guillet B, et al. Recombinant factor VIII products and inhibitor development in previously untreated boys with severe hemophilia A. Blood. 2014;124:3398–408. doi: 10.1182/blood-2014-07-586347. [DOI] [PubMed] [Google Scholar]