

ABSTRACT
Many cellular functions, such as translation, require ribonucleoproteins (RNPs). The biogenesis of RNPs is a multi-step process that, depending on the RNP, can take place in many cellular compartments. Here we examine 2 different RNPs: telomerase and small Cajal body-specific RNPs (scaRNPs). Both of these RNPs are enriched in the Cajal body (CB), which is a subnuclear domain that also has high concentrations of another RNP, small nuclear RNPs (snRNPs). SnRNPs are essential components of the spliceosome, and scaRNPs modify the snRNA component of the snRNP. The CB contains many proteins, including WRAP53, SMN and coilin, the CB marker protein. We show here that coilin, SMN and coilp1, a newly identified protein encoded by a pseudogene in human, associate with telomerase RNA and a subset of scaRNAs. We also have identified a processing element within box C/D scaRNA. Our findings thus further strengthen the connection between the CB proteins coilin and SMN in the biogenesis of telomeras e and box C/D scaRNPs, and reveal a new player, coilp1, that likely participates in this process.
KEYWORDS: Coilin, Cajal body, pseudogene, SMN, scaRNA, telomerase
Introduction
Ribonucleoproteins (RNPs) are involved in essential cellular functions such as translation and pre-mRNA splicing. Owing to the fact that RNPs contain both RNA and protein components, their assembly is highly ordered and may contain steps that take place in the cytoplasm, nucleus or nucleolus. Small nuclear RNPs (snRNPs), which are crucial components of the spliceosome, are one such RNP that requires cytoplasmic and nuclear steps for its formation.1 SMN, the survival of motor neuron protein, governs many of these biogenesis steps.2-7 Most cases of spinal muscular atrophy (SMA), the leading genetic cause of infant mortality,8,9 are caused by deletion of the SMN1 gene.10 There are point mutations within SMN that also cause SMA,10-12 demonstrating that specific disruption of SMN function is also pertinent for the SMA phenotype. Although the snRNP biogenesis-promoting role of SMN is clear and well documented,2-7 there is some controversy in the field as to whether the disruption of this aspect of SMN function leads to SMA 13,14,15 Indeed, functions for SMN in neuromuscular junctions and muscle formation as well as in the afferent nerves may involve other activities of SMN besides that centered upon snRNP formation.16,17 For example, SMN may take part in the formation of messenger RNPs comprised of mRNA and mRNA binding proteins.18 Additionally, when considering that tissues outside the nervous system, from muscle to liver to bone (and others) are sites of pathology in SMA,19-21 it is important that a full understanding of SMN function in the cell is elucidated.
Regarding the well-studied contribution of SMN to snRNP formation, an important nuclear step in snRNP biogenesis is the modification of the small nuclear RNA (snRNA) component of snRNPs, which takes place in the Cajal body (CB), a subnuclear domain.1 In addition to being localized to the cytoplasm, SMN is also enriched within the CB.22 Thus it is possible that SMN participates in the modification of snRNAs. These modifications (pseudouridylation and 2′-O-methylation) of snRNA are conducted by another type of RNP, small Cajal-body specific RNPs (scaRNPs). There are 3 types of scaRNPs, as determined by the type of small Cajal-body specific RNA (scaRNA) present in the scaRNP. The different classes of scaRNAs each recruit a set of core proteins. Box C/D scaRNPs contain the core proteins Nop56, Nop58, 15.5 kDa and fibrillarin (the methyltransferase) while box H/ACA scaRNPs include the core proteins Nop10, Nhp2, Gar1 and dyskerin (the pseudouridylase). The third type of scaRNP contains a mixed domain scaRNA that has both box C/D and box H/ACA motifs.1
Another type of RNP is telomerase. Disruptions in telomerase activity result in the disease dyskeratosis congenita.23 Mature telomerase contains a box H/ACA RNA (hTR if referring to that in human) and the core proteins Nop10, Nhp2, Gar1, and dyskerin along with the telomerase reverse transcriptase (hTERT).24 During the early stages of telomerase biogenesis, the complex contains an assembly protein, NAF1, which is replaced by Gar1 later in the assembly process.25 Part of telomerase biogenesis includes the processing of hTR, which is generated as a long transcript that requires processing at the 3′-end by an unknown mechanism.25 Mature telomerase and box H/ACA scaRNPs share many of the same proteins due to the presence of the H/ACA motif present in the RNA moiety of the RNP. Interestingly, hTR and box H/ACA scaRNAs share another motif, the CAB box.26 This cis element is bound by the protein WRAP53 (TCAB1/WDR79),27,28 which facilitates telomerase and box H/ACA scaRNP localization to the CB. No CAB motif is present in human box C/D scaRNPs, and WRAP53 does not interact very strongly with this type of scaRNA,29 leaving open the question as to how box C/D scaRNPs are targeted to the CB. We have previously showed that coilin, the CB marker protein, interacts very strongly with box C/D scaRNAs thus providing a potential pathway whereby this type of scaRNP can accumulate in CBs.30 Another report has observed that the G.U/U.G wobble stem of intron-encoded box C/D scaRNAs is required for their targeting to CBs and in vivo association with WRAP53.29 Since coilin has been shown to associate directly with WRAP53 30,31 in addition to box C/D scaRNAs,30 it is possible that the localization of box C/D scaRNPs to CBs is more dependent on coilin than box H/ACA scaRNPs, which require WRAP53 for their CB accumulation. We have also observed that coilin associates with hTR and small nucleolar RNAs.30 Another report 32 has confirmed and extended our observations, elegantly demonstrating by UV crosslinking/immunoprecipitation (iCLIP) of a coilin-GFP fusion protein that hundreds of small RNAs associate with the CB marker protein. Clearly, therefore, coilin may participate more directly in the biogenesis of these RNPs than previously imagined. In support of this hypothesis, we have found that coilin has RNA processing activity with specificity toward the 3′-end of pre-processed hTR.33-35
In addition to coilin, the involvement of SMN in scaRNP and telomerase biogenesis is likely, but not well defined. SMN has been shown to associate with hTERT.36 This interaction is not mediated by RNA, suggesting that SMN directly associates with hTERT or a mediator protein. Moreover, telomerase activity can be detected in SMN immunoprecipitations, indicating that SMN is associated with the telomerase holoenzyme.36 Another line of evidence supporting a role for SMN in telomerase biogenesis comes from studies showing that SMN interacts with the GAR1 protein.37 GAR1 binds H/ACA motifs present in hTR (and some scaRNAs and small nucleolar RNAs) and also associates with dyskerin.38,39 Since SMN interacts directly with hTERT and indirectly interacts with hTR via GAR1, this leads to the hypothesis that SMN may facilitate telomerase holoenzyme formation. Interestingly, both hTERT and hTR accumulate in CBs that are associated with telomeres during S phase.26,40-42 A final bit of evidence in support of a role for SMN in telomerase formation comes from studies showing that SMN associates with WRAP53.31 Reduction of WRAP53 abolishes CBs and mislocalizes both SMN and coilin to the nucleolus, clearly indicating that the nuclear fraction of SMN is influenced by WRAP53.31 As mentioned above, WRAP53 interacts with the CAB motif present within hTR to target this RNA to the CB. Furthermore, SMN and coilin directly interact via symmetrically dimethylated arginines present within coilin.43,44 All these findings strongly suggest that SMN may participate in telomerase holoenzyme assembly. Since snRNP, telomerase, and scaRNP biogenesis are similar in that they require the assembly of proteins onto a non-coding RNA, and SMN associates with factors required for the formation of each class of RNPs, it is logical to investigate if SMN also contributes to telomerase and scaRNP biogenesis. This idea is further strengthened when considering that SMN directly interacts with fibrillarin,45 the methyltransferase component of box C/D scaRNPs. Given that SMN does not readily localize to the nucleolus in normal conditions, it is unlikely that SMN directly participates in small nucleolar RNP (snoRNP) biogenesis. However, the interaction of SMN with both Gar1 and fibrillarin, which are also part of snoRNPs (H/ACA class and C/D class, respectively), indicates that SMN may indirectly impact the formation of the rRNA modification machinery.
To further clarify how telomerase and box C/D scaRNPs are generated, we conducted experiments designed to identify cis elements and factors that impact the processing of the RNA components of these RNPs. Our studies demonstrate that the CB proteins SMN and coilin may directly participate in the formation of telomerase and box C/D scaRNPs. We also report the identification of coilp1, a protein encoded by a coilin pseudogene, and provide evidence that this novel protein likely contributes to telomerase and box C/D scaRNP biogenesis. Since coilp1 is also detected in mouse, we discuss the possibility that this protein contributes to the viability of the coilin knockout mouse.
Results
The GU repeat region of box C/D scaRNA 2 and 9 impacts their processing
ScaRNA 2, 9 and 17, which are box C/D class scaRNAs, were first identified in 2004.46 While scaRNA9 (also known as mgU2-19/30) is intron encoded, scaRNA 2 (mgU2-25/61) and 17 (mgU12-22/U4-8) are derived from independently transcribed genes. Full-length scaRNA 2, 9 and 17 are enriched in the nucleoplasm (likely in CBs). However, these RNAs can also be processed into smaller, nucleolus-enriched guide RNAs.46 Hence scaRNA 2, 9 and 17, when full-length, are speculated to take part in the modification of snRNAs, but, when processed, may contribute to the modification of RNA in the nucleolus. The factors responsible for processing scaRNA 2, 9 and 17 are largely unknown.47 Our previous work has shown that coilin has RNA degradation/processing activity, and can process scaRNA9 in vitro, but is unlikely to be the sole factor responsible for the processing of scaRNA 2 and 9.30,33,34,48 Both scaRNA 2 and 9 have a region enriched in GU repeats (Fig. 1A), and we have previously shown that deletion of the GU repeat region in scaRNA9 decreases its in vitro degradation/processing by coilin.48 This finding indicates that the GU repeat region of scaRNA 2 and 9 may function as a cis element that influences the processing of these scaRNAs.
Figure 1.
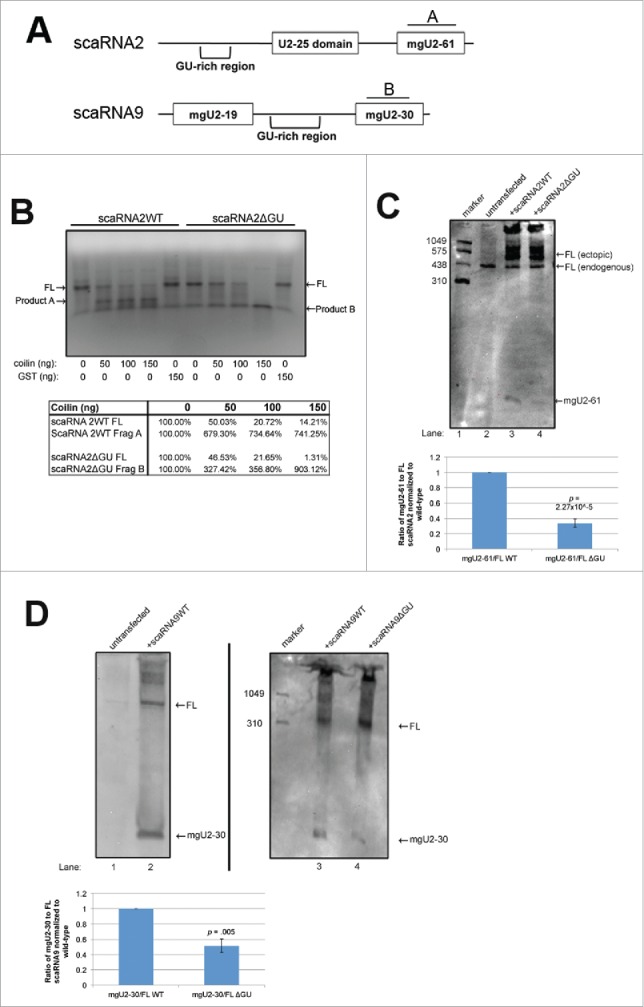
Deletion of GU-rich regions in scaRNA2 and 9 results in a decrease in processing. (A) Schematic of scaRNA2 and scaRNA9 used in subsequent experiments. A and B indicate the binding location for probes used in Northern detection. (B) RNA degradation assay using scaRNA2 wildtype (WT) and scaRNA2 with its GU-rich region deleted (ΔGU). RNAs were treated with either no protein; 10ng, 100ng, 150ng bacterially purified coilin; or 150ng GST as a control. The location of full-length (FL) scaRNA2 or scaRNA2ΔGU is indicated, as are their degradation products (A for scaRNA2 and B for scaRNA2ΔGU). The quantification of this data is shown below gel. (C,D) ScaRNA2WT, scaRNA2ΔGU, scaRNA9, or scaRNA9ΔGU pcDNA 3.1+ constructs were expressed in HeLa cells for 24 hours. Isolated RNA was then subjected to Northern blotting and probing with probe A (for scaRNA2 RNAs) or probe B (for scaRNA9 RNAs). The mgU2-61 signal for scaRNA2 is significantly reduced in ΔGU expressing cells compared to WT (n = 3 experimental repeats, p = 0.005) (histogram). The mgU2-30 signal for scaRNA9 is significantly reduced in ΔGU expressing cells compared to WT (n = 3 experimental sets, p = 2.27×10−5) (histogram). For panel D, scaRNA9 can be observed in untransfected cells, but the mgU2-30 fragment is difficult to detect (lane 1).
To extend these studies, we examined if, like scaRNA9, deletion of the GU rich region in scaRNA2 would also decrease the processing/degradation of this RNA by coilin in vitro (Fig. 1B). This was not observed. Instead, the processing of both wild-type (WT) and GU deletion scaRNA2 still occurred in reactions containing purified coilin, but the resultant fragments are different (WT scaRNA2 processing results in product A, while scaRNA2 GU deletion processing generates product B, with very little detected product A). As expected, negative control reactions containing purified GST did not show any processing or degradation of either RNA. These findings support the hypothesis that the GU rich region of scaRNA 2 and 9 impacts or otherwise alters processing. We next examined the processing of WT and GU deletion scaRNA 2 and 9 in vivo by conducting Northern blots on RNA isolated from cells transfected with normal and mutant constructs. The detection of endogenous processed fragments of scaRNA2 (mgU2-61) and scaRNA9 (mgU2-19 and mgU2-30) using non-radioactive methods is difficult, but we have previously published that endogenous mgU2-61 can be detected with a sufficiently long exposure.48 However, transient transfection of scaRNA2 easily allows for the detection of the mgU2-61 processed fragment. The generation and initial characterization of the construct to ectopically express scaRNA2 was described previously.48 For scaRNA9 ectopic expression, a construct was generated that allowed for the expression of this RNA from the intron of the host gene. As shown in Fig. 1C and D, full-length scaRNA 2 and 9 can be detected using the indicated probes (denoted in Fig. 1A). Additionally, the processed, nucleolus-enriched, guide RNAs (mgU2-61 for scaRNA2 and mgU2-30 for scaRNA9) are also detected with these probes. Notably, the amount of these smaller guide RNAs is significantly decreased (60% for scaRNA2 and 50% for scaRNA9, histograms) upon deletion of the GU rich region. These results clearly indicate that the GU rich region of scaRNA 2 and 9 serves as a cis element that positively regulates processing.
Association of SMN, coilin and the newly defined coilin derivative coilp1 with scaRNA2, scaRNA9 and hTR
Our previously published results have showed that the endogenous coilin immunoprecipitation complex is highly enriched for scaRNA2, scaRNA9 and hTR.30 Moreover, another group has shown that a coilin-GFP fusion protein associates with hundreds of small RNAs.32 Since hTR, scaRNA2 and scaRNA9 are processed, and the factors responsible for this processing are unknown, we initiated studies designed to identify these processing components. As a first step toward this goal, we conducted RNA pulldown experiments in which cell lysate was incubated with biotinylated scaRNA2, scaRNA9 or hTR, followed by capture of complexes onto avidin beads. Control reactions lacked labeled RNA. We then monitored the recovery of candidate proteins by Western blotting of the avidin bead complexes. Both WT and fragments of scaRNA 2 and 9 were used for RNA pulldowns in order to demarcate which region of these RNAs mediates interaction (Fig. 2A). Note that mgU2-61 is processed from scaRNA2 and mgU2-19 and mgU2-30 are derived from scaRNA9.46 When examining WT scaRNA 2 and 9 in the pulldown assay, we observed that coilin recovery is significantly increased over reactions containing beads alone (2.5-fold over beads for scaRNA2 and 3-fold over beads for scaRNA9, Fig. 2B). Note that the increased recovery of coilin in reactions containing WT scaRNA 2 and 9 compared to reactions with beads alone can also be observed in Fig. 2D, E, F, H and I. These findings thus verify our previous RNA sequencing data of coilin immunoprecipitation complexes.30 In addition to coilin, the anti-coilin antibody used here also recognizes a smaller 28-kDa species, and this protein is recovered by scaRNA 2 and 9 but not by beads alone. This 28-kDa fragment is denoted as coilp1. We have previously reported that coilin antibodies can recognize a smaller 28-kDa species.49 It is possible that this smaller protein is a fragment generated from full-length coilin by calpain.49 As detailed below, however, we show that a coilin pseudogene, COILP1, may also account for the presence of the coilin derivative that is reactive to coilin antibodies and associates with scaRNA 2 and 9.
Figure 2.

Interaction profile of scaRNA 2 and 9. (A) Schematic of scaRNA constructs used for subsequent experiments to determine the interaction profile of the 5′ and 3′ ends. Dotted line indicates a 3′ extension representing an immature form of the scaRNA9. (B) RNA pulldown using in vitro transcribed biotin labeled wild type, 5′, and 3′ constructs of scaRNA2 and scaRNA9. Beads indicate a control pulldown containing no RNA. Coilin, and coilp1 interaction was assessed using Western blotting of the RNA-bead complexes and detection with anti-coilin antibodies. Fibrillarin and SMN detection was used positive control, and β-Tubulin detection was used as a negative control. Coilin detection is enriched 2.5-fold over beads for scaRNA2 (n = 3 experimental sets, p = 4.51×10−3) and 3-fold over beads for scaRNA9 (n = 3 experimental repeats, p = 1.49×10−3). Input represents 1.5% of lysate used in pulldown reactions. (C) Histogram analysis of data in Fig. B. Data was generated by normalizing either coilin or coilp1 to fibrillarin signal (n = 3 experimental sets for each construct, and * indicates p < 0.05). Error bars represent standard error. Also shown is the quantification of the coilin/coilp1 ratio for each construct, with the WT value for scaRNA2 or 9 normalized to 1. (D-G) In vitro transcribed biotin labeled RNAs were used to pulldown coilin and coilp1 from HeLa lysate. Beads indicate a control pulldown containing no RNA. The coilin/coilp1 ratio, with the WT value normalized to 1, is shown below each lane in (d) and (e). For (f), coilin recovery by scaRNA9 3′ extension is enriched 1.86-fold over scaRNA9WT (n = 3 experimental sets and p = 2.02×10−4). For (g), coilin recovery with hTR pulldown was 3-fold over beads (n = 5 experimental sets and p = 1.5×10−3). Fibrillarin detection was used as a positive control for scaRNA2 and 9 pulldowns, and dyskerin detection was used as a positive control for hTR pulldowns. Input represents 1.5% of lysate used in pulldown. (H) Western blot of RNA pulldowns using scaRNA9WT. Beads indicate a control pulldown containing no RNA. Bead complexes were washed with either PBS or RIPA prior to analysis. Coilin and coilp1, SMN and fibrillarin were detected with the appropriate antibodies. Input represents 1.5% of lysate used in the pulldown reactions. (I) RNA pulldown using HeLa lysate and beads only, scaRNA2, or the negative control Xef RNA. Recovery was determined by Western blotting and detection with the indicated antibodies.
SMN is also enriched in reactions containing scaRNA 2 and 9 compared to beads alone, as is the positive control box C/D core protein fibrillarin (Fig. 2B). In contrast, the negative control protein tubulin is not recovered in the RNA pulldown reactions. Pulldown assays using both 5′ and 3′ fragments of scaRNA 2 and 9 (Fig. 2B) demonstrate that the recovery of coilp1 (relative to fibrillarin) is significantly higher with WT scaRNA9 compared to the amount of coilp1 recovered by the scaRNA9 fragments (Fig. 2C). We also observed that the relative recovery of coilin is significantly greater for the scaRNA2 fragments compared to WT scaRNA2 (Fig. 2C). The ratio of coilin to coilp1 was also observed to change when using the different RNA fragments, most notably when comparing the coilin/coip1 ratio for WT scaRNA9 versus the scaRNA9 fragments. These findings indicate that coilin and coilp1 differentially associate with scaRNA 2 and 9, and possibly require different binding elements or association partners. Given that the GU rich region of scaRNA 2 and 9 impact the processing of these RNAs, we also examined if coilin and coilp1 association with scaRNA2 and scaRNA9 is likewise affected by the GU rich region. No differences in the amount (relative to fibrillarin) of coilin or coilp1 recovered by WT or GU deletion scaRNA2 (Fig. 2D) or scaRNA9 (Fig. 2E) were observed, suggesting that the GU rich motif does not mediate coilin or coilp1 association with scaRNA 2 or 9. We also examined a pre-processed scaRNA9, which contains an additional 134 nucleotides at the 3′ end (dashed line, Fig. 2A) that are normally removed in order to generate the mature scaRNA9 (Fig. 2F). Relative to the amount of fibrillarin present, no changes in the recovery of coilp1 in the scaRNA9 3′ extension compared to that obtained for WT scaRNA9 were observed. However, the relative amount of coilin was significantly increased 1.86-fold in reactions containing scaRNA9 3′ extension compared to the amount of coilin recovered by WT scaRNA9. Regarding the doublet seen for coilp1 in Fig. 2F, we observe this on occasion and believe that this is a consequence of over-sonication of cell lysate during harvesting. RNA pulldown experiments were also conducted using hTR (Fig. 2G). As with scaRNA2 and scaRNA9, both coilin and coilp1 are recovered in significantly greater amounts (3-fold increase for coilin) when hTR is present compared to that obtained with beads alone. SMN is also recovered by hTR, as is the positive control protein dyskerin. As expected, the negative control protein tubulin is not enriched in the RNA pulldown reactions.
We also examined the stability of the association between coilin, SMN and coilp1 with scaRNA9 by modifying the pulldown protocol to include washes using the more stringent RIPA buffer compared to the less stringent PBS. As shown in Fig. 2H, coilin, coilp1, fibrillarin and SMN recovery on scaRNA9 beads is greater than that obtained for beads alone, regardless of wash condition. To further demonstrate that coilin, coilp1 and SMN are interacting with specificity toward scaRNA2, scaRNA9 and hTR, we have conducted additional RNA pulldowns using a control RNA (Xenopus elongation factor 1alpha) generated from the in vitro transcription of the pTRI-Xef control template. These data show that coilin, coilp1, SMN and fibrillarin binding to the control RNA is reduced (approximately 2 to 3-fold) relative to that observed for reactions using scaRNA2 (Fig. 2I). Further evidence for the specificity of coilp1 is found in Fig. 2B, which shows that coilp1 recovery with scaRNA9 fragments is less robust than that found for WT scaRNA9. We would not expect to see any changes in the binding of coilp1 with scaRNA9 fragments if this protein was non-specifically interacting with any RNA. Collectively, the data shown in Fig. 2 demonstrate that both SMN and coilin associate with scaRNA2, scaRNA9 and hTR. These data also show that a new protein, coilp1, likewise interacts with these RNAs.
Coilin, SMN and fibrillarin association with scaRNA2 is affected by knockdown of CB proteins
To examine if the reduction of coilin, WRAP53 or SMN alters the association of coilin, coilp1, SMN or fibrillarin with scaRNA2, RNA pulldowns were conducted using lysate obtained from cells treated with siRNA for 48 hrs. As shown in Fig. 3, the amount of coilin recovered in the RNA pulldown from coilin siRNA lysate was, as expected, reduced compared to that obtained using control siRNA lysate. No change in the amount of coilin recovered was observed with SMN knockdown. However, WRAP53 reduction significantly increased (1.65-fold) the amount of coilin associated with scaRNA2 relative to control (Fig. 3B). This finding suggests that WRAP53 negatively regulates the association of coilin with scaRNA2. Coilin, WRAP53 or SMN reduction did not significantly impact the binding of coilp1 to scaRNA2 relative to that seen with control siRNA. It should be pointed out that the level of coilp1 does not significantly decrease with coilin siRNA at 48 hrs knockdown, which is the time point of this experiment. Hence the amount of coilp1 recovered in coilin siRNA lysate is, as expected, approximately equal to the amount recovered with control siRNA lysate. Probing of this membrane with SMN reveals that coilin and WRAP53 knockdown correlate with significantly increased SMN association with scaRNA2, relative to that obtained with control siRNA. Specifically, coilin reduction results in a 2.2-fold increase in the amount of SMN recovered in the RNA pulldown reaction relative to control siRNA, and WRAP53 reduction results in a 2.5-fold increase in SMN (Fig. 3B). These findings suggest that coilin and WRAP53 negatively regulate SMN interaction with scaRNA2. As expected, SMN knockdown decreased the amount of SMN recovered by scaRNA2. Upon detection of fibrillarin, we observed that coilin and WRAP53 knockdown both result in a significant increase (2.6- and 2.8-fold, respectively) in the amount of fibrillarin recovered, but SMN knockdown correlates with reduced (50%) fibrillarin association. Typical knockdowns of coilin, WRAP53 and SMN upon 48 hr siRNA treatment are shown in Fig. 3C and D. Collectively, the data shown in Fig. 3 clearly demonstrate that proteins enriched within the CB modulate the association of other CB-enriched proteins with scaRNA2.
Figure 3.

ScaRNA2 differentially binds coilin, SMN and fibrillarin when the Cajal body proteins coilin, WRAP53 and SMN are transiently knocked down. (A) Western blot of RNA pulldown using scaRNA2WT with coilin, coilp1 and SMN detection. Fibrillarin detection was used as a positive control. (B) Histogram representation of the data in Fig. 3A (n = 4 experimental sets, * indicates p < 0.05). Error bars represent standard error. All knockdown data was normalized to the values obtained with control knockdown. (C,D) Verification of coilin, SMN, and WRAP53 knockdowns via Western blotting. β-Tubulin detection was used as a loading control.
Coilp1, a product of a pseudogene, is detected in coilin knockout mouse cells
As mentioned above, we have previously published a paper describing the coilin derivative coilp1 and showed that this protein could be the result of calpain processing of full-length coilin.49 We noted in this manuscript that several different anti-coilin antibodies detect this coilin derivative. Moreover, the first paper to characterize coilin antibodies in 1993 observed this and other coilin derivatives and suggested that the identity of “these reactivities needs to be determined.”50 Considering the strong association of the coilin derivative coilp1 with scaRNA2, scaRNA9 and hTR in the RNA pulldown assays, indicating a potential function of this protein, we next made a concerted effort to further characterize this protein. We first examined if cell lines derived from WT (MEF26) or coilin knockout (KO, MEF42) mice contain the coilin derivative coilp1 (Fig. 4A). Based on the targeting construct used to generate the coilin KO mouse, the N-terminal 82 aa of coilin can potentially be expressed in this model.51 The coilin antibody used here, however, would not detect this product as it was raised against a C-terminal human coilin fragment from aa 276–576. As shown in lanes 1 and 3 of Fig. 4A, both full-length coilin and the coilin derivative coilp1 are detected in lysate from the WT mouse line (MEF26) and HeLa. Coilin, as expected, is not detected in coilin KO cells (MEF42, lane 2), but the coilin derivative coilp1 is still detected in this line. If the coilin derivative were generated only by calpain processing of full-length coilin, we would not expect to see the coilp1 band in coilin KO cells. This observation forced us to reconsider the origin of the coilin derivative. Toward this end, we proved that the antibody we are using to detect coilin and coilp1 is specific to coilin sequences. This was accomplished by immunodepleting the coilin antibody with GST-coilin beads. Coilin and coilp1 signal is drastically reduced when GST-coilin is used for immunodepletion compared to GST (Fig. 4B). Immunodepletion of the coilin antibody with GST-coilin was also sufficient to reduce the coilin and coilp1 signal in WT (26) and coilin KO (42) MEF cells (Fig. 4C). This finding demonstrates that the detection of the coilin derivative coilp1 is due to the antibody binding coilin sequences, or very closely related sequences, and not due to a non-specific antibody reaction. Since the antibody we used here was raised against a C-terminal fragment of coilin (from aa 276–576), coilp1 must contain some of these amino acids in order to be detected by this antibody. We then tested if mouse coilp1 could associate with human scaRNA9 using the RNA pulldown assay. As shown in Fig. 4D, when using the WT MEF line (MEF26) the amount of coilin is significantly increased in reactions containing scaRNA9 compared to that observed with beads alone. Additionally, coilp1 is also recovered in pulldown reactions with scaRNA9 compared to beads alone, and this is observed using lysate derived from both WT and coilin KO (MEF42) cell lines. Both SMN and fibrillarin are also enriched in reactions with scaRNA9 over beads alone using lysate from WT or coilin KO cell lines. Mouse coilp1, therefore, associates with human scaRNA9, like human coilp1.
Figure 4.
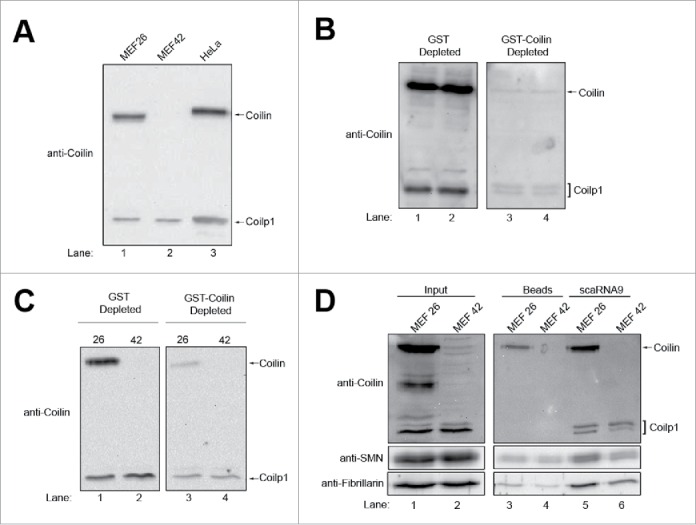
The coilin derivative coilp1 is detected in mouse coilin knockout cells. (A) Western blotting of lysate from wild-type (MEF26) or coilin knockout (MEF42) mouse embryonic fibroblast (MEF) cells and HeLa cells. The location of full-length coilin and the coilin derivative coilp1 are shown, as detected by anti-coilin antibodies. (B-C) Coilin antibody was incubated with purified GST or GST-coilin bound to glutathione beads. After incubation, the samples were centrifuged and the supernatant was then used to probe a membrane with HeLa (b) or MEF26 and MEF42 (c) lysate. Inputs represent 1.5% of lysate used in pulldowns. (D) Western blot of scaRNA9WT pulldowns in MEF26 and MEF42, with beads alone reactions serving as controls. Coilin, coilp1, SMN and fibrillarin were detected with the appropriate antibodies. Inputs represent 1.5% of lysate used in pulldowns.
Because mouse coilin KO cells contain coilp1, and the targeting strategy for deleting Coil eliminates the possibility that a C-terminal protein product could be generated,51 we speculated that coilp1 could be encoded by a coilin pseudogene. No coilin pseudogene is annotated for mouse, but in human there are 2 coilin pseudogenes (COILP1 and COILP2). As with most pseudogenes, it is not expected that these pseudogenes give rise to full-length proteins. Indeed, COILP1 was identified in 1994 when screening for a full-length coilin cDNA.52 The sequence of this pseudogene contains numerous insertions, deletions, and frameshifts that would be expected to preclude the formation of a full-length coilin protein product. A DNA alignment of COILP1 and COIL is shown in Fig. S1. In the last 10–15 years it has been found that some pseudogenes are transcribed and some produce proteins.53 The presence of a pseudogene that encodes the coilin derivative coilp1 in mouse would explain why we can detect this protein in coilin KO cells. According to NCBI AceView, COILP1 has the potential to encode a 203 aa protein that would be expected to react with the anti-coilin antibody used here. This protein would have an expected size of 22.6 kDa but would, like full-length coilin, contain a high percentage of intrinsic disorder that may slow its mobility on SDS-PAGE.33 An alignment of the putative human COILP1 protein product with human coilin is shown in Fig. 5D. Incidentally, the best predicted protein for COILP2 according to NCBI AceView is 39 aa. To definitively prove that the coilin derivative coilp1 is in fact encoded by the COILP1 pseudogene, we modified a purification strategy we developed previously 49 which fractionates the coilin derivative away from full-length coilin (Fig. 5AB). This partially purified coilin derivative (Fig. 5B, lane 1) was then put over a HPLC column, and fractions from this step were screened by Western blotting and probing with anti-coilin antibodies (Fig. 5C). The fraction with the highest concentration of the coilin derivative (fraction 8) was then subjected to tryptic digestion and Liquid Chromatography/Mass Spectrometry (LC/MS) analysis. Preliminary Discovery Proteomics experiments yielded peptides corresponding to COILP1. However, the low signal/noise of the transitions were not suitable for confirmation by this approach due to the abundance of other peptides (derived from keratins and other common abundant protein contaminants). These empirically detected peptides were used in Multiple Reaction Monitoring (MRM) LC/MS experiments. In addition, peptides derived from the predicted COILP1 amino acid sequence using peptide transitions and MS settings predicted using Skyline Targeted Proteomics Environment, version 3.1 (freeware) were also used. The MRM method greatly improves the signal/noise ratio by selecting specific peptide transitions instead of sifting through all possible peptides. A 5500 QTRAP Mass Spectrometer (SCIEX, Inc. Framingham, MA) coupled to a Dionex Ultimate 3000 HPLC running in capillary flow mode and all under the control of Analyst 1.5.2 software was used in the analyses. Several peptides (7 out of 8) match to the putative COILP1 product (Fig. 5D, yellow, orange or pink shaded boxes; Fig. S2, Tables S1 and S2). One identified peptide matches to both coilin and coilp1 proteins. This finding clearly demonstrates that the COILP1 pseudogene encodes a protein product, which we term coilp1. This 203 aa protein shares many of the same aa with coilin starting after residue 77 (matching coilin residues 194–311, with a few changes). Interestingly, we have shown that coilin aa 121–291 are important for RNA binding 34 and coilp1 shares many of these same amino acids. Our RNA pulldown assays clearly show that coilp1 interacts with scaRNA2, scaRNA9 and hTR (Fig. 2), suggesting that coilp1, like coilin, contains a RNA binding domain.
Figure 5.
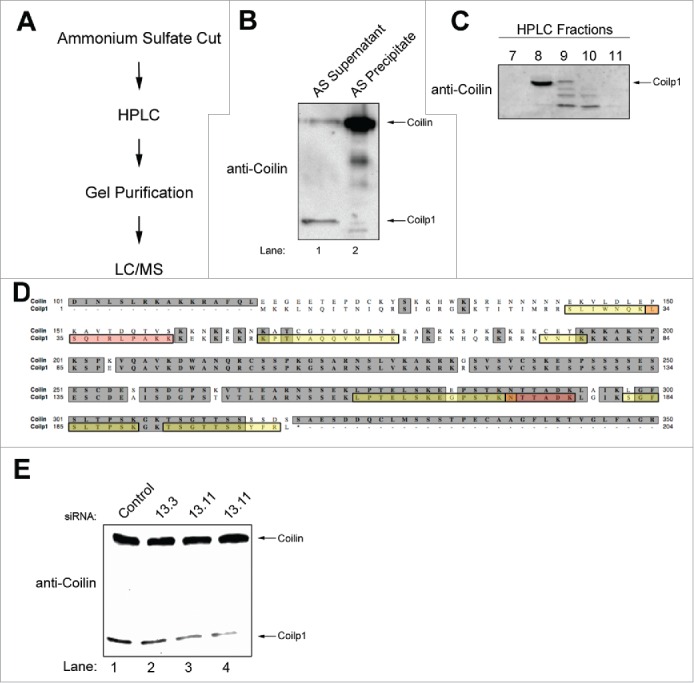
Purification and identification of coilp1. (a) A flow chart depicting the overall process used to purify and identify the peptide sequence of the coilin derivative coilp1. (b) Western blot showing that coilp1 can be enriched in the ammonium sulfate supernatant fraction (lane 1) compared to coilin, which is enriched in the ammonium sulfate precipitant (lane 2). (c) The ammonium sulfate supernatant fraction shown in lane 1 of (b) was subjected to HPLC. Western blotting of some of the fractions from the HPLC run was conducted and the membrane probed with anti-coilin antibodies. The location of coilp1 is indicated and found to be enriched in fraction 8. Fraction 8 was subjected to tryptic digestion and LC/MS to identify peptide sequences. (d) Alignment of coilin and coilp1 amino acid sequence. Peptides identified by mass spectrometry are highlighted in yellow or pink. Regions of overlap between peptides is indicated by an orange color. Note that 7 out of the 8 peptides unambiguously show that the COILP1 pseudogene can encode the coilp1 protein. One peptide contains sequence (NTTADK) that is shared between coilp1 and coilin (Fig. S2 and Tables S1 and S2). Peptide SLIWNQKLSQIRLPAKK has low confidence (Table S1), but complementary MRM evidence (Table S2) support the existence of this and other peptides. (e) Reduction of coilp1 protein via RNAi. Cells were transfected for 48 hrs with control or coilp1 (13.3 and 13.11) siRNAs, followed by lysate generation, SDS-PAGE, Western transfer and probing with anti-coilin antibodies.
As expected for a pseudogene, COILP1 (located on chromosome 14) is highly identical to COIL (located on chromosome 17) at the DNA level, with the exception that COIL contains introns (Fig. S1). Likewise, coilp1 and coilin mRNA are also very similar. We have tested several siRNAs that are fully complementary to both coilp1 and coilin message, but only observe a decrease in coilin protein and not coilp1 protein at 48 hrs treatment. At 72 hrs treatment, we do observe a slight decrease in coilp1 protein using these cross-reacting siRNAs.49 We initially suspected that this lack of knockdown indicates that coilp1 protein has a longer half-life than coilin, but data shown below refute this hypothesis. A more probable explanation is that coilin message is more abundant than coilp1 message, and thus will be more efficiently reduced by siRNAs. To test this possibility, we determined the relative amount of coilin and coilp1 mRNA. To specifically amplify coilp1 message, we took advantage of the fact that the 5′ UTR of coilp1 and coilin are completely different (Fig. S1). Primers were designed to specifically amplify the 5′ UTR of coilp1, and reverse transcriptase qRT-PCR was conducted using RNA isolated from HeLa cells. The resultant PCR product was sequenced and found to be strictly coilp1, with no coilin sequences present. These primers, therefore, are effective in specifically amplifying the coilp1 message. Further analysis shows that coilin mRNA is at least 36 times more abundant than coilp1 mRNA. Taking advantage of the difference between the 5′ UTRs, we designed siRNAs that should target coilp1 and not coilin mRNA (Fig. S1). Relative to control siRNA, the level of coilp1 message is reduced 65% using the 13.11 siRNA and reduced 35% using the 13.3 siRNA, upon 48 hrs treatment. Coilin message levels were unchanged with the 13.11 and 13.3 siRNA treatment compared to that obtained with control siRNA. In addition to coilp1 message levels, 13.11 siRNA treatment also reduces coilp1 proteins levels approximately 50% compared to that obtained with control siRNA treated lysate, and coilin protein levels are unaffected (Fig. 5E). Hence, these siRNAs, 13.3 and 13.11, can be used to reduce coilp1 message and protein levels but these siRNAs will not impact coilin mRNA or protein amounts.
Transiently expressed coilp1 fusion proteins localize to the nucleoplasm and nucleolus
We next cloned the COILP1 pseudogene region expected to encode for coilp1 from genomic DNA into bacterial and mammalian expression vectors. Transient expression of GFP- or Myc-tagged coilp1 demonstrates that a protein of the expected size can be immunoprecipitated by GFP or Myc antibodies, respectively, and detected by anti-coilin antibodies (Fig. 6A). Localization studies show that transiently expressed coilp1 accumulates in the nucleus, with strong accumulation in the nucleolus (Fig. 6BC). No obvious enrichment in CBs is observed, as monitored by using anti-SMN (Fig. 6B) or anti-coilin antibodies (Fig. 6C). Anti-coilin does, however, detect GFP- and myc-tagged coilp1, as expected. We also examined the stability of endogenous coilp1 by conducting studies with the translation inhibitor cycloheximide. As shown in Fig. 6D, coilp1 is more unstable than coilin, with a half-life of approximately 10 hours.
Figure 6.
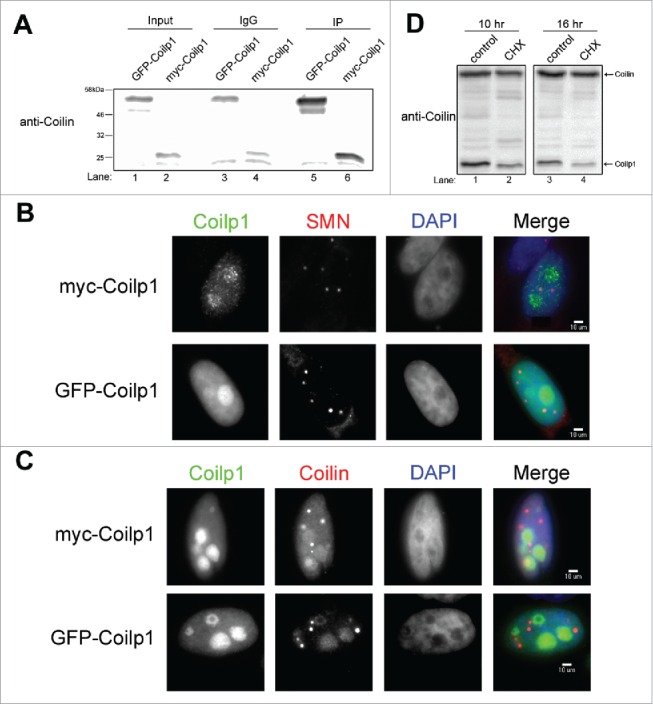
Generation and localization of coilp1 fusion proteins, and stability of endogenous coilp1. (A) Western blot of ectopically expressed GFP-coilp1 and myc-coilp1. IPs were conducted using either anti-GFP or anti-myc antibody and then detected using coilin polyclonal antibody. Normal Mouse IgG was used as a negative control. Input represents 1.5% of lysate used in IPs. (B) Immunofluorescence of coilp1 fusion proteins in order to assess co-localization with Cajal bodies by using SMN monoclonal antibody. (C) Immunofluorescence of coilp1 fusion proteins and co-staining with coilin antibody. (D) Cyclohexamide treatment of HeLa cells to determine the half-life of coilp1. At 10 hours, coilp1 is reduced 51.9% relative to coilin (n = 3 experimental sets, p = 1.9×10−4). At 16 hours, coilp1 is reduced 77.9% relative to coilin (n = 3 experimental sets, p = 5×10−4).
Preferential binding of coilin, coilp1 and SMN to scaRNA 2 and 9 in vitro
To monitor the direct association of SMN, coilin, coilp1 and WRAP53 to scaRNA2, scaRNA9 and hTR, we conducted RNA pulldown assays using bacterially purified protein. In addition, we employed an hTR construct with an extended 3′ end to determine if this pre-processed RNA would interact differently compared to the mature RNA found in telomerase. This data is shown in Fig. 7. To ascertain the preference of a given protein to a given RNA, the signal obtained in the pulldown reaction was normalized to the input signal (Fig. 7F). Based on these studies, we conclude that coilin, coilp1, SMN and WRAP53 all directly interact with scaRNA2, scaRNA9, hTR and the hTR 3′ extension. When comparing binding preferences, coilin interacts more strongly with scaRNA2 and scaRNA9 than hTR, which is agreement with our previous analysis of the coilin immunoprecipitation complex.30 Coilp1 and SMN have a similar binding preference as observed for coilin. Somewhat surprisingly, we do not observe high levels of interaction between WRAP53 and the CAB-box containing hTR and hTR 3′ extension. In fact, WRAP53 interaction with these RNAs is lower than that observed for scaRNA2 and scaRNA9. Interestingly, coilp1 interacts most strongly with the hTR containing the 3′ extension (Fig. 7D), possibly indicating that this protein may take part in the processing complex that generates mature hTR. As negative controls, pulldown reactions were also conducted with purified GST or a C-terminal construct of coilin (from aa 362–576, C214) that does not contain RNA binding activity (Fig. 7GH). Compared to the positive control proteins GST-coilp1 (Fig. 7G) and coilin (Fig. 7H), GST and the coilin fragment C214 are not significantly increased in pulldown reactions containing scaRNA9 vs. that obtained using beads alone. A control pullldown using Xef RNA shows that GST-coilp1 recovery is greater (approximately 2-fold) in reactions containing scaRNA2 compared to Xef RNA (Fig. 2I).
Figure 7.

ScaRNA2, scaRNA9, hTR, and hTR 3′ extension differentially bind His-T7 fusion proteins. (A-E) In vitro pulldowns using biotin labeled scaRNAs (scaRNA2, 9, hTR, and hTR 3′ extension) and bacterially purified His-T7 tagged proteins (coilin, coilp1, SMN, and WRAP53). His-WRAP53 and His-SMN were detected using T7 monoclonal antibody. His-coilin and His-coilp1 were detected using coilin polyclonal antibody. Inputs represent 10% of the protein used in the pulldown reactions. Beads indicate pulldown reactions lacking RNA. Note that the input lane order for a-d is coilp1, WRAP53 then SMN, but the pulldown order is coilp1, SMN, then WRAP53. (F) Histogram representation of the preceding data. Protein recovered from the pulldown was analyzed in terms of input recovery. Error bars represent standard error. (G) In vitro RNA pulldown using biotin labeled scaRNA9 and GST-Coilp1. Recovery of GST-Coilp1 from the pulldown was analyzed by Western blotting and detected using coilin polyclonal antibody. GST serves as a negative control and was detected using GST monoclonal antibody. Inputs represent 10% of protein used in pulldowns. Beads indicate a pulldown using no RNA. (H) In vitro pulldown using scaRNA9 with either His-coilin 362–576 (C214), which serves as a negative control, or His-coilin. Recovery was determined through Western blotting and detection with coilin polyclonal antibody. Inputs represent 10% of protein used in pulldowns, beads indicate a pulldown lacking RNA. (I) In vitro RNA pulldown using GST-coilp1 plus beads only, scaRNA2 or the negative control Xef RNA. Recovery was determined by Western blotting and detection with coilin polyclonal antibody.
Discussion
To more fully understand the mechanisms by which scaRNA2, scaRNA9 and hTR are processed and incorporated into functional RNPs, we conducted a series of experiments designed to identify processing elements and factors that may take part in the biogenesis of these RNPs. Our previous work has shown that ectopically expressed scaRNA2 is most likely processed in the nucleoplasm.48 We also showed in this work that deletion of the GU rich region in scaRNA9 decreases the in vitro processing of this RNA by coilin. Here we show that deletion of the GU rich region in both scaRNA2 and scaRNA9 alters their processing (Fig. 1). Hence the GU rich region of scaRNA2 and scaRNA9 serves as a cis element that positively regulates the processing of these scaRNAs. We are currently investigating if the deletion of this GU rich cis element alters the localization of scaRNA 2 and 9.
Coilp1, a newly identified interactor of hTR and box C/D scaRNA
Previous results have shown that SMN and coilin associate with RNA.30,32,33,54,55 In regards to coilin, we have shown that scaRNA2, scaRNA9 and hTR are present in coilin immunocomplexes.30 Here we describe the use of RNA pulldown in order to monitor association with candidate proteins and map the region of the RNA that mediates these interactions (Fig. 2). It is important to note that the RNA pulldown assay we employ here most likely does not fully recapitulate the in vivo interaction observed between the proteins we investigate with a given RNA. For example, we would expect that fibrillarin, which is a known component of scaRNP 2 and 9, would more strongly interact with scaRNA9 in RIPA conditions than we observed (Fig. 2H). Nonetheless, the RNA pulldown method does allow us to identify, in general, binding regions within a given RNA for the proteins we are investigating. Additionally, the use of bacterially purified proteins allows us to determine if the interactions between protein and RNA are direct (Fig. 7). From these studies, we show that coilin, SMN and WRAP53 all interact with scaRNA2, scaRNA9 and hTR. Considering that WRAP53 does not interact very strongly in vivo with box C/D scaRNAs, which lack a CAB motif, but does associate with H/ACA scaRNAs and hTR, which have a CAB box motif,29 we expected that WRAP53 would associate most strongly with hTR in our in vitro pulldown reactions. This was not observed, however. Our study is the first to investigate the binding properties of WRAP53 using purified components, so it is possible that bacterially generated WRAP53 does not have the same binding activity as endogenous WRAP53. Alternatively, it is possible that, in vivo, additional factors contribute to the binding specificity of WRAP53 to H/ACA RNAs with CAB boxes.
The most striking finding from the RNA pulldown assays is the observation that a coilin derivative, we term coilp1, associates with the RNAs tested. Mapping experiments show that coilp1 binding to scaRNA9 is reduced when using fragments containing the 5′ or 3′ regions of this RNA (Fig. 2). This finding would seem to indicate that coilp1 interacts within the GU rich region of this RNA, but RNA pulldowns using GU deletion constructs of scaRNA 2 and 9 did not alter the relative amount of coilp1 recovered compared to that observed with WT RNA. Interestingly, the relative recovery of coilin increased with 4 constructs (scaRNA2 5′ region, scaRNA2 3′ region, scaRNA9 5′ region and scaRNA 9 3′ extension) compared to that observed with WT scaRNA 2 or 9. One possible interpretation of these results is that the binding of other proteins is disrupted in these constructs, allowing for coilin to have more accessibility to the RNAs. Thus there may be a dynamic balance between the proteins that interact with these RNAs, and altering the levels of one protein may change the binding of another protein. The RNA pulldown data using lysate treated with siRNAs to coilin, WRAP53 and SMN (Fig. 3) support this hypothesis. Further support for this hypothesis comes from our previous work which found, using an qRT-PCR approach, that reduction of WRAP53 impacts the in vivo association of coilin with scaRNA 2 and 9.30 In summary, therefore, we believe that coilin and coilp1 RNA binding specificity in vivo is influenced by their localization, their protein interactions and their access to RNA. Given that coilin, SMN and WRAP53 are enriched within the CB, which contains high concentrations of scaRNAs, it seems obvious that these proteins may interact with these RNAs. As we have found previously, however, RNA sequencing of the endogenous coilin IP complex demonstrates that scaRNA2 and scaRNA9 are highly represented.30 Thus additional factors, such as interaction with other proteins, contribute to the highly specific interaction of coilin (and possibly coilp1) with scaRNA2 and scaRNA9 in vivo.
A pseudogene encodes human coilp1
We have previously suggested that the smaller coilin bands first detected in 1993 with coilin antibodies 50 could be the result of specific processing of coilin by calpain.49 Although this hypothesis is still valid, the data presented here clearly and unambiguously demonstrate that at least one of these coilin derivatives, the 28-kDa coilp1, arises from a pseudogene in human. This pseudogene, COILP1, was first identified in 1994 but not expected to generate a full-length coilin protein considering the numerous mutations present.52 However, our data in Fig. 4 plainly show that coilp1 signal can be reduced when anti-coilin antibodies are depleted by GST-coilin, which provides evidence that this protein is highly related to coilin. The presence of coilp1 in coilin knockout MEF cells (Fig. 4) suggests that another mechanism must exist to generate coilp1 besides calpain processing of full-length coilin. The data we show in Fig. 5 conclusively demonstrate that coilp1 is encoded by COILP1 in human. Traditionally, pseudogenes have not been thought to be transcribed or translated. However, a recent analysis of the human proteome identified 140 translated pseudogenes,53 clearly indicating these pseudogene-derived proteins are present. The contribution of these proteins to cellular activity and organismal function will be an exciting avenue of future investigations. In regards to coilin specifically, the presence of coilp1 in coilin knockout mouse cells strongly suggests that a mouse coilin pseudogene is present. Identification of this pseudogene by database mining has not been successful, so our future work will seek to identify this pseudogene by purifying mouse coilp1 and conducting LC/MS as we did for human coilp1. In so doing, we should be able to identify peptide sequences unique to the mouse coilin pseudogene. The identification of the origin of mouse coilp1 will greatly aid in the understanding of the contribution of coilin and coilp1 to cell and organismal function. Generally, the characterization of coilp1 is very important to the nuclear organization and RNA biology fields. For example, a recent report 32 has confirmed our finding30 that coilin associates with many scaRNAs and snoRNAs. For this work, Machyna and colleagues used a C-terminal GFP fusion of coilin (coilin-GFP) and UV crosslinking/immunoprecipitation (iCLIP) to identify coilin interactions. Anti-GFP was used to pulldown the coilin complexes. In contrast, our study30 utilized an antibody to coilin, and thus obtained both coilin and, presumably, coilp1 complexes. The presence of coilp1 complexes in our analysis of RNAs associated with the coilin IP complex may explain why we observed such a large enrichment for scaRNA 2 and 9 compared to the Machyna study, which examined coilin-GFP complexes.32
Localization studies (Fig. 6) show that ectopically expressed coilp1 is enriched in the nucleoplasm and nucleoli. We have recently begun the process to generate coilp1 antibodies. If we can generate coilp1 antibodies, we will be able to see if endogenous coilp1 is found in the nucleoplasm and nucleoli like overexpressed coilp1. Coilp1 does not contain the coilin self-association domain (aa 1–92 of coilin), and this might be an explanation as to why ectopically expressed coilp1 does not localize to CBs. The lack of overexpressed coilp1 in CBs, and its enrichment in the nucleoplasm and nucleoli, may indicate a possible role for this protein in the processing of scaRNA2 and 9 and the subsequent chaperoning of the processed fragments (mgU2-61, mgU2-19 and mgU2-30) to the nucleolus where these fragments are enriched. The majority of coilin is actually nucleoplasmic, so it could be that coilin and coilp1 work together to regulate the ratio between full-length and processed scaRNA2 and 9. It is also important to note that CBs are not present in every cell type, and cell types that lack CBs still have scaRNPs and functional snRNPs. In cell types that lack CBs, therefore, the RNP promoting biogenesis activities that take place in the CB must take place in the nucleoplasm.
Overall, the data presented here provide new insights into the role that Cajal body proteins SMN, WRAP53 and coilin may have on telomerase and box C/D scaRNP biogenesis, and introduce a new player, coilp1, as a likely participant in this process. Deciphering these roles, and identifying the cis elements within hTR, scaRNA2 and scaRNA9 that mediate these protein interactions, will be the subject of future studies. Further clarification of telomerase and box C/D scaRNP formation may demonstrate that SMN contributes to these processes more than previously appreciated. Additionally, our previous work 48 suggests that SMN may contribute toward the processing of box C/D scaRNA 2, 9 and 17, generating nucleolus-enriched guide RNAs of unknown function. It is possible that these guide RNAs contribute to the modification of rRNA. If true, this would mean that SMN contributes to snRNP and scaRNP formation, which are important for pre-mRNA splicing, as well as rRNA modification, which is necessary for functional ribosomes and translation. Disruptions in SMN function, either by reduced levels of full-length protein, or point mutation, may thus have far-reaching effects on fundamental cellular events such as splicing and translation. Considering the motor neuron-centered alterations observed in SMA, however, other cell types must be able to somewhat compensate for the reduced levels of functional SMN. Nonetheless, other tissues besides anterior horn motor neurons are impacted in SMA,19 and it is possible that defects in the scaRNP-promoting activity of SMN contribute to the SMA disease state.
Materials and methods
Cell lines and cell culture
HeLa cells were obtained from the American Type Culture Collection (Manassas, VA, USA). Mouse embryonic fibroblast (MEF) cell lines derived from wild-type (MEF26) or coilin knockout (MEF42) mice 56 were a gift from Greg Matera (University of North Carolina at Chapel Hill, USA). Cell lines were cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Invitrogen, Carlsbad, CA, USA) containing 10% fetal calf serum (Atlanta Biologicals, Flowery Branch, GA, USA) and 1% penicillin/streptomycin (ThermoFisher, Waltham, MA, USA) in a 5% CO2 incubator at 37°C.
RNAi
HeLa cells were transfected with siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer's suggested protocol, and all knockdowns were for 48 hours. The following siRNAs, purchased from Integrated DNA Technologies (Coralville, IA, USA), were used:
Control siRNA:
Forward 5′ – UAAGGCUAUGAAGAGAUACUU – 3′
Reverse 5′ – AAGUAUCUCUUCAUAGCCUUA – 3′
Coilin siRNA:
Forward 5′ – GAGAGAACCUGGGAAAUUUUU – 3′
Reverse 5′ – AAAUUUCCCAGGUUCUCUCUU – 3′
SMN siRNA:
Forward 5′ – UCUGUGAAGUAGCUAAUAAUAUAGA – 3′
Reverse 5′ – UCUAUAUUAUUAGCUACUUCACAGAUU – 3′
WRAP53 siRNA:
Forward 5′ – GUGAUACCAUCUAUGAUUACUGCTG – 3′
Reverse 5′ – CAGCAGUAAUCAUAGAUGGUAUCACCU – 3′
Coilp1 siRNA (13.3):
Forward 5′ – CCUUAAACAAUAAGGCUUGAAAGTC – 3′
Reverse 5′ – GACUUUCAAGCCUUAUUGUUUAAGGAA – 3′
Coilp1 siRNA (13.11):
Forward 5′ – GAUGAAAUACUAUCUAUGGUAGCTA – 3′
Reverse 5′ – UAGCUACCAUAGAUAGUAUUUCAUCUG – 3′
Transfection, plasmids and reverse transcriptase RT-PCR
HeLa cells were transfected with DNA using FuGENE HD (Promega, Madison, WI, USA) following the manufacturer's suggested protocol. His-T7-WRAP53,30 His-T7-SMN,43 and His-T7-Coilin 57 cloned in pET28a were described previously. Coilp1 was cloned from genomic DNA via thermocycling using standard molecular biology techniques. The following primers were used to clone coilp1 with its flanking genomic sequence:
Forward: 5′ - ATGCTTCGGCTTCATTTCTGG – 3′
Reverse: 5′ – CTAGAGTCTGAAATAGGAAGATGT – 3′
The product was then TA cloned (into pCR2.1, Invitrogen, Carlsbad, CA, USA) following the manufacturer's suggest protocol. The TA clones were then transformed into One Shot MAX Efficiency DH5α-T1R competent cells (Invitrogen, Carlsbad, CA, USA) following the manufacturer's suggested protocol. The DNA was then isolated using a MiniPrep kit (Qiagen, Hilden, Germany) and amplified with thermocycling using the following primers targeted to COILP1's proposed sequence with EcoR1 and SalI restriction sites (underlined):
Forward 1: 5′ – TATGGAATTCATGAAGAAACTGAACCAGATTACAAATA – 3′
Forward 2: 5′ – TATGGAATTCACATGAAGAAACTGAACCAGAT – 3′
Reverse: 5′ – CATAGTCGACCTAGAGTCTGAAATAGGAAGA – 3′
These products and vectors were then double restriction digested with either EcoR1 and SalI using standard molecular biology techniques, and gel purified using a Gel Extraction kit (Qiagen, Hilden, Germany). The product of Forward 1 and Reverse was inserted into pET28a, pGEX-6P-1, and pEGFP-C2, and the product of Forward 2 and Reverse was inserted into pCMV-myc using standard molecular biology techniques. These products were then transformed into One Shot MAX Efficiency DH5α-T1R competent cells and sequence verified. pET28a and pGEX-6P-1 coilp1 clones were subsequently transformed into One Shot BL21 (DE3) pLysS chemically competent E. coli cells (Invitrogen, Carlsbad, CA, USA) following the manufacturer's suggested protocol. All His-T7 tagged proteins were purified as previously described.34
ScaRNA2, scaRNA9, scaRNA 9 3′ extension,30 and scaRNA9ΔGU 48 in pBluescript KS+ were described previously. ScaRNA2 in pCDNA3.1 was also described previously.48 ScaRNA2 5′, scaRNA2 3′, scaRNA9 5′, and scaRNA9 3′ were all cloned from the aforementioned scaRNA2 and scaRNA9 pBluescript KS+ constructs using standard molecular biology techniques for cloning, using the following primers with a BamHI restriction site on the forward primers and an EcoRI restriction site on the reverse primers:
scaRNA2 5′
Forward: 5′ – GGCGGATCCGTTTTAGGGAGGGAGAGCGG – 3′
Reverse: 5′ – GGCGAATTCGGCTTCGCAGGAGGAGAGCTT – 3′
scaRNA2 3′
Forward: 5′ – GGCGGATCCCCCTCGGGGCGCTGTGCAGCG – 3′
Reverse: 5′ – GGCGAATTCCCAGATCAGAATCGCCTCGAT – 3′
scaRNA9 5′
Forward: 5′ – GGCGGATCCTTTCTGAGATCTGCTTTTAG - 3′
Reverse: 5′ – GGCGAATTCGACATATGCCCTTATTGTTTT – 3′
scaRNA9 3′
Forward: 5′ – GGCGGATCCTACGCACATGTGTTTATAAAG – 3′
Reverse: 5′ – GGCGAATTCTGAGCTCAGGTCAAGTGTAGA – 3′
These products were then put through a PCR Clean up kit (Qiagen, Hilden, Germany) and subsequently cloned into pBluescript SK+ cut with the appropriate enzymes.
To generate an hTR RNA with a 415 nucleotide 3′ extension, genomic DNA was amplified using thermocycling with the following primers with EcoRI and SacI restriction sites:
Forward: 5′ – GCCGAGCTCCGGGTTGCGGAGGGTGGGCCTGGGAG – 3′
Reverse: 5′ – GCCGAATTCGCTTGTGGGGGTTATATCCTACTG – 3′
These primers were used to amplify hTR 3′ extension through thermocycling using standard molecular biology techniques. Following amplification, the product was put through a PCR Clean-up Kit (Qiagen, Hilden, Germany) and cloned into pBluescript KS+ cut with the appropriate enzymes. These ligations were then transformed into One Shot MAX Efficiency DH5α-T1R competent cells following the manufacturer's suggested protocol and sequence verified.
ScaRNA2ΔGU was generated by site directed mutagenesis in both scaRNA2 pcDNA 3.1+ and pBluescript KS+ vectors using standard molecular biology techniques. Mutants were generated by replacing the GU-rich region upstream of the U2-25 domain with a 15-base linker. The primers used for this mutagenesis were as follows (with the linker sequence underlined):
Forward: 5′ – ATCGTCGCAGGATCCGCGCGCTTGGAGCGTG – 3′
Reverse: 5′ – GGATCCTGCGACGATCCTGCACGCG – 3′
ScaRNA9, which is encoded within an intron, was amplified from genomic DNA using the following primers (which partially bind to the exonic sequences of the host gene). EcoRI and NotI restriction sties present within the primers are underlined:
Forward: 5′ – GGCGAATTCTTAAGTTATGCTGTGGAGGAAG – 3′
Reverse: 5′ – GGCGCGGCCGCTTTCATAACTTAAAAGGCTCC – 3′
The product of this reaction was TA cloned into pCR4-TOPO (Invitrogen, Carlsbad, CA, USA), subsequently cloned cloned into pcDNA 3.1+ digested with EcoRI and NotI and sequenced verified. To delete the GU region of scaRNA9 in pcDNA 3.1+, the following mutagenesis primers, with linker sequenced underlined, were used:
Forward: 5′ – ATCGTCGCAGGATCCACGCACATGTGTTTATAAAGATAACAGC – 3′
Reverse: 5′ – GGATCCTGCGACGATTATGCCCTTATTGTTTTACATTGTTTTATAGTTTTGC – 3′
In pBluescript, the size of each RNA clone is: hTR – 451nt, hTR 3′ extension – 966nt, scaRNA9 – 352nt, scaRNA9 5′ – 149nt, scaRNA9 3′ – 166nt, scaRNA9ΔGU – 314nt, scaRNA9 3′ extension - 486nt, scaRNA2 – 420nt, scaRNA2 5′ – 253nt, scaRNA2 3′ – 167nt, and scaRNA2ΔGU – 401nt. Note that this size does not include the actual transcript size, which includes upstream and downstream sequences.
For reverse transcriptase RT-PCR, total RNA was isolated using the Macherey-Nagel NucleoSpin kit (Bethlehem, PA, USA). This RNA was then used as a template in a reaction using the Brilliant II SYBR Green qRT-PCR Master Mix from Agilent (Santa Clara, CA, USA). Primers to amplify coilin mRNA have been described previously (22). The primers used to amplify coilp1 mRNA are indicated in Fig. S1, and are:
Coilp1 For:
5′ – GTTTTCACTTTACTTTTCCC – 3′
Coilp1 Rev:
5′ – CGGAAGCCGACTTTCAAGCC – 3′
Antibodies
The following antibodies were used in this study: GFP monoclonal (11814460001, Roche, Indianapolis, IN, USA), SMN monoclonal (610646, BD Transduction Laboratories, Franklin Lakes, NJ, USA), Coilin polyclonal (sc-32860, Santa Cruz Biotechnology, Santa Cruz, CA, USA), Dyskerin polyclonal (sc-48794, Santa Cruz Biotechnology, Santa Cruz, CA, USA), β-Tubulin (T5201, Sigma Aldrich, St. Louis, MO, USA), Fibrillarin polyclonal (sc-25397, Santa Cruz Biotechnology, CA, USA), Normal Mouse IgG (sc-2025, Santa Cruz Biotechnology, Santa Cruz, CA, USA), T7 monoclonal antibody (ab50545, Cambridge, MA, USA), Goat Anti-Mouse Alexa Fluor 488 (A11001, Invitrogen, Carlsbad, CA, USA), Goat Anti-Rabbit Alexa Fluor 488 (A11008, Invitrogen, Carlsbad, CA, USA), and Goat Anti-Rabbit Alexa Fluor 594 (A11012, Invitrogen, Carlsbad, CA, USA).
Immunoprecipitation
HeLa cells were lysed in RIPA buffer (50 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% Sodium deoxycholate, 1 mM EDTA, 0.1% SDS) containing protease inhibitor cocktail (Roche, Indianapolis, IN, USA) on ice for 10 minutes. The lysates were sonicated 3 times in 5-second intervals at 5W using the Fisher Scientific Sonic Dismembrator Model 100. The lysates were then clarified by centrifugation at 16,000 g for 15 minutes at 4°C. 15 μl of the cleared lysate from each sample was saved for input and the remainder was incubated for one hour at 4°C while rocking with 2 μg of antibody. Then, the cleared lysates were incubated for 2 hours at 4°C while rocking with 40μl 50% Protein G Sepharose 4 Fast Flow beads (GE Healthcare, Pittsburg, PA, USA). The antibody-bead complexes were then washed 3 times with 1 mL mRIPA buffer following centrifugation at 4,500 g. Beads were resuspended in 2X-SDS loading buffer and subjected to SDS-PAGE and Western transfer.
Western blot
Cell lysate or immunocomplexes were re-suspended in 2x SDS LB and boiled at 95°C for 5 minutes. Then, the samples were subjected to SDS-PAGE and transferred to a nitrocellulose membrane. The membrane was then blocked with 5% non-fat milk and TBST (50 mM Tris, 150 mM NaCl, 0.05% Tween 20). Membranes were then immunoblotted with antibodies in 2.5% non-fat milk and TBST. Bands were detected by incubating the membranes with species-specific HRP conjugated antibodies in 2.5% non fat milk and TBST for 1 hour followed by 5 minute incubation with SuperSignal West Pico Chemiluminescent Substrate (Life Technologies, Grand Island, NY, USA). All membranes were imaged using a ChemiDoc imager with Quantity One software (BioRad, Hercules, CA, USA). Any adjustments made to images (using the high and low settings within QuantityOne software) were made across the entire gel.
RNA pulldown
To generate in vitro transcription products used for the RNA pulldown assays, pBluescript vectors were linearized with PstI, BamH1 or HindIII (depending on the plasmid used) and gel purified using standard molecular biological techniques. The linear DNA was next used to generate Biotin-labeled RNA probes by using either T3 Megascript kit (all scaRNA2 constructs and all scaRNA9 constructs except scaRNA9 3′ extension). ScaRNA9 3′ extension and hTR transcripts were generated using the T7 Megascript kit (Ambion by Life Technologies, Pittsburgh, PA, USA) and Biotin-16-UTP (Roche, Indianapolis, IN, USA) in a ratio of 2:3 Biotin-16-UTP:UTP. Reactions were performed as suggested by the manufacturer's suggested protocol. For a negative control RNA, the pTRI-Xef linear template provided in the Megascript kit was in vitro transcribed by T7 RNA polymerase, resulting in Xenopus elongation factor 1 α RNA. Following the in vitro transcription reaction, the resultant RNAs were DNase I treated for 30 minutes at 37°C to remove template DNA, and then purified using the Zymo RNA Clean and Concentrator (Zymo Research, Irvine, CA, USA) following the manufacturer's suggested protocol. In vivo pulldown: HeLa cells were lysed using RIPA buffer (50 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% Sodium deoxycholate, 1 mM EDTA, 0.1% SDS). Pulldowns were performed by the addition of 10 μg RNA probe to clarified cell lysate from 20 × 106 cells and incubation for 1 hour while at 4°C with rocking, followed by the addition of 100 μL 50% streptavidin beads and 1 hour incubation with rocking at 4°C for 1 hour. In vitro pulldown: 30 μL of His-tagged protein, described above, was used for each pulldown with 3 μL of the His-tagged protein used as input for analysis of recovery. 5 μg of RNA probe was added to 1 mL PBS containing His-tagged protein and nutated for 1 hour at 4°C. Then, 50 μL 50% streptavidin beads were added and incubated while nutating for 1 hour at 4°C. For both in vitro and in vivo pulldowns, the resulting bead complexes were then washed 3 times in PBS, except where noted, and boiled in 30 μl 2x SDS LB before being subjected to Western Blotting using methods described above. To verify that equivalent amounts of RNA were used in the pulldown reactions, RNAs were quantified using a NanoDrop spectrophotometer and agarose gel electrophoresis using a Bio-Rad Chemi-Doc and QuantityOne software.
Coilin antibody depletion
30 μl of coilin polyclonal antibody (sc-32860) was incubated with 10 μg GST or GST-coilin, both of which were bound to glutathione beads, in 1 ml PBS for 1.5 hrs while rocking at 4°C. The plasmids used to generate GST and GST-coilin, and the protein purification protocol, have been published previously (Hebert et al., 2001; Toyota et al., 2010). After incubation, the tubes were centrifuged at 16,000 g in a microcentrifuge for 2 minutes. The supernatants containing GST depleted or GST-coilin depleted coilin antibody solution was then diluted into 15 mls of TBST with 2.5% non-fat milk and used to probe Western transfer membranes.
Ammonium sulfate fractionation
8.8 × 106 HeLa cells were detached from dishes using trypsin, followed by resuspension in DMEM and centrifugation to collect cell pellet. The cell pellet was then washed twice with cold PBS and then resuspended in 1 ml Buffer N (15 mM Tris-HCl pH = 7.5, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, 1 mM DTT). After the pellet was resuspended in Buffer N, 1 ml 0.6% NP-40 in Buffer N was added. Nuclei were then isolated by centrifugation at 2,000 g for 5 minutes at 4°C. The supernatant was saved as the cytoplasmic fraction. The nuclei pellet was washed twice with Buffer N. The pellet was then re-suspended with 1 ml Buffer B (10 mM PIPES pH = 6.8, 100 mM KCl, 300 mM Sucrose, 3 mM MgCl2, 1 mM EGTA) and incubated with 2μl DNase AT1 for 45 minutes at 37°C. The solution was then centrifuged at 1,500 g for 10 minutes at 4°C. The pellet was then re-suspended in 250 mM Ammonium Sulfate in Buffer B. The solution was centrifuged at 4,000 g for 5 minutes at 4°C. The supernatant and pellet Ammonium Sulfate fractions were utilized for subsequent isolation steps.
HPLC
Ammonium sulfate supernatant cuts were fractionated using standard HPLC techniques on an Ultimate 3000 Chromatography System consisting of a LPG-3400SD pump, ACC3000T autosampler/column heater, and VWD-3400RS multi-wavelength detector all controlled by Chromeleon software, version 6.80 (Thermo Fisher Scientific, Inc. Waltham, MA 02451). The samples were adjusted to 10% acetonitrile, 0.1% TFA final concentration and 25 μg of total protein was applied to a BetaBasic-18 reversed-phase HPLC column (150 × 2.1 mm; 3.5 micron pore size; Thermo Fisher Scientific, Inc. Waltham, MA 02451) temperature controlled at 35°C. A binary solvent system consisting of 0.1% trifluoroacetic acid in water (Buffer A) and 95% acetonitrile/0.1% trifluoroacetic acid in water (Buffer B) was used. Proteins were eluted with a multi-segmented gradient created by mixing Buffers A and B: Equilibration 5 minutes at 10% Buffer B; ramp to 70% Buffer B over 27 minutes (resolving phase); ramp to 98% Buffer B over 2 minutes; hold at 98% Buffer B for 2 minutes; ramp to 10% Buffer B over 2 minutes; re-equilibrate at 10% B for 15 minutes. The flow rate was 0.243 mL/minute. The elution of proteins was monitored simultaneously at 205, 230, 260, and 280 nm. Fraction collection was facilitated with a Gilson 203B Fraction Collector (Gilson, Inc. Middleton, WI 53562).
Mass spectrometry
Discovery and targeted proteomics experiments on gel bands and fractionated samples were accomplished using an LC/MS system comprised of an ABSCIEX 5500 QTRAP Mass Spectrometer with a Turbo V electrospray ionization source (ABSCIEX, Framingham, MA 01701) coupled to a Dionex Ultimate 3000 Nano/Cap NCS 3500RS HPLC system with an Aquasil C18 reversed-phase column (150 × 0.32 mm, 3 micron particle size) and WPS-3000TBFC autosampler (Thermo Fisher Scientific, Inc. Waltham, MA 02451) under the control of Analyst 1.5.2 software. A binary buffer system (see HPLC method section) was used except formic acid replaced trifluoroacetic acid, equilibration was at 2% Buffer B, and the flow rate was 8 μL/minute. Samples were trypsinized using standard in-gel or solution digest techniques and Proteomics grade trypsin following the manufacturer's instructions (Pierce Trypsin Protease, MS Grade, product number 90057, ThermoFisher Scientific, Inc. Waltham, MA 02451). After speed vacuuming to dryness, samples were resuspended in 0.1% formic acid/water, and analyzed via LC/MS. For Discovery experiments, information dependent data acquisition with ion intensity as the trigger was used to collect sequence information from peptides eluting over a 3-hour resolving phase for the HPLC gradient. The raw data was then transferred to a Workstation running the Protein Pilot 4.0 analysis package (ABSCIEX, Framingham MA 01701) for protein identification. For targeted proteomics, Skyline version 3.1 (MacCoss Lab, Dept. of Genome Sciences, Univ. of Washington) was used to build multiple reaction monitoring (MRM) methods based on the known or predicted amino acid sequence of candidates. Using this information, samples were analyzed by LC/MS in MRM mode to confirm the presence of specific tryptic peptide sequences and thus protein identity and presence. These general protocols are standard procedures used in the Mass Spectrometry Core Facility (MSCF) at UMMC (https://www.umc.edu/Education/Schools/Medicine/Basic_Science/Pharmacology_and_Toxicology/Facilities.
Cyclohexamide treatment
Cyclohexamide solution (D4540, Sigma Aldrich, St. Louis, MO, USA) was used to treat HeLa cells. Cells were treated with vehicle (DMSO) or 30μg/mL cyclohexamide for 2, 4, 6, 8, 10, 16, and 24-hours. Following treatment, protein stability of coilin and coilp1 was analyzed by Western blotting as described above.
RNA degradation assay
ScaRNA2 and scaRNA2 ΔGU clones in pBluescript KS were transcribed in vitro as previously described.30 100 ng of each transcript was incubated at 37°C for 30 minutes with no protein, nucleic acid free purified coilin (50 ng, 100 ng, or 150 ng) or purified GST (150 ng). Coilin was purified as previously described 34 with modifications mentioned in.30 The reactions were then subjected to agarose gel electrophoresis and stained with ethidium bromide.
Northern blotting
HeLa cells were transfected with scaRNA9, scaRNA9ΔGU, scaRNA2, or scaRNA2ΔGU clones in pcDNA 3.1+ vector for 24 hrs. The cells were then harvested and RNA was isolated using TRI Reagent (Molecular Research Center, Cincinnati, OH, USA) according to the manufacturer's suggested protocol. 20 μg of each RNA sample was then run on a 6% denaturing polyacrylamide gel in 1X TBE at 200 V for 30 – 40 minutes. Each gel was washed in 200 ml 1x TBE for 10 minutes with gentle shaking. The gel was then transferred onto a positively charged nylon membrane using DNA Stacks (Invitrogen, Carlsbad, CA, USA) and the iBlot Gel Transfer device (Life Technologies, Grant Island, NY, USA) using program P8 for 13 minutes for transfer. After transfer, the blot was rinsed quickly in distilled water and allowed to dry. The RNA was then cross-linked to the membrane using a UV cross-linker at a setting of 120,000μJ/cm2. The blot was then placed in a hybridization bottle and pre-hybridized using 15–20 ml Ultrahyb Ultrasensitive Hybridization buffer per blot for 30 minutes at 37°C in a hybridization oven. The following DNA oligo probes was used for detection of scaRNA9 and scaRNA9ΔGU:
5′ – TAGAAACCATCATAGTTACAAAGATCAGTAGTAAAACCTTTTCATCATTGCCC – 3′
The following DNA probe was used for the detection of scaRNA2 and scaRNA2ΔGU:
5′ – CTCGTCTATCTGATCAATTCATCACTTCT – 3′
The probes were labeled using the DIG Oligonucleotide Tailing Kit, 2nd Generation (Roche, Indianapolis, IN, USA) according to the manufacturer's protocol. After pre-hybridization, 22 μl of tailed probe was added to 20 ml of Ultrahyb Ultrasensitive Hybridization buffer to a final concentration of 4.5 nM. The blot was then hybridized overnight at 37°C with slow rotation in a hybridization oven. Washes were conducted according to protocol from,58 using the DIG Wash and Block Buffer Set (Roche, Indianapolis, IN, USA), with a few modifications. DIG antibody was used at 1:10,000 and DIG Wash buffer steps were reduced to two 15-minute washes. Signal was generated using CSPD (Roche, Indianapolis, IN, USA) in a 1:100 dilution with detection buffer. The blot was incubated with this substrate for 5 minutes then placed between 2 plastic transparencies and incubated for 15 minutes at 37°C for 15 minutes in the dark. Blots were imaged using a ChemiDoc imager, and processed using QuantityOne software.
Immunofluorescence
Cells were grown on a glass chamber slide and fixed using 4.0% paraformaldehyde followed by permeabilization with 0.5% Triton. The cells were then blocked with 10% normal goat serum for 30 minutes. For detection of protein, cells were incubated for 30 minutes with either 250 μg SMN monoclonal, 200 μg myc monoclonal, and/or 200 μg coilin polyclonal primary antibodies followed by a 30 minute incubation with either Alexa Fluor 488 or Alexa Fluor 594 secondary antibodies. Where indicated, cells were transfected with plasmid DNA using FuGene (For coilp1 fusion proteins, 125 ng DNA was used). Cells were observed using a Nikon Eclipse E600 epiflourescence microscope, and digital images were taken using Photometics CoolSnap HQ2 CCD camera and processed using MetaView software. PowerPoint and Adobe Photoshop Elements 7 were used in the preparation of images for this manuscript.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The Intramural Research Support Program of the University of Mississippi Medical Center supported this work. Jackson Coole was supported by the Summer Undergraduate Research Experience (SURE) program of the School of Graduate Studies at the University of Mississippi Medical Center.
References
- 1.Kiss T. Biogenesis of small nuclear RNPs. J Cell Sci 2004; 117:5949-51; PMID:15564372; http://dx.doi.org/ 10.1242/jcs.01487 [DOI] [PubMed] [Google Scholar]
- 2.Fischer U, Liu Q, Dreyfuss G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell 1997; 90:1023-9; PMID:9323130; http://dx.doi.org/ 10.1016/S0092-8674(00)80368-2 [DOI] [PubMed] [Google Scholar]
- 3.Pellizzoni L, Charroux B, Dreyfuss G. SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proc Natl Acad Sci U S A 1999; 96:11167-72; PMID:10500148; http://dx.doi.org/ 10.1073/pnas.96.20.11167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pellizzoni L, Yong J, Dreyfuss G. Essential Role for the SMN Complex in the Specificity of snRNP Assembly. Science 2002; 298:1775-9; PMID:12459587; http://dx.doi.org/ 10.1126/science.1074962 [DOI] [PubMed] [Google Scholar]
- 5.Meister G, Eggert C, Fischer U. SMN-mediated assembly of RNPs: a complex story. Trends Cell Biol 2002; 12:472-8; PMID:12441251; http://dx.doi.org/ 10.1016/S0962-8924(02)02371-1 [DOI] [PubMed] [Google Scholar]
- 6.Paushkin S, Gubitz AK, Massenet S, Dreyfuss G. The SMN complex, an assemblyosome of ribonucleoproteins. Curr Opin Cell Biol 2002; 14:305-12; PMID:12067652; http://dx.doi.org/ 10.1016/S0955-0674(02)00332-0 [DOI] [PubMed] [Google Scholar]
- 7.Coady TH, Lorson CL. SMN in spinal muscular atrophy and snRNP biogenesis. Wiley Interdiscip Rev RNA 2011; 2:546-64; PMID:21957043; http://dx.doi.org/ 10.1002/wrna.76 [DOI] [PubMed] [Google Scholar]
- 8.Pearn J. Classification of spinal muscular atrophies. Lancet 1980; 8174:919-22; PMID:6103267; http://dx.doi.org/7813012 10.1016/S0140-6736(80)90847-8 [DOI] [PubMed] [Google Scholar]
- 9.Gitlin JM, Fischbeck K, Crawford TO, Cwik V, Fleischman A, Gonye K, Heine D, Hobby K, Kaufmann P, Keiles S, et al.. Carrier testing for spinal muscular atrophy. Genetics Med 2010; 12:621-2; PMID:20808230; http://dx.doi.org/7813012 10.1097/GIM.0b013e3181ef6079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al.. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995; 80:155-65; PMID:7813012; http://dx.doi.org/ 10.1016/0092-8674(95)90460-3 [DOI] [PubMed] [Google Scholar]
- 11.Jedrzejowska M, Gos M, Zimowski JG, Kostera-Pruszczyk A, Ryniewicz B, Hausmanowa-Petrusewicz I. Novel point mutations in survival motor neuron 1 gene expand the spectrum of phenotypes observed in spinal muscular atrophy patients. Neuromuscul Disord 2014; 24:617-23; PMID:24844453; http://dx.doi.org/ 10.1016/j.nmd.2014.04.003 [DOI] [PubMed] [Google Scholar]
- 12.Praveen K, Wen Y, Gray KM, Noto JJ, Patlolla AR, Van Duyne GD, Matera AG. SMA-causing missense mutations in survival motor neuron (Smn) display a wide range of phenotypes when modeled in Drosophila. PLoS Genet 2014; 10:e1004489; PMID:25144193; http://dx.doi.org/ 10.1371/journal.pgen.1004489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lotti F, Imlach WL, Saieva L, Beck ES, Hao le T, Li DK, Jiao W, Mentis GZ, Beattie CE, McCabe BD, et al.. An SMN-dependent U12 splicing event essential for motor circuit function. Cell 2012; 151:440-54; PMID:23063131; http://dx.doi.org/ 10.1016/j.cell.2012.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Praveen K, Wen Y, Matera AG. A Drosophila model of spinal muscular atrophy uncouples snRNP biogenesis functions of survival motor neuron from locomotion and viability defects. Cell Reports 2012; 1:624-31; PMID:22813737; http://dx.doi.org/ 10.1016/j.celrep.2012.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia EL, Lu Z, Meers MP, Praveen K, Matera AG. Developmental arrest of Drosophila survival motor neuron (Smn) mutants accounts for differences in expression of minor intron-containing genes. RNA 2013; 19:1510-6; PMID:24006466; http://dx.doi.org/ 10.1261/rna.038919.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goulet BB, Kothary R, Parks RJ. At the “junction” of spinal muscular atrophy pathogenesis: the role of neuromuscular junction dysfunction in SMA disease progression. Curr Mol Med 2013; 13:1160-74; PMID:23514457; http://dx.doi.org/ 10.2174/15665240113139990044 [DOI] [PubMed] [Google Scholar]
- 17.Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, Alvarez FJ, Sumner CJ, O'Donovan MJ. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 2011; 69:453-67; PMID:21315257; http://dx.doi.org/ 10.1016/j.neuron.2010.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Donlin-Asp PG, Bassell GJ, Rossoll W. A role for the survival of motor neuron protein in mRNP assembly and transport. Curr Opin Neurobiol 2016; 39:53-61; PMID:27131421; http://dx.doi.org/ 10.1016/j.conb.2016.04.004 [DOI] [PubMed] [Google Scholar]
- 19.Hamilton G, Gillingwater TH. Spinal muscular atrophy: going beyond the motor neuron. Trends Mol Med 2013; 19:40-50; PMID:23228902; http://dx.doi.org/ 10.1016/j.molmed.2012.11.002 [DOI] [PubMed] [Google Scholar]
- 20.Shababi M, Lorson CL, Rudnik-Schoneborn SS. Spinal muscular atrophy: a motor neuron disorder or a multi-organ disease? J Anat 2014; 224:15-28; PMID:23876144; http://dx.doi.org/ 10.1111/joa.12083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci 2009; 10:597-609; PMID:19584893; http://dx.doi.org/ 10.1038/nrn2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carvalho T, Almeida F, Calapez A, Lafarga M, Berciano MT, Carmo-Fonseca M. The spinal muscular atrophy disease gene product, SMN: A link between snRNP biogenesis and the Cajal (coiled) body. J Cell Biol 1999; 147:715-28; PMID:10562276; http://dx.doi.org/ 10.1083/jcb.147.4.715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandez Garcia MS, Teruya-Feldstein J. The diagnosis and treatment of dyskeratosis congenita: a review. J Blood Med 2014; 5:157-67; PMID:25170286; http://dx.doi.org/ 10.2147/JBM.S47437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trahan C, Dragon F. Dyskeratosis congenita mutations in the H/ACA domain of human telomerase RNA affect its assembly into a pre-RNP. Rna 2009; 15:235-43; PMID:19095616; http://dx.doi.org/ 10.1261/rna.1354009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Egan ED, Collins K. Biogenesis of telomerase ribonucleoproteins. Rna 2012; 18:1747-59; PMID:22875809; http://dx.doi.org/ 10.1261/rna.034629.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jady BE, Bertrand E, Kiss T. Human telomerase RNA and box H/ACA scaRNAs share a common Cajal body-specific localization signal. J Cell Biol 2004; 164:647-52; PMID:14981093; http://dx.doi.org/ 10.1083/jcb.200310138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tycowski KT, Shu MD, Kukoyi A, Steitz JA. A conserved WD40 protein binds the Cajal body localization signal of scaRNP particles. Mol Cell 2009; 34:47-57; PMID:19285445; http://dx.doi.org/ 10.1016/j.molcel.2009.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Venteicher AS, Abreu EB, Meng Z, McCann KE, Terns RM, Veenstra TD, Terns MP, Artandi SE. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 2009; 323:644-8; PMID:19179534; http://dx.doi.org/ 10.1126/science.1165357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marnef A, Richard P, Pinzon N, Kiss T. Targeting vertebrate intron-encoded box C/D 2′-O-methylation guide RNAs into the Cajal body. Nucleic Acids Res 2014; 42:6616-29; PMID:24753405; http://dx.doi.org/ 10.1093/nar/gku287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Enwerem II, Velma V, Broome HJ, Kuna M, Begum RA, Hebert MD. Coilin association with Box C/D scaRNA suggests a direct role for the Cajal body marker protein in scaRNP biogenesis. Biol Open 2014; 3:240-9; PMID:24659245; http://dx.doi.org/ 10.1242/bio.20147443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahmoudi S, Henriksson S, Weibrecht I, Smith S, Soderberg O, Stromblad S, Wiman KG, Farnebo M. WRAP53 is essential for Cajal body formation and for targeting the survival of motor neuron complex to Cajal bodies. PLoS Biol 2010; 8:e1000521; PMID:21072240; http://dx.doi.org/25514182 10.1371/journal.pbio.1000521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Machyna M, Kehr S, Straube K, Kappei D, Buchholz F, Butter F, Ule J, Hertel J, Stadler PF, Neugebauer KM. The coilin interactome identifies hundreds of small noncoding RNAs that traffic through Cajal bodies. Mol Cell 2014; 56:389-99; PMID:25514182; http://dx.doi.org/ 10.1016/j.molcel.2014.10.004 [DOI] [PubMed] [Google Scholar]
- 33.Broome HJ, Hebert MD. In vitro RNase and nucleic acid binding activities implicate coilin in U snRNA processing. PLoS One 2012; 7:e36300; PMID:22558428; http://dx.doi.org/ 10.1371/journal.pone.0036300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Broome HJ, Hebert MD. Coilin displays differential affinity for specific RNAs in vivo and is linked to telomerase RNA biogenesis. J Mol Biol 2013; 425:713-24; PMID:23274112; http://dx.doi.org/ 10.1016/j.jmb.2012.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Broome HJ, Carrero ZI, Douglas HE, Hebert MD. Phosphorylation regulates coilin activity and RNA association. Biol Open 2013; 2:407-15; PMID:23616925; http://dx.doi.org/ 10.1242/bio.20133863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bachand F, Boisvert FM, Cote J, Richard S, Autexier C. The product of the survival of motor neuron (SMN) gene is a human telomerase-associated protein. Mol Biol Cell 2002; 13:3192-202; PMID:12221125; http://dx.doi.org/ 10.1091/mbc.E02-04-0216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pellizzoni L, Baccon J, Charroux B, Dreyfuss G. The survival of motor neurons (SMN) protein interacts with the snoRNP proteins fibrillarin and GAR1. Curr Biol 2001; 11:1079-88; PMID:11509230; http://dx.doi.org/ 10.1016/S0960-9822(01)00316-5 [DOI] [PubMed] [Google Scholar]
- 38.Wang C, Meier UT. Architecture and assembly of mammalian H/ACA small nucleolar and telomerase ribonucleoproteins. Embo J 2004; 23:1857-67; PMID:15044956; http://dx.doi.org/ 10.1038/sj.emboj.7600181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li S, Duan J, Li D, Ma S, Ye K. Structure of the Shq1-Cbf5-Nop10-Gar1 complex and implications for H/ACA RNP biogenesis and dyskeratosis congenita. EMBO J 2011; 30:5010-20; PMID:22117216; http://dx.doi.org/ 10.1038/emboj.2011.427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu Y, Tomlinson RL, Lukowiak AA, Terns RM, Terns MP. Telomerase RNA accumulates in Cajal bodies in human cancer cells. Mol Biol Cell 2004; 15:81-90; PMID:14528011; http://dx.doi.org/ 10.1091/mbc.E03-07-0525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tomlinson RL, Ziegler TD, Supakorndej T, Terns RM, Terns MP. Cell cycle-regulated trafficking of human telomerase to telomeres. Mol Biol Cell 2006; 17:955-65; PMID:16339074; http://dx.doi.org/ 10.1091/mbc.E05-09-0903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jady BE, Richard P, Bertrand E, Kiss T. Cell Cycle-dependent recruitment of telomerase RNA and Cajal bodies to human telomeres. Mol Biol Cell 2006; 17:944-54; PMID:16319170; http://dx.doi.org/ 10.1091/mbc.E05-09-0904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hebert MD, Szymczyk PW, Shpargel KB, Matera AG. Coilin forms the bridge between Cajal bodies and SMN, the spinal muscular atrophy protein. Genes Dev 2001; 15:2720-9; PMID:11641277; http://dx.doi.org/ 10.1101/gad.908401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hebert MD, Shpargel KB, Ospina JK, Tucker KE, Matera AG. Coilin methylation regulates nuclear body formation. Dev Cell 2002; 3:329-37; PMID:12361597; http://dx.doi.org/ 10.1016/S1534-5807(02)00222-8 [DOI] [PubMed] [Google Scholar]
- 45.Jones KW, Gorzynski K, Hales CM, Fischer U, Badbanchi F, Terns RM, Terns MP. Direct interaction of the spinal muscular atrophy disease protein SMN with the small nucleolar RNA-associated protein fibrillarin. J Biol Chem 2001; 276:38645-51; PMID:11509571; http://dx.doi.org/ 10.1074/jbc.M106161200 [DOI] [PubMed] [Google Scholar]
- 46.Tycowski KT, Aab A, Steitz JA. Guide RNAs with 5′ caps and novel box C/D snoRNA-like domains for modification of snRNAs in metazoa. Curr Biol 2004; 14:1985-95; PMID:15556860; http://dx.doi.org/ 10.1016/j.cub.2004.11.003 [DOI] [PubMed] [Google Scholar]
- 47.Gerard MA, Myslinski E, Chylak N, Baudrey S, Krol A, Carbon P. The scaRNA2 is produced by an independent transcription unit and its processing is directed by the encoding region. Nucleic Acids Res 2010; 38:370-81; PMID:19906720; http://dx.doi.org/ 10.1093/nar/gkp988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Enwerem II, Wu G, Yu YT, Hebert MD. Cajal body proteins differentially affect the processing of box C/D scaRNPs. PLoS One 2015; 10:e0122348; PMID:25875178; http://dx.doi.org/ 10.1371/journal.pone.0122348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Velma V, Broome HJ, Hebert MD. Regulated specific proteolysis of the Cajal body marker protein coilin. Chromosoma 2012; 121:629-42; PMID:23064547; http://dx.doi.org/ 10.1007/s00412-012-0387-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andrade LE, Tan EM, Chan EK. Immunocytochemical analysis of the coiled body in the cell cycle and during cell proliferation. Proc Natl Acad Sci USA 1993; 90:1947-51; PMID:8446613; http://dx.doi.org/ 10.1073/pnas.90.5.1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tucker KE, Berciano MT, Jacobs EY, LePage DF, Shpargel KB, Rossire JJ, Chan EK, Lafarga M, Conlon RA, Matera AG. Residual Cajal bodies in coilin knockout mice fail to recruit Sm snRNPs and SMN, the spinal muscular atrophy gene product. J Cell Biol 2001; 154:293-307; PMID:11470819; http://dx.doi.org/ 10.1083/jcb.200104083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chan EK, Takano S, Andrade LE, Hamel JC, Matera AG. Structure, expression and chromosomal localization of human p80-coilin gene. Nucleic Acids Res 1994; 22:4462-9; PMID:7971277; http://dx.doi.org/ 10.1093/nar/22.21.4462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al.. A draft map of the human proteome. Nature 2014; 509:575-81; PMID:24870542; http://dx.doi.org/ 10.1038/nature13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lorson CL, Androphy EJ. The domain encoded by exon 2 of the survival motor neuron protein mediates nucleic acid binding. Hum Mol Genet 1998; 7:1269-75; PMID:9668169; http://dx.doi.org/ 10.1093/hmg/7.8.1269 [DOI] [PubMed] [Google Scholar]
- 55.Bertrandy S, Burlet P, Clermont O, Huber C, Fondrat C, Thierry-Mieg D, Munnich A, Lefebvre S. The RNA-binding properties of SMN: deletion analysis of the zebrafish orthologue defines domains conserved in evolution. Hum Mol Genet 1999; 8:775-82; PMID:10196366; http://dx.doi.org/ 10.1093/hmg/8.5.775 [DOI] [PubMed] [Google Scholar]
- 56.Tucker KE. Residual Cajal bodies in coilin knockout mice fail to recruit Sm snRNPs and SMN, the spinal muscular atrophy gene product. J Cell Biol 2001; 154:293-308; PMID:11470819; http://dx.doi.org/ 10.1083/jcb.200104083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toyota CG, Davis MD, Cosman AM, Hebert MD. Coilin phosphorylation mediates interaction with SMN and SmB'. Chromosoma 2010; 119:205-15; PMID:19997741; http://dx.doi.org/ 10.1007/s00412-009-0249-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim SW, Li Z, Moore PS, Monaghan AP, Chang Y, Nichols M, John B. A sensitive non-radioactive northern blot method to detect small RNAs. Nucleic Acids Res 2010; 38:e98; PMID:20081203; http://dx.doi.org/ 10.1093/nar/gkp1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.