

ABSTRACT
Because low levels of DNA double strand breaks (DSBs) appear not to activate the ATM-mediated prophase I checkpoint in full-grown oocytes, there may exist mechanisms to protect chromosome integrity during meiotic maturation. Using live imaging we demonstrate that low levels of DSBs induced by the radiomimetic drug Neocarzinostatin (NCS) increase the incidence of chromosome fragments and lagging chromosomes but do not lead to APC/C activation and anaphase onset delay. The number of DSBs, represented by γH2AX foci, significantly decreases between prophase I and metaphase II in both control and NCS-treated oocytes. Transient treatment with NCS increases >2-fold the number of DSBs in prophase I oocytes, but less than 30% of these oocytes enter anaphase with segregation errors. MRE11, but not ATM, is essential to detect DSBs in prophase I and is involved in H2AX phosphorylation during metaphase I. Inhibiting MRE11 by mirin during meiotic maturation results in anaphase bridges and also increases the number of γH2AX foci in metaphase II. Compromised DNA integrity in mirin-treated oocytes indicates a role for MRE11 in chromosome integrity during meiotic maturation.
KEYWORDS: double strand DNA breaks, DNA damage, MRE11, meiotic maturation, mouse oocytes
Introduction
Cells possess mechanisms that protect genome integrity. Double-strand DNA breaks (DSBs) are the most serious DNA lesions that can lead, if left unrepaired, to loss of genetic information, or if incorrectly repaired, to chromosome rearrangements. DSBs arise from cellular metabolism (e.g., oxygen radicals), DNA processes (replication, transcription), cell cycle deregulation or exogenous genotoxic stress (e.g.,, ionizing irradiation). To protect genome integrity cells have evolved DNA damage response (DDR) mechanisms that ensure DNA damage detection, arrest of cell cycle progression (by activating DNA damage checkpoints) and trigger the repair process. When the DNA damage cannot be efficiently repaired cells enter senescence or apoptosis.1
The complex of MRE11, RAD50 and NBS1 (MRN) plays a central role in sensing and repairing DSBs.2 MRN exhibits several enzymatic activities including 3´-5´ exonuclease and endonuclease activity of MRE11, and ATPase and adenylate kinase activity of RAD50. Although NBS1 lacks enzymatic activity it provides regulatory functions through protein-protein interactions.3 MRN is an early DSBs sensor that binds to DSBs and promotes Ataxia telangiectasia mutated (ATM) kinase activation.4,5 ATM phosphorylates histone H2AX at serine 139 (γH2AX).6 Consequently, MDC1 (mediator DNA damage checkpoint protein), a predominant γH2AX recognition factor in mammalian cells, interacts with γH2AX. MDC1 recruits further MRN complexes that stimulate ATM, thereby initiating a positive feedback loop that leads to γH2AX propagation around DSBs.7 MRN promotes repair of DSBs through homology recombination (HR), non-homologous end-joining (NHEJ) and alternative non-homologous end-joining (a-NHEJ) pathways. MRN also forms a bridge between broken DNA ends and stimulates a resection of DNA ends that is essential for DSBs repair.2 Single-strand DNA generated by DNA resection activates ataxia telangiectasia and RAD3-related (ATR) kinase. Locally active sensor kinases ATR and ATM phosphorylate and activate transducer checkpoint kinases 1 and 2 (CHK1 and CHK2, respectively). CHK1 and CHK2 then phosphorylate many effector proteins involved in cell cycle checkpoint and DNA repair.8
Natural formation of DSBs in zygotene stage by SPO11 endonuclease is essential for meiotic recombination of homologous chromosomes that results in chiasmata formation.9 Ionizing irradiation causes loss of primordial follicles through TAp63-dependent expression of proapoptotic proteins PUMA and NOXA in oocytes. Interestingly, the fertility of Puma−/− Noxa−/− mice exposed to ionizing radiation is the same as wild type non-irradiated controls,10 which suggests that oocytes can repair DSBs during the long prophase I arrest.11
Mouse oocytes from preovulatory follicles can detect DSBs as documented by γH2AX foci formation after exogenous DSBs induction.12-15 Full-grown oocytes express a relatively low level of ATM when compared to growing oocytes or blastocysts, which likely accounts for the limited activation of the ATM-CHK1 pathway after inducing DSBs in full-grown oocytes.14 Such limited ATM activation, however, does not prevent H2AX phosphorylation in these oocytes, suggesting that H2AX phosphorylation is independent of ATM, at least when DNA damage is low. Only a high level of DSBs triggers CHK1-dependent inhibitory phosphorylation of CDC25B on S323 and DNA damage checkpoint activation that results in a delay in resumption of meiosis.14 For example, severe DSBs induced by drugs of the bleomycin family delay meiotic resumption, anaphase promoting complex (APC/C) activation, and first polar body emission. Although the incidence of oocytes resuming meiosis is decreased when DSBs are numerous, those oocytes that successfully resume meiosis mature to and arrest at metaphase II. Nevertheless, unaligned chromosome fragments arise in metaphase I and the spindle assembly checkpoint (SAC) activity is prolonged as evidenced from the persistent localization of BUB3 to kinetochores and delayed cyclin B destruction.12,13 In addition, severe DSBs induced by neocarzinostatin (NCS) (≥300 ng/ml) prevent first polar body emission and cause chromosomal fragmentation.15
DNA integrity in oocytes is essential for reproduction.11,16,17 Thus, it is unclear why the DNA damage threshold for checkpoint activation in full-grown prophase I oocytes seems elevated. A possible explanation is that there may be a selective advantage to repair the damage later during the cell cycle rather than activate the checkpoint during resumption of meiosis. Oocytes can repair previously induced minor DNA damage during the lengthy meiotic M phases 11,14; note that UV irradiation or treatment with mutagenic drugs induces unscheduled DNA synthesis in prophase I oocytes, and metaphase I and II eggs.18,19 Similarly, although freshly isolated mouse metaphase II eggs exhibit substantial DNA damage, most of the damage is repaired in RAD51-dependent manner following 6 h of in vitro culture.20 Furthermore, microinjection recombinant RAD51 or pharmacological RAD51 inhibition enhances or decreases DNA repair, respectively, after IR irradiation or doxorubicin treatment.20,21
There is little, if any, knowledge about the activity of DDR signaling during meiotic maturation. Here, we use H2AX phosphorylation, together with MDC1 co-localization, as a marker of DSBs and DDR activity. By employing immunofluorescence and live confocal microscopy, we find that MRE11 is involved in H2AX phosphorylation in meiosis I not only under normal conditions but also after inducing exogenous DSBs. Moreover, we find that MRE11 is important for maintaining chromosome integrity during meiotic maturation.
Results
Double strand DNA breaks (DSBs) induced by Neocarzinostatin in GV-stage oocytes result in chromosome segregation problems in anaphase I
To study the response of oocytes following increased DNA damage during meiotic maturation we used the radiomimetic drug neocarcinostatin (NCS), which effectively induces DSBs in mouse oocytes.15 Oocytes arrested in GV-stage by milrinone were exposed to increasing concentrations of NCS (5–100 ng/ml) for 1 h and then analyzed for γH2AX/MDC1 foci (Fig. 1A-B), which are sensitive markers of DSBs.6 We identified a NCS concentration-dependent increase in DSBs as documented by the increased number of γH2AX/MDC1 foci (Fig. 1A-B).
Figure 1.
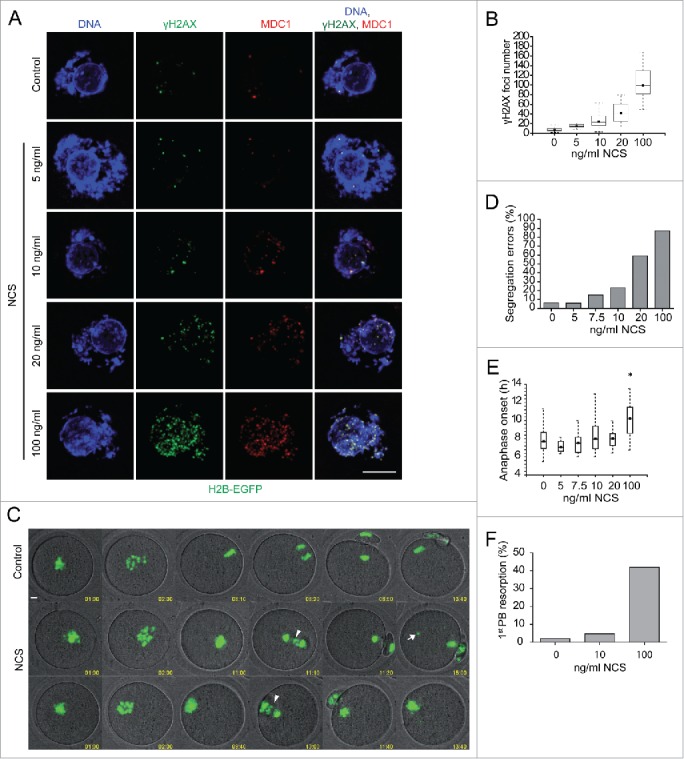
DSBs induced by Neocarzinostatin in GV-stage oocytes result in chromosome segregation problems in anaphase I. (A) Immunofluorescence of control and NCS treated GV-stage oocytes labeled with pS139 H2AX (γH2AX) and MDC1 antibodies. Maximum z-projection of confocal section across nucleus is shown. (B) Number of γH2AX foci in control and NCS treated GV-stage oocytes (n=137, 22, 118, 29, 21). (C) Selected time frames of live H2B-EGFP control and NCS treated oocytes imaged by confocal microscopy. Arrowheads show chromosome segregation errors in anaphase and arrow shows diffusing chromosome fragment in MII. Maximum z-projection of H2B-EGFP and single bright filed section is shown. Time in hh:mm. (D) Percentage of segregation errors in anaphase I and (E) timing of anaphase onset in control and NCS treated oocytes (for C and D n=98, 17, 20, 56, 47, 113). (F) Percentage of 1st polar body resorption after anaphase onset in control, 10 and 100 ng/ml NCS treated oocytes (n=98, 55, 76). Data in D), E) and F) are from analysis of time lapse movies of control and NCS treated H2B-EGFP oocytes as shown in C).
To analyze the effects of increased DSBs on cell cycle progression and chromosome segregation we treated oocytes isolated from transgenic mice constitutively expressing H2B-EGFP22 with NCS as described above and after transfer to milrinone-, NCS-free medium, the oocytes were examined for meiotic progression and chromosome segregation by live-cell confocal imaging (Fig. 1C and Movie 1). Increasing the number of DSBs led to a dose-dependent increase in chromosome segregation problems in anaphase I (Fig. 1D). NCS treatment had no effect on the incidence of resumption of meiosis and time to onset of anaphase (data not shown and Fig. 1E). The highest NCS concentration, however, significantly delayed anaphase onset (Fig. 1E) and caused resorption of the first polar body in ˜40% of oocytes (Fig. 1F).
In summary, these data demonstrate that we can induce and readily quantify the number of new (extra) DSBs in GV-stage oocytes. At a low concentration of NCS (10 ng/ml) GV-stage oocytes showed ˜2.5-fold increase in γH2AX foci but only about a quarter of these oocytes exhibited chromosome segregation problems (Fig. 1D). In principle, using this low NCS concentration would permit studying a physiologically relevant response to DSBs and moreover, may be low enough to enable DSBs repair or other DNA processes that protect chromosome integrity.
Segregation problems in anaphase I following NCS treatment are predominantly due to chromosome fragmentation
Detailed examination of segregation problems in NCS treated oocytes suggested the presence of lagging chromosomes at anaphase I and free diffusion of several chromosomes in the cytoplasm of metaphase II oocytes (Fig. 1C). To understand how these abnormal chromosomes are generated, we monitored the dynamics of individual chromosomes throughout meiotic maturation in oocytes expressing the chromosome marker H2B-mCHERRY and the kinetochore marker 2mEGFP-CENPC23 after treatment of NCS at the GV-stage (Fig. 2A and Movie 2). No significant differences in chromosome congression and alignment were observed between control and NCS-treated oocytes. However, we found that in NCS-treated oocytes at metaphase, a substantial number of chromosomes were abnormally stretched as indicated by a significantly longer distance between homologous kinetochores (Fig. 2B). The hyperstretched chromosomes often had a region with undetectable H2B-mCHERRY, although the physical connection between homologous kinetochores was unlikely to be completely lost because the hyperstretched chromosomes remained aligned on the metaphase plate (Movie 2). At anaphase I, NCS-treated oocytes exhibited 1–6 lagging chromosomes, whereas no control oocytes did. The vast majority of the lagging chromosomes carried no kinetochores (Fig. 2C), indicating that they are chromosome fragments. These results suggest that a higher number of DSBs leads to the loss of chromosome integrity and results in chromosome fragmentation at anaphase I.
Figure 2.
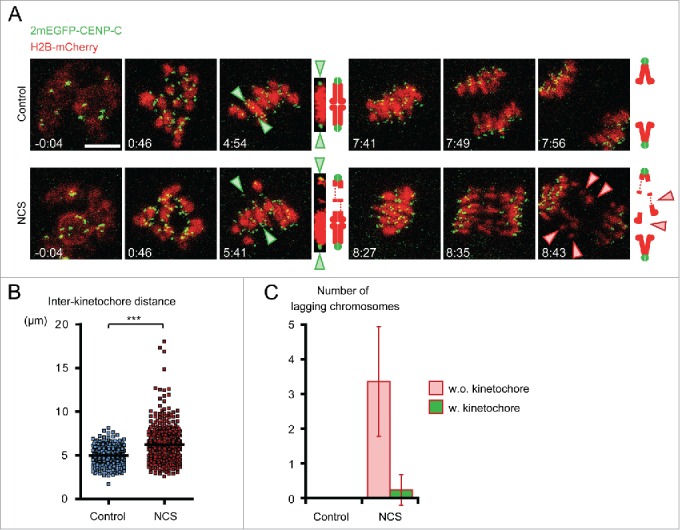
Chromosomes are fragmented in NCS-treated oocytes. (A) Time-lapse imaging of NCS-treated oocytes. Oocytes expressing 2mEGFP-CENP-C (green) and H2B-mCherry (red) were treated with NCS at 100 ng/ml. Green arrowheads indicate the homologous kinetochores of a bivalent, which is magnified on the right. Red arrowheads indicate lagging chromosomes. Time after NEBD (h:mm). Scale bar, 10 μm. (B) Chromosome hyperstretching in NCS-treated oocytes. The distance between homologous kinetochores was measured in 3D in oocytes 6 h after NEBD (n=252, 409 bivalents). ***p < 0.0001. (C) NCS-treated oocytes exhibit lagging chromosomes without kinetochores. The number of lagging chromosomes at anaphase was counted (n=11, 17 oocytes). The lagging chromosomes were categorized. Error bars, s.d.
Low levels of DSBs cause chromosome segregation problems but do not delay APC/C activation and anaphase onset
Inducing DSBs in mouse oocytes by bleomycin or zeocin treatment leads to a substantial delay in emission of the first polar body, and a delayed and reduced activation of APC/C.12,13 Treating oocytes with a very high concentration of NCS (1µg/ml) inhibits emission of the first polar body.15 Severe DSBs also indirectly activate the SAC by disrupting kinetochore-microtubule attachment.12 As shown above we found that a low NCS concentration (10 ng/ml) led to chromosome segregation problems in about 25% oocytes but a high NCS concentration (100 ng/ml) resulted in about 90% of the oocytes displaying problems with chromosome segregation (Fig. 1D).
In light of these findings we monitored APC/C activation by expressing securin-EGFP, which is a well-established marker of APC/C activation,24 in oocytes treated with a low or high NCS concentration (Fig. S1A and Movie 3). The time of anaphase onset (Fig. S1B) and securin-EGFP destruction (Fig. S1C) was not altered either in control oocytes not exhibiting chromosome segregation problems or oocytes treated with the 10 ng/ml NCS that had or did not have such problems. We observed delayed securin-EGFP destruction and anaphase onset only in oocytes treated with 100 ng/ml of NCS in which all oocytes exhibited chromosome segregation problems (Fig. S1B, C). These data suggest that a low level of DSBs (close to a physiological level) does not delay APC/C activation and anaphase onset, although it generates errors in chromosome segregation.
H2AX phosphorylation changes during meiotic maturation and phosphorylated H2AX co-localizes with MDC1
We analyzed H2AX phosphorylation on S139 (γH2AX) in control and oocytes treated with a low concentration of NCS (10 ng/ml) during meiotic maturation (Fig. 3A). In control prophase I arrested GV-stage oocytes γH2AX localized in small nuclear foci. After meiotic resumption γH2AX spread along the entire condensed chromosomes although some chromosome domains with a locally higher γH2AX signal intensity were distinguishable. The γH2AX signal associated with chromosomes reached maximum intensity at metaphase I and was distributed homogenously along the chromosome. The intensity of the γH2AX signal was reduced in MII eggs when compared to MI eggs (Fig. 3B).
Figure 3.
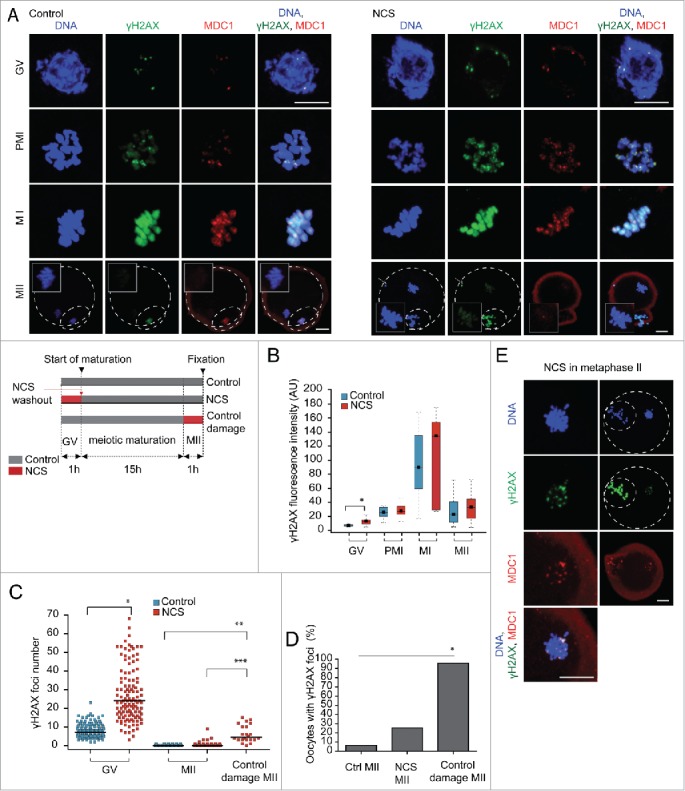
H2AX phosphorylation changes during meiotic maturation. (A) Immunofluorescence of control and NCS treated GV-stage, prometaphase I (PMI), metaphase I (MI) and metaphase II (MII) oocytes labeled with γH2AX and MDC1 antibodies. Maximum z-projection of confocal section across chromatin region is shown. In the case of MII oocytes, maximum z-projection for DNA and γH2AX but single confocal sections for MDC1 are used. Insets show maximum z-projection of metaphase II plate area. (B) Quantification of γH2AX during meiotic maturation in control and NCS treated oocytes (n = 26, 28, 26, 27, 43, 8, 28, 23). (C) Quantification of γH2AX foci number in GV-stage and MII oocytes treated with NCS in GV stage before meiotic maturation. Acute NCS treatment of metaphase II eggs (control damage MII) is also shown (n = 137, 118, 98, 84, 24). (D) Percentage of MII eggs with at least one γH2AX focus (n = 106, 55, 21). (E) Immunofluorescence of MII eggs treated acutely with NCS and labeled with γH2AX and MDC1 antibodies. Maximum z-projection of confocal sections is shown.
As expected, NCS increased the staining intensity of chromosome-associated γH2AX in GV-stage oocytes, but once the oocytes entered meiosis I, similar amounts of chromosome-associated γH2AX were observed in control and NCS-treated oocytes at metaphase I and the same was noted for MII (Fig. 3B). As described above, the number of γH2AX foci in GV-stage oocytes was increased ˜2.5 times after NCS treatment when compared to controls (Fig. 3C). The significant decrease in chromosome-associated γH2AX signal during the metaphase I – metaphase II transition allowed us to analyze γH2AX foci formation in MII eggs. The number of γH2AX foci significantly decreased both in control and NCS-treated oocytes during maturation from GV stage to MII. The small increase in the median number of γH2AX foci of NCS MII eggs when compared to controls was not significant (Fig. 3C). When we counted the number of MII oocytes with at least one γH2AX focus we found that about 30% of NCS-treated oocytes exhibited at least one γH2AX focus in MII compared to only about 5% of control oocytes, whereas 95% of the control oocytes did not have any γH2AX foci (Fig. 3D). Interestingly, the number of oocytes entering anaphase I with segregation errors after NCS treatment was consistent with the number of oocytes exhibiting γH2AXfoci in MII (Fig. 1D).
The low number of γH2AX foci in MII eggs could reflect lower accessibility of the of γH2AX antibody to detect γH2AX foci on the highly condensed MII chromosomes. Accordingly we induced DSBs in MII eggs (control damage MII) (Fig. 3E) and observed an increase in the number of γH2AX foci when compared to MII control or MII NCS eggs (Fig. 3D); this treatment was sufficient to generate at least one γH2AX focus > 90% of the oocytes (Fig. 3D). The reduced number of γH2AX foci in control damage MII eggs compared to GV-stage NCS-treated oocytes (Fig. 3C) probably reflects lower NCS accessibility due to the condensed structure of metaphase chromosomes.
In both control and NCS-treated oocytes MDC1, which acts as γH2AX sensor, completely co-localized with γH2AX until MI. Nevertheless, we observed a reduced MDC1 association with γH2AX foci and no association with the weak chromosomal γH2AX signal on MII chromosomes (Fig. 3A). Interestingly, induction of new DSBs in MII eggs (control damage MII) led to formation of DSBs foci containing both γH2AX and MDC1 (Fig. 3E).
These data suggest that H2AX is phosphorylated during meiotic maturation in a cell cycle-dependent manner. Because induction of new DSBs in both GV- and MII-stage oocytes increased γH2AX/MDC1 foci formation we propose that γH2AX foci in GV- and MII-stages reflect DSBs. However, because γH2AX did not localize in small foci and total H2AX phosphorylation was not significantly affected by induced DSBs (in contrast to GV-stage oocytes, Fig. 3B) during metaphase I, we suggest that this M-phase associated chromosomal γH2AX increase is not connected to DNA damage. This suggestion is consistent with the finding that in somatic cells H2AX is phosphorylated during mitosis in cell cycle-dependent but DNA damage-independent manner.25-27 Most importantly, the decrease in the number of both endogenous and NCS-induced γH2AX foci between GV-stage and MII implies that DNA repair or other DNA processes, such as DNA break tethering, occur during meiotic maturation.
ATR, but not ATM, is involved in H2AX phosphorylation during meiotic maturation
We next explored what signaling pathway was responsible for H2AX phosphorylation during meiotic maturation. ATM is the principal kinase that phosphorylates H2AX in the region of DSBs in somatic cells.6 ATM is expressed in mouse oocytes but is activated only after extensive DSBs occur.14 As anticipated, pharmacological ATM inhibition by KU55933 had no effect on γH2AX/MDC1 foci number in GV-stage oocytes (Fig. S2A, B) and also no significant effect on H2AX phosphorylation and MDC1 chromatin association in MI (Fig. S2A, C). The lack of an effect of inhibiting ATM on γH2AX phosphorylation in MI was confirmed by using the more potent ATM inhibitor KU 60019 28 (Fig. S2 D, E). Moreover, KU55933 treatment did not reduce the number of γH2AX foci induced by NCS in GV-stage oocytes (Fig. S2A, B) nor did induce any chromosome segregation problems in anaphase I (Fig. S2F). Furthermore, we did not observe an increase in the number of oocytes with at least 1 γH2AX focus after separate treatment with two different ATM inhibitors (Fig. S2G). In contrast, KU55933 efficiently inhibited H2AX phosphorylation in NCS-treated mouse NIH3T3 fibroblasts, demonstrating that the drug was active (Fig. S2 H, I). In summary, ATM has no impact on H2AX phosphorylation either during normal meiotic maturation or after induction of a low number of DSBs, which is in agreement with a previous report.14 We conclude that ATM is dispensable for meiotic maturation.
We also ascertained whether ATR mediates H2AX phosphorylation during meiotic maturation. ATR phosphorylates H2AX after stalled DNA replication in somatic cells29 and during meiotic sex chromosome inactivation in the male germ line.30 Pharmacological inhibition of ATR by VE821 significantly decreased H2AX phosphorylation on chromosomes in MI (Fig. S2D, E) and also supressed γH2AX foci in MII (Fig. S2G). On the other hand VE821 treatment had no effect on segregation errors in anaphase I (Fig. S2F). We conclude that although ATR is involved in H2AX phosphorylation during meiotic maturation, it is not essential for chromosome integrity during chromosome segregation. Because we were unable to block completely H2AX phosphorylation it is still possible that the residual H2AX phosphorylation has some other function.
MRE11 is essential for H2AX phosphorylation in MI but not MII and inhibiting MRE11 increases the number of DSBs in MII eggs
We next determined whether MRE11, which is involved in sensing of DSBs, DDR signal amplification, and DNA repair,2 is involved in H2AX phosphorylation in oocytes. MRE11 localized in the nucleus of GV-stage oocytes (Fig. 4A) and MRE11 protein is expressed during meiotic maturation (Fig. 4B). To establish a role for MRE11 in H2AX phosphorylation in mouse oocytes, we first employed a morpholino-mediated MRE11 knockdown approach (Fig. 4B). This approach, however, was unsuccessful due to the stability of MRE11 during the 24-h culture period, which is about the maximum length that can be employed before oocyte health starts to deteriorate. As an alternative approach we used the small molecule inhibitor mirin, which inhibits nuclease activity of MRE11 and prevents MRN-dependent signal amplification in somatic cells.31 Treatment of prophase I arrested GV-stage oocytes with 100 µM mirin for 1 h did not affect the number of γH2AX/MDC1 foci (Fig. 4C-D), whereas when DNA damage was induced by NCS in the presence of mirin the number of γH2AX/MDC1 foci was significantly reduced compared to NCS only (Fig. 4C-E). Inhibition of MRE11 during meiotic maturation by mirin substantially decreased the amount of chromatin-associated phosphorylated H2AX and MDC1 binding in MI oocytes (Fig. 4C-F), followed by the increase in γH2AX foci number on metaphase II chromosomes (Fig. 4C-D). The number of MII eggs with at least one γH2AX focus was highly increased after mirin treatment during meiotic maturation (Fig. 4G).
Figure 4.
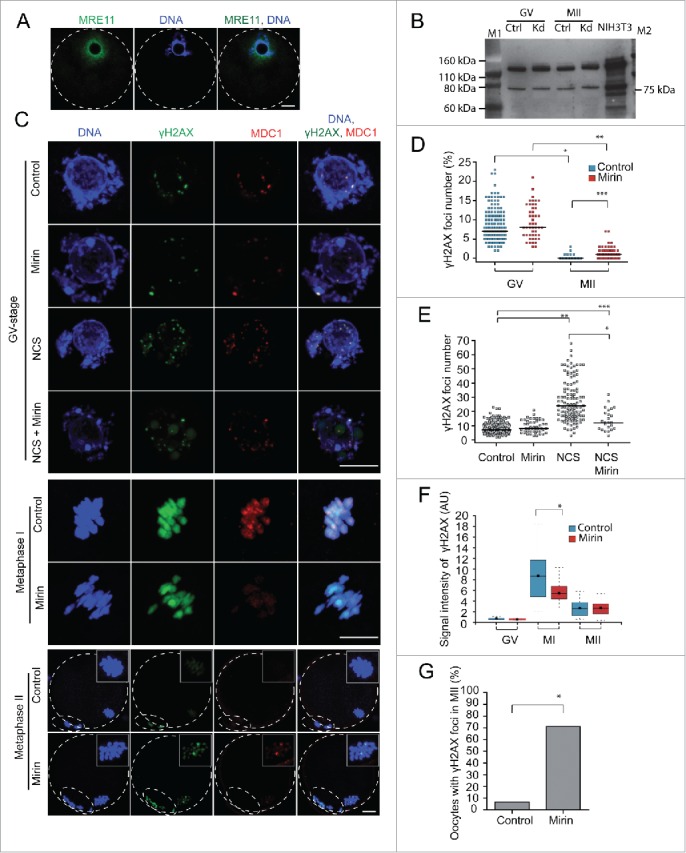
MRE11 is essential for H2AX phosphorylation in meiosis I and its inhibition during meiotic maturation causes the increase in DSBs in metaphase II. (A) Imunofluorescence of GV-stage oocytes stained with MRE11 antibody. Maximum z-projection is shown. (B) Expression of MRE11 (81 kDa) in control morpholino (ctrl) and Mre11 morpholino (kd, knock down) injected GV-stage and MII oocytes detected by western blot (n=200 oocytes/lane). M1 and M2 are protein markers of indicated MW. Control NIH3T3 cells were used as a positive control (12 µg/lane). We have seen similar expression of MRE11 in noninjected GV and MII oocytes (data not shown). (C) Immunofluorescence of control, mirin and NCS treated GV-stage, metaphase I and metaphase II oocytes labeled with γH2AX and MDC1 antibodies. Maximum z-projection of confocal section across chromatin region is shown. GV-stage oocytes were treated with Mirin, NCS or both for 1 h in milrinone supplemented medium before fixation. Metaphase I and II oocytes matured in the presence of Mirin for 7 or 16 h before fixation. (D) Quantification of γH2AX foci number in GV-stage and MII oocytes treated with mirin in GV stage before meiotic maturation (n=137, 33, 200, 109). E) Quantification of γH2AX foci number in GV-stage oocytes non-treated (control) or treated with mirin, NCS or both for 1 h in milrinone-supplemented medium before fixation (n=153, 48, 118, 24). (F) Quantification of γH2AX during meiotic maturation in control and mirin-treated oocytes (n=26, 8, 15, 21, 14, 32). G) Percentage of oocytes with at least one γH2AX focus matured into the metaphase II in control or mirin-supplemented medium (n=31, 58).
The increased number of γH2AX foci in MII eggs after maturation in mirin-containing medium raised the question whether MRE11 is essential for regulation of H2AX phosphorylation in MII eggs. Accordingly, we isolated in vivo matured MII eggs that were then treated in vitro with NCS and cultured in the presence or absence of mirin (Fig. S3A). Treatment with mirin alone had no effect on the number of γH2AX foci, whereas NCS induced a significant increase in the number of γH2AX foci. Although treatment with both NCS and mirin decreased the number of γH2AX foci (p=0.025) this decrease was small when compared to dramatic effect of mirin on γH2AX foci induced by NCS in GV-stage oocytes (p < 0.0001, Fig. 4E). Moreover, mirin did not affect the number of MII eggs with at least 3 γH2AX foci (Fig. S3B), suggesting that H2AX phosphorylation is less dependent on MRE11 at MII. In contrast, when control oocytes or oocytes treated with NCS at GV stage and then matured in mirin-free or mirin-containing medium the number of γH2AX foci in MII eggs was significantly higher in the mirin or NCS+mirin groups when compared to controls (Fig. S3C,D). Thus, MRE11 is not essential for γH2AX foci formation in MII eggs when the oocytes were previously treated with NCS in the GV stage.
Collectively we found that MRE11 regulates H2AX phosphorylation in MI that is not associated with DSBs (see above), but the increased number of γH2AX foci in MII eggs after maturation in mirin suggests an increase in the number of DSBs during maturation and that H2AX phosphorylation in MII eggs is partly independent of MRE11 activity.
MRE11 controls chromosome segregation and integrity
Inducing new DSBs by NCS leads to chromosome segregation problems in anaphase I (see Figs. 1C, D, 2A). Because we observed an increased number of DSBs in MII eggs after mirin treatment during meiotic maturation, we used live imaging to monitor chromosome segregation in H2B-EGFP-expressing oocytes to gain better insight into chromosome integrity (Fig. 5A and Movie 4). Interestingly, addition of mirin (100 µM) to the culture medium significantly increased chromosome segregation errors from 3.8% in control to 30.8% in mirin (p=0.017, n=38, 28, respectively) during anaphase I. This finding, together with increased DSBs in MII eggs, suggests that chromosome integrity is compromised when MRE11 is inhibited. In mirin-treated oocytes we also observed a delay in both resumption of meiosis and anaphase onset (Fig. S4A, B). Mirin inhibited resumption of meiosis in ˜10% of treated oocytes and increased the portion of oocytes arrested in MI from 10% in control to 30% in mirin-treated (Fig. S4C). This effect of mirin on cell cycle progression is probably not a direct consequence of increased DNA damage because induction of DSBs by NCS has no effect on both resumption of meiosis (data not shown) and anaphase entry, except at the highest concentration of NCS (100 ng/ml) (Fig. 1E).
Figure 5.
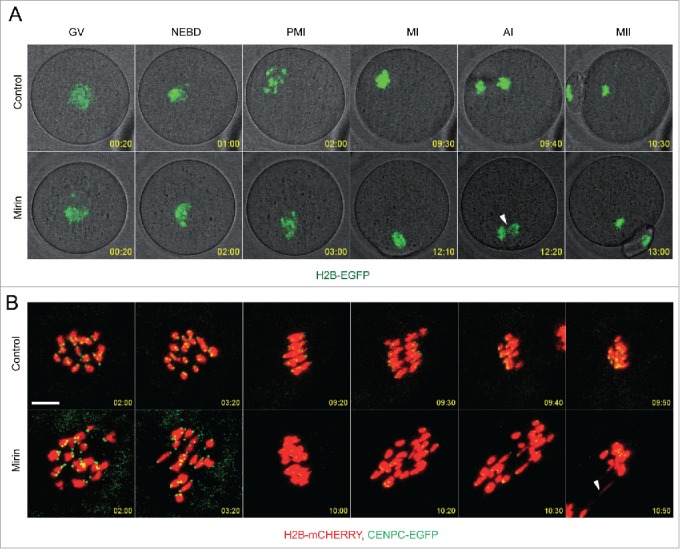
MRE11 controls chromosome segregation and integrity.(A) Time-lapse imaging of control and mirin treated H2B-EGFP oocytes. Maximum intensity z-projection of H2B-EGFP and single section of bright-field images in selected time frames are shown. (B) Imaging of chromosome dynamics and integrity in live oocytes expressing H2B-mCHERRY and CENPC-EGFP and maturing in control or Mirin supplemented medium. Maximum z-projection of confocal section of selected time intervals is shown. Arrowheads show DNA anaphase bridges.
In somatic cells and mitotic Xenopus extracts MRE11 is involved in spindle formation and chromosome alignment in part by regulating RAN-GTP signaling.32 We also used live imaging to monitor spindle formation in oocytes expressing MAP4-EGFP and H2B-CHERRY (Fig. S4D and Movie 5). Mirin-treated oocytes exhibited normal spindle formation including spindle volume and normal chromosome alignment (Fig. S4D, E) demonstrating that MRE11 does not regulate spindle formation in mouse oocytes.
To characterize the types of segregation errors in mirin-treated oocytes in more detail we analyzed chromosome segregation and integrity in oocytes expressing H2B-mCHERRY and the kinetochore marker CENPC-EGFP (Fig. 5B and Movie 6). Anaphase bridges represented almost all segregation problems. In contrast to NCS-treated oocytes (Fig. 2) we did not detect chromosome fragment formation during anaphase (Fig. 5B).
Taken together, these results suggest that MRE1 is involved in correct chromosome segregation and preservation of chromosome integrity.
Discussion
Results of experiments reported here extend the previous finding that phosphorylation of H2AX in mouse prophase I oocytes increases in response to new DSBs induced exogenously by radiomimetic drugs 12,13,15 by using lower concentrations of NCS that generate a physiological number of DSBs and finding an increase in the number of chromosome segregation errors without activation of the APC/C and any delay in onset of anaphase I. These findings contrast with previous results that used high concentrations of NCS that induce an excessive increase in the number of DSBs and result in delayed resumption of meiosis and MI arrest with fragmented chromosomes.15 In addition, such oocytes with high level of DSBs also exhibit a delay in the time of emission of the first polar body, lower APC/activation but greater SAC activation as a consequence of disrupted microtubule-kinetochore attachments.12,13
High-resolution imaging of live oocytes treated with low concentrations of NCS reveal that chromosome structure is altered shortly after resumption of meiosis as evidenced by stretched homologs. Chromosome fragments, however, appear only after anaphase onset, which implies that mechanical forces during chromosome segregation and cohesion loss are the source of chromosome breakage for chromosomes that harboured a higher number of DSBs. Before chromosome segregation DSBs are already present on chromosomes but cohesion between sister chromatids may help support chromosome structure. We also observe instances in what appears to be intact lagging chromosomes, suggesting that microtubule-kinetochore attachment is affected by DSBs. In cancer cells when DNA damage is induced in mitosis, DDR selectively stabilizes microtubule-kinetochore attachments through AURKA and PLK1 kinases, which increases the frequency of lagging chromosomes in anaphase.33 Thus, this phenomenon appears to operate in MI when homologous chromosomes segregate. An interesting question for future investigation is whether the increased level of DSBs in GV-stage oocytes from both older human females or mice 17 contributes to the higher incidence of aneuploidy associated with age; loss of cohesion is the primary source.34
H2AX is phosphorylated during DDR in interphase cells and forms typical γH2AX foci.6 However, during mitosis γH2AX does not form foci but rather spreads over the condensed chromosomes and H2AX phosphorylation increases in PIKKs (ATM, DNA-PKcs)-dependent manner even in the absence of exogenous DNA damage.25,27 In oocytes we find that H2AX phosphorylation increases after resumption of meiosis in a cell –cycle dependent manner: it reaches a maximum at MI and then decreases by MII. Our data indicate that H2AX phosphorylation in MI is ATM-independent, which is in agreement with low ATM expression and activation in mouse oocytes,14 and that MRE11 and ATR, not ATM, are involved in cell-cycle dependent regulation of H2AX phosphorylation in MI. Similar to somatic cells, whole chromosome H2AX phosphorylation during MI is likely regulated independently of the DNA damage response because NCS-treated oocytes do not exhibit significantly higher levels of γH2AX in MI. Inhibiting either MRE11 or ATR results in decreased H2AX phosphorylation in MI but only MRE11 inhibition results in chromosome segregation problems. Thus, MRE11 function in chromosome integrity during meiotic maturation appears independent of H2AX phosphorylation whose function in MI remains unknown.
Immunofluorescence γH2AX foci detection is widely used as a sensitive marker for DSBs.6,35-36 Accordingly, we used γH2AX/MDC1 foci formation as the main read-out for DSBs. It should be noted, however, that H2AX phosphorylation does not always depend on DSBs, e.g., it may by induced by hypoxia37 or in cell cycle-dependent manner in mitosis.25-27 We find, however, that inducing new DSBs by NCS results in a concentration-dependent increase in γH2AX/MDC1 foci in GV-stage oocytes. This increase correlates with the increase in chromosome segregation errors (mainly chromosome fragments) during anaphase I and thus suggests that these foci are a reliable means to estimate DSBs in GV-stage oocytes. A different situation exists in MI when γH2AX increases in cell cycle-dependent manner but DSBs-independent manner.
Whereas γH2AX spreads over chromosomes in MI during maturation under normal conditions and after inducing DSBs, in prophase I and MII γH2AX forms separate foci. The increase in γH2AX foci formation enhanced by NCS treatment in prophase I arrested oocytes is prevented by MRE11 inhibition. In addition, when normal ovulated MII eggs are treated with NCS together with mirin, we observe only reduced number of γH2AX foci, i.e., there still is an increase in the number of γH2AX foci. These data suggest that DSBs recognition in prophase I arrested oocytes is MRE11-dependent, whereas in MII DSBs recognition is not or at least partly independent of MRE11 activity. DSBs repair in MII eggs depends on the homologous recombination factor RAD51 20,38 and MRE11 is involved in homologous recombination in somatic cells.2 A question for future investigation is whether MRE11 is essential for DNA repair although it is not essential for DSBs recognition in MII.
MRE11 is active and involved in H2AX phosphorylation during meiotic maturation without exogenous DSBs. Inhibiting MRE11 during meiotic maturation increases the number of γH2AX foci in MII eggs and imaging live oocytes reveals more stretched homologous chromosomes in prometaphase and MI, which is similar to the effect of NCS treatment. Spindle formation and chromosome alignment is normal but DNA anaphase bridges are visible when MRE11 is inhibited. Anaphase bridges may be associated with incomplete or incorrect DSBs repair.39 Interestingly, MRE11 and CtIP direct repair of meiotic DSBs toward error-free HR and supress Ku70 - DNA ligase IV-mediated NHEJ repair during Caenorhabditis elegans gametogenesis. MRE11 or CtIP inhibition suppresses inter-homolog crossovers and leads to chromosomal fragmentation and aggregation that can be rescued by NHEJ repair inhibition.40,41 Consistent with these findings, it is possible that MRE11 is not only involved in potential RAD51-dependent HR in MI but also that it may be essential to suppress NHEJ repair that otherwise may lead to erroneous repair and chromosome fusion. In conclusion, our data suggest that MRE11 is essential to prevent structural instability but is dispensable for spindle formation during meiotic maturation in mouse oocytes.
Somatic cells in mitosis can detect DSBs as evident from γH2AX/MDC1 and MRE11 foci formation. Downstream events, however, such as RNF8 and RNF16 binding and recruitment of repair factors 53BP and BRCA1, are suppressed by CDK1 and PLK1 that phosphorylate RNF8, 53BP1 and a regulatory subunit of the DNA ligase IV (XRCC4).42 Artificial restoration of 53BP binding to DSBs in mitotic cells by expressing a non-phorphorylatable 53BP1 mutant leads to sister telomere fusion due to NHEJ repair followed by chromosome segregation errors.43,44 Although the efficiency of DSB repair during mitosis is much lower than in interphase, a substantial proportion of the DSBs is repaired during mitosis and the repair process is regulated by mechanisms other than those active in other cell-cycle phases. XRCC4 does not localize to mitotic chromosomes but DNA ligase IV associates with chromosomes.45 CtIP facilitates DSB-end resection by MRN, which is involved in both HR and A-NHEJ repair pathways.46 Interestingly, down-regulation of CtIP in mitotic cells significantly increases incidence of anaphase bridges,45 which are marker of genome instability.47
Our findings indicate that DNA repair operates during meiotic maturation. In particular, we observe that the number of γH2AX foci significantly decreases between prophase I and MII during oocyte maturation. Oocytes transiently treated with NCS exhibit a 2.5-fold increase in DSBs number in prophase I, but following maturation to MII only about 30% of the MII eggs had > 1 DSB. Interestingly, this number is similar to the number of oocytes entering anaphase with chromosome segregation errors after transient NCS treatment in prophase I. Additionally, inhibiting MRE11 during meiotic maturation increases the number of γH2AX foci in MII eggs and also results in anaphase bridges. Because mirin inhibits nuclease activity (DSB-end resection), but not DNA tethering activity of MRE11,31 compromised DNA integrity in mirin-treated oocytes implicates involvement of MRE11 in the DNA repair during meiotic maturation. Moreover, DNA damage induced by UV irradiation leads to detectable DNA synthesis in maturing oocytes.18,19 Alternatively, the anaphase bridges observed in mirin-treated oocytes could be a consequence of telomere dysfunction; in primary human telomerase negative G2 cells, MRE11 and phosphorylated NBS1 are recruited to telomeres to ensure their protection.48 Interestingly, expression of several players in DSBs repair (e.g., BRCA1, MRE11 and RAD51) decreases with age in human and mouse oocytes.17 This decrease could compromise DNA repair and genome integrity in both growing and maturing oocytes and thereby result in reduced fertility.
Collectively, our data suggest that DNA repair occurs after resumption of meiosis during the prolonged MI phase and that MRE11 is involved in such DNA repair. Future investigations will address the mechanisms of DNA repair, which appear to differ in interphase cells, mitosis or meiosis and which repair pathway(s) operate(s) in maturing oocytes, noting that MRE11 is involved in all types of repair2 and RAD51 (HR factor) is involved in repair in MII eggs.20,38
Materials and methods
Animal use ethical statement
The mice in this study were used in accordance with the Act No 246/1992 of the Czech Republic on the protection of animals against cruelty. The Projects of Experiments on animals and animal welfare were approved by The Expert Committee of the Academy of Sciences of the Czech Republic and by The Animal Care and Use Committee of RIKEN CDB. Mice were housed in a temperature-controlled room with proper 12/12 darkness-light cycles, fed with a regular ad libitum diet.
Culture of oocytes and mRNA microinjection
The oocytes were obtained from 8–12-week-old CD1 or H2B-EGFP transgenic mice22 on CD1 genetic background (CD1-Tg(HIST1H2BB/EGFP)) sacrificed by cervical dislocation 46 h after the injection of 5 IU equine serum gonadotropin (eCG). To obtain CD1-Tg(HIST1H2BB/EGFP) we repeatedly (>10 times) backcrossed transgenic and H2B-EGFP-expressing males with wild-type CD1 females. Oocytes were collected in M2 medium and cultured in MEM medium (Sigma-Aldrich) supplemented with 0.22 mM sodium pyruvate, 4 mg/ml BSA (Sigma-Aldrich) and 50 μg/ml gentamicin (Sigma-Aldrich) in a 5% CO2 atmosphere. The culture medium was supplemented with 2.5 μM milrinone (Sigma-Aldrich) or 200 nM 3-isobutyl-1-metyl-xanthine (IBMX, Sigma-Aldrich) to prevent meiotic maturation.49 The MRE11 inhibitor mirin (Sigma-Aldrich) was used at concentration 100 µM, the radiomimetic drug NCS (neocarzinostatin, Sigma-Aldrich) at 5 - 100 ng/ml, the ATM inhibitor KU55933 (Sigma-Aldrich) at 10 µM, the ATM inhibitor KU60019 (Selleckchem) at 1 µM and the ATR inhibitor VE821 (Selleckchem) at 1 µM.
To obtain in vivo matured MII eggs mice were stimulated by injection of 5 IU human chorionic gonadotropin (hCG) 44 h after eCG stimulation. MII eggs were isolated 12 h post-hCG administration in M2 medium and the expanded cumulus cells surrounding MII eggs were removed by hyaluronidase (Sigma Aldrich, H4272) treatment.
Oocytes were microinjected as previously described50 in M2 medium with 5 pl of 80 ng/µl H2b-mCherry,51 125 ng/µl Egfp-Cenp-C,51 150 ng/µl 2mEGFP-CENP-C23, 50 ng/µl Securin-Egfp52 and 125 ng/µl Map4-Egfp53 cRNAs. Oocytes were cultured for 2 h in MEM medium supplemented with milrinone or IBMX to allow protein expression after mRNAs microinjection.
Morpholino oligonucleotide mediated MRE11 knock down
The antisense morpholino oligonucleotides for MRE11 knockdown (5′ATAGTAGAGTTCCTCTGTACCAGGT-3′) and standard control oligo were designed and obtained from Gene Tools, LLC, Philomath, OR. The antisense or standard oligo were diluted to 1.5mM and injected into GV-stage arrested oocytes in M2 medium supplemented with 2.5 µM milrinone. 70-kDa dextran conjugated with tetramethylrhodamine (TAMRA) (Sigma Aldrich, D-1819) was co-injected with morpholino oligonucleotides to estimate the efficiency of microinjection. After microinjection, the oocytes were arrested at the GV-stage in MEM medium supplemented with 2.5 µM milrinone for 24 h to prevent meiosis resumption. After 24 h GV oocytes were collected and MRE11 expression was analyzed by Western blot (200oocytes/lane). To obtain MII eggs, after 24 h of GV-stage arrest the oocytes were cultured for 18 h in MEM medium.
Live confocal microscopy
Time-lapse image acquisitions in Figure 1C, S1A and S3A, B were performed using Leica TCS SP5 with an HCX PL Apo Lambda Blue 40x 1.25 oil objective with 1x zoom; 12 confocal 7.5-µm optical sections were taken with a1024 × 1024 pixel image resolution using 10 min time intervals. For kinetochore imaging in EGFP-CENP-C expressing oocytes (Fig. 6E) we used the tracking function of the Matrix Screen module in LAS AF software (Leica Microsystems). The area of the chromosomes of individual oocytes expressing H2B-mCHERRY and EGFP-CENP-C was irradiated with 6% of the 488 nm and 3% of the 561 nm lasers and 14 confocal 2-µm optical sections scanned by HyD detectors with 12x zoom in 256 × 256 or 512 × 512 pixel image resolution by 1000Hz speed bi-directional scan were acquired. Kinetochores imaging in live oocytes in Fig. 2A was done as described previously.23
Immunofluorescence confocal microscopy
After a brief wash in PBS the oocytes were fixed in 3.7% paraformaldehyde in PBS for 30 min at room temperature; the zona pellucida was not removed. After washing with PBS (3X for 10 min), the oocytes were permeabilized for 15 min with 0.5% Triton X-100 in PBS and incubated with 2% normal donkey serum (Jackson Immuno Research, 017-000-121) in PBS for 2h at room temperature. The oocytes were then incubated overnight at 4°C with primary antibodies diluted in PBS/0.2% normal donkey serum. After washing the oocytes in PBS (3X for 20 min), the cells we incubated for 1 h at room temperature with secondary antibodies. γH2AX was detected by rabbit monoclonal antibody (Cell Signaling Technology, 9718), MDC1 by mouse monoclonal antibody, clone P2B11 (Millipore, 05–1572), MRE11 by rabbit polyclonal antibody (Novus Biologicals, NB100-142). All primary antibodies were used at a dilution 1:200. Anti-mouse antibody conjugated with TRITC or anti-rabbit antibody conjugated with FITC (Jackson ImmunoResearch) were used at a dilution 1:100. After a final wash in PBS/BSA, the oocytes were mounted in Mowiol (with 0.25µg/ml DAPI for DNA staining) between a slide and cover glass using a spacer to preserve the three-dimensional (3D) structure of the oocytes.
Oocytes were scanned using Leica TCS SP5 with HCX Pl Apo Lambda Blue 63x 1.4 oil objective. A sequential scan was applied in the 16-bit image depth with 1024 × 1024 pixel image resolution for 4x zoom on the entire oocytes or 512 × 512 pixel image resolution for 8x zoom on the chromosome area. Three-dimensional scanning was performed using 1-µm optical sections through the oocyte volume.
Image analysis
Image analysis was performed using Fiji software.54 For γH2AX foci number analysis we segmented γH2AX signal by applying an intensity threshold on 3D images previously denoised using PureDenoise plugin.55 γH2AX foci number was measured by 3D Object Counter56 on the segmented 3D images. γH2AX fluorescence associated with chromatin was measured by redirection of 3D mask of intensity-thresholded DNA signal to γH2AX channel.
We denoised the original time-lapse images in Figures S1A and S3B by applying Gaussian blur (r=1). Securin-EGFP intensity in Figure S1C was measured on maximum intensity z-projection images of the oocytes. Spindle volume in Fig. S3B was measured from 3D+t reconstructed images when EGFP-MAP4 signal was intensity-thresholded.
Western blot
For immunoblotting, 200 oocytes per sample were collected at GV stage (0 h) or MII stage (7 h). Oocytes were lysed in 10 μl of 1 × Reducing SDS Loading Buffer (Cell Signaling) and heated at 100°C for 3 min. Proteins were separated by gradient 4–12% SDS–PAGE and transferred to a nitrocellulose membrane using a dry blotting system iBlot (Life Technologies). The membrane was blocked in 5% milk in 0.05% Tris-buffer saline-Tween (TBST), pH 7.4 for 1 h. The membrane was then washed in TBST and incubated at 4°C overnight in the presence of the MRE11 antibody (Cell Signaling Technology, 4895) at dilution 1:1000. To visualize the proteins an ECL kit (Amersham) was used and films were scanned with a GS-800 densitometer (Bio-Rad).
Statistical analysis
The data for most of the results were obtained from at least 3 independent experiments. The data are pooled. NCSS2007 software (NCSS, LLC., Kaysville Utah) was used for statistical analysis. Differences in foci number and immunofluorescence intensity were evaluated by the Student t-test. Differences in medians were measured by Mann-Whitney test when the data showed non-normal distribution. Samples with P<0.05 were considered as statistically significant.
Supplementary Material
Abbreviations
- 53BP1
tumor protein p53 binding protein 1
- a-NHEJ
alternative non-homologous end-joining
- APC/C
anaphase - promoting complex/cyclosome
- ATM
Ataxia telangiectasia mutated
- AURKA
Aurora kinase A
- BRCA1
breast cancer 1, early onset
- CDC25B
cell division cycle 25B
- CDK1
cyclin-dependent kinase 1
- CENPC
centromere protein C
- CHK (1,2)
checkpoint kinase (1,2)
- CtIP
C-terminal binding protein interacting protein
- DDR
DNA damage response
- DNA-PKcs
DNA-dependent protein kinase catalytic subunit
- DSBs
DNA double strand breaks
- EGFP
enhanced green fluorescent protein
- GTP
guanosine triphosphate
- GV
germinal vesicle
- hCG
human chorionic gonadotropin
- HR
homologous recombination
- Kd
knock down
- MDC1
mediator of DNA damage checkpoint protein 1
- MI
metaphase I
- MII
metaphase II
- MRE11
meiotic recombination 11 homolog
- MRN
complex MRE11, RAD50 and NBS1 complex
- NBS1
Nijmegen breakage syndrome 1
- NCS
Neocarzinostatin
- NHEJ
non-homologous end-joining
- PIKKs
phosphatidylinositol 3-kinase-related kinases
- PLK1
polo-like kinase 1
- PMI
propmetaphase I
- PUMA
p53 up-regulated modulator of apoptosis
- RAN
RAS-related nuclear protein
- RNF
(8, 16) ring finger protein (8, 16)
- SAC
spindle assembly checkpoint
- UV
ultraviolet
- XRCC 4
X-ray repair cross-complementing protein 4
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This study was supported by grant LH12057 (Czech-US scientific program) to P.S. Work at IAPG was supported by Institutional Research Concept 67985904. This work benefited from European Regional Development Fund, project ExAM - CZ.1.05/2.1.00/03.0124 to. J.M. Y.S. was supported by a RIKEN Special Postdoctoral Fellowship. T.S.K. was supported by a grant for JSPS Bilateral Joint Research projects with P.S and by RIKEN CDB. V.B was supported by a grant from SRDA (APVV-0237-10). R.M.S. was supported by a grant from NIH (HD022681).
References
- [1].Altmeyer M, Lukas J. To spread or not to spread-chromatin modifications in response to DNA damage. Curr Opin Genet Dev 2013; 23:156–65; PMID:23312207; http://dx.doi.org/ 10.1016/j.gde.2012.11.001 [DOI] [PubMed] [Google Scholar]
- [2].Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol 2011; 12:90–103; PMID:21252998; http://dx.doi.org/ 10.1038/nrm3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rein K, Stracker TH. The MRE11 complex: an important source of stress relief. Exp Cell Res 2014; 329:162–9; PMID:25447316; http://dx.doi.org/ 10.1016/j.yexcr.2014.10.010 [DOI] [PubMed] [Google Scholar]
- [4].Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J 2003; 22:6610–20; PMID:14657032; http://dx.doi.org/ 10.1093/emboj/cdg630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 2003; 22:5612–21; PMID:14532133; http://dx.doi.org/ 10.1093/emboj/cdg541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 2001; 276:42462–7; PMID:11571274; http://dx.doi.org/ 10.1074/jbc.C100466200 [DOI] [PubMed] [Google Scholar]
- [7].Jungmichel S, Stucki M. MDC1: The art of keeping things in focus. Chromosoma 2010; 119:337–49; PMID:20224865; http://dx.doi.org/ 10.1007/s00412-010-0266-9 [DOI] [PubMed] [Google Scholar]
- [8].Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179–204; PMID:20965415; http://dx.doi.org/ 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Baudat F, Manova K, Yuen JP, Jasin M, Keeney S. Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell 2000; 6:989–98; PMID:11106739; http://dx.doi.org/ 10.1016/S1097-2765(00)00098-8 [DOI] [PubMed] [Google Scholar]
- [10].Kerr JB, Hutt KJ, Michalak EM, Cook M, Vandenberg CJ, Liew SH, Bouillet P, Mills A, Scott CL, Findlay JK, et al.. DNA damage-induced primordial follicle oocyte apoptosis and loss of fertility require TAp63-mediated induction of Puma and Noxa. Mol Cell 2012; 48:343–52; PMID:23000175; http://dx.doi.org/ 10.1016/j.molcel.2012.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Carroll J, Marangos P. The DNA damage response in mammalian oocytes. Front Genet 2013; 4:117; PMID:23805152; http://dx.doi.org/ 10.3389/fgene.2013.00117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lin F, Ma XS, Wang ZB, Wang ZW, Luo YB, Huang L, Jiang ZZ, Hu MW, Schatten H, Sun QY. Different fates of oocytes with DNA double-strand breaks in vitro and in vivo. Cell Cycle 2014; 13:2674–80; PMID:25486355; http://dx.doi.org/ 10.4161/15384101.2015.945375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ma JY, Ou Yang YC, Wang ZW, Wang ZB, Jiang ZZ, Luo SM, Hou Y, Liu ZH, Schatten H, Sun QY. The effects of DNA double-strand breaks on mouse oocyte meiotic maturation. Cell Cycle 2013; 12:1233–41; PMID:23518501; http://dx.doi.org/ 10.4161/cc.24311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Marangos P, Carroll J. Oocytes progress beyond prophase in the presence of DNA damage. Curr Biol 2012; 22:989–94; PMID:22578416; http://dx.doi.org/ 10.1016/j.cub.2012.03.063 [DOI] [PubMed] [Google Scholar]
- [15].Yuen WS, Merriman JA, O'Bryan MK, Jones KT. DNA double strand breaks but not interstrand crosslinks prevent progress through meiosis in fully grown mouse oocytes. PLoS One 2012; 7:e43875; PMID:22928046; http://dx.doi.org/ 10.1371/journal.pone.0043875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ashwood-Smith MJ, Edwards RG. DNA repair by oocytes. Mol Hum Reprod 1996; 2:46–51; PMID:9238657; http://dx.doi.org/ 10.1093/molehr/2.1.46 [DOI] [PubMed] [Google Scholar]
- [17].Titus S, Li F, Stobezki R, Akula K, Unsal E, Jeong K, Dickler M, Robson M, Moy F, Goswami S, et al.. Impairment of BRCA1-related DNA double-strand break repair leads to ovarian aging in mice and humans. Sci Transl Med 2013; 5:172ra21; PMID:23408054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Brazill JL, Masui Y. Changing levels of uv light and carcinogen-induced unscheduled DNA synthesis in mouse oocytes during meiotic maturation. Exp Cell Res 1978; 112:121–5; PMID:415889; http://dx.doi.org/ 10.1016/0014-4827(78)90532-3 [DOI] [PubMed] [Google Scholar]
- [19].Masui Y, Pedersen RA. Ultraviolet light-induced unscheduled DNA synthesis in mouse oocytes during meiotic maturation. Nature 1975; 257:705–6; PMID:1237799; http://dx.doi.org/ 10.1038/257705a0 [DOI] [PubMed] [Google Scholar]
- [20].Kujjo LL, Laine T, Pereira RJ, Kagawa W, Kurumizaka H, Yokoyama S, Perez GI. Enhancing survival of mouse oocytes following chemotherapy or aging by targeting Bax and Rad51. PLoS One 2010; 5:e9204; PMID:20169201; http://dx.doi.org/ 10.1371/journal.pone.0009204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Perez GI, Acton BM, Jurisicova A, Perkins GA, White A, Brown J, Trbovich AM, Kim MR, Fissore R, Xu J, et al.. Genetic variance modifies apoptosis susceptibility in mature oocytes via alterations in DNA repair capacity and mitochondrial ultrastructure. Cell Death Differ 2007; 14:524–33; PMID:17039249; http://dx.doi.org/ 10.1038/sj.cdd.4402050 [DOI] [PubMed] [Google Scholar]
- [22].Hadjantonakis AK, Papaioannou VE. Dynamic in vivo imaging and cell tracking using a histone fluorescent protein fusion in mice. BMC Biotechnol 2004; 4:33; PMID:15619330; http://dx.doi.org/ 10.1186/1472-6750-4-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sakakibara Y, Hashimoto S, Nakaoka Y, Kouznetsova A, Hoog C, Kitajima TS. Bivalent separation into univalents precedes age-related meiosis I errors in oocytes. Nat Commun 2015; 6:7550; PMID:26130582; http://dx.doi.org/ 10.1038/ncomms8550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Herbert M, Levasseur M, Homer H, Yallop K, Murdoch A, McDougall A. Homologue disjunction in mouse oocytes requires proteolysis of securin and cyclin B1. Nat Cell Biol 2003; 5:1023–5; PMID:14593421; http://dx.doi.org/ 10.1038/ncb1062 [DOI] [PubMed] [Google Scholar]
- [25].Ichijima Y, Sakasai R, Okita N, Asahina K, Mizutani S, Teraoka H. Phosphorylation of histone H2AX at M phase in human cells without DNA damage response. Biochem Biophys Res Commun 2005; 336:807–12; PMID:16153602; http://dx.doi.org/ 10.1016/j.bbrc.2005.08.164 [DOI] [PubMed] [Google Scholar]
- [26].McManus KJ, Hendzel MJ. ATM-dependent DNA damage-independent mitotic phosphorylation of H2AX in normally growing mammalian cells. Mol Biol Cell 2005; 16:5013–25; PMID:16030261; http://dx.doi.org/ 10.1091/mbc.E05-01-0065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tu WZ, Li B, Huang B, Wang Y, Liu XD, Guan H, Zhang SM, Tang Y, Rang WQ, Zhou PK. gammaH2AX foci formation in the absence of DNA damage: mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett 2013; 587:3437–43; PMID:24021642; http://dx.doi.org/ 10.1016/j.febslet.2013.08.028 [DOI] [PubMed] [Google Scholar]
- [28].Golding SE, Rosenberg E, Valerie N, Hussaini I, Frigerio M, Cockcroft XF, Chong WY, Hummersone M, Rigoreau L, Menear KA, et al.. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Therapeutics 2009; 8:2894–902; PMID:19808981; http://dx.doi.org/ 10.1158/1535-7163.MCT-09-0519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 2001; 276:47759–62; PMID:11673449; http://dx.doi.org/ 10.1074/jbc.M009785200 [DOI] [PubMed] [Google Scholar]
- [30].Royo H, Prosser H, Ruzankina Y, Mahadevaiah SK, Cloutier JM, Baumann M, Fukuda T, Hoog C, Toth A, de Rooij DG, et al.. ATR acts stage specifically to regulate multiple aspects of mammalian meiotic silencing. Genes Dev 2013; 27:1484–94; PMID:23824539; http://dx.doi.org/ 10.1101/gad.219477.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dupre A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, et al.. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat Chem Biol 2008; 4:119–25; PMID:18176557; http://dx.doi.org/ 10.1038/nchembio.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rozier L, Guo Y, Peterson S, Sato M, Baer R, Gautier J, Mao Y. The MRN-CtIP pathway is required for metaphase chromosome alignment. Mol Cell 2013; 49:1097–107; PMID:23434370; http://dx.doi.org/ 10.1016/j.molcel.2013.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bakhoum SF, Kabeche L, Murnane JP, Zaki BI, Compton DA. DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov 2014; 4:1281–9; PMID:25107667; http://dx.doi.org/ 10.1158/2159-8290.CD-14-0403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chiang T, Duncan FE, Schindler K, Schultz RM, Lampson MA. Evidence that weakened centromere cohesion is a leading cause of age-related aneuploidy in oocytes. Curr Biol 2010; 20:1522–8; PMID:20817534; http://dx.doi.org/ 10.1016/j.cub.2010.06.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kuo LJ, Yang LX. Gamma-H2AX - a novel biomarker for DNA double-strand breaks. In Vivo 2008; 22:305–9; PMID:18610740 [PubMed] [Google Scholar]
- [36].Mah LJ, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 2010; 24:679–86; PMID:20130602; http://dx.doi.org/ 10.1038/leu.2010.6 [DOI] [PubMed] [Google Scholar]
- [37].Wrann S, Kaufmann MR, Wirthner R, Stiehl DP, Wenger RH. HIF mediated and DNA damage independent histone H2AX phosphorylation in chronic hypoxia. Biol Chem 2013; 394:519–28; PMID:23241668; http://dx.doi.org/ 10.1515/hsz-2012-0311 [DOI] [PubMed] [Google Scholar]
- [38].Kujjo LL, Ronningen R, Ross P, Pereira RJ, Rodriguez R, Beyhan Z, Goissis MD, Baumann T, Kagawa W, Camsari C, et al.. RAD51 plays a crucial role in halting cell death program induced by ionizing radiation in bovine oocytes. Biol Reprod 2012; 86:76; PMID:22190703; http://dx.doi.org/ 10.1095/biolreprod.111.092064 [DOI] [PubMed] [Google Scholar]
- [39].Acilan C, Potter DM, Saunders WS. DNA repair pathways involved in anaphase bridge formation. Genes Chromosomes Cancer 2007; 46:522–31; PMID:17366618; http://dx.doi.org/ 10.1002/gcc.20425 [DOI] [PubMed] [Google Scholar]
- [40].Lemmens BB, Johnson NM, Tijsterman M. COM-1 promotes homologous recombination during Caenorhabditis elegans meiosis by antagonizing Ku-mediated non-homologous end joining. PLoS Genetics 2013; 9:e1003276; PMID:23408909; http://dx.doi.org/ 10.1371/journal.pgen.1003276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yin Y, Smolikove S. Impaired resection of meiotic double-strand breaks channels repair to nonhomologous end joining in Caenorhabditis elegans. Mol Cell Biol 2013; 33:2732–47; PMID:23671188; http://dx.doi.org/ 10.1128/MCB.00055-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Terasawa M, Shinohara A, Shinohara M. Double-strand break repair-adox: Restoration of suppressed double-strand break repair during mitosis induces genomic instability. Cancer Sci 2014; 105:1519–25; PMID:25287622; http://dx.doi.org/ 10.1111/cas.12551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lee DH, Acharya SS, Kwon M, Drane P, Guan Y, Adelmant G, Kalev P, Shah J, Pellman D, Marto JA, et al.. Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol Cell 2014; 54:512–25; PMID:24703952; http://dx.doi.org/ 10.1016/j.molcel.2014.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, Escribano-Diaz C, Durocher D. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 2014; 344:189–93; PMID:24652939; http://dx.doi.org/ 10.1126/science.1248024 [DOI] [PubMed] [Google Scholar]
- [45].Terasawa M, Shinohara A, Shinohara M. Canonical non-homologous end joining in mitosis induces genome instability and is suppressed by M-phase-specific phosphorylation of XRCC4. PLoS Genetics 2014; 10:e1004563; PMID:25166505; http://dx.doi.org/ 10.1371/journal.pgen.1004563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang Y, Jasin M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct Mol Biol 2011; 18:80–4; PMID:21131978; http://dx.doi.org/ 10.1038/nsmb.1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Montgomery E, Wilentz RE, Argani P, Fisher C, Hruban RH, Kern SE, Lengauer C. Analysis of anaphase figures in routine histologic sections distinguishes chromosomally unstable from chromosomally stable malignancies. Cancer Biol Ther 2003; 2:248–52; PMID:12878857; http://dx.doi.org/ 10.4161/cbt.2.3.362 [DOI] [PubMed] [Google Scholar]
- [48].Verdun RE, Crabbe L, Haggblom C, Karlseder J. Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol Cell 2005; 20:551–61; PMID:16307919; http://dx.doi.org/ 10.1016/j.molcel.2005.09.024 [DOI] [PubMed] [Google Scholar]
- [49].Tsafriri A, Chun SY, Zhang R, Hsueh AJ, Conti M. Oocyte maturation involves compartmentalization and opposing changes of cAMP levels in follicular somatic and germ cells: studies using selective phosphodiesterase inhibitors. Dev Biol 1996; 178:393–402; PMID:8812137; http://dx.doi.org/ 10.1006/dbio.1996.0226 [DOI] [PubMed] [Google Scholar]
- [50].Solc P, Saskova A, Baran V, Kubelka M, Schultz RM, Motlik J. CDC25A phosphatase controls meiosis I progression in mouse oocytes. Dev Biol 2008; 317:260–9; PMID:18367163; http://dx.doi.org/ 10.1016/j.ydbio.2008.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kitajima TS, Ohsugi M, Ellenberg J. Complete kinetochore tracking reveals error-prone homologous chromosome biorientation in mammalian oocytes. Cell 2011; 146:568–81; PMID:21854982; http://dx.doi.org/ 10.1016/j.cell.2011.07.031 [DOI] [PubMed] [Google Scholar]
- [52].Kudo NR, Wassmann K, Anger M, Schuh M, Wirth KG, Xu H, Helmhart W, Kudo H, McKay M, Maro B, et al.. Resolution of chiasmata in oocytes requires separase-mediated proteolysis. Cell 2006; 126:135–46; PMID:16839882; http://dx.doi.org/ 10.1016/j.cell.2006.05.033 [DOI] [PubMed] [Google Scholar]
- [53].Schuh M, Ellenberg J. Self-organization of MTOCs replaces centrosome function during acentrosomal spindle assembly in live mouse oocytes. Cell 2007; 130:484–98; PMID:17693257; http://dx.doi.org/ 10.1016/j.cell.2007.06.025 [DOI] [PubMed] [Google Scholar]
- [54].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al.. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9:676–82; PMID:22743772; http://dx.doi.org/ 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Luisier F, Vonesch C, Blu T, Unser M. Fast interscale wavelet denoising of Poisson-corrupted images. Signal Proc 2010; 90:415–27; http://dx.doi.org/ 10.1016/j.sigpro.2009.07.009 [DOI] [Google Scholar]
- [56].Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 2006; 224:213–32; PMID:17210054; http://dx.doi.org/ 10.1111/j.1365-2818.2006.01706.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.