

Abstract
Primary biliary cirrhosis (PBC) is a progressive autoimmune liver disease with a long natural history. The pathogenesis of PBC is thought to be orchestrated by Th1 and/or Th17. In this study, we investigated the role of CD4+ helper T subsets and their cytokines on PBC using our previous established murine model of 2-OA-OVA immunization. We prepared adeno-associated virus (AAV)-IFN-γ and AAV-IL-4 and studied their individual influences on the natural history of autoimmune cholangitis in this model. Administration of IFN-γ significantly promotes recruitment and lymphocyte activation in the earliest phases of autoimmune cholangitis but subsequently leads to downregulation of chronic inflammation through induction of the immunosuppressive molecule IL-30. In contrast, the administration of IL-4 does not alter the initiation of autoimmune cholangitis, but does contribute to the exacerbation of chronic liver inflammation and fibrosis. Thus Th1 cells and IFN-γ are the dominant contributors in the initiation phase of this model but clearly may have different effects as the disease progress. In conclusion, better understanding of the mechanisms by which helper T cells function in the natural history of cholangitis is essential and illustrates that precision medicine may be needed for patients with PBC at various stages of their disease process.
Primary biliary cholangitis (PBC), hitherto called primary biliary cirrhosis, is considered a Th1 or Th17 disease with significant increases in IFN-γ and IL-171,2,3,4,5,6. Within the liver, there is a significantly higher frequency of IFN-γ mRNA-positive cells1. In addition, there is an increased frequency of IL-17+ liver infiltrating lymphocytes3,5. Two independent genome-wide association studies have demonstrated that Th1 related IL-12A and IL-12RB2 variants are strongly associated with PBC7,8. Collectively, the data suggests an important role of CD4+ T helper subsets and their cytokines in disease pathogenesis. However, PBC has a long natural history and it is logical to consider that different mechanisms are in play at different stages of disease.
Our laboratory has attempted to define the natural history of biliary lesions by focusing on a molecular mimicry murine model of PBC induced following immunization with a mimotope of the highly conserved inner lipoyl domain of PDC-E2, the major mitochondrial autoantigen. Essentially mice immunized with either of two mimotopes, 2-octynoic acid-BSA (2-OA-BSA) or 2-octynoic acid-OVA (2-OA-OVA) develop high titer anti-mitochondrial antibodies (AMAs) and autoimmune cholangitis with lymphocytic infiltrates, portal inflammation, granuloma formation, and bile duct damage. In addition, if mice are stimulated with α-galactosylceramide (α-GalCer), which activates iNKT cells, the biliary damage is exacerbated and includes the appearance of fibrosis9,10,11,12. We report herein that administration of IFN-γ significantly promotes recruitment and lymphocyte activation in the earliest phases of autoimmune cholangitis but subsequently leads to downregulation of chronic inflammation through induction of the immunosuppressive molecule IL-30. In contrast, the administration of IL-4 does not alter the initiation of autoimmune cholangitis, but does contribute to the exacerbation of chronic liver inflammation and fibrosis. Better understanding of the mechanisms by which helper T cells function in the natural history of cholangitis is essential and illustrates that precision medicine, i.e. different therapies, may be needed for patients with PBC at various stages of their disease process.
Results
The cytokine profiles of 2-OA-OVA immunized model
To understand the roles of Th1/IFN-γ and Th17/IL-17 in the pathogenesis of autoimmune cholangitis PBC, we first determined basal expression levels of IFN-γ, IL-4, and IL-17 in 2-OA-OVA immunized mice. As shown in Fig. 1a, IFN-γ was readily detected in serum and peaked at approximately 4 weeks post 2-OA-OVA immunization, while neither IL-17 nor IL-4 was detectable. There were no detectable levels of IFN-γ, IL-4 and IL-17 at any time points in control mice immunized with adjuvant alone (data not shown). IFN-γ mRNA expression was significantly increased in the liver of mice immunized with 2-OA-OVA, while neither IL-17 nor IL-4 was increased (Fig. 1b). We then determined cytokine expression in hepatic lymphocytes by stimulating cells with phorbol-myristate acetate (PMA) and ionomycin; 2-OA-OVA immunized mice had a higher frequency of IFN-γ expressing lymphocytes compared to the control group immunized with adjuvant without 2-OA-OVA, an increased frequency of IL-17 expressing lymphocytes was also noted, but the frequency was low in both groups. The percentage of IL-4+ in hepatic lymphocytes of both groups was also low (Fig. 1c,d). Strikingly, there was a markedly elevated frequency of IFN-γ expressing CD4+ T cells (Th1 cells) in 2-OA-OVA immunized mice, but no differences in the percentage of IFN-γ expression in hepatic CD8+ T, NK (CD3− NK1.1+), and NKT (CD3+ NK1.1+) cells (Fig. 1e). These results suggested that the IFN-γ and Th1 mediated immune response was dominant in 2-OA-OVA immunized mice.
Figure 1. Cytokine profiles in serum and liver of mice immunized with 2-OA-OVA.

Mice were immunized with 2-OA-OVA as described in the Material and Methods. (a) Serum levels of IFN-γ, IL-4, and IL-17 were detected at different time points post 2-OA-OVA immunization by ELISA. (b–e) Mice were sacrificed at 5 weeks and liver specimens were prepared individually. (b) Liver IFN-γ, IL-4, and IL-17 expressions were measured by qRT-PCR analysis. Cytokine expressions were normalized to β-actin and relative expression levels were shown. (c,d) The expressions of IFN-γ, IL-4 and IL-17 in liver lymphocytes were measured by flowcytometry. (e) The percentages of IFN-γ+ in CD4+ T, CD8+ T, NK (CD3−NK1.1+), and NKT (CD3+NK1.1+) cells were detected by flowcytometry. Control indicates results from mice immunized with adjuvant while without 2-OA-OVA. n = 8–11 mice per group. **p < 0.01; ***p < 0.001.
Increased liver inflammation in AAV-IFN-γ treated 2-OA-OVA immunized mice
To investigate the mechanism of how IFN-γ affected autoimmune cholangitis, we administered AAV-IFN-γ before 2-OA-OVA immunization. First, we examined mice for serum IFN-γ following treatment with a single intravenous injection of AAV- IFN-γ or mock virus and then immunized with 2-OA-OVA. As shown in Fig. 2a, IFN-γ was highly expressed in the serum of AAV- IFN-γ injected mice at all time points but basal expression only levels of IFN-γ were noted in controls injected with either 2-OA-OVA and mock virus or normal saline (Fig. 2a). At 5 weeks post 2-OA-OVA immunization, there was a significant increase in the titers of anti-PDC-E2 in AAV-IFN-γ treated 2-OA-OVA immunized mice (Fig. 2b). Total liver lymphocytes were increased 2.5-fold in AAV-IFN-γ treated 2-OA-OVA immunized mice (Fig. 2c). Lymphocyte subsets including T, B, NK, and NKT cells were all significantly increased. There was approximately a two-fold increase of T, B and NKT cells, and a 10-fold increase of NK cells in mice treated with IFN-γ (Fig. 2d). There was a 1.3 fold increase in CD4+ T cells but a 3-fold increase of CD8+ T cells (Fig. 2e). Of note, the numbers of professional antigen presenting cells, including B cells, macrophages and dendritic cells, in the liver were all significantly increased in AAV-IFN-γ treated 2-OA-OVA immunized mice (Fig. 2d,f). The expression of MHC class II on macrophages and B cells were also significantly increased (Fig. 2g). The frequencies of activated CD8+ T cells and NK cells were increased (Fig. 2h). In addition, the frequencies of IFN-γ secreting CD4+ and CD8+ T cells were significantly increased in AAV-IFN-γ treated 2-OA-OVA immunized mice (Fig. 2i).
Figure 2. Increased AMA IgG and liver inflammation in AAV-IFN-γ treated 2-OA-OVA immunized mice.

Mice were injected with AAV-IFN-γ, AAV mock or normal saline three days before the first 2-OA-OVA immunization. (a) Serum levels of IFN-γ were detected by ELISA. (b–i) Mice were sacrificed at 5 weeks and sera and liver specimens were prepared individually. (b) Serum levels of anti-PDC-E2 IgM and IgG were measured by ELISA. IgM, 1:150 dilution. IgG 1:300 dilution. O.D., optical density. (c) Liver lymphocytes were counted. (d) The numbers of T, B, NK, and NKT cells in the liver were measured. (e) The numbers of CD4+ and CD8+ T cells in the liver were measured. (f) The numbers of macrophages and dendritic cells in the livers were measured. (g) IAb (MHC class II) expressions in macrophages, dendritic cells, and B cells were measured by flowcytometry. MFI, mean fluorescence intensity. (h) The percentages of CD69+ in CD8+ T cells and NK cells were measured. (i) The percentages of IFN-γ+ in CD4+ and CD8+ T cells were determined. n = 10–11 mice per group. *p < 0.05; **p < 0.01; ***p < 0.001.
At 10 weeks post 2-OA-OVA immunization, an increase in lymphocytic infiltrate in the AAV-IFN-γ treated 2-OA-OVA immunized mice could still be found. However, compared with a 2.5 fold increase at 5 weeks, there was only a 1.4 fold increase at 10 weeks (Fig. 3a). Threefold increase of NK cells and 1.5 fold increase of NKT cells were observed following IFN-γ administration. Neither T cells nor B cells were increased after 10 weeks of IFN-γ administration (Fig. 3b). However, the numbers of CD4+ memory T cells were decreased in the AAV-IFN-γ treated 2-OA-OVA immunized mice (Fig. 3c). As expected control mice immunized with 2-OA-OVA without α-GalCer as well as treated with AAV-mock and normal saline induced a slight increase of expression of collagen I and III, there were no changes in the expressions of collagen I and III in IFN-γ treatment (Fig. 4a). However, mice immunized with 2-OA-OVA and stimulated with α-GalCer had an approximate 6-fold expression of collagen I and III compared to naïve mice. 2-OA-OVA/α-GalCer immunized mice treated with AAV-IFN-γ had a lower expression of collagen I and III than controls (Fig. 4b). These results suggest that IFN-γ increased liver inflammation but subsequently led to reduced fibrosis.
Figure 3. Increased NK and NKT cells at 10 weeks of AAV-IFN-γ treated 2-OA-OVA immunized mice.

Mice were injected with AAV- IFN-γ, AAV mock or normal saline three days before the first 2-OA-OVA immunization and sacrificed at week 10. (a) Liver lymphocytes were counted. (b) The numbers of T, B, NK, and NKT cells in the liver were measured. (c) The numbers of memory T cells (CD3+ CD4+ CD44+ CD62L− or CD3+CD8+CD44+ CD62L−) were measured. n = 10–11 mice per group. *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 4. Decreased collagen levels in AAV-IFN-γ treated 2-OA-OVA immunized mice.

(a) Mice were injected with AAV- IFN-γ, AAV mock or normal saline three days before the first 2-OA-OVA immunization and sacrificed at week 10. The expressions of collagen I and collagen III mRNA in the liver were detected by qRT-PCR. (b) Mice were injected with AAV- IFN-γ, AAV mock or normal saline three days before the first 2-OA-OVA/α-GalCer immunization and sacrificed at week 10. The expressions of collagen I and collagen III mRNA in the liver were detected by qRT-PCR. n = 10–15 mice per group. *p < 0.05.
IL-30 expression in the liver correlated with IFN-γ expression in the liver
IFN-γ induces IL-30 expression in macrophages and dendritic cells13,14 and IL-30 inhibits IL-12-, IFN-γ-, and Con A- mediated hepatotoxicity by suppression of endogenous IFN-γ expression13. As shown in Fig. 5a, a 10-15-fold increase of IL-30 was found at 5 weeks post 2-OA-OVA immunization, while a 65-fold increase was noted in AAV-IFN-γ treated 2-OA-OVA immunization. However, there was only a 3-fold increase at 10 weeks post 2-OA-OVA immunization and a 26-fold increase in mice treated with AAV-IFN-γ. The expression level of the IL-27 subunit EBI3 was up-regulated in the liver with a similar but much lower pattern compared to p28 (IL-30) (Fig. 5a), suggesting that IL-30, not IL-27, was largely secreted following IFN-γ treatment. In addition, in 2-OA-OVA immunized mice, IL-30 expression in the liver significantly correlated with IFN-γ expression in the liver (r = 0.647, p < 0.01) (Fig. 5b). Taken together, IFN-γ induces IL-30 production which suppresses IFN-γ mediated inflammation.
Figure 5. Increased IL-30 (IL-27 p28) in the liver of AAV-IFN-γ treated 2-OA-OVA immunized mice.
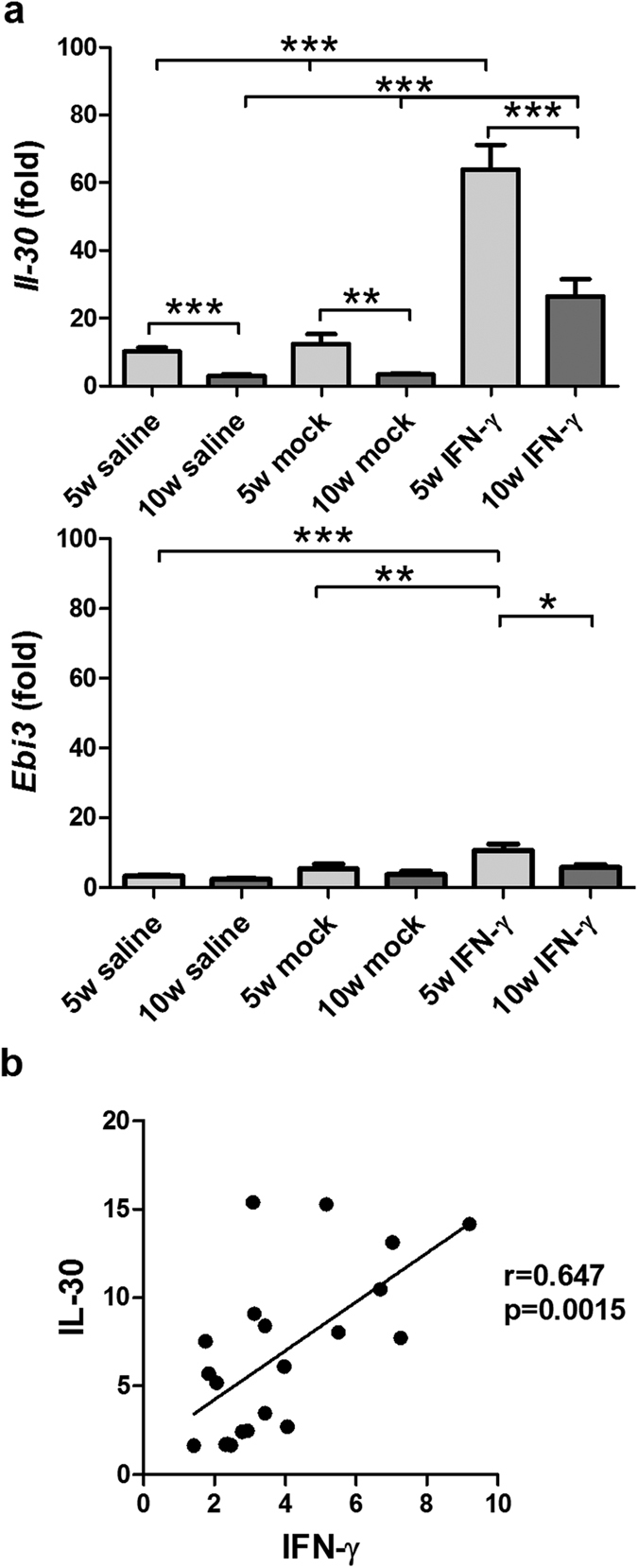
Mice were injected with AAV- IFN-γ, AAV mock or normal saline three days before the first 2-OA-OVA immunization and sacrificed at weeks 5 and 10. (a) The expressions of IL-30 and EBi3 in liver were measured by qRT-PCR. This data represents the fold change of data normalized to the naïve mice. n = 10–11 mice per group. **p < 0.01; ***p < 0.001. (b) The relationship between the liver expression of IFN-γ and IL-30 in the normal saline treated 2-OA-OVA immunized mice. Each dot represents an individual mouse.
Administration of IL-4 did not reduce Th1 mediated autoimmune cholangitis but markedly exacerbated liver inflammation and fibrosis
We then investigated whether the Th2 cytokine IL-4 could suppress Th1 IFN-γ mediated autoimmune cholangitis and fibrosis. We treated mice as above with AAV-IL-4 and then immunized with 2-OA-OVA/α-GalCer using the protocol outlined above. At 5 weeks post immunization, there were no increases in liver lymphocyte infiltration (Fig. 6a). However, T cells, including CD4+ and CD8+ T cells, and NK cells were significantly decreased in IL-4 administered 2-OA-OVA/α-GalCer mice. In contrast, B cells in IL-4 administered 2-OA-OVA/α-GalCer mice were significantly increased (Fig. 6b,c). Further, we noted that the numbers of macrophages and granulocytes in the liver of IL-4 administered 2-OA-OVA/α-GalCer immunized mice were greatly increased compared to either the AAV mock or the normal saline control groups (Fig. 6a). Moreover, mice injected with AAV-IL-4 developed a significant induction of fibrosis 10 weeks following immunization (Fig. 7a). Consistent with this data, there were higher expressions of collagen I and III in mice treated with AAV-IL-4 compared to the AAV-mock or normal saline controls (Fig. 7b). These results suggest that administration of IL-4 decreased T cell infiltration but increased macrophages and granulocytes and exacerbated fibrosis.
Figure 6. Increased macrophages and granulocytes but decreased T cells in AAV-IL-4 treated 2-OA-OVA/α-GalCer immunized mice.

Mice were injected with AAV-IL-4, AAV mock or normal saline three days before the first 2-OA-OVA/α-GalCer immunization and sacrificed at week 5. (a) The numbers of lymphocytes, macrophages and granulocytes in the liver were measured. (b) The numbers of T, B, NK, and NKT cells in the liver were measured. (c) The numbers of CD4+ and CD8+ T cells in the liver were measured. n = 5 mice per group. *p < 0.05; **p < 0.01.
Figure 7. Augmented collagen levels in AAV-IL-4 treated 2-OA-OVA/α-GalCer immunized mice.

Mice were injected with AAV-IL-4, AAV mock or normal saline three days before the first 2-OA-OVA/α-GalCer immunization and sacrificed at week 10. (a) Representative stained liver sections of Masson’s trichrome stain (x100 and magnification). The collagen fibers are stained blue. (b) The expressions of collagen I and collagen III mRNA in the liver were detected by qRT-PCR. n = 14–16 mice per group. ***p < 0.001.
To investigate whether administration of IL-4 suppressed Th1 cells in this model, we analyzed the Th1 cells in liver at 5 weeks post 2-OA-OVA/α-GalCer immunization. Interestingly, we found that there were no differences in the frequency of IFN-γ producing CD4+ T cells and the expression level of IFN-γ in CD4+ T cells among three groups of 2-OA-OVA/α-GalCer immunized mice. Surprisingly, the expression level of IFN-γ in CD8+ T cells was increased in IL-4 injected mice. Furthermore, the frequencies and expression level of IFN-γ in NK and NKT cells of IL-4 administered 2-OA-OVA/α-GalCer mice were significantly higher than that of controls (Fig. 8). Hence, administration of IL-4 activates CD8+ T, NK and NKT cells to secrete more IFN-γ and exacerbate cholangitis.
Figure 8. Increased IFN-γ production in CD8+ T, NK and NKT cells in AAV-IL-4 treated 2-OA-OVA/α -GalCer immunized mice.

Mice were injected with AAV-IL-4, AAV mock or normal saline three days before the first 2-OA-OVA/α-GalCer immunization and sacrificed at week 5. The expression of IFN-γ in liver CD4+ T, CD8+ T, NK and NKT cells was measured by flowcytometry. MFI indicates mean fluorescence intensity of positive cells. n = 5 mice per group. *p < 0.05; **p < 0.01.
Discussion
The cellular immune response is critical in the natural history of pathology of PBC3,4,5,15,16,17,18,19,20,21. CD4+ T cells include at least three distinct subsets based on their cytokine production profile and function: Th1, Th2, and Th17. Th1 cells, characterized by the secretion of IFN-γ in response to IL-12, are a major defense in the eradication of intracellular pathogens, while Th2 cells, characterized by production of IL-4, IL-5 and IL-13, are activators of B cells for IgE production, eosinophil recruitment and mucosal expulsion mechanisms. Th17 cells, which secrete IL-17, IL-21, and IL-22, mediate host defensive mechanisms to various infections, especially extracellular bacterial infections22. The pathogenesis of organ-specific autoimmune diseases including multiple sclerosis, rheumatoid arthritis, type I diabetes, and Hashimoto’s thyroiditis are thought to be orchestrated and augmented by Th1 and/or Th1723,24. In contrast Th2 cells may exert down regulation25,26,27,28,29.
Autoreactive CD4+ T cells have been linked to the pathogenesis of PBC. Th1 cells and their signature cytokine, IFN-γ, and Th17 cells and IL-17 are thought to be the main pathogenic mediators in PBC1,2,3,4,5,6,7,8. In the 2-OA-OVA mimicry model, the numbers and levels of Th1 cells and IFN-γ are significantly increased and appear to correlate with the onset of disease30. Overexpression of IFN-γ enhances liver inflammation by increasing immune cell infiltrates, upregulating MHC class II expression of antigen presenting cells, promoting anti-PDC-E2 production, and activating CD4+ and CD8+ T and NK cells in initiation of disease. However, IFN-γ also protects the portal inflammation through IFN-γ-IL-30 axis at the effector phase. In contrast, IL-17 is not considered critical in this model; there are low levels of IL-17 in serum and liver of 2-OA-OVA immunized mice.
IFN-γ expression is readily detected in the serum and liver of patients with PBC1,2,6. Genome-wide association studies reflect that Th1 related IL-12A and IL-12RB2 variants are strongly associated with PBC7,8 and previous studies have demonstrated that deletion of IFN-γ in 2-OA-BSA-immunized mice reduced inflammatory portal infiltrates associated with prevention of bile duct damage30. In the study herein, we demonstrated that IFN-γ was significantly increased and IFN-γ enhanced liver inflammation in 2-OA-OVA immunization. In addition, we note that IL-12p40−/− dnTGFβRII mice manifest a dramatic reduction in histological autoimmune cholangitis and significant decreases in levels of intrahepatic proinflammatory cytokines31. These results suggest that Th1 cells and IFN-γ are the dominant contributors in the initiation of the disease by increasing numerous immune cell infiltrates, upregulating MHC class II expression of antigen presenting cells, promoting anti-PDC-E2 antibodies production, and activating CD4+ and CD8+ T cells and NK cells. Indeed our data is consistent with recent data in another murine model that mice with a deletion of the IFN-γ 3’ untranslated region adenylate-uridylate–rich element have prolong and chronic expression of IFN-γ and develop a primary biliary cholangiopathy similar to PBC, which likewise proposes a key role of IFN-γ in disease initiation32,33.
IFN-γ is regarded traditionally as a critical proinflammatory mediator in several Th1-driven autoimmune disease model systems23. However, despite the expected overall proinflammatory and disease-enforcing role in some models, there is also evidence for protection against inflammation in other models34,35,36,37,38. The latter data is consistent our thesis that IFN-γ has a dual role in the natural history of an autoimmune disease, particularly a disease with a long natural history39,40. Our data also reveals a potential role of IL-30. IL-30, a subunit p28 of IL-27, has been detected in macrophages, dendritic cells, and hepatocytes13,41. Induction of high expression of IL-30 occurs with the coordination of activated T cells42. In liver injury, IL-30 suppresses the intrinsic ability of CD4+ T cells to produce IFN-γ in acute liver inflammation13,41 and IL-30 attenuates liver fibrosis by recruiting natural-killer–like T cells to the liver to remove activated hepatic stellate cells43. IL-30 has also been shown to inhibit central nervous system autoimmunity via antagonizing Th1 and Th17 responses in experimental autoimmune uveitis44,45. More studies are needed to understand how IL-30 inhibits autoimmune cholangitis but we submit that IFN-γ promotes disease by initially activating and recruiting immune cells and thence triggers robust immunosuppressive IL-30 production which reduces the liver inflammation.
Consistent with earlier data, mice immunized with 2-OA-OVA without α-GalCer did not induce collagen production and fibrosis while mice injected with 2-OA-OVA and α-GalCer manifest collagen production and fibrosis9,10. The administration of IFN-γ did not induce collagen production in 2-OA-OVA immunized mice. In fact, overexpression of IFN-γ decreases collagen production in 2-OA-OVA/α-GalCer immunized mice. α-GalCer is a well-defined potent and specific ligand for iNKT cell activation in both humans and mice. Upon ligation of iNKT cell receptors with α-GalCer, iNKT cells rapidly produce large amounts of cytokines, including IFN-γ and IL-411.
IL-4 is not normally considered to play a major role in autoimmune disease, i.e. experimental autoimmune encephalomyelitis (EAE), multiple sclerosis (MS), rheumatoid arthritis (RA), inflammatory bowel disease (IBD), or psoriasis46,47,48. However, systemic IL-4 immunotherapy has been shown to improve some Th1/Th17-associated autoimmune diseases25,26,27,28,29. In our study, IL-4 levels in 2-OA-OVA immunized autoimmune cholangitis remained at basal values. Nonetheless the administration of IL-4 increased liver inflammation with marked increases in macrophage, granulocyte and IFN-γ secreting CD8+ T, NK and NKT cell populations. Gene transfer of IL-4 in the liver of naïve mice induces hepatitis characterized by hepatocyte apoptosis and a massive macrophage infiltrate49,50 and IL-4 administration in primates is associated with hepatic damage51,52. Overall our data demonstrates Th1 cells and IFN-γ are the dominant contributors in the initiation phase of this model but clearly may have different effects as the disease evolves PBC. Delineating the varying activities of IFN-γ during the course of disease provides insight into the complex role of IFN-γ in autoimmunity and illustrates the complexities of treating a disease like PBC with biologics and the concept that one size may not fit all patients.
Methods
Experimental mice
Female C57BL/6 mice aged 7–9 weeks were obtained from the National Laboratory Animal Center, Taiwan and mice maintained in the Animal Center of the College of Medicine, National Taiwan University. This study was approved by the Institutional Animal Care and Use Committee (IACUC) of National Taiwan University College of Medicine and College of Public Health, and performed in accordance with the approved guidelines.
Preparation of AAV-IFN-γ and AAV-IL-4
cDNA containing murine IFN-γ or IL-4 were inserted into a recombinant adeno-associated viral vector (pAAV-IRES-GFP) (Cell Biolabs, San Diego, CA, USA). The cytokine genes were inserted using a pAAV-IRES-GFP plasmid which was co-transfected with pAAV-DJ and pHelper at a ratio of 1:1:1 into the adenovirus packaging AD293 cell line. Viruses were purified from infected cells 42–48 hours after infection by three freeze-thaw cycles followed by a Hi-Trap Heparin column. Viral titers (transduction unit, TU) were measured by GFP expression in infected 293T cells using flow cytometry. Throughout these studies a mock AAV was used as a control; it did not contain a transgene in the expression cassette. Details of the preparation of these viral vectors, including positive and negative controls, are described elsewhere12.
Experimental protocol
Female C57BL/6 mice, at 7–9 weeks of age, were intraperitoneally immunized with 2-OA-OVA in the presence of complete Freund’s adjuvant (CFA, Sigma-Aldrich, St. Louis, MO, USA) and subsequently boosted at weeks 2, 4, 6 and 8 with 2-OA-OVA in incomplete Freund’s adjuvant (IFA, Sigma-Aldrich). Mice were sacrificed at 5 weeks post 2-OA-OVA immunization for determining of IFN-γ, IL-4 and IL-17 in liver tissues by quantitative real-time RT-PCR analysis and cytokine expressing immune cells by flow cytometry. In a subsequent set of experiments, and based on our data on the influence of α-GalCer on fibrosis in this model9,10,11,12, two μg of α-GalCer (Funakoshi, Tokyo, Japan) were injected with the first and second 2-OA-OVA immunizations. AAV-IFN-γ and AAV-IL-4 were administered to mice at 3 days before the first mimeotope immunization. Mice were sacrificed at 5 or 10 weeks post-immunization for liver histopathology, definition of mononuclear cell phenotypes, assay of IFN-γ production, and titers of AMAs.
Isolation of mRNA and real-time PCR
Total RNA from liver specimens was obtained by the TRIzol method (Invitrogen Life Technologies, Carlsbad, CA, USA). The cDNA was generated by oligonucleotide priming using High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems, Foster City, CA, USA). Amplification was performed with SYBR Green MasterMix (Thermo Scientific, USA) using the 7500 Real-Time PCR System (Applied Biosystems). Results were analyzed by 2−ΔΔCt relative quantification method and normalized to β-actin.
Liver mononuclear cell quantitation and cytokine detection
Livers were perfused with PBS containing 0.2% BSA (PBS/0.2% BSA), passed through a 100 μm nylon mesh, and re-suspended in PBS/0.2% BSA. The parenchymal cells were removed as pellets after centrifugation at 50 g for 5 minute and the non-parenchymal cells isolated using 40% and 70% Percoll (GE HealthCare Biosciences, Quebec, Canada). After centrifugation, collected cells were washed with PBS/0.2% BSA and the viability confirmed by trypan blue dye exclusion. Subsets of liver mononuclear cells were measured by flow cytometry. Before staining cells, with a previously defined optimal dilution of monoclonal antibodies (Abs), the cells were pre-incubated with anti-CD16/32 (clone 93) to block non-specific FcRγ binding. The following mAbs were used in this study: anti-CD3, anti-CD4, anti-CD8a, anti-CD19, anti-NK1.1, anti-CD69, anti-CD44, anti-CD62L, anti-IAb, anti-CD11b, anti-CD11c, anti-F4/80, anti-Gr1, anti-Ly6G (Biolegend, San Diego, CA, USA). For cytokine detection, liver mononuclear cells were stimulated with phorbol-myristate acetate (PMA, 1 ng/ml, Sigma-Aldrich) and ionomycin (1 μg/ml, Sigma-Aldrich) for 4 or 18 hours and IFN-γ, IL-4, or IL-17 intracellular staining was performed after cell surface staining. Stained cells were measured with a flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star, Inc., Ashland, OR, USA). Optimal concentrations of the mAbs were used throughout and all assays included positive and negative controls.
Serum AMAs and cytokine detection
Serum titers of IgM and IgG anti-PDC-E2 autoantibodies were measured by ELISA using our well standardized recombinant mouse PDC-E2 as described previously10,11,12. Serum levels of IFN-γ, IL-4 and IL-17 were assayed by ELISA (R&D Systems, Minneapolis, MN, USA).
Histopathology
Portions of liver were excised and immediately fixed with 10% buffered formalin solution for 2 days at room temperature. Paraffin-embedded tissue sections were then cut into 4-μm slices for Masson’s trichrome staining.
Statistical analysis
Mann-Whitney U analysis was used to determine significant differences between groups. The Spearman test was used to evaluate correlations (Prism 5; Graph-Pad Software, La Jolla, CA, USA). Results are expressed as the mean ± standard error of the mean (SEM).
Additional Information
How to cite this article: Syu, B.-J. et al. Dual Roles of IFN-γ and IL-4 in the Natural History of Murine Autoimmune Cholangitis: IL-30 and Implications for Precision Medicine. Sci. Rep. 6, 34884; doi: 10.1038/srep34884 (2016).
Acknowledgments
We thank Yu-Ping Chiang at the National Taiwan University Hospital for providing technical advice and Patrick Leung at the University of California at Davis for providing reagents. This work was supported by grants from the Ministry of Science and Technology, Taiwan, MOST 104-2320-B-002-033-MY3 (YHC) and National Taiwan University, NTU-CDP-104R7880, 101R39012, 102R39012 (YHC) and in part by a grant from the National Institutes of Health, DK067003 (MEG).
Footnotes
Author Contributions Conceived and designed the experiments: B.-J.S., C.-E.L. and Y.-H.C. Performed the experiments: B.-J.S., C.-E.L., Y.-H.H. and Y.-H.C. Analyzed the data: B.-J.S., C.-E.L., M.E.G. and Y.-H.C. Wrote the paper: M.E.G. and Y.-H.C. All authors reviewed the manuscript.
References
- Harada K. et al. In situ nucleic acid hybridization of cytokines in primary biliary cirrhosis: predominance of the Th1 subset. Hepatology 25, 791–796 (1997). [DOI] [PubMed] [Google Scholar]
- Nagano T. et al. Cytokine profile in the liver of primary biliary cirrhosis. J Clin Immunol 19, 422–427 (1999). [DOI] [PubMed] [Google Scholar]
- Lan R. Y. et al. Hepatic IL-17 responses in human and murine primary biliary cirrhosis. Journal of autoimmunity 32, 43–51 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong G. et al. Imbalance between T helper type 17 and T regulatory cells in patients with primary biliary cirrhosis: the serum cytokine profile and peripheral cell population. Clin Exp Immunol 156, 217–225 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada K. et al. Periductal interleukin-17 production in association with biliary innate immunity contributes to the pathogenesis of cholangiopathy in primary biliary cirrhosis. Clin Exp Immunol 157, 261–270 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krams S. M., Cao S., Hayashi M., Villanueva J. C. & Martinez O. M. Elevations in IFN-gamma, IL-5, and IL-10 in patients with the autoimmune disease primary biliary cirrhosis: association with autoantibodies and soluble CD30. Clin Immunol Immunopathol 80, 311–320 (1996). [DOI] [PubMed] [Google Scholar]
- Hirschfield G. M. et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med 360, 2544–2555 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat Genet 42, 658–660 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. J. et al. Innate immunity and primary biliary cirrhosis: activated invariant natural killer T cells exacerbate murine autoimmune cholangitis and fibrosis. Hepatology 53, 915–925 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. H. et al. Innate immunity drives xenobiotic-induced murine autoimmune cholangitis. Clin Exp Immunol 177, 373–380 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. H. et al. Innate immunity drives the initiation of a murine model of primary biliary cirrhosis. PLoS One 10, e0121320 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh Y. H., Chang Y. N., Loh C. E., Gershwin M. E. & Chuang Y. H. AAV-IL-22 modifies liver chemokine activity and ameliorates portal inflammation in murine autoimmune cholangitis. Journal of autoimmunity 66, 89–97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibra D. et al. Interleukin-30: a novel antiinflammatory cytokine candidate for prevention and treatment of inflammatory cytokine-induced liver injury. Hepatology 55, 1204–1214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Guan X. & Ma X. Regulation of IL-27 p28 gene expression in macrophages through MyD88- and interferon-gamma-mediated pathways. The Journal of experimental medicine 204, 141–152 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda S. et al. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. The Journal of clinical investigation 102, 1831–1840 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita H. et al. Analysis of TCR antagonism and molecular mimicry of an HLA-A0201-restricted CTL epitope in primary biliary cirrhosis. Hepatology 36, 918–926 (2002). [DOI] [PubMed] [Google Scholar]
- Kita H. et al. Quantitation and phenotypic analysis of natural killer T cells in primary biliary cirrhosis using a human CD1d tetramer. Gastroenterology 123, 1031–1043 (2002). [DOI] [PubMed] [Google Scholar]
- Yang G. X. et al. CD8 T cells mediate direct biliary ductule damage in nonobese diabetic autoimmune biliary disease. Journal of immunology 186, 1259–1267 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhirapong A. et al. Therapeutic effect of cytotoxic T lymphocyte antigen 4/immunoglobulin on a murine model of primary biliary cirrhosis. Hepatology 57, 708–715 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda S. et al. Natural killer cells regulate T cell immune responses in primary biliary cirrhosis. Hepatology 62, 1817–1827 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W. et al. Murine autoimmune cholangitis requires two hits: cytotoxic KLRG1(+) CD8 effector cells and defective T regulatory cells. Journal of autoimmunity 50, 123–134 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Yamane H. & Paul W. E. Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 28, 445–489 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardalhon V., Korn T., Kuchroo V. K. & Anderson A. C. Role of Th1 and Th17 cells in organ-specific autoimmunity. Journal of autoimmunity 31, 252–256 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberal R. et al. Cutting edge issues in autoimmune hepatitis. Journal of autoimmunity (2016). [DOI] [PubMed] [Google Scholar]
- Racke M. K. et al. Cytokine-induced immune deviation as a therapy for inflammatory autoimmune disease. The Journal of experimental medicine 180, 1961–1966 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller R., Krahl T. & Sarvetnick N. Pancreatic expression of interleukin-4 abrogates insulitis and autoimmune diabetes in nonobese diabetic (NOD) mice. The Journal of experimental medicine 184, 1093–1099 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarner I. H. et al. Retroviral gene therapy of collagen-induced arthritis by local delivery of IL-4. Clin Immunol 105, 304–314 (2002). [DOI] [PubMed] [Google Scholar]
- Ghoreschi K. et al. Interleukin-4 therapy of psoriasis induces Th2 responses and improves human autoimmune disease. Nat Med 9, 40–46 (2003). [DOI] [PubMed] [Google Scholar]
- Guenova E. et al. IL-4 abrogates T(H)17 cell-mediated inflammation by selective silencing of IL-23 in antigen-presenting cells. Proceedings of the National Academy of Sciences of the United States of America 112, 2163–2168 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawata K. et al. Identification of potential cytokine pathways for therapeutic intervention in murine primary biliary cirrhosis. PLoS One 8, e74225 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K. et al. Deletion of interleukin-12p40 suppresses autoimmune cholangitis in dominant negative transforming growth factor beta receptor type II mice. Hepatology 50, 1494–1500 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae H. R. et al. Chronic Expression of Interferon Gamma Leads to Murine Autoimmune Cholangitis with a Female Predominance. Hepatology (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani D. & Mieli-Vergani G. Mouse model of primary biliary cholangitis with a striking female predominance: A new powerful research tool. Hepatology (2016). [DOI] [PubMed] [Google Scholar]
- Wang Z. et al. Role of IFN-gamma in induction of Foxp3 and conversion of CD4+ CD25− T cells to CD4+ Tregs. The Journal of clinical investigation 116, 2434–2441 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debray-Sachs M. et al. Prevention of diabetes in NOD mice treated with antibody to murine IFN gamma. Journal of autoimmunity 4, 237–248 (1991). [DOI] [PubMed] [Google Scholar]
- Jones L. S. et al. IFN-gamma-deficient mice develop experimental autoimmune uveitis in the context of a deviant effector response. Journal of immunology 158, 5997–6005 (1997). [PubMed] [Google Scholar]
- Kelchtermans H. et al. Protective role of IFN-gamma in collagen-induced arthritis conferred by inhibition of mycobacteria-induced granulocyte chemotactic protein-2 production. Journal of leukocyte biology 81, 1044–1053 (2007). [DOI] [PubMed] [Google Scholar]
- Hu X. & Ivashkiv L. B. Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity 31, 539–550 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelchtermans H., Billiau A. & Matthys P. How interferon-gamma keeps autoimmune diseases in check. Trends in immunology 29, 479–486 (2008). [DOI] [PubMed] [Google Scholar]
- Arellano G., Ottum P. A., Reyes L. I., Burgos P. I. & Naves R. Stage-Specific Role of Interferon-Gamma in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Front Immunol 6, 492 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S. et al. High susceptibility to liver injury in IL-27 p28 conditional knockout mice involves intrinsic interferon-gamma dysregulation of CD4+ T cells. Hepatology 57, 1620–1631 (2013). [DOI] [PubMed] [Google Scholar]
- Dibra D., Cutrera J. J. & Li S. Coordination between TLR9 signaling in macrophages and CD3 signaling in T cells induces robust expression of IL-30. Journal of immunology 188, 3709–3715 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A. et al. IL-30 (IL27p28) attenuates liver fibrosis through inducing NKG2D-rae1 interaction between NKT and activated hepatic stellate cells in mice. Hepatology 60, 2027–2039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumhofer J. S. et al. A role for IL-27p28 as an antagonist of gp130-mediated signaling. Nat Immunol 11, 1119–1126 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong W. P. et al. IL-27p28 inhibits central nervous system autoimmunity by concurrently antagonizing Th1 and Th17 responses. Journal of autoimmunity 50, 12–22 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland H. F. & Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol 8, 913–919 (2007). [DOI] [PubMed] [Google Scholar]
- Nair R. P. et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet 41, 199–204 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonel G. et al. Cutting edge: A critical functional role for IL-23 in psoriasis. Journal of immunology 185, 5688–5691 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- David A. et al. Adenovirus-mediated gene transfer in rat liver of interleukin 4 but not interleukin 10 produces severe acute hepatitis. Cytokine 9, 818–829 (1997). [DOI] [PubMed] [Google Scholar]
- Guillot C. et al. Lethal hepatitis after gene transfer of IL-4 in the liver is independent of immune responses and dependent on apoptosis of hepatocytes: a rodent model of IL-4-induced hepatitis. Journal of immunology 166, 5225–5235 (2001). [DOI] [PubMed] [Google Scholar]
- Prendiville J. et al. Recombinant human interleukin-4 (rhu IL-4) administered by the intravenous and subcutaneous routes in patients with advanced cancer–a phase I toxicity study and pharmacokinetic analysis. Eur J Cancer 29A, 1700–1707 (1993). [DOI] [PubMed] [Google Scholar]
- Leach M. W., Snyder E. A., Sinha D. P. & Rosenblum I. Y. Safety evaluation of recombinant human interleukin-4. I. Preclinical studies. Clin Immunol Immunopathol 83, 8–11 (1997). [DOI] [PubMed] [Google Scholar]