

Abstract
Background
The aim of this study was to evaluate the cardio-protective roles of glaucocalyxin A (GLA) in myocardial ischemia-reperfusion injury and to explore the underlying mechanism.
Material/Methods
Myocardial ischemia-reperfusion in wild-type C57BL/6J mice was induced by transient ligation of the left anterior descending artery. GLA or vehicle (solvent) was administrated intraperitoneally to the mice before reperfusion started. After 24 h of myocardial reperfusion, ischemic size was revealed by Evans blue/TTC staining. Cardiac function was evaluated by echocardiography and microvascular thrombosis was assessed by immunofluorescence staining of affected heart tissue. We also measured the phosphorylation of AKT, ERK, P-GSK-3β, and cleaved caspase 3 in the myocardium.
Results
Compared to the solvent-treated control group, GLA administration significantly reduced infarct size (GLA 13.85±2.08% vs. Control 18.95±0.97%, p<0.05) and improved left ventricular ejection fraction (LVEF) (GLA 53.13±1.11% vs. Control 49.99±1.25%, p<0.05) and left ventricular fractional shortening (LVFS) (28.34±0.71% vs. Control 25.11±0.74%, p<0.05) in mice subjected to myocardial ischemia-reperfusion. GLA also attenuated microvascular thrombosis (P<0.05) and increased the phosphorylation of pro-survival kinase AKT (P<0.05) and GSK-3β (P<0.05) in the myocardium upon reperfusion injury.
Conclusions
Administration of GLA before reperfusion ameliorates myocardial ischemia-reperfusion injury in mice. The cardio-protective roles of GLA may be mediated through the attenuation of microvascular thrombosis.
MeSH Keywords: Diterpenes, Kaurane; Myocardial Reperfusion Injury; Thrombosis
Background
Coronary heart disease is becoming a major public health burden of both developed and developing countries, with acute myocardial infarction (AMI) as the leading cause of premature mortality [1]. In most cases, AMI is initiated by the disruption of an unstable atherosclerotic plaque that leads to coronary thrombosis and occlusion of the artery [2]. Sustained loss of perfusion causes ischemic injury to the myocardium, leading to irreversible damage to the heart, including cell death and reconstitution of cardiomyocytes by fibrotic tissue [3]. Early reperfusion is currently the preferred therapy to salvage acutely ischemic myocardium and improve overall prognosis [4]. Paradoxically, additional injuries associated with coronary reperfusion, which may cause cardiogenic shock and arrhythmias, have been noticed since the beginning of the thrombolytic era [5]. For decades, a variety of methods and drugs have been investigated to seek a way to protect the heart from myocardial reperfusion injury (RI), although few of them are practical [6].
The major mechanisms involved in RI are oxidative stress, neutrophil-endothelium infiltration, and apoptosis. When the occluded artery is revascularized, abundant reactive oxygen species derived from endothelial cells during ischemia result in regional redox imbalance, giving rise to inflammation [7,8]. The resulting inflammatory cytokines promote the adherence of activating leukocytes to endothelium, thus inducing in situ thrombosis inside the lumen of microarteries and exacerbating the ischemic damage [9]. The priming step of thrombosis requires activation of platelets, which are recruited to the region, where prothrombogenic substances are exposed to circulating blood. At the site of endothelial damage, the binding of platelet GPVI receptor to collagen activates platelet-adhesion receptors, including integrins αIIbβ3 and α2β1, through phosphorylation of Fc receptor γ-chain and Src kinase/Syk/PLCγ2/PI3K/MAPK axis. Activated αIIbβ3 then binds to collagen, eliciting platelet activation and aggregation [10].
Due to its crucial role in the pathogenesis of microvascular lesions and thrombosis, platelets have been studied as major targets of interest for the management of myocardial RI. Of note, antagonism of platelet acting factor (PAF), a well-studied molecule in RI, has been reported to ameliorate RI through attenuation of oxidative stress and activation of platelets and neutrophils in animal models of myocardial RI [11]. Other clinically available agents inhibiting multiple steps of platelet activation, such as irreversible cyclooxygenase antagonists, adenosine diphosphate (ADP) receptor inhibitors, and glycoprotein IIb/IIIa inhibitors, showed only moderate therapeutic effects on RI, while their application results in a substantially increased risk of bleeding [12]. Although ischemia preconditioning has been experimentally shown to be capable of reducing RI, its clinical use remains elusive. Thus, the use of effective agents to attenuate myocardial RI is becoming a major challenge in combating coronary heart disease.
The plant Rabdosia japonica (Burm. f.) var. glaucocalyx (Maxim.) Hara was traditionally used for the management of gastrointestinal disease, inflammation, and tumors in Asian folk medicine. Initially isolated from this perennial herb in the early 1990s, a diterpenoid compound named glaucocalyxin A (GLA) was shown to have therapeutic effect in ischemic disease, leukemia, neurodegenerative disorders, and breast cancer [14]. We recently reported that GLA inhibits platelet activation and aggregation, with minimal bleeding risk [13]. Moreover, GLA was also shown to protect cardiomyocytes against oxidative stress and to ameliorate neuroinflammation [15,16]. Notwithstanding these pleiotropic effects of GLA, its potential therapeutic effect in myocardial RI has not previously been explored. In the current study, we tested the hypothesis that GLA attenuates myocardial RI after acute myocardial ischemia-reperfusion. Our results show, for the first time, that administration of GLA reduces infarct size and improves cardiac function in mice subjected to myocardial RI, and these beneficial effects are likely to be mediated through the suppression of microvascular thrombosis.
Material and Methods
Animals
Wild-type (WT) C57BL/6J mice were purchased from the Jackson Laboratory, Bar Harbor, MA. All animals used in this work were 10 to 12 weeks old. The mice were kept under normal laboratory conditions (20~26°C, 40~60% humidity, 12/12 h day/night light cycle) and given free access to food and water. Animal studies were carried out in compliance with Institutional Animal Care and Use Committee-approved protocols of Soochow University, and conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978).
Experiment protocols in vivo
Ten- to 12-week-old WT mice were weighed and anesthetized with 2% isoflurane via inhalation using a ventilation delivery system (Matrx, MIDMAEK), then placed in a supine position on a heating pad to maintain normal body temperature. Following left thoracotomy, the left anterior descending coronary artery (LAD) was ligated with a slipknot using 6-0 silk sutures transiently at 2 to 3 mm beneath the lower border of the left atria. The thorax was then closed with purse-string suture after manually squeezing the chest wall to prevent pneumothorax [17]. Transthoracic echocardiography was carried out immediately to verify successful ligation. After 1 h of prolonged ischemia, the slipknot was released to allow coronary reperfusion and the mice were observed for another 24 h before the infarct size was determined. Sham operation was performed similarly in spite of the ligation. A single dose of GLA (10 mg/kg) was administrated to mice via intraperitoneal injection before the reperfusion started, with the solvent-treated (10% DMSO in PBS) (DMSO, cat. no. V900090, Sigma, Shanghai, China) animals as the control group (Figure 1).
Figure 1.
Schematic study design of mice undergoing ischemia reperfusion. GLA or solvent (Control) was administrated intraperitoneally just before reperfusion started.
Extraction and isolation of GLA
Glaucocalyxin A (GLA) [7a,14b-dihydroxy-ent-kaur-16-en-3,15-dione], a natural ent-kaurane diterpenoid with a molecular formula of C20H28O4, was isolated from Rabdosia japonica, as previously described [13]. Briefly, stems and leaves of Rabdosia japonica (Burm. f.) var. glaucocalyx (Maxim.) Hara were collected in Tieli, Heilongjiang province in September 2009 and air-dried. The dried and powdered stems and leaves (900 g) were chopped and extracted with 80% EtOH (cat. no. 10009259, Sinoreagent, Shanghai, China) twice under reflux and concentrated under vacuum to yield an EtOH extract. The extract (74.0 g) was applied to column chromatography over a silica gel (200–300 mesh, 1200 g) column eluted with a system of petroleum ether: ethyl acetate (pet.et: EtOAc) (8:1; 4:1; 2:1; 1:1) to obtain fractions A–D. GLA (0.09 g) was obtained by recrystallization in EtOH from the fraction C (10.0 g).
Echocardiography
Cardiac function was evaluated by echocardiography after 24 h of reperfusion (n=8 for each group). Echocardiographic measurements were performed using the Vevo 2100 System (VisualSonics, Toronto, Canada) by an experienced technician who was blinded to the treatments. Two-dimensional and M mode views at the level of the papillary muscle were captured and analyzed. Interventricular septum thickness diameter (IVSTd), left ventricular internal dimension in diastole (LVDd), left ventricular posterior wall thickness diameter (LVPWd), and left ventricular internal dimension in systole (LVDs) were measured. Left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) were then calculated using Vevostrain software.
Myocardial infarct size
After 24 h of reperfusion, mice underwent thoracotomy and the untied knot was retied followed by intravenous administration of 5% Evans blue dye (cat. no. E2129, Sigma, Shanghai, China) (Control group: n=9, GLA group: n=12). Ten minutes later, mice were sacrificed and the hearts were excised. The left ventricle was weighed before being frozen for another 10 min at −80°C. After that, the left ventricle was sectioned transversely into 2-mm slices and incubated in 1.0% 2,3,5-triphenyltetrazolium chloride (TTC; cat. no. T8877, Sigma, Shanghai, China) dissolved in PBS (pH 7.4 at 37°C) for 15 min. Each section was then weighed and immersed in 4% formaldehyde (cat. no. 80096675, Sinoreagent, Shanghai, China) for 24 h before being photographed with a high-resolution cooled CCD camera (cat. no. SZX16, Olympus, Japan). Pictures were analyzed using ImageJ software (NIH, Bethesda, MD), and the area at risk (AAR) and infarct area (IA) were determined. The infarct size was calculated as the ratio of IA/AAR weighted by the mass of each section and the ischemic size was defined as the ratio of AAR/LV (weight of left ventricle) [18].
Western blot
By the end of each distinct protocol, mice were sacrificed and hearts harvested through excision (Control group: n=4, GLA group: n=6). The left ventricle was cut and homogenized using a Bio-Gen PRO200 blade rotor homogenizer (cat. no. 01-01200, Oxford, CT), then lysed with radio-immunoprecipitation assay cell lysis buffer (cat. no. R0010-100, Solarbio, Beijing, China) containing 1% phenylmethylsulfonyl fluoride (PMSF, cat. no. ST506, Beyotime, Shanghai, China), proteinase inhibitors (cat. no. ab65621, Abcam, Cambridge, UK) and phosphatase inhibitors (cat. no. ab201112, Abcam, Cambridge, UK). The lysate was cleared by centrifugation at 4°C and 14000× rpm for 10 min to collect the supernatants. The protein concentration was measured using a BCA protein assay kit (cat. no. P0012, Beyotime, Shanghai, China). A total of 20 μg of homogenate sample protein from each subject was separated on SDS/PAGE gel, then transferred to a nitrocellulose membrane and blocked in 5% skim milk for 1 h before being probed with the following primary antibodies: mouse-anti-phospho-Akt (1:1000, Cat. no. 4051, CST, Danvers, MA), mouse-anti-Akt (1:1000, cat. no. 9272, CST, Danvers, MA), mouse-anti-phospho-ERK1/2 (1:1000, cat. no. 4376, CST, Danvers, MA) and mouse-anti-ERK1/2 (1:1000, cat. no. 9102, CST, Danvers, MA). After washing 3 times, membranes were incubated with Goat-anti-Rabbit IRDye 800CW (1: 10000, cat. no. 926-32211, Lincoln, NE) secondary antibody for 1 h, washed with PBS-T 3 times, and scanned using an Odyssey reader (Licor, Lincoln, NE) at 800 nm. Densitometry analysis using ImageJ software (NIH, Bethesda, MD) was performed to quantify the protein expression [19]. Data were displayed as normalized-fold changes over controls.
Immunohistochemistry
After dissection, the left ventricle was put in a tissue block, immersed in cryo-embedding media (OCT, cat. no. 4583, Sakura Finetek, Torrance, CA), frozen in liquid nitrogen, and stored at −80° (n=5 for each group). One day before the sectioning, the frozen tissue block was transferred to another freezer at −20°C. The frozen tissue block was sectioned into 5-μm-thick slides using a cryostat cryotome, and placed onto glass slides for drying overnight. Next, the slides were fixed in 4% paraformaldehyde for 10 min, followed by permeabilization with 0.2% Triton-X100 (Cat. no. T8200, Solarbio, Beijing, China), and were incubated in 0.3% H2O2 (cat. no. 10011208, Sinoreagent, Shanghai, China) at room temperature for 10 min. Then the slides were washed with PBS and blocked in 1% BSA (cat. no. BS043F, Biosharp, Hefei, China) for 1 h, washed with PBS, and incubated in rat-anti-mouse CD41 antibody (1:100, cat. no. 553847, BD, Franklin Lakes, NJ) at 4°C overnight. After that, the slides were washed and incubated in 647 donkey-F(ab)2-anti-rat IgG (H+L) (1:200, cat. no. ab150151, Abcam, Cambridge, UK) for 1 h at room temperature, covered with VECTASHIELD Hard Set Mounting Medium with DAPI (cat. no. C1002, Beyotime, Shanghai, China), and observed under an IX-81 laser confocal microscope (Olympus, Japan). Pictures were taken at 200× magnification and 5 views were averaged for each sample. Microvascular thrombosis was quantified as the average fluorescence intensity of CD41 per high-power view [20]. Data were expressed as normalized-fold changes over controls.
Statistical analysis
All data are shown as Mean ±SEM (standard error of mean), and statistical analyses were performed using GraphPad Prism software (GraphPad Software, LA Jolla, CA). Data included in the statistics satisfied criteria for normal distribution and homogeneity of variance as determined by Kolmogorov-Smirnov test and F test, respectively. For continuous data, the 2-tailed t test was used to compare the difference between 2 groups. Differences among multiple groups (more than 2) were compared using one-way analysis of variance (ANOVA) with Bonferroni’s post hoc test for inter-group comparisons. P<0.05 was considered statistical significance for all tests.
Results
Administration of GLA reduces infarct size in mice subjected to myocardial I/R
To evaluate the effect of GLA on infarct size in myocardial RI, mice were given an intraperitoneal injection of GLA before reperfusion after 1 h of LAD occlusion. After 24 h of reperfusion, mice treated with GLA showed significantly decreased infarct sizes (IA/AAR) compared to those treated with vehicle (GLA 13.85±2.08% vs. Control 18.95±0.97%, p<0.05, Figure 2A, 2B), while GLA- and vehicle-treated mice had similar ischemic areas (AAR/LV) (Figure 2A, 2C).
Figure 2.

GLA reduces infarct size in mice subjected to myocardial RI. Male wild-type C57BL/6J mice were administrated solvent (Control) or GLA (10 mg/kg) intraperitoneally before reperfusion. (A) Representative photographs of heart sections double-stained with Evans blue and TTC from solvent- or GLA-treated mice. (B) Quantification of infarct size (IA/AAR) and (C) ischemic size (AAR/LV). N.S. – no significance. * P<0.05 v.s control group (n=9–12 per group).
GLA improves cardiac function in mouse MI/R
Mice subjected to sham surgery underwent transthoracic echocardiography to yield baseline parameters of cardiac function at the end of the observation period. As expected, two-dimensional and M mode echocardiography showed a decreased LVEF (Control 49.99±1.25% vs. Sham 75.39±1.21%, p<0.05) and a reduced LVFS (Control 25.11±0.74% vs. Sham 35.36±0.74%, p<0.05) in the control group compared to the mice in the sham surgery group at 24 h after myocardial I/R. Importantly, intraperitoneal injection of GLA prior to myocardial reperfusion significantly improved LVEF (GLA 53.13±1.11% vs. Control 49.99±1.25%, p<0.05) and LVFS (GLA 28.34±0.71% vs. Control 25.11±0.74%, p<0.05) in myocardial I/R-stressed mice (Figure 3A–3C).
Figure 3.

GLA improves cardiac function as revealed by echocardiography after myocardial I/R. Male wild-type C57BL/6J mice were intraperitoneally administrated solvent (Control) or GLA before myocardial reperfusion. (A) Characteristic echocardiograms of the mice in the sham, control, and GLA groups are displayed. (B) Left ventricular ejection fraction (LVEF), and (C) left ventricular fractional shortening (LVFS) for each group were calculated. * P<0.05 vs. sham group, ** P<0.05 vs. control group (n=8 per group).
GLA enhances the phosphorylation of Akt and GSK-3β but not ERK1/2
To explore the molecular mechanism underlying the cardio-protective role of GLA, we measured the activation of the reperfusion injury salvage kinase (RISK) pathway. Interestingly, phosphorylation of the pro-survival kinase Akt (Figure 4A, 4B) and GSK-3β (Figure 4E, 4F), but not ERK1/2 (Figure 4C, 4D), was significantly increased by GLA in mice subjected to cardiac I/R (P<0.05). However, the cardiac expression of cleaved caspase 3, an established marker of apoptosis, was not affected by GLA treatment following myocardial I/R (Figure 4G, 4H).
Figure 4.

GLA promotes Akt phosphorylation but not ERK1/2 phosphorylation without affecting apoptosis in mouse heart after I/R. (A) Representative Western blots of phosphorylated/total Akt, (C) phosphorylated/total ERK1/2, (E) GSK-3β, and (G) cleaved caspase 3 are shown. (B) Phosphorylation of AKT, (D) ERK1/2, (F) GSK-3β, and (H) cleavage of caspase 3 were quantified by densitometry analysis. * P<0.05 vs. control. (n=4–6 per group), N.S. – no significance.
GLA attenuates microvascular thrombosis after cardiac I/R
According to our previous findings, GLA is able to inhibit the GPVI pathway of platelet activation, and reduces arterial thrombosis in vivo [13]. Therefore, we speculated that GLA, due to its antiplatelet potential, may also reduce microvascular thrombosis, which is an active mediator of myocardial RI. To further delineate how GLA confers protection against cardiac RI, we performed immunohistochemistry studies to quantify microvascular thrombosis in the heart. Our results show that GLA administration before reperfusion significantly decreased the degree of microvascular thrombosis, indicated by CD41 staining in the mouse hearts after myocardial I/R (P<0.05) (Figure 5A, 5B).
Figure 5.
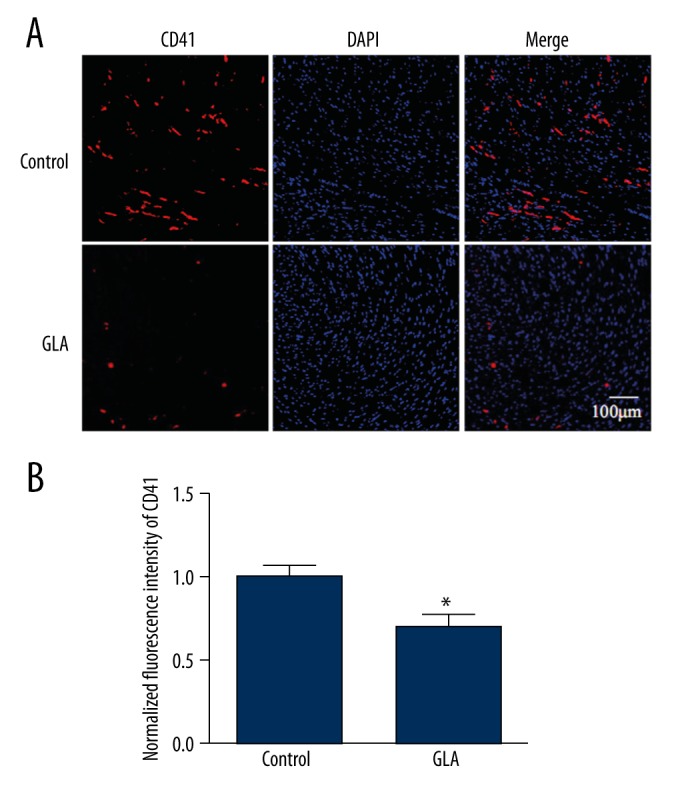
(A, B) GLA attenuates microvascular thrombosis in mouse hearts after I/R. Sections of hearts were stained with CD41 (red) and DAPI (blue). The burden of thrombosis was calculated according to the average fluorescence intensity of CD41 per high-power field. * P<0.05 vs. control group. (n=5 per group).
Discussion
Glaucocalyxin A is a novel diterpenoid isolated from a plant previously used as traditional medicine for gastrointestinal and malignant disorders in Asia. The pharmacological mechanism of GLA has been studied mostly in platelets and cancer cell lines, uncovering its role in regulating platelet activation, cell death, redox signaling, and inflammation. Previous evidence shows that GLA inhibits platelet activation and thrombosis via the GPVI-dependent pathway [13,14]. Nevertheless, the role of GLA in cardiac protection remains largely unknown. A recent report that GLA confers protection in ischemic brain injury led us to explore its putative therapeutic role in cardiac RI [21]. As we expected, amelioration of myocardial RI was achieved by administration of GLA in mice subjected to myocardial I/R, indicated by improved cardiac function and reduced infarct size. Further mechanistic studies revealed that GLA treatment attenuated microvascular thrombosis and activated a pro-survival pathway in mouse hearts subjected to myocardial I/R.
Myocardial reperfusion has been used as the most effective approach for myocardial infarction for the last 3 decades. Paradoxically, inevitable cardiac damages secondary to reperfusion substantially compromise the benefits of percutaneous coronary intervention therapy. Thus, developing optimal therapeutic options for myocardial reperfusion injury remains the most challenging area in invasive cardiology. Extensive studies on reperfusion injury have shed light on its underlying mechanism, although few approaches have been determined to be effective in RI protection [22,23]. Microvascular thrombosis due to aggregation of activated platelets spectacularly exacerbates ischemic and hypoxic damage of the heart upon myocardial I/R. In an early study, the pernicious role of platelets during RI was shown by improved cardiac function in a rabbit I/R model with physical platelet depletion [24]. Further investigation demonstrated that platelet activating factor (PAF) could promote myocardial infarction upon myocardial IR, while inhibition of PAF has emerged as a potent therapy for RI. Accordingly, pretreatment with PAF antagonists before cardiac reperfusion preserves platelet counts, reduces infarct size, and prevents arrhythmias and hypotension. Moreover, the accumulation of activated platelets was also suppressed by PAF inhibition in the ischemic area [25,26]. GPIIb/IIIa, an important receptor responsible for platelet activation, was reported to be a potent target for reducing myocardial RI [27]. Direct inhibition of p-selectin was also shown to compromise platelet function, reduce thrombus formation, and rescue the myocardium from RI [28]. Similar improvements were observed in I/R-stressed ex vivo rodent hearts due to a P2Y12 receptor antagonist that disrupts platelet function [29]. These findings suggest that inhibition of platelet activation provides an effective way to reduce cardiac RI. Accordingly, our previous finding that GLA suppresses platelet activation through GPVI-mediated signals suggests its potential role in protection against myocardial RI. Consistent with this, we confirmed the attenuation of microvascular thrombosis in mouse hearts by GLA, which may be mediated by its anti-platelet activity. Decreased thrombosis by GLA in the myocardium may contribute to its cardio-protection against RI.
Inflammatory response, featuring activation of neutrophils that promote platelet adhesion to endothelium, has been associated with myocardial RI. Previous reports state that GLA protects against neurodegeneration through inhibition of the inflammatory pathway [15]. However, whether these beneficial effects are involved in myocardial protection against RI requires further investigation. Given that the improvement of microcirculation by GLA alone may reduce hypoxia and alleviate inflammation, a platelet-free ex vivo perfused heart model would further facilitate the clarification of the potential anti-inflammatory role of GLA in cardiomyocytes. Considering its great similarity to the human heart, the porcine heart may provide an ideal model for future mechanistic studies of GLA in myocardial RI [30].
Another important aspect of RI is the disturbance of cellular calcium handling. Upon cardiac I/R, the activity of sarcoplasmic reticulum Ca2+-ATPase (SERCA) is suppressed, which subsequently leads to insufficient transport of Ca2+ to ER from cytosol. The resulting hyper-physiological calcium level in the cytoplasmic compartment is called calcium overload, a pathological status that may lead to cell death. Furthermore, excessive calcium accumulation in cardiomyocytes promotes cardiac hyper-contracture, depletion of energy reservoir, and ER stress-mediated cell apoptosis [31]. Interestingly, GLA has been demonstrated to maintain cytoplasmic calcium levels in lymphocytes [14]. These findings suggest that GLA may counteract RI through inhibition of apoptosis, but GLA is known to induce apoptosis in cancer cell lines [32–34]. Our finding that GLA treatment did not affect the expression of cleaved caspase 3 in mouse hearts subjected to myocardial I/R may be because we used a drug concentration that differed from that used in other studies. In addition, in vivo vs. in vitro administration of GLA may lead to different pleiotropic effects, which should also be considered when interpreting the present findings.
Activation of pro-survival protein kinases has been found to participate in myocardial I/R and to be protective against RI. The canonical signaling pathways involve PI3K/Akt-ERK1/2 and JAK2/STAT3. Activation of either pathway will inhibit the opening of mPTP, and alleviate mitochondria-mediated energy depletion and apoptosis in RI [31]. Inhibition of GSK-3β by phosphorylation at Ser-9 was shown to ameliorate cardiac ischemic-reperfusion injury via inhibiting mTOR-mediated autophagy [35]. Taken together, our results show that treatment with GLA upregulated AKT and GSK-3β phosphorylation in the myocardium, while ERK1/2 phosphorylation was not significantly changed. Nonetheless, Xiao et al. demonstrated that GLA suppressed AKT phosphorylation and promoted apoptosis in a malignant glioma cell line-U87MG in vitro. This differential regulation of AKT by GLA is likely mediated through cellular specificity. Future research using cultured cardiomyocytes is needed to elucidate the modulation of protein kinase signaling by GLA.
Generation of ROS in I/R is linked to oxidative stress-mediated RI [36]. Treatments for RI decrease oxidative stress, as revealed by a lower malondialdehyde (MDA) level. Considering the improvement of microcirculation by GLA, it is speculated to mitigate oxidative damage in cardiac RI through attenuation of ischemia. Of note, GLA protects H9c2 cardiomyoblast cells from oxidative stress induced by H2O2 [16]. Intriguingly, GLA exhibits pleiotropic effects on redox signal among different cell types. For instance, GLA induces oxidative stress in leukemia cells [32,37], despite its protective role in cardiac RI. However, only a higher concentration of GLA induces ROS in plant cells, while a lower concentration, similar to that used in our study, displays a pro-survival role [37]. Whether and how GLA modulates the redox signal in native cardiomyocytes requires further investigation.
Recently, the cardiac RI process was found to have 2 phases – the first phase is from 2 through 20 min and the second from 30 through 120 min after reperfusion starts. Targeting the later phase is more effective in reducing RI [38]. In our study, a single dose of GLA was given to the mice just before reperfusion. Of note, a recent pharmacokinetic study showed a very short half-life of GLA following intravenous injection in rats. In this model, the elimination of GLA follows a two-compartment open model, with a t(1/2 alpha) of 4.327 min and t(1/2 beta) of 28.56 min, respectively [39]. Considering the relatively slower drug kinetics following intraperitoneal compared to intravenous administration, GLA is likely to regulate both early and later phases of cardiac RI in our model. However, no study so far has addressed the pharmacokinetics and pharmacodynamics features of GLA after intraperitoneal injection in rodents. The temporal role of GLA during early and later phase of cardiac RI remains to be discovered. Additional pharmacological studies will help answer these questions.
Conclusions
Our findings suggest that GLA improves cardiac function, decreases infarcted myocardium, and activates pro-survival signals in mouse hearts subjected to myocardial ischemia-reperfusion injury. These cardioprotective roles of GLA may be mediated by the reduction of microvascular thrombosis due to its anti-platelet effect. With low bleeding risk, GLA may be a promising therapeutic option for ameliorating myocardial RI during cardiac revascularization. Nevertheless, the complexity of signaling crosstalk underlying the cardio-protective effect of GLA against RI remains to be elucidated, and future studies are warranted to delineate the specific cellular and temporal roles of GLA.
Acknowledgments
We thank Dr. Chaojun Tang (Soochow University, China) for assisting in preparation of this manuscript.
Footnotes
Source of support: Natural Science Foundation of China [grants 81370373, 81170132, and 91439112 to LZ] and the Priority Academic Development Program of Jiangsu Higher Education Institutions (PAPD) of China [to LZ] and Specialized Research Fund for the Doctoral Program of Higher Education [20113201110012 to LZ]
Conflict of interest
The authors declare that they have no conflicts of interest related to this work.
References
- 1.Ambrose JA, Singh M. Pathophysiology of coronary artery disease leading to acute coronary syndromes. F1000Prime Rep. 2015;7:08. doi: 10.12703/P7-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsujita K, Kaikita K, Soejima H, et al. Acute coronary syndrome-initiating factors. Nihon Rinsho. 2010;68(4):607–14. [in Japanese] [PubMed] [Google Scholar]
- 3.Xu L, Yates CC, Lockyer P, et al. MMI-0100 inhibits cardiac fibrosis in myocardial infarction by direct actions on cardiomyocytes and fibroblasts via MK2 inhibition. J Mol Cell Cardiol. 2014;77:86–101. doi: 10.1016/j.yjmcc.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gottlieb S, Boyko V, Harpaz D, et al. Long-term (three-year) prognosis of patients treated with reperfusion or conservatively after acute myocardial infarction. Israeli Thrombolytic Survey Group. J Am Coll Cardiol. 1999;34(1):70–82. doi: 10.1016/s0735-1097(99)00152-7. [DOI] [PubMed] [Google Scholar]
- 5.Hausenloy DJ, Yellon DM. Time to take myocardial reperfusion injury seriously. N Engl J Med. 2008;359(5):518–20. doi: 10.1056/NEJMe0803746. [DOI] [PubMed] [Google Scholar]
- 6.Lonborg JT. Targeting reperfusion injury in the era of primary percutaneous coronary intervention: Hope or hype? Heart. 2015;101(20):1612–18. doi: 10.1136/heartjnl-2015-307804. [DOI] [PubMed] [Google Scholar]
- 7.Sanada S, Komuro I, Kitakaze M. Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am J Physiol Heart Circ Physiol. 2011;301(5):H1723–41. doi: 10.1152/ajpheart.00553.2011. [DOI] [PubMed] [Google Scholar]
- 8.Liao YH, Xia N, Zhou SF, et al. Interleukin-17A contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and neutrophil infiltration. J Am Coll Cardiol. 2012;59(4):420–29. doi: 10.1016/j.jacc.2011.10.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verrier E. The microvascular cell and ischemia-reperfusion injury. J Cardiovasc Pharmacol. 1996;27(Suppl 1):S26–30. doi: 10.1097/00005344-199600001-00007. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Andrews M, Yang Y, et al. Platelets in thrombosis and hemostasis: Old topic with new mechanisms. Cardiovasc Hematol Disord Drug Targets. 2012;12(2):126–32. doi: 10.2174/1871529x11202020126. [DOI] [PubMed] [Google Scholar]
- 11.Morgan EN, Boyle EM, Jr, Yun W, et al. Platelet-activating factor acetylhydrolase prevents myocardial ischemia-reperfusion injury. Circulation. 1999;100(19 Suppl):II365–68. doi: 10.1161/01.cir.100.suppl_2.ii-365. [DOI] [PubMed] [Google Scholar]
- 12.Moeckel D, Jeong SS, Sun X, et al. Optimizing human apyrase to treat arterial thrombosis and limit reperfusion injury without increasing bleeding risk. Sci Transl Med. 2014;6(248):248ra105. doi: 10.1126/scitranslmed.3009246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li W, Tang X, Yi W, et al. Glaucocalyxin A inhibits platelet activation and thrombus formation preferentially via GPVI signaling pathway. PLoS One. 2013;8(12):e85120. doi: 10.1371/journal.pone.0085120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiang Z, Wu X, Liu X, Jin Y. Glaucocalyxin A: A review. Nat Prod Res. 2014;28(24):2221–36. doi: 10.1080/14786419.2014.934235. [DOI] [PubMed] [Google Scholar]
- 15.Kim BW, Koppula S, Hong SS, et al. Regulation of microglia activity by glaucocalyxin-A: attenuation of lipopolysaccharide-stimulated neuroinflammation through NF-kappaB and p38 MAPK signaling pathways. PLoS One. 2013;8(2):e55792. doi: 10.1371/journal.pone.0055792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu MJ, Sun Q, Yu LJ, et al. Protective effect of glaucocalyxin A inhibits H2O2-induced H9c2 cardiomyocytes injury. Lat Am J Pharm. 2014;33(7):1216–20. [Google Scholar]
- 17.Gao E, Lei YH, Shang X, et al. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res. 2010;107(12):1445–53. doi: 10.1161/CIRCRESAHA.110.223925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suematsu Y, Miura S, Takata K, et al. A novel inducible cholesterol efflux peptide, FAMP, protects against myocardial ischemia reperfusion injury through a nitric oxide pathway. Int J Cardiol. 2016;202:810–16. doi: 10.1016/j.ijcard.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 19.Kim BJ, Hur JW, Park JS, et al. Expression of matrix metalloproteinase-2 and -9 in human ligamentum flavum cells treated with tumor necrosis factor-alpha and interleukin-1beta. J Neurosurg Spine. 2016;24(3):428–35. doi: 10.3171/2015.6.SPINE141271. [DOI] [PubMed] [Google Scholar]
- 20.Galindo M, Gonzalo E, Martinez-Vidal MP, et al. Immunohistochemical detection of intravascular platelet microthrombi in patients with lupus nephritis and anti-phospholipid antibodies. Rheumatology (Oxford) 2009;48(8):1003–7. doi: 10.1093/rheumatology/kep152. [DOI] [PubMed] [Google Scholar]
- 21.Zhu L, Tang X, Li Q, et al. The role of glaucocalyxin A in inhibiting platelet activation and protecting against hypoxic ischemic brain injury. Journal of Thrombosis and Haemostasis. 2013;11:445–45. [Google Scholar]
- 22.Sharma V, Bell Rm, Yellon DM. Targeting reperfusion injury in acute myocardial infarction: a review of reperfusion injury pharmacotherapy. Expert Opin Pharmacother. 2012;13(8):1153–75. doi: 10.1517/14656566.2012.685163. [DOI] [PubMed] [Google Scholar]
- 23.Michel SG, La Muraglia GM, II, Madariaga LM, et al. Twelve-hour hypothermic machine perfusion for donor heart preservation leads to improved ultrastructural characteristics compared to conventional cold storage. Ann Transplant. 2015;20:461–68. doi: 10.12659/AOT.893784. [DOI] [PubMed] [Google Scholar]
- 24.Kutsumi Y, Misawa T, Tada H, et al. Effect of a leukocyte-platelet removal filter on ischemia induced reperfusion injury. ASAIO Trans. 1990;36(3):M723–26. [PubMed] [Google Scholar]
- 25.Montrucchio G, Alloatti G, Mariano F, et al. Role of platelet-activating factor in the reperfusion injury of rabbit ischemic heart. Am J Pathol. 1990;137(1):71–83. [PMC free article] [PubMed] [Google Scholar]
- 26.Qayumi AK, English JC, Godin DV, et al. The role of platelet-activating factor in regional myocardial ischemia-reperfusion injury. Ann Thorac Surg. 1998;65(6):1690–97. doi: 10.1016/s0003-4975(98)00275-6. [DOI] [PubMed] [Google Scholar]
- 27.Chang ST, Chung CM, Chu CM, et al. Platelet glycoprotein IIb/IIIa inhibitor tirofiban ameliorates cardiac reperfusion injury. Int Heart J. 2015;56(3):335–40. doi: 10.1536/ihj.14-322. [DOI] [PubMed] [Google Scholar]
- 28.Oostingh GJ, Pozgajova M, Ludwig RJ, et al. Diminished thrombus formation and alleviation of myocardial infarction and reperfusion injury through antibody- or small-molecule-mediated inhibition of selectin-dependent platelet functions. Haematologica. 2007;92(4):502–12. doi: 10.3324/haematol.10741. [DOI] [PubMed] [Google Scholar]
- 29.Barrabes JA, Inserte J, Mirabet M, et al. Antagonism of P2Y12 or GPIIb/IIIa receptors reduces platelet-mediated myocardial injury after ischaemia and reperfusion in isolated rat hearts. Thromb Haemost. 2010;104(1):128–35. doi: 10.1160/TH09-07-0440. [DOI] [PubMed] [Google Scholar]
- 30.Weymann A, Sabashnikov A, Patil NP, et al. Eprosartan improves cardiac function in swine working heart model of ischemia-reperfusion injury. Med Sci Monit Basic Res. 2014;20:55–62. doi: 10.12659/MSMBR.890444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang M, Sun GB, Zhang JY, et al. Elatoside C protects the heart from ischaemia/reperfusion injury through the modulation of oxidative stress and intracellular Ca(2)(+) homeostasis. Int J Cardiol. 2015;185:167–76. doi: 10.1016/j.ijcard.2015.03.140. [DOI] [PubMed] [Google Scholar]
- 32.Yang WH, Zhang Z, Sima YH, et al. Glaucocalyxin A and B-induced cell death is related to GSH perturbation in human leukemia HL-60 Cells. Anticancer Agents Med Chem. 2013;13(8):1280–90. doi: 10.2174/18715206113139990200. [DOI] [PubMed] [Google Scholar]
- 33.Xiao X, Cao W, Jiang X, et al. Glaucocalyxin A, a negative Akt regulator, specifically induces apoptosis in human brain glioblastoma U87MG cells. Acta Biochim Biophys Sin (Shanghai) 2013;45(11):946–52. doi: 10.1093/abbs/gmt097. [DOI] [PubMed] [Google Scholar]
- 34.Gao LW, Zhang J, Yang WH, et al. Glaucocalyxin A induces apoptosis in human leukemia HL-60 cells through mitochondria-mediated death pathway. Toxicol In Vitro. 2011;25(1):51–63. doi: 10.1016/j.tiv.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 35.Zhai P, Sciarretta S, Galeotti J, Volpe M, Sadoshima J. Differential roles of GSK-3beta during myocardial ischemia and ischemia/reperfusion. Circ Res. 2011;109(5):502–11. doi: 10.1161/CIRCRESAHA.111.249532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Misra MK, Sarwat M, Bhakuni P, et al. Oxidative stress and ischemic myocardial syndromes. Med Sci Monit. 2009;15(10):RA209–19. [PubMed] [Google Scholar]
- 37.Yang WH, Zheng LP, Yuan HY, Wang JW. Glaucocalyxin A and B regulate growth and induce oxidative stress in lettuce (Lactuca sativa L.) roots. J Plant Growth Regul. 2014;33(2):384–96. [Google Scholar]
- 38.Povlsen JA, Lofgren B, Dalgas C, et al. Frequent biomarker analysis in the isolated perfused heart reveals two distinct phases of reperfusion injury. Int J Cardiol. 2014;171(1):9–14. doi: 10.1016/j.ijcard.2013.11.035. [DOI] [PubMed] [Google Scholar]
- 39.Cao L, Sun J, Shen Y, et al. Pharmacokinetic analysis of glaucocalyxin A in rat plasma by liquid-liquid extraction and HPLC. Planta Med. 2009;75(6):629–31. doi: 10.1055/s-0029-1185401. [DOI] [PubMed] [Google Scholar]