

Abstract
ERK5, encoded by MAPK7, has been proposed to play a role in cell proliferation, thus attracting interest as a cancer therapeutic target. While oncogenic RAS or BRAF cause sustained activation of the MEK1/2-ERK1/2 pathway, ERK5 is directly activated by MEK5. It has been proposed that RAS and RAF proteins can also promote ERK5 activation. Here we investigated the interplay between RAS-RAF-MEK-ERK and ERK5 signaling and studied the role of ERK5 in tumor cell proliferation in 2 disease-relevant cell models. We demonstrate that although an inducible form of CRAF (CRAF:ER*) can activate ERK5 in fibroblasts, the response is delayed and reflects feed-forward signaling. Additionally, oncogenic KRAS and BRAF do not activate ERK5 in epithelial cells. Although KRAS and BRAF do not couple directly to MEK5-ERK5, ERK5 signaling might still be permissive for proliferation. However, neither the selective MEK5 inhibitor BIX02189 or ERK5 siRNA inhibited proliferation of colorectal cancer cells harbouring KRASG12C/G13D or BRAFV600E. Furthermore, there was no additive or synergistic effect observed when BIX02189 was combined with the MEK1/2 inhibitor Selumetinib (AZD6244), suggesting that ERK5 was neither required for proliferation nor a driver of innate resistance to MEK1/2 inhibitors. Finally, even cancer cells with MAPK7 amplification were resistant to BIX02189 and ERK5 siRNA, showing that ERK5 amplification does not confer addiction to ERK5 for cell proliferation. Thus ERK5 signaling is unlikely to play a role in tumor cell proliferation downstream of KRAS or BRAF or in tumor cells with ERK5 amplification. These results have important implications for the role of ERK5 as an anti-cancer drug target.
Keywords: BIX02189, ERK5, MEK5, RAS, RAF, selumetinib, tumor cell proliferation
Introduction
The mitogen-activated protein kinase (MAPK) signaling cascades transduce extracellular signals within the cell, regulating cytosolic and nuclear factors to influence cell fate, proliferation, survival and motility. There are 4 MAPK kinase pathways; extracellular-signal regulated kinase (ERK) 1/2,1 c-Jun N-terminal kinase (JNK),2 p38 MAPK3 and ERK5 (also known as Big MAP Kinase 1 (BMK1)) pathways, the latter of which is the least well characterized.4,5 While ERK1/2 are strongly activated by growth factors and JNK and p38 by cellular stresses, ERK5 is responsive to both growth factor and stresses. The ERK1/2 pathway comprises the small GTPase RAS, which activates the RAF protein kinases (MKKKs), which in turn activate MEK1/2 (MAPK or ERK kinase, a MKK), which finally activates ERK1/2 (the MAPK).6 The ERK5 pathway comprises MEKK2 and MEKK3 (the MKKKs), MEK5 (MKK) and finally ERK5 (MAPK).4,5 The ERK5 protein is encoded by the MAPK7 gene.4 It contains an N-terminal kinase domain that shares 50% identity with ERK2 and a large, unique C-terminal extension that contains a transactivation domain, nuclear localization and export sequences and 2 proline-rich regions. Owing to its large size, ERK5 is sometimes referred to as BMK1 (big MAPK1).5
The RAS-RAF-MEK1/2-ERK1/2 pathway is frequently de-regulated in human cancer due to amplification or mutation of receptor tyrosine kinases and mutations in RAS (especially KRAS) or BRAF (e.g. BRAFV600E).7 These mutations drive hyperactivation of ERK1/2 which in turn promotes tumor cell proliferation and survival. Many tumor cells become addicted to ERK1/2 signaling providing an opportunity for tumor-selective therapeutic intervention.7 Indeed, the highly selective BRAFV600E inhibitor vemurafenib8 is now approved for the treatment of BRAFV600E mutant melanoma, while MEK1/2 inhibitors such as trametinib9 or selumetinib (AZD6244/ARRY-142886)10 are either approved or in late stage clinical development. However, the success of such targeted therapies has been limited by the emergence of acquired resistance11,12 so there is an urgent need to identify other disease driving pathways that can be targeted in drug combination strategies.
Since ERK5 signaling is activated by growth factors, it is possible that it too is hyper-activated in cancer and may serve as a drug target. Indeed, ERK5 signaling has been proposed to play a role in receptor tyrosine kinase driven proliferation of the cervical cancer cell line HeLa,13 the breast cancer cell lines MCF7 and BT474,14 and the immortalised breast epithelial cell line MCF10A.13
In contrast, the role of ERK5 downstream of RAS or RAF or in RAS- or BRAF-dependent tumors is less clear and is subject to conflicting results. Early studies indicated that oncogenic HRASG12V could activate a co-expressed mutant form of ERK5 consisting of only the kinase domain in HEK293 cells.15 Subsequently HRASG12V was shown to activate ERK5 in transfected PC12 cells but not in COS7 cells, indicating that Ras-ERK5 coupling might be cell type specific,16 Crosstalk also exists between the ERK1/2 and ERK5 pathways; MEK5D, an active form of MEK5, co-operated with CRAF to transform NIH 3T3 cells.15 Conversely, ERK1/2 signaling can also inhibit ERK5 signaling, since selective inhibition of ERK1/2 enhanced and sustained activation of ERK5.17,18 The relationship between ERK1/2 and ERK5 signaling is clearly complex and these studies suggest that ERK5 may lie downstream of RAS and RAF or ERK5 may be subject to negative-feedback regulation by strong ERK1/2 activation.
Other studies implicated increased ERK5 protein levels in tumor progression as high ERK5 expression was associated with decreased disease-free survival in breast cancer,19,20 while in prostate cancer elevated MEK5 levels correlated with the presence of bone metastases and less favorable disease-specific survival.21 Indeed, over-expression of MEK5 induces proliferation of the prostate cancer cell line LNCaP.21 Finally, the ERK5 locus is amplified in approximately 50% of primary hepatocellular carcinomas.22
Here we investigated the interplay between RAF-MEK1/2-ERK1/2 signaling and the MEK5-ERK5 pathway and assessed the role of ERK5 signaling in 2 relevant cancer cell models; colorectal cancer cells harbouring mutant KRAS or BRAF and cancer cells that express high levels of ERK5 due to MAPK7 amplification. We show that in fibroblasts, ERK5 can be activated downstream of an inducible CRAF:ER* construct; however, this response was delayed, resulting from ERK1/2 activation and required new protein synthesis. We find no evidence of ERK5 activation by mutant KRAS or BRAF in epithelial cells, even upon overexpression and even when the ERK1/2 pathway is inhibited to remove any inhibitory cross talk. Proliferation of a panel of CRC cells lines with either KRAS or BRAF mutation was refractory to inhibition by the MEK5 inhibitor BIX02189, and siRNA-mediated knockdown of ERK5 had no effect on the proliferation of HCT116 cells, arguing against a role for ERK5 in promoting tumor cell proliferation downstream of RAS or BRAF. Finally, the proliferation of multiple cancer cell lines harbouring MAPK7 amplification was insensitive to BIX02189 or siRNA to ERK5, suggesting that even ERK5 amplification does not make a strong contribution to tumor cell proliferation.
Results
Sustained CRAF:ER activity leads to a delayed activation of ERK5 downstream of ERK1/2 that requires new protein synthesis in fibroblasts
To determine if activation of the RAF-MEK1/2-ERK1/2 pathway could influence activation of ERK5 we used CR1–11 cells, a stable clone of CCl39 fibroblasts that stably express the conditional kinase ΔCRAF:ER*.23 Treatment of these cells with 4-hydroxytamoxifen (4HT) resulted in the rapid (within 15 mins) and sustained activation of ERK1 (Fig. 1A). Interestingly, ERK5 activity was also substantially increased, but its activation was delayed by 2–3 hours compared to that of ERK1. To determine if the delayed activation of ERK5 was dependent upon prior ERK1/2 activation we used PD184352, an allosteric MEK1/2-selective inhibitor.24 Although this drug can also inhibit ERK5 signaling at doses above 10 µM, we previously showed that at low doses it abolishes ERK1/2 signaling with no effect on ERK5.18 Pre-treatment with 100nM PD184352 before ΔCRAF:ER* activation with 4-HT prevented activation of both ERK1 and ERK5, demonstrating that the delayed ΔCRAF:ER*-driven ERK5 activation was dependent on prior activation of ERK1/2 (Fig. 1A). Next we assessed the requirement of ERK1/2 signaling for ERK5 activity. To do this we first activated ΔCRAF:ER* with 4-HT then treated with PD184352 for the times indicated. ERK1 activity declined immediately, whereas ERK5 activity persisted for at least 4h before gradually declining (Fig. 1B). Together these results demonstrate that ERK5 can be activated as a delayed consequence of ERK1/2 activation downstream of CRAF:ER in fibroblasts.
Figure 1.
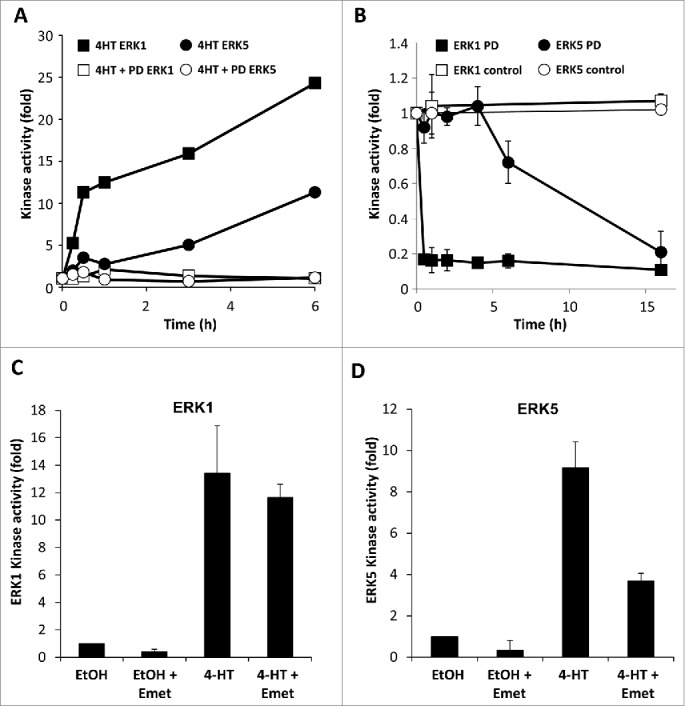
Sustained CRAF:ER activity leads to the delayed activation of ERK5 downstream of ERK1/2. (A) CR1-11 cells were pre-treated for 90min with 0.1µM PD184352 or vehicle then treated with 100nM 4-HT for the times indicated. ERK1 and ERK5 were immune-precipitated and kinase activity determined as described in materials and methods. (B) CR1-11 cells were treated with 100nM 4-HT for 6 hours prior to the addition of 0.1µM PD184352 for the times indicated. ERK1 and ERK5 activity were determined as in (A). (C) and (D) CR1-11 cells were pre-treated with 10µM emetine (Emet) for 20min prior to an 8h treatment with 100nM 4HT. ERK1 and ERK5 activity were determined as in (A).
The slow increase in ERK5 activity compared with the rapid increase in ERK1 prompted us to determine if expression of a new protein was required for ERK5 activation. To this end CR1–11 cells were pre-treated with the protein synthesis inhibitor emetine prior to activation of CRAF:ER* with 4-HT. Activation of ERK1 proceeded normally in the presence of emetine (Fig. 1C) whereas ERK5 activation was inhibited by up to 60% in the presence of emetine (Fig. 1D), showing that synthesis of new protein(s) was required for ERK5 activation. The delayed kinetics and sensitivity to low doses of PD184352 and emetine suggest that in CCl39 fibroblasts CRAF can activate ERK5 indirectly, through an ERK1/2 dependent feed forward mechanism requiring protein neosynthesis.
Transient expression of oncogenic KRAS and BRAF does not activate ERK5 signaling in epithelial cells
Prompted by these results we sought to investigate if ERK5 might be activated downstream of ERK1/2 activation arising from mutant KRAS and BRAF in epithelial cells. To this end we employed HEK293 cells and transiently transfected ERK5 and its upstream activator MEK5 (wt), together with either the active forms of KRAS (KRASG12V) or BRAF (BRAFV600E) or the kinase dead form of BRAF (BRAFD594A) and KRASG12V (as they are known to co-operate with CRAF in ERK1/2 signaling25). ERK5 activation was assessed by immuno-blotting for dual-phosphorylation of the activation-loop (T218, Y220) and by the reduction in mobility that is observed following kinase activation and auto-phosphorylation. Co-expression of MEK5D (a constitutively active form of MEK5 where the activation-loop phosphorylation sites are mutated to aspartic acid) strongly induced phosphorylation of the T218EY220 motif in ERK5 and caused a decrease in ERK5 mobility (Fig. 2A). In contrast, the oncogenic forms of KRAS and BRAF did not induce phosphorylation of ERK5. KRASG12V, BRAFV600E and BRAFD549A co-expressed with KRASG12V all induced substantial phosphorylation of endogenous ERK1/2, clearly demonstrating that these mutants were active in HEK293 cells (Fig. 2A). As an alternative and more sensitive measure of ERK5 activity in cells, we used a GAL4 UAS:luciferase reporter system which is driven by the transactivation domain of MEF2D (an ERK5 substrate that is transcriptionally active when phosphorylated) fused to the GAL4 DNA binding domain.26 The ERK5 MEF2D:GAL4 UAS:luciferase reporter system was transfected into HEK293 cells with either wild type MEK5 and the oncogenic forms of KRAS or BRAF or MEK5D for 24h. Whereas we detected strong activation of MEF2D-mediated luciferase activity with MEK5D, we were unable to detect any increase in luciferase activity over the small increase observed upon expression of wild type MEK5 following co-transfection with KRASG12V, BRAFV600E or BRAFD594A with KRASG12V (Fig. 2B). Together these results demonstrate that oncogenic forms of BRAF and KRAS can activate ERK1/2 but cannot activate ERK5 in HEK293 cells.
Figure 2.
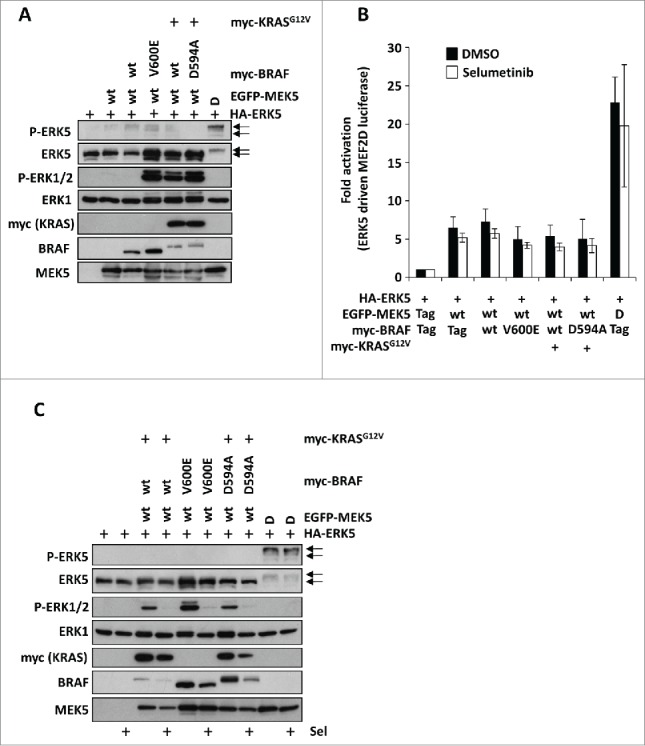
Oncogenic KRAS and BRAF do not activate ERK5 signaling when transiently expressed in HEK293 cells. (A) HEK293 cells were transfected with HA-ERK5 and either EGFP-MEK5 wt, 5D or vector control; with either myc-BRAF wt, V600E, D594A or vector control; and with myc-KRASG12V or vector control as indicated. 24h post-transfection cells were lysed and whole cell lysates were separated by SDS-PAGE then immunoblotted with the antibodies indicated. (B) HEK293 cell were transfected as in (A) together with GAL4-MEF2D, GAL4:LUC and a Renilla construct. 6h post transfection cell were treated with 1µM selumetinib (sel) or vehicle control as indicated. Following 24h cells were then lysed and firefly luciferase activity was measured and normalized to Renilla. A representative experiment of 3 is shown and values are expressed as the mean of triplicate transfections ± SD (C) HEK293 cells were transfected as in (A) then 6h post transfection cell were treated with 2 µM selumetinib (sel) or vehicle control as indicated. Following 24h cells were lysed and whole cell lysates were separated by SDS-PAGE then immunoblotted with the antibodies indicated.
Strong ERK1/2 activation by oncogenic KRAS and BRAF does not repress activation of ERK5 in HEK293 cells
ERK1/2 signaling is known to inhibit ERK5 activation since treatment of HeLa or CCL39 fibroblasts with the MEK1/2 inhibitor PD184352 enhances MEK5-ERK5 signaling.17,18 Therefore we speculated that the lack of ERK5 activation seen following co-expression of oncogenic KRAS or BRAF was due to the strong activation of ERK1/2 signaling that was driving a persistent or tonic inhibition of the ERK5 pathway. To address this we transfected cells with oncogenic KRAS or BRAF with MEK5 and ERK5 in the presence or absence of the highly selective allosteric MEK1/2 inhibitor selumetinib (also known as AZD6244/ARRY-142886) to repress ERK1/2 signaling and monitored the activation and activity of ERK5. Immuno-blotting for phospho-ERK1/2 demonstrated that selumetinib inhibited the ERK1/2 phosphorylation induced by KRASG12V, BRAFV600E or co-expression of BRAFD594A and KRASG12V. However, even when ERK1/2 signaling was inhibited, the oncogenic forms of KRAS and BRAF did not stimulate ERK5 phosphorylation (Fig. 2C). Furthermore, the presence of selumetinib failed to increase ERK5-dependent activation of the MEF2D luciferase reporter (Fig. 2B). Thus the failure of oncogenic KRAS and BRAF to activate ERK5 was not due to chronic inhibition of the ERK5 pathway by ERK1/2 signaling.
MEK5-ERK5 signaling is dispensable for cell proliferation of colorectal cancer cells
ERK5 has been proposed to play a role in the proliferation and survival of many cancer cells lines.13,14,21,27,28 Thus, although KRAS and BRAF failed to activate ERK5 it remained possible that basal ERK5 signaling was permissive for proliferation in tumor cells harbouring these oncogenes. We therefore investigated the role of MEK5-ERK5 signaling in a panel of CRC cells. We approached this both chemically, using the MEK5 selective small molecule inhibitor BIX0218929 and genetically, using siRNA mediated knock-down of ERK5. We previously showed that BIX02189 selectively inhibits ERK5 activation in HEK293 cells and fibroblasts with no effect on ERK1/2 activity.30 We first defined the effective concentration range for inhibition of ERK5 signaling by BIX02189 in the CRC cell lines HCT116 (KRASG13D) and HT29 (BRAFV600E) using the MEK5D-driven MEF2D reporter system. BIX02189 caused a dose-dependent complete inhibition of ERK5 signaling with IC50 values of 0.3–1µM (Fig. 3A and 3B), as previously reported.29 When we assayed the effect of BIX02189 on cell proliferation we observed a substantial right shift in the dose response curves so that inhibition of cell proliferation was only observed at doses of 10µM or above either after 24 or 48h treatment, at which ERK5 activity was inhibited by >95%; furthermore, inhibition of cell proliferation was not always complete (Fig. 3A and 3B). The data suggested that doses of BIX02189 that completely inhibited ERK5 signaling had no effect on cell proliferation. We then determined the concentrations inhibiting cell proliferation by 50% (GI50) for BIX02189-dependent inhibition of proliferation in a panel of CRC cells and consistently determined a value >10µM (Fig. 3C) and yet ERK5-MEF2D activity was completely repressed at 1–5µM in all cell lines tested (Fig. 3D). To determine if cells grown in 3D culture, to more closely mimic tumor conditions in vivo, were sensitive to MEK5-ERK5 inhibition we treated HCT116 and HT29 cell spheroids with BIX02189, drawing comparisons with selumetinib. Selumetinib inhibits the proliferation of HCT116 and HT29 cells grown in 2D31 and also inhibited the viability of HCT116 and HT29 spheroids in a dose-dependent manner (Fig. 4A and 4B); in contrast, HCT116 and HT29 spheroids were insensitive to MEK5-ERK5 pathway inhibition by BIX02189 (Fig. 4A and 4B). Finally, siRNA mediated knock-down of ERK5 protein levels in HCT116 cells showed similar results; there was no effect on cell proliferation even when ERK5 levels were substantially knocked down (Fig. 5A and 5B). Taken together these results demonstrate that although ERK5 activity can be completely inhibited in a number of cell lines by BIX02189, the MEK5-ERK5 pathway is not required for proliferation of CRC cells with KRAS or BRAF mutations, either in 2D or 3D culture conditions.
Figure 3.

Inhibition of the MEK5-ERK5 signaling pathway by BIX02189 in colorectal cancer cells does not inhibit cell proliferation. (A and B) Subconfluent cultures of (A) HCT116 harbouring KRASmut or (B) HT29 harbouring BRAFmut cells were maintained in 10% FBS then treated with increasing concentrations of BIX02189 (100 nM to 30 µM) for 24 or 48 hours, and DNA synthesis was assayed by [3H]thymidine incorporation; the results are presented as an average of 3 experiments ± SD. Alternatively, cells were transfected as in Fig. 2(C), 6h post-transfection cell were treated with increasing concentrations of BIX02189 (100 nM to 30 µM) for 24 hours. Cells were then lysed and firefly luciferase activity was measured and normalized to Renilla. Results are presented as the average of 3 experiments ± SD. (C) Sub-confluent cultures of colorectal cancer cell lines harbouring KRASmut, BRAFmut or WT for both were maintained in 10% FBS and treated with increasing concentrations of BIX02189 for 24 hr. DNA synthesis was assayed by [3H]-thymidine incorporation. IC50 values for each cell line are shown; the results are presented as the mean of 3 experiments ± SD (D) Sub-confluent cultures of colorectal cancer cell lines harbouring KRASmut, BRAFmut or WT for both were transfected as in Fig. 2(C), 6h post-transfection cell were treated with either 1 or 5 µM BIX02189. Following 24h the cells were lysed and firefly luciferase activity was measured and normalized to Renilla. A representative experiment of 3 is shown and values are expressed as the mean of triplicate transfections ± SD.
Figure 4.
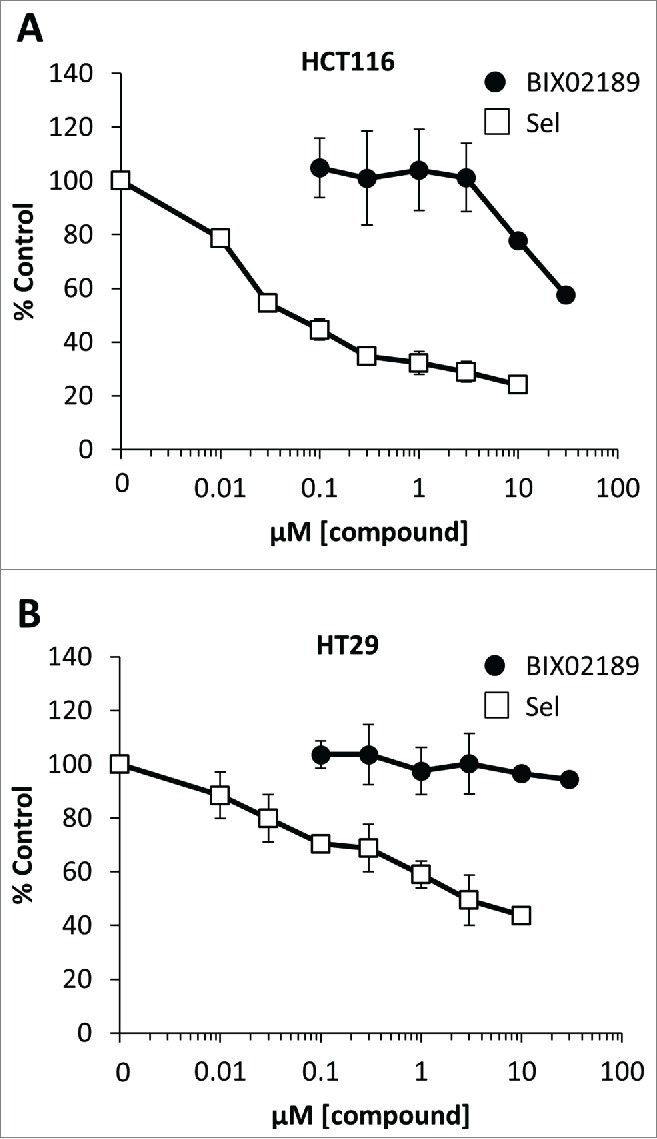
Inhibition of the MEK5-ERK5 signaling pathway by BIX02189 in colorectal cancer cell spheroids does not inhibit cell viability. (A) HCT116 harbouring KRASmut or (B) HT29 harbouring BRAFmut cells were gown in 10% FBS for 96h in ultra-low attachment plates to allow the formation of cell spheroids. The spheroids were then treated with either selumetinib (sel) or BIX02189 at the concentrations indicated for 72 h. Cell viability was determined using CellTitre Glo. The results are presented as the mean of 3 experiments ± the SD.
Figure 5.
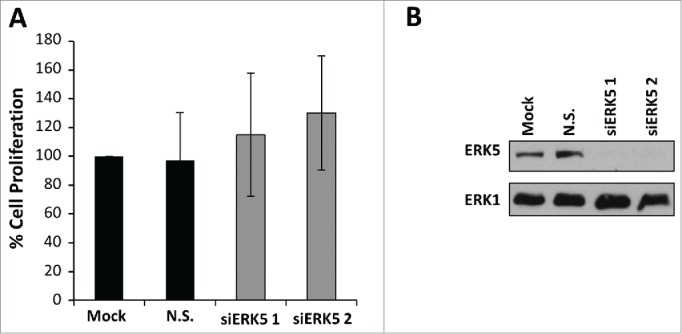
siRNA mediated knockdown of ERK5 in colorectal cancer cells does not inhibit cell proliferation. (A) HCT116 cells were transfected with ERK5-specific (siERK5 1 and 2) or non-silencing (N.S.) siRNA oligos. Mock cells were left untransfected. Forty-eight hours post-transfection, DNA synthesis was assayed by [3H]thymidine incorporation; the results are presented as an average of 3 experiments ± SD (B) HCT116 cells were transfected as in (A) ERK5 and ERK1 abundances were determined by Western blot analysis of whole-cell extracts.
MEK5-ERK5 signaling does not mediate intrinsic resistance to Selumetinib (AZD6244) in colorectal cancer cells
Intrinsic sensitivity or resistance of cancer cells that harbour KRAS or BRAF mutations to MEK1/2 inhibitors such as selumetinib can be predicted, in part, by a biochemical signature; cells with high ERK1/2 activity tend to be addicted to ERK1/2 signaling and are more responsive. In some instances intrinsic resistance is driven by the parallel PI3-kinase signaling pathway31; however in other instances the mechanism of intrinsic resistance is not known. To investigate whether ERK5 signaling might compensate for ERK1/2 and be a driver of intrinsic resistance we tested the effects of selumetinib in combination with BIX02189. The CRC cell lines showed varying sensitivities to selumetinib with respect to proliferation: HCT116, COLO205 and HT29 being the most sensitive as reported previously.31 As seen in Figure 3C, BIX02189 had no effect on proliferation of any of the cell lines tested. When the CRC cell lines were treated with the combination of selumetinib and BIX02189, there were no additive or synergistic effects observed (Fig. 6); BIX02189 could not overcome intrinsic resistance to selumetinib in cells such as CaCo-2 or DLD-1. This demonstrates that MEK5-ERK5 signaling plays no active role in the proliferation of CRC cells, even when ERK1/2 signaling is inhibited.
Figure 6.
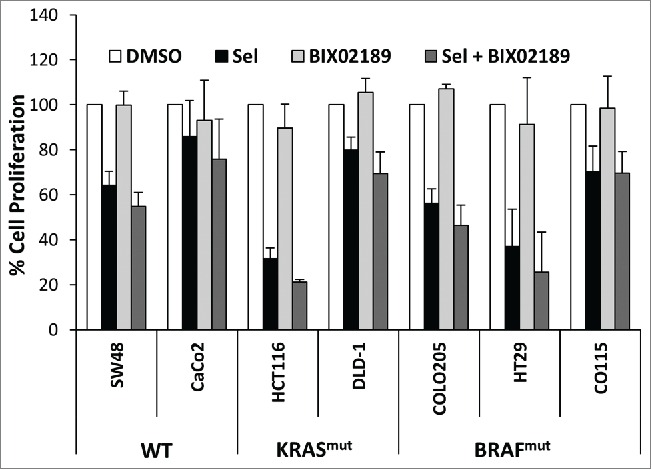
Inhibitors of ERK1/2 and ERK5 signaling do not synergise to inhibit colorectal cancer cell proliferation. Sub-confluent cultures of colorectal cancer cell lines harbouring KRASmut, BRAFmut or WT for both were maintained in 10% FBS and treated with 2 µM selumetinib (sel) and/or 1 µM BIX02189 for 24 hr. DNA synthesis was assayed by [3H]-thymidine incorporation; the results are presented as the mean of 3 experiments ± SD.
Amplification of the MAPK7 gene does not predict a requirement for ERK5 activity for cancer cell proliferation
An amplification at 17p11, which contains the MAPK7 gene (encoding ERK5 protein), is present at high frequency in hepatocellular carcinoma (HCC).22 The HCC cell line SNU449 contains this amplification, and expresses high levels of ERK5 compared to BT474 cells (also known to have high ERK5 expression) (Fig. 7A). We reasoned that cells with amplification of ERK5 were most likely to show strong ‘ERK5 addiction’ for proliferation. Indeed, siRNA-mediated knockdown of ERK5 has previously demonstrated a role for ERK5 in cell proliferation of SNU449 cells.22 We therefore tested the effects of BIX02189 on the proliferation of SNU449 cells. First we confirmed the efficacy of BIX02189 in SNU449 cells. Using the MEK5D-driven ERK5-MEF2D:GAL4 luciferase reporter, we demonstrated that ERK5 activity was inhibited following BIX02189 treatment with an IC50 of 0.2µM (Fig. 7B), similar to that observed in other cell types (Fig. 3A and 3B).29 However, there was no effect of BIX02189 on the proliferation of SNU449 at concentrations where ERK5 was fully inhibited (Fig. 7B). For example, 3 µM BIX02189 abolished ERK5 signaling but had no effect on SNU449 proliferation at 24 or 48 hours. To investigate this further we tested a panel of 4 different siRNA duplexes targeted toward ERK5 but found no effect on SNU449 cell proliferation (Fig. 7C) even though substantial knockdown of ERK5 protein levels was demonstrated (Fig. 7D).
Figure 7.
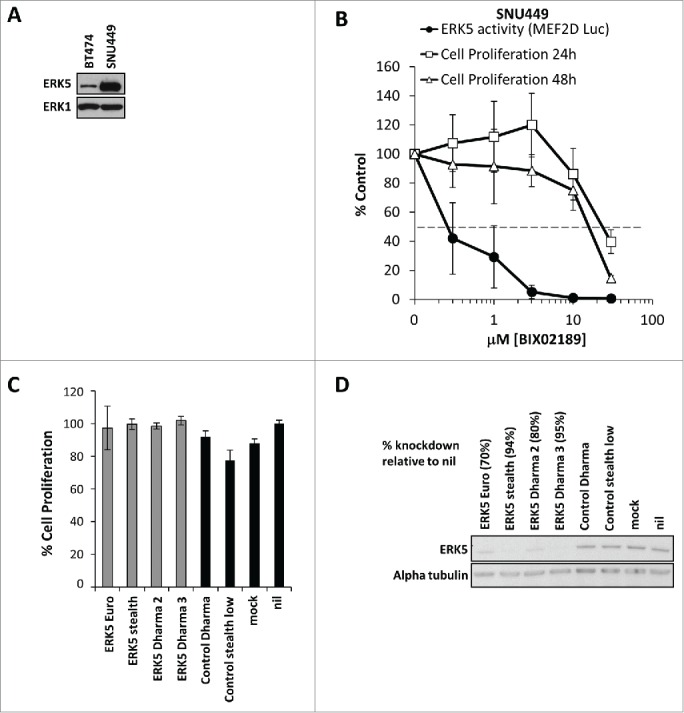
The hetatocellular carcinoma cell line SNU449 expresses high levels of ERK5 but is not dependent on MEK5-ERK5 signaling for proliferation. (A) Subconfluent cultures of BT474 and the liver hepatocellular carcinoma cell line SNU449 harbouring an amplification containing the ERK5 gene were maintained in 10% FBS. Cells were lysed, whole cell lysates were separated by SDS-PAGE and immunoblotted with the antibodies indicated. (B) Subconfluent cultures of SNU449 cells were maintained in 10% FBS then treated with increasing concentrations of BIX02189 (100 nM to 30 µM) for 24 or 48 hours, and DNA synthesis was assayed by [3H]thymidine incorporation; the results are presented as an average of 3 experiments ± SD. Alternatively, cells were transfected as in Fig. 2(C), 6h post-transfection cell were treated with increasing concentrations of BIX02189 (100 nM to 30 µM) for 24 hours. Cells were then lysed and firefly luciferase activity was measured and normalized to Renilla. A representative experiment of 3 is shown and values are expressed as the mean of triplicate transfections ± SD (C) SNU449 cells were transfected with ERK5-specific as indicated or non-silencing (control) siRNA oligos. Mock cells were left untransfected. Seventy-two hours post-transfection, ERK5 and α-tubulin abundances were determined by Western blot analysis of whole-cell extracts. (D) SNU449 cells were transfected as in (C) cell viability was determined by MTT assay. A representative experiment of 2 is shown and values are expressed as the mean of 8 values ± CoV.
Finally, we extended this study to investigate the effects of BIX02189 on a panel of cancer cell lines that had high ERK5 protein expression (Fig. 8A). We examined the esophageal cancer cell line KYSE30,32 a lung cancer cell line NCI-H838 (76th highest MAPK7 mRNA expression in the CCLE panel) and a renal cell line A498 (very high level of ERK5 protein expression, LG personal communication) (Fig. 8A). In these cell lines there was no clear correlation between ERK5 protein expression and sensitivity to BIX02189; all cell lines were comparatively refractory to treatment (Fig. 8B). In addition, the small cell lung cancer cell lines, NCI-H1436 and NCI-H1105, have the 6th and 7th highest MAPK7 mRNA expression in the Cancer Cell Line Encyclopaedia (CCLE; www.broadinstitute.org/ccle), respectively. However, the GI50 for BIX02189 was in excess 100μM (data not shown). Collectively, these data suggest that high ERK5 protein expression, including that arising from MAPK7 gene amplification, does not drive a dependency upon ERK5 activity for cancer cell proliferation.
Figure 8.
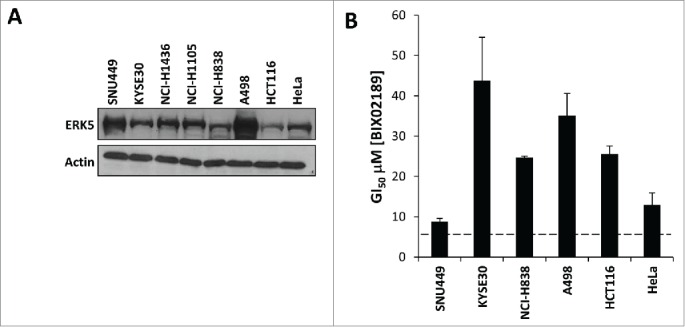
Tumor cell sensitivity to the MEK5-ERK pathway inhibitor BIX02189 does not correlate with ERK5 protein expression. (A) Relative ERK5 protein expression in a panel of tumor cell lines determined by immuno-blot analysis of whole-cell extracts. (B) GI50 data generated with BIX02189 in adherent tumor cells lines following 72 h of incubation. Data represent the mean ± SD of three independent experiments.
Discussion
There is an emerging interest in targeting therapeutics against the ERK5 pathway in the treatment of cancer.19,28,33-39 Clinical evidence associates high ERK5 and MEK5 levels with poor prognosis, whereas biochemical and cell biology studies have begun to investigate the molecular mechanisms by which the MEK5-ERK5 pathway contributes to cell transformation (reviewed in33,34,36). The MEK5-ERK5 pathway is proposed to be de-regulated by oncogenes (RAS,13,40 RAF,15 COT41 and SRC42-44) leading to an increase in activity, or by an increase in protein level due to copy number gain of the MAPK7/ERK5 gene22; thus highlighting ERK5 as a possible drug target. In this study we show that: (i) oncogenic KRAS and BRAF do not activate the MEK5-ERK5 pathway; (ii) cancer cells harbouring these mutations are not sensitive to MEK5-ERK5 pathway inhibition for proliferation; (iii) MEK5-ERK5 signaling does not mediate intrinsic resistance to MEK1/2-ERK1/2 pathway inhibitors and finally (iv) cancer cells that over-express ERK5 are not addicted to the MEK5-ERK5 pathway for proliferation.
We tested 3 clinically relevant oncogenic drivers of the RAS-RAF pathway; BRAFV600E, KRASG12V and the kinase dead form of BRAF (D594A) which cooperates with KRASG12V and CRAF. None of these oncogenic proteins or complexes could activate full-length ERK5 (Fig. 2). In some cell types the use of a dominant negative form of HRAS (N17) blocks growth factor induced ERK5 activation e.g., EGF and NGF simulated ERK5 activation in rat phaeochromocytoma PC12 cells and in mouse myoblast C2C12 cells.16 Perhaps this is an isoform specific role of HRAS or reflects the cell type specific expression of a particular adaptor or coupling protein; it will however be of interest to confirm these results with siRNA. We did find that CRAF:ER* could drive a feed forward activation of the ERK5 pathway downstream of ERK1/2 in CCl39 fibroblasts but this was dependent on de novo protein expression (Fig. 1) and probably reflects the ERK1/2-dependent expression of growth factors or cytokines that then act in an autocrine manner. There is strong precedent for ERK1/2 driving expression of growth factors and cytokines45-47 which are biologically active and can drive activation of other pathways downstream of ERK1/2 such as PI3K.48 The profile of such factors will no doubt be different in different cell types. Therefore, it is possible that some cells such as fibroblasts (Fig. 1) may exhibit indirect RAS or RAF driven ERK5 activation through downstream autocrine signaling, whereas HEK293 cells clearly do not (Fig. 2). Future studies should aim to determine the nature of this feed forward activation of ERK5 by ERK1/2.
Although we could not detect any direct or indirect activation of ERK5 by oncogenic KRAS or BRAF over-expressed in HEK293 cells it remained possible that ERK5 could be a permissive signal for KRAS or BRAF-driven proliferation or could be a driver of innate resistance to MEK1/2 inhibitors. For example, Mulloy et al49 demonstrated that expression of the G1 cyclin, CCND1, was stimulated by the MEK5-ERK5 pathway independently of the MEK1/2-ERK1/2 pathway. Since expression of CCND1 can confer resistance to ERK1/2 pathway inhibitors50 this might be a plausible role for ERK5 signaling. It is also consistent with the observation that constitutively active MEK5 can drive the proliferation of HEK293 and LNCaP cells.21 However, in colon cancer cells harbouring KRAS or BRAF mutations chemical or genetic inhibition of the MEK5-ERK5 pathway did not inhibit proliferation (Figs. 3, 4 and 5) demonstrating that this pathway was not important for proliferation in these tumor cells. MEK1/2-ERK1/2 signaling is known to inhibit ERK5 signaling in some cells,17,18 thus it was possible that treatment of KRAS- or BRAF-driven tumor cells with MEK1/2 inhibitors might activate the MEK5-ERK5 pathway and reveal a dependency on this pathway for cell proliferation. Indeed, not all KRAS and BRAF driven tumor cells undergo cell cycle inhibition upon MEK1/2 inhibitor treatment.31 However, we did not observe ERK5 activation in transient transfection experiments with oncogenic KRAS or BRAF when we inhibited the canonical ERK1/2 pathway (Fig. 2) and there were no synergistic or additive effects of combining selumetinib with BIX02189 to inhibit both MEK1/2 and MEK5 in selumetinib-resistant KRAS- or BRAF-driven colorectal cancer cell lines (Fig. 6).
Finally, many driving oncogenes are overexpressed as a result of gene amplifications e.g. the receptor tyrosine kinase EGFR.51 Cancer cells become addicted to the amplified oncoprotein and this provides a therapeutic window in which the cancer cells are hypersensitive to inhibition of the elevated protein. MAPK7, the gene encoding ERK5, is amplified in approximately 50% of hepatocellular carcinomas.22 Nevertheless, inhibition of the MEK5-ERK5 pathway either chemically or genetically in SNU449 cells – a hepatocellular cancer cell line with an ERK5 amplification – or in a panel of cancer cell lines with MAPK7 copy number gain and elevated ERK5 protein levels did not reduce cell proliferation (Figs. 7 and 8) suggesting that even high levels of ERK5 do not confer ERK5 addiction for tumor cell proliferation.
Notably our results contrast the strong anti-proliferative effects observed in triple negative breast cancer cells treated with TG02, a clinical stage inhibitor which targets ERK5.19 However, TG02 is 10-fold more potent against CDK1, CDK2, CDK3, CDK5 and CDK9 than ERK552 and this likely accounts for the inhibition of proliferation in these studies; indeed, the mixed arrest of cells in G1 and G2 is consistent with CDK1, CDK2 and CDK9 inhibition. Taken together, our results argue that the role of the MEK5-ERK5 pathway in tumor development is not through driving tumor cell proliferation and suggests that anti-tumor effects seen with ERK5 pathway targeted therapies28 are likely through other mechanisms. Given the strength of the clinical evidence suggesting a role for MEK5 and ERK5 in cancer development,20-22 it will be of great interest to investigate the role of this pathway in other aspects of tumor cell biology.
Materials and methods
Materials
Cell culture reagents were purchased from Invitrogen and Sigma. Selumetinib (AZD6244; ARRY142886) was supplied by P. Smith, AstraZeneca, Cambridge, UK. An initial small quantity of BIX02189 was kindly provided by R. Snow, Boehringer-Ingelheim and used to confirm efficacy, potency and selectivity; subsequently a stock was prepared by Dr Jonathan Clark, Babraham Biological Chemistry Facility for this study. PD184352 was from Selleck. Mouse anti-HA was provided by the Babraham Institute Monoclonal facility. Phospho-ERK5 TEY (3371, http://www.cellsignal.com/products/primary-antibodies/3371), myc 9B11 (2276, http://www.cellsignal.com/products/primary-antibodies/2276) and phospho-ERK1/2 (9106, http://www.cellsignal.com/products/primary-antibodies/9106) antibodies were from Cell Signaling Technology. Total ERK5 antibodies were from Santa Cruz (sc1284, Figures 2, 4 and 6, http://www.scbt.com/datasheet-1284-erk-5-c-20-antibody.html) and Cell Signaling Technology (3372S, Figure 7, http://www.cellsignal.com/products/primary-antibodies/3372?hit=productId &Ntte=3372S). A total Raf-B antibody (sc-5284, http://www.scbt.com/datasheet-5284-raf-b-f-7-antibody.html) and tubulin (sc-8035, http://www.scbt.com/datasheet-8035-α-tubulin-tu-02-antibody.html) were obtained from Santa Cruz and total MEK5 antibody (AB3184, http://www.merckmillipore.com/GB/en/product/Anti-MEK5-Antibody,MM_NF-AB3184) from Chemicon. Total ERK1 antibody was from BD Biosciences (610031, http://www.bdbiosciences.com/ds/pm/tds/610031.pdf). Antibodies for actin were from Sigma (A4700, http://www.sigmaaldrich.com/catalog/product/sigma/a4700?lang=enandregion=GB). SRB and XTT were purchased from Roche. CellTitre-Glo® 3D Cell Viability Assay was purchased from Promega (https://www.promega.co.uk/products/cell-health-and-metabolism/cell-viability-assays/celltiter_glo-3d-cell-viability-assay/). Emetine (http://www.sigmaaldrich.com/catalog/product/sigma/e2375?lang=en andregion=GB) and 4HT (http://www.sigmaaldrich.com/catalog/product/sial/t176?lang=en andregion=GB) purchased from Sigma.
Cell culture
CR1-11 cells are a stable clone of CCl39 fibroblasts expressing the conditional kinase ∆CRAF:ER*. Culture of CR1-11, HEK293, SW48, CaCo2, HCT116, DLD-1, COLO205, HT29 and CO115 cells has been described previously.23,26,31 BT474 were a kind gift from C. Watson, University of Cambridge and were cultured as described.20 Hela, A-498, SNU449, NCI-H838, NCI-H1105, and NCI-H1436 cells were obtained from ATCC (Rockvill, MD). HeLa, A-498, SNU449 and NCI-H838 cells were maintained in RPMI-1640 medium supplemented with 10% FBS and NCI-H1105 and NCI-H1436 cells in DMEM-Ham's F12 medium with 5% FBS. KYSE30 cells were purchased from DSMZ (Germany) and maintained in DMEM Ham's F12 medium supplemented with 10% FBS. All cell lines were cultured at 37oC in an atmosphere of 5% CO2 and 95% humidity.
HCT116 and HT29 cell spheroids
HCT116 and HT29 cells were cultured in round bottom, ultra-low attachment 96-well plate tissue culture plates (http://www.sigmaaldrich.com/catalog/product/sigma/cls4520?lang=enandregion=GB) in 200 µl culture medium (described above) using the method described by Vinci et al.53
Plasmids
The HA-ERK5 and EGFP-MEKwt and 5D constructs have been described previously26 and were derived from constructs provided by J.D. Lee. Myc-BRAF constructs were a kind gift from R. Marais, Cancer Research UK Manchester Institute, Manchester. Myc-KRASG12V construct was from A. Cox UNC, Chapel Hill, North Carolina. GAL4-MEF2D was a kind gift from J.C.McDermott, Center for Research in Biomolecular Interactions, York University, Toronto, Canada; GAL4:LUC reporter from D. Gillespie, University of Glasgow, Glasgow, UK.
Immune complex kinase assays
Following stimulation, as described, active kinase complexes were immunoprecipitated from cell extracts normalized for protein content. The extracts were mixed, for 3 h at 4°C, with a 10 μl bed volume of Protein A–Sepharose (ERK1) (Sigma, http://www.sigmaaldrich.com/catalog/product/sigma/p9424?lang=en andregion=GB) or Protein G–Sepharose (ERK5) (GE Healthcare, http://www.gelifesciences.com/webapp/wcs/stores/servlet/ProductDisplay?categoryId=11504 andcatalogId=10101 andproductId=21386 andstoreId=12751 andlangId=−1) and antibody; 5 μl of rabbit anti-ERK1 antibody (BD Biosciences, http://www.bdbiosciences.com/eu/applications/research/stem-cell-research/stem-cell-signaling/human/purified-mouse-anti-erk1-mk12/p/610031), or 10μl of goat anti-ERK5 antibody (Santa Cruz Biotechnology, http://www.scbt.com/datasheet-1284-erk-5-c-20-antibody.html). The bead complexes were washed twice with lysis buffer and once in kinase buffer (30 mM Tris, pH 8, 20 mM MgCl2, 2 mM MnCl2, 25 mM β-glycerol phosphate and 0.1 mM sodium vanadate). Kinase activities were assessed by incubating the drained beads in 30 μl of kinase buffer supplemented with 10 μM ATP, 3 μCi of [γ-32P]ATP and either 7 μg of myelin basic protein (ERK1 assays), or 8 μg of glutathione S-transferase (GST)-MEF2C (175–327) (ERK5 assays) for 30 min at 30 °C in a shaking incubator. Kinase reactions were terminated by boiling samples in 4xLaemmli SDS sample buffer before resolving by SDS-PAGE. Gels were fixed and dried and incorporation of 32P into respective substrates quantified by Phosphorimager (Fuji). Unless otherwise indicated, data are either combined from independent experiments or are from a single experiment on triplicate dishes, representative of at least 2 others giving similar results.
Luciferase assays
HEK293, SW48, CaCo2, HCT116, DLD-1, COLO205 and CO115 cells were transfected using Lipofectamine 2000 (Life Technologies, https://www.lifetechnologies.com/uk/en/home/brands/product-brand/lipofectamine/lipofectamine-2000.html), according to the manufacturer's instructions. HT29 and SNU449 cells were transfected using JetPEI (PolyPlus, http://www.polyplus-transfection.com/2009/08/high-throughput-screening-jetpei/) according to the manufacturer's instructions. BIX02189 was added at concentrations indicated, 6–h post transfection. After a further 16–18–h cells were harvested and processed for firefly and renilla luciferase activity using the Dual Luciferase Reporter (Promega, http://www.promega.co.uk/products/reporter-assays-and-transfection/reporter-assays/dual_luciferase-reporter-assay-system/) assay according to the manufacturer's instructions.
Western blot analysis
For western blotting 6 cm dishes were routinely transfected as described or 10 cm dishes were routinely used to prepare whole cell extracts of cancer cell lines. Cells were treated BIX02189 or Selumetinib at the concentrations and for the indicated times prior to harvesting and western blotted as previously described.54,55
Assay of cell proliferation
Cell proliferation were measured by [3H]thymidine incorporation and were performed as described previously.31 To determine the sensitivity of additional tumor cell lines with high ERK5 expression to ERK5 kinase inhibition, the effect of continuous treatment over a 72 h period was ascertained using either a Sulforhodamine B (SRB) assay for adherent lines or XTT assay for non-adherent lines, as previously described.42,56,57 Spheroid cell viability was measured using CellTitre-Glo® 3D Cell Viability Assay reagent as previously described.53
siRNA sequences and RNAi
For transient RNAi, the following oligos were used: siERK5 1, GACCCACCUUUCAGCCUUA; siERK5 2, GGAUGGCCAGGCAGAUUCA; and siN.S. (non-silencing) as described by Roberts et al58; ERK5 Euro, GGUGUUGGCUUUGACCUGGAGGAAU; ERK5 Dharma 2 (J-003513–09); ERK5 Dharma 3 (J-003513-08); Dharma control 1 (D-001810-01-20) from Dharmacon; control stealth (46-2002) from Invitrogen. For HCT116 cell transfection 100pmol of the appropriate oligo was mixed with 1ml opti-MEM and 10µl lipofectamine2000, and then incubated at room temperature for 20 min. 5 ml of 2 × 105 cells/ml in penicillin/steptamycin free media were then added and mixed well. 6 h post transfection the media was changed to normal media and the cell incubated overnight. The following day the cells were trypsinised and counted and plated in the appropriate vessel. For SNU449 2×105 cells were seeded per well of a 6 well plate 24h prior transfection. siRNA was added to 1.25 ml opti-MEM to give a final concentration of 10nM, and 25µl of lipofectamine2000 was added to 1.25 ml opti-MEM, these were then mixed and incubated at room temperature for 20min. 510 µl aliquots of mix were added/ well and the media replaced after 6h. The following day the cells split into 96 well plates containing 3 × 103 cells/ well and the remaining cells were seeded into 6 cm plates for immunoblotting.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank Paul Smith for provision of selumetinib and Roger Snow for initial provision of BIX02189.
Funding
This work was funded by a project grant from the Association of International Cancer Research, a program grant from Cancer Research UK and an MRC DPFS Industry Collaboration Agreement. The Cook lab receives strategic support from the BBSRC in the form of an Institute Strategic Program grant.
References
- [1].Johnson GL. ERK1/ERK2 MAPK Pathway. (Connections Map in the Database of Cell Signaling, as seen 27 February 2014), http://stke.sciencemag.org/cgi/cm/stkecm;CMP_10705. Sci Signal 2005 [Google Scholar]
- [2].Johnson GL. JNK MAPK Pathway. (Connections Map in the Database of Cell Signaling, as seen 27 February 2014) http://stke.sciencemag.org/cgi/cm/stkecm;CMP_10827. Sci Signal 2003 [Google Scholar]
- [3].Gary L. Johnson. p38 MAPK Pathway. (Connections Map in the Database of Cell Signaling, as seen 27 February 2014), http://stke.sciencemag.org/cgi/cm/stkecm;CMP_10958. Sci Signal 2008 [Google Scholar]
- [4].Zhou G, Bao ZQ, Dixon JE. Components of a new human protein kinase signal transduction pathway. J Biol Chem 1995; 270:12665-9; PMID:7759517; http://dx.doi.org/ 10.1074/jbc.270.21.12665 [DOI] [PubMed] [Google Scholar]
- [5].Lee JD, Ulevitch RJ, Han J. Primary structure of BMK1: a new mammalian map kinase. Biochem Biophys Res Commun 1995; 213:715-24; PMID:7646528; http://dx.doi.org/ 10.1006/bbrc.1995.2189 [DOI] [PubMed] [Google Scholar]
- [6].Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 2006; 24:21-44; PMID:16393692; http://dx.doi.org/ 10.1080/02699050500284218 [DOI] [PubMed] [Google Scholar]
- [7].Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007; 26:3291-310; PMID:17496923; http://dx.doi.org/ 10.1038/sj.onc.1210422 [DOI] [PubMed] [Google Scholar]
- [8].Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363:809-19; PMID:20818844; http://dx.doi.org/ 10.1056/NEJMoa1002011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang J, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res 2011; 17:989-1000; PMID:21245089; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2200 [DOI] [PubMed] [Google Scholar]
- [10].Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, Cockerill M, Cartlidge S, Smith PD. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther 2007; 6:2209-19; PMID:17699718; http://dx.doi.org/ 10.1158/1535-7163.MCT-07-0231 [DOI] [PubMed] [Google Scholar]
- [11].McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M, Candido S, Malaponte G, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget 2012; 3:1068-111; PMID:23085539; http://dx.doi.org/ 10.18632/oncotarget.659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Little AS, Smith PD, Cook SJ. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene 2013; 32:1207-15; PMID:22562245; http://dx.doi.org/ 10.1038/onc.2012.160. [DOI] [PubMed] [Google Scholar]
- [13].Kato Y, Tapping RI, Huang S, Watson MH, Ulevitch RJ, Lee JD. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 1998; 395:713-6; PMID:9790194; http://dx.doi.org/ 10.1038/27234 [DOI] [PubMed] [Google Scholar]
- [14].Esparis-Ogando A, Diaz-Rodriguez E, Montero JC, Yuste L, Crespo P, Pandiella A. Erk5 participates in neuregulin signal transduction and is constitutively active in breast cancer cells overexpressing ErbB2. Mol Cell Biol 2002; 22:270-85; PMID:11739740; http://dx.doi.org/ 10.1128/MCB.22.1.270-285.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].English JM, Pearson G, Hockenberry T, Shivakumar L, White MA, Cobb MH. Contribution of the ERK5/MEK5 pathway to Ras/Raf signaling and growth control. J Biol Chem 1999; 274:31588-92; PMID:10531364; http://dx.doi.org/ 10.1074/jbc.274.44.31588 [DOI] [PubMed] [Google Scholar]
- [16].Kamakura S, Moriguchi T, Nishida E. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J Biol Chem 1999; 274:26563-71; PMID:10473620; http://dx.doi.org/ 10.1074/jbc.274.37.26563 [DOI] [PubMed] [Google Scholar]
- [17].Mody N, Leitch J, Armstrong C, Dixon J, Cohen P. Effects of MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett 2001; 502:21-4; PMID:11478941; http://dx.doi.org/ 10.1016/S0014-5793(01)02651-5 [DOI] [PubMed] [Google Scholar]
- [18].Squires MS, Nixon PM, Cook SJ. Cell-cycle arrest by PD184352 requires inhibition of extracellular signal-regulated kinases (ERK) 1/2 but not ERK5/BMK1. Biochem J 2002; 366:673-80; PMID:12069688; http://dx.doi.org/ 10.1042/bj20020372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ortiz-Ruiz MJ, Alvarez-Fernandez S, Parrott T, Zaknoen S, Burrows FJ, Ocana A, Pandiella A, Esparis-Ogando A. Therapeutic potential of ERK5 targeting in triple negative breast cancer. Oncotarget 2014; 5:11308-18; PMID:25350956; http://dx.doi.org/ 10.18632/oncotarget.2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Montero JC, Ocana A, Abad M, Ortiz-Ruiz MJ, Pandiella A, Esparis-Ogando A. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS One 2009; 4:e5565; PMID:19440538; http://dx.doi.org/ 10.1371/journal.pone.0005565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mehta PB, Jenkins BL, McCarthy L, Thilak L, Robson CN, Neal DE, Leung HY. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene 2003; 22:1381-9; PMID:12618764; http://dx.doi.org/ 10.1038/sj.onc.1206154 [DOI] [PubMed] [Google Scholar]
- [22].Zen K, Yasui K, Nakajima T, Zen Y, Gen Y, Mitsuyoshi H, Minami M, Mitsufuji S, Tanaka S, Itoh Y, et al. ERK5 is a target for gene amplification at 17p11 and promotes cell growth in hepatocellular carcinoma by regulating mitotic entry. Genes Chromosomes Cancer 2009; 48:109-20; PMID:18973138; http://dx.doi.org/ 10.1002/gcc.20624 [DOI] [PubMed] [Google Scholar]
- [23].Weston CR, Balmanno K, Chalmers C, Hadfield K, Molton SA, Ley R, Wagner EF, Cook SJ. Activation of ERK1/2 by deltaRaf-1:ER* represses Bim expression independently of the JNK or PI3K pathways. Oncogene 2003; 22:1281-93; PMID:12618753; http://dx.doi.org/ 10.1038/sj.onc.1206261 [DOI] [PubMed] [Google Scholar]
- [24].Allen LF, Sebolt-Leopold J, Meyer MB. CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK). Semin Oncol 2003; 30:105-16; PMID:14613031; http://dx.doi.org/ 10.1053/j.seminoncol.2003.08.012 [DOI] [PubMed] [Google Scholar]
- [25].Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010; 140:209-21; PMID:20141835; http://dx.doi.org/ 10.1016/j.cell.2009.12.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gilley R, March HN, Cook SJ. ERK1/2, but not ERK5, is necessary and sufficient for phosphorylation and activation of c-Fos. Cell Signal 2009; 21:969-77; PMID:19249353; http://dx.doi.org/ 10.1016/j.cellsig.2009.02.006 [DOI] [PubMed] [Google Scholar]
- [27].Carvajal-Vergara X, Tabera S, Montero JC, Esparis-Ogando A, Lopez-Perez R, Mateo G, Gutierrez N, Parmo-Cabanas M, Teixido J, San Miguel JF, et al. Multifunctional role of Erk5 in multiple myeloma. Blood 2005; 105:4492-9; PMID:15692064; http://dx.doi.org/ 10.1182/blood-2004-08-2985 [DOI] [PubMed] [Google Scholar]
- [28].Yang Q, Deng X, Lu B, Cameron M, Fearns C, Patricelli MP, Yates JR 3rd, Gray NS, Lee JD. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell 2010; 18:258-67; PMID:20832753; http://dx.doi.org/ 10.1016/j.ccr.2010.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tatake RJ, O'Neill MM, Kennedy CA, Wayne AL, Jakes S, Wu D, Kugler SZ Jr., Kashem MA, Kaplita P, Snow RJ. Identification of pharmacological inhibitors of the MEK5/ERK5 pathway. Biochem Biophys Res Commun 2008; 377:120-5; PMID:18834865; http://dx.doi.org/ 10.1016/j.bbrc.2008.09.087 [DOI] [PubMed] [Google Scholar]
- [30].Gilley R, Lochhead PA, Balmanno K, Oxley D, Clark J, Cook SJ. CDK1, not ERK1/2 or ERK5, is required for mitotic phosphorylation of BIMEL. Cell Signal 2012; 24:170-80; PMID:21924351; http://dx.doi.org/ 10.1016/j.cellsig.2011.08.018 [DOI] [PubMed] [Google Scholar]
- [31].Balmanno K, Chell SD, Gillings AS, Hayat S, Cook SJ. Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int J Cancer 2009; 125:2332-41; PMID:19637312; http://dx.doi.org/ 10.1002/ijc.24604 [DOI] [PubMed] [Google Scholar]
- [32].Gavine PR, Wang M, Yu D, Hu E, Huang C, Xia J, Su X, Fan J, Zhang T, Ye Q, et al. Identification and validation of dysregulated MAPK7 (ERK5) as a novel oncogenic target in squamous cell lung and oesophageal carcinoma. AACR Annual Meeting 2014: Abstract 2923 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Drew BA, Burow ME, Beckman BS. MEK5/ERK5 pathway: the first fifteen years. Biochim Biophys Acta 2012; 1825:37-48; PMID:22020294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lochhead PA, Gilley R, Cook SJ. ERK5 and its role in tumour development. Biochem Soc Trans 2012; 40:251-6; PMID:22260700; http://dx.doi.org/ 10.1042/BST20110663 [DOI] [PubMed] [Google Scholar]
- [35].Nithianandarajah-Jones GN, Wilm B, Goldring CE, Muller J, Cross MJ. ERK5: structure, regulation and function. Cell Signal 2012; 24:2187-96; PMID:22800864; http://dx.doi.org/ 10.1016/j.cellsig.2012.07.007. [DOI] [PubMed] [Google Scholar]
- [36].Yang Q, Lee JD. Targeting the BMK1 MAP kinase pathway in cancer therapy. Clin Cancer Res 2011; 17:3527-32; PMID:21385929; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Deng X, Elkins JM, Zhang J, Yang Q, Erazo T, Gomez N, Choi HG, Wang J, Dzamko N, Lee JD, et al. Structural determinants for ERK5 (MAPK7) and leucine rich repeat kinase 2 activities of benzo[e]pyrimido-[5,4-b]diazepine-6(11H)-ones. Eur J Med Chem 2013; 70:758-67; PMID:24239623; http://dx.doi.org/ 10.1016/j.ejmech.2013.10.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Deng X, Yang Q, Kwiatkowski N, Sim T, McDermott U, Settleman JE, Lee JD, Gray NS. Discovery of a benzo[e]pyrimido-[5,4-b][1,4]diazepin-6(11H)-one as a Potent and Selective Inhibitor of Big MAP Kinase 1. ACS Med Chem Lett 2011; 2:195-200; PMID:21412406; http://dx.doi.org/ 10.1021/ml100304b [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Elkins JM, Wang J, Deng X, Pattison MJ, Arthur JS, Erazo T, Gomez N, Lizcano JM, Gray NS, Knapp S. X-ray crystal structure of ERK5 (MAPK7) in complex with a specific inhibitor. J Med Chem 2013; 56:4413-21; PMID:23656407; http://dx.doi.org/ 10.1021/jm4000837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].English JM, Pearson G, Baer R, Cobb MH. Identification of substrates and regulators of the mitogen-activated protein kinase ERK5 using chimeric protein kinases. J Biol Chem 1998; 273:3854-60; PMID:9461566; http://dx.doi.org/ 10.1074/jbc.273.7.3854 [DOI] [PubMed] [Google Scholar]
- [41].Chiariello M, Marinissen MJ, Gutkind JS. Multiple mitogen-activated protein kinase signaling pathways connect the cot oncoprotein to the c-jun promoter and to cellular transformation. Mol Cell Biol 2000; 20:1747-58; PMID:10669751; http://dx.doi.org/ 10.1128/MCB.20.5.1747-1758.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Abe J, Takahashi M, Ishida M, Lee JD, Berk BC. c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1. J Biol Chem 1997; 272:20389-94; PMID:9252345; h`ttp://dx.doi.org/ 10.1074/jbc.272.33.20389 [DOI] [PubMed] [Google Scholar]
- [43].Barros JC, Marshall CJ. Activation of either ERK1/2 or ERK5 MAP kinase pathways can lead to disruption of the actin cytoskeleton. J Cell Sci 2005; 118:1663-71; PMID:15797923; http://dx.doi.org/ 10.1242/jcs.02308 [DOI] [PubMed] [Google Scholar]
- [44].Schramp M, Ying O, Kim TY, Martin GS. ERK5 promotes Src-induced podosome formation by limiting Rho activation. J Cell Biol 2008; 181:1195-210; PMID:18573916; http://dx.doi.org/ 10.1083/jcb.200801078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gangarosa LM, Sizemore N, Graves-Deal R, Oldham SM, Der CJ, Coffey RJ. A raf-independent epidermal growth factor receptor autocrine loop is necessary for Ras transformation of rat intestinal epithelial cells. J Biol Chem 1997; 272:18926-31; PMID:9228072; http://dx.doi.org/ 10.1074/jbc.272.30.18926 [DOI] [PubMed] [Google Scholar]
- [46].McCarthy SA, Samuels ML, Pritchard CA, Abraham JA, McMahon M. Rapid induction of heparin-binding epidermal growth factor/diphtheria toxin receptor expression by Raf and Ras oncogenes. Genes Dev 1995; 9:1953-64; PMID:7649477; http://dx.doi.org/ 10.1101/gad.9.16.1953 [DOI] [PubMed] [Google Scholar]
- [47].Schulze A, Nicke B, Warne PH, Tomlinson S, Downward J. The transcriptional response to Raf activation is almost completely dependent on Mitogen-activated Protein Kinase Kinase activity and shows a major autocrine component. Mol Biol Cell 2004; 15:3450-63; PMID:15090615; http://dx.doi.org/ 10.1091/mbc.E03-11-0807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schulze A, Lehmann K, Jefferies HB, McMahon M, Downward J. Analysis of the transcriptional program induced by Raf in epithelial cells. Genes Dev 2001; 15:981-94; PMID:11316792; http://dx.doi.org/ 10.1101/gad.191101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mulloy R, Salinas S, Philips A, Hipskind RA. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene 2003; 22:5387-98; PMID:12934098; http://dx.doi.org/ 10.1038/sj.onc.1206839 [DOI] [PubMed] [Google Scholar]
- [50].Smalley KS, Lioni M, Dalla Palma M, Xiao M, Desai B, Egyhazi S, Hansson J, Wu H, King AJ, Van Belle P, et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol Cancer Ther 2008; 7:2876-83; PMID:18790768; http://dx.doi.org/ 10.1158/1535-7163.MCT-08-0431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Grandis JR, Sok JC. Signaling through the epidermal growth factor receptor during the development of malignancy. Pharmacol Ther 2004; 102:37-46; PMID:15056497; http://dx.doi.org/ 10.1016/j.pharmthera.2004.01.002 [DOI] [PubMed] [Google Scholar]
- [52].Goh KC, Novotny-Diermayr V, Hart S, Ong LC, Loh YK, Cheong A, Tan YC, Hu C, Jayaraman R, William AD, et al. TG02, a novel oral multi-kinase inhibitor of CDKs, JAK2 and FLT3 with potent anti-leukemic properties. Leukemia 2012; 26:236-43; PMID:21860433; http://dx.doi.org/ 10.1038/leu.2011.218 [DOI] [PubMed] [Google Scholar]
- [53].Vinci M, Gowan S, Boxall F, Patterson L, Zimmermann M, Court W, Lomas C, Mendiola M, Hardisson D, Eccles SA. Advances in establishment and analysis of three-dimensional tumor spheroid-based functional assays for target validation and drug evaluation. BMC Biol 2012; 10:29; PMID:22439642; http://dx.doi.org/ 10.1186/1741-7007-10-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Balmanno K, Cook SJ. Sustained MAP kinase activation is required for the expression of cyclin D1, p21Cip1 and a subset of AP-1 proteins in CCL39 cells. Oncogene 1999; 18:3085-97; PMID:10340380; http://dx.doi.org/ 10.1038/sj.onc.1202647 [DOI] [PubMed] [Google Scholar]
- [55].Garner AP, Weston CR, Todd DE, Balmanno K, Cook SJ. Delta MEKK3:ER* activation induces a p38 α/β 2-dependent cell cycle arrest at the G2 checkpoint. Oncogene 2002; 21:8089-104; PMID:12444545; http://dx.doi.org/ 10.1038/sj.onc.1206000 [DOI] [PubMed] [Google Scholar]
- [56].Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990; 82:1107-12; PMID:2359136; http://dx.doi.org/ 10.1093/jnci/82.13.1107 [DOI] [PubMed] [Google Scholar]
- [57].Willmore E, Elliott SL, Mainou-Fowler T, Summerfield GP, Jackson GH, O'Neill F, Lowe C, Carter A, Harris R, Pettitt AR, et al. DNA-dependent protein kinase is a therapeutic target and an indicator of poor prognosis in B-cell chronic lymphocytic leukemia. Clin Cancer Res 2008; 14:3984-92; PMID:18559621; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-5158 [DOI] [PubMed] [Google Scholar]
- [58].Roberts OL, Holmes K, Muller J, Cross DA, Cross MJ. ERK5 is required for VEGF-mediated survival and tubular morphogenesis of primary human microvascular endothelial cells. J Cell Sci 2010; 123:3189-200; PMID:20736307; http://dx.doi.org/ 10.1242/jcs.072801 [DOI] [PubMed] [Google Scholar]