

ABSTRACT
Among all tissues of the metazoa, the transcritpome of testis displays the highest diversity and specificity. However, its composition and dynamics during spermatogenesis have not been fully understood. Here, we have identified 20,639 message RNAs (mRNAs), 7,168 long non-coding RNAs (lncRNAs) and 15,101 circular RNAs (circRNAs) in mouse spermatogenic cells, and found many of them were specifically expressed in testes. lncRNAs are significantly more testis-specific than mRNAs. At all stages, mRNAs are generally more abundant than lncRNAs, and linear transcripts are more abundant than circRNAs. We showed that the productions of circRNAs and piRNAs were highly regulated instead of random processes. Based on the results of a small-scale functional screening experiment using cultured mouse spermatogonial stem cells, many evolutionarily conserved lncRNAs are likely to play roles in spermatogenesis. Typical classes of transcription factor binding sites are enriched in the promoters of testis-specific m/lncRNA genes. Target genes of CREM and RFX2, 2 key TFs for spermatogenesis, were further validated by using ChIP-chip assays and RNA-seq on RFX2-knockout spermatogenic cells. Our results contribute to the current understanding of the transcriptomic complexity of spermatogenic cells and provide a valuable resource from which many candidate genes may be selected for further functional studies.
KEYWORDS: circRNAs, expression, lncRNAs, mRNAs, spermatogenesis, testis, transcription
Abbreviations
- SPC
spermatogenic cell
- SSC
spermatogonial stem cells
- TFBS
transcription factor binding site
- priSG-A
primitive type A spermatogonia
- plpSC
preleptotene spermatocytes
- pacSC
pachytene spermatocytes
- rST
round spermatids
- mRNA
messenger RNA
- lncRNA
long noncoding RNA
- circRNA
circular RNA
- piRNA
PIWI-interacting RNA
- FPKM
fragments per kilobase per million
- KO
knockout
- KD
knockdown
- ts
testis-specific
Introduction
The majority of mammalian genomes generate a large diversity of RNA species via alternative splicing and other forms of post-transcriptional processings and modifications.1 Historically, the transcriptomic complexity has been investigated with different methods such as Expressed Sequence Tags (EST), Serial Analysis of Gene Expression (SAGE) and microarray technologies. Recently, as a highly sensitive and quantitative method, high throughput RNA-sequencing technology (RNA-seq) has been widely used to address this question. The testis contains highly diverse and specific transcriptomes in comparison with other tissues. For example, more than 15,000 mRNA genes are expressed in this small organ,2,3 and it has been estimated that ∼4.8% of the protein-coding genes are specifically expressed in the mouse testis based on EST and microarray expression data of mRNAs.4
While the involvement of lncRNAs in X chromosome inactivation, genetic imprinting and cell development has long been recognized, the catalogs and expression patterns of lncRNAs have been revealed only in the past several years.5,6 Cabili et al. reported that the human long intergenic noncoding RNAs (lincRNAs) are much more tissue-specific than mRNAs and a third of the 8,000 human lincRNAs are specifically expressed in the testis.7 Soumillon et al. systematically studied the transcriptomes in multiple species and found that the testes of avians and mammals have the highest degree of complexity for both coding and non-coding RNAs.3 The same group further indicated that the mouse genome encodes about 15,000 lncRNAs.8
It has been estimated that more than 92% of human genes are alternatively spliced,9 and each gene may simultaneously produce up to 12 isoforms.10 Alternative splicing has been proposed to be a primary driving force in the evolution of mammalian phenotypic complexity, as significant differences in alternative splicing complexity, the highest being in primates, are observed among vertebrates spanning ∼350 million years of evolution, and the brain has the most abundant alternative splicing followed by the testis in all examined species.11
Circular RNAs (circRNAs) are the products of a unique type of alternative splicing, by which the 3′-end of an exon is spliced to the 5´-end of an upstream exon resulting in a circular RNA molecule.12,13 Although a few examples of circRNAs were reported more than 2 decades ago, the ubiquitous presence and function of circRNAs as a novel form of RNAs remained unknown until recently, partially due to the low abundances of circRNAs.14,15 Deep sequencing of the transcriptomes of various tissues/cell types from different animal species identified a large number of circRNAs, which are developmentally regulated and conserved.16-20 A number of circRNAs have been reported to be sponge-like miRNA sinks,19,21 or to promote the transcription of parent genes.20,22 Further, circRNAs have been shown to be mRNA traps by competing with linear splicing,23,24 and to serve as templates for translation.25,26 It remains a matter of debate whether most low-abundant non-translatable circRNAs have any function or are simply inconsequential side-products of noisy splicing.27
The testis is the male gonad, in which highly specialized haploid sperm are produced from the diploid spermatogonial stem cells (SSCs) through meiosis, a key step in the multistep process known as spermatogenesis. Many types of morphologically distinguishable spermatogenic cells (SPCs), which dominate the testicular cell population, are generated sequentially during spermatogenesis. The mammalian spermatogenesis occurs in a cyclic manner and the development of SPCs in the first cycle was synchronized. Taking advantage of this fact, various types of SPCs can be isolated at different timepoints after birth when they are first generated for genomic, transcriptomic and proteomic studies.28-30 The primitive type A spermatogonia (priSG-A) are earliest stage spermatogonia which undergo mitosis; the preleptotene spermatocytes (plpSC) have completed meiotic DNA replication and are ready for subsequent meiotic events such as DNA recombination and synapsis. The pachytene spermatocytes (pacSC) are meiotic cells, in which synapses are fully established; the round spermatids (rST) are early stage haploids, and undergo a lengthy postmeiotic developmental process to finally generate mature sperm. SPCs are unique in that they specifically express a unique type of small RNA, piRNAs.31 Spermatogonia (SG) mainly express piRNAs that map to mRNAs and retrotransposons, while SC and ST mainly generate piRNAs that map to intergenic regions.28 It has been shown recently that 95% of the adult mouse piRNAs are generated from only 214 genic and intergenic loci.32
We were interested in how the complex mammalian transcriptomes are established. Using spermatogenesis as a model system, we address this question first by profiling the mRNAs, lncRNAs and circRNAs and examining their relative abundances in 4 types of SPCs (priSG-A, plpSC, pacSC, rST). By reanalyzing public datasets, we also identified a large number of testis-specific mRNAs and lncRNAs, and studied their distributions in the 4 SPC types. We further compared the expression patterns of related RNAs and carried out a small-scale functional screen for lncRNAs that are preferentially expressed in priSG-A and conserved across mice and humans. We scanned the promoters of the mRNA and lncRNA genes for putative transcription factor (TF) binding sites (TFBSs) in order to understand how their transcription is regulated. Several families of key TFs such as the CREM family and the RFX family were identified as regulators for transcription of both mRNAs and lncRNAs. The predicted targets of CREM were validated using the ChIP-chip experiment. Moreover, the expression of many mRNAs and lncRNAs were changed when RFX2 was knocked out in mice. These results help us to understand how the complex mammalian transcriptomes are established at the transcription and posttranscription levels.
Results
Comparative analysis of m/lnc/circRNA expression in mouse SPCs
We applied strand-specific, rRNA-depleted, and paired-end RNA-seq to profile the transcriptomes of priSG-A, plpSC, pacSC, and rST. For each cell type, 2 biological replicates were prepared, and the reads of each duplicate were pooled together for further analysis since their Pearson correlation coefficient in terms of mRNA abundances were all above 0.90. As a result, 20,639 mRNAs, 7,168 lncRNAs, and 15,101 circRNAs were found to be expressed in these male germ cells, and they were named g-mRNAs, g-lncRNAs, and g-circRNAs (Table 1, Table 2, Table S1). 59% g-lncRNAs are intergenic, 21% are antisense to coding exons, 11% are intronic, and 9% adopt a head-to-head transcription direction relative to the protein coding genes. Of the g-circRNAs, 71%, 12%, and 17% are exonic, intronic, and intergenic, respectively. By comparing the distributions of their log2-transformed FPKMs (Fragments Per Kilobase of exon model per Million mapped fragments), the overall abundances of g-mRNAs were higher than those of g-lncRNAs by at least one order of magnitude in different cell types (Fig. 1A). The relative levels of circRNAs to linear RNAs were shown by the distributions of log2-transformed ratios of junction reads representing circRNAs to junction reads representing linear RNAs, and the average abundances of circRNAs are 7.1%∼9.8% of the linear transcripts in each cell type (Fig. 1B). Similar to lncRNAs, the average relative abundance of circRNAs in rST is higher than in other cell types.
Table 1.
Nomenclature and counts of g-m/lncRNAs and the testis-specific sets.
| g-RNAs |
|||||
|---|---|---|---|---|---|
| g-mRNAs(20639) | g-lncRNAs (7168) | ||||
| ts-mRNA(332 + 2745 = 3077) | ts-mRNAs-1(JS = 1; 332) | g-ts-mRNAs-1(274) | NA | ||
| ts-RNAs | ts-mRNAs-0.5(0.5 < JS < 1; 2745) | g-ts-mRNAs-0.5(2545) | NA | ||
| ts-lncRNA (3281 + 3426 = 6707) | ts-lncRNAs-1(JS = 1; 3281) | NA | g-ts-lncRNAs-1(2108) | ||
| ts-lncRNAs-0.5(0.5 < JS < 1; 3426) | NA | g-ts-lncRNAs-0.5(2786) | |||
| non-ts-RNAs | non-ts-mRNAs(15932) | g-non-ts-mRNAs(15416) | NA | ||
| non-ts-lncRNAs(1942) | NA | g-non-ts-lncRNAs(1184) | |||
| Others(detected in spermatogenic cells but undetected in tissues) | g-unclassified-mRNAs(2404) | g-unclassified-lncRNAs(1090) | |||
g, germline; ts, testis-specific; JS, Jensen-Shannon divergence 7.
Table 2.
CircRNAs expressed in spermatogenic cells (SPCs).
| circRNAs | overlapping with SSC-R+ (31461) | Non-overlapping with SSC-R+ | |
|---|---|---|---|
| SSC | 5573 | 4376(79%a, 14%b) | 1197(21%c) |
| priSG-A | 5596 | 3948(71%a, 13%b) | 1648(29%c) |
| plpSC | 6689 | 3679(55%a,12%b) | 3010(45%c) |
| pacSC | 4677 | 2490(53%a, 8%b) | 2187(47%c) |
| rST | 7220 | 3162(44%a, 10%b) | 4058(56%c) |
| 4 cell type subtotal | 15101 | 6867(45%a, 22%b) | 8234(55%c) |
denotes the percentages of circRNAs identified in each sample, which overlap circRNAs in the RNase R-treated SSC sample (SSC-R+);
denotes the percentages of circRNAs identified in SSC-R+, which overlap the circRNAs from each cell type;
denotes the percentages of circRNAs identified in each sample, which do not overlap circRNAs in SSC-R+.
Figure 1.
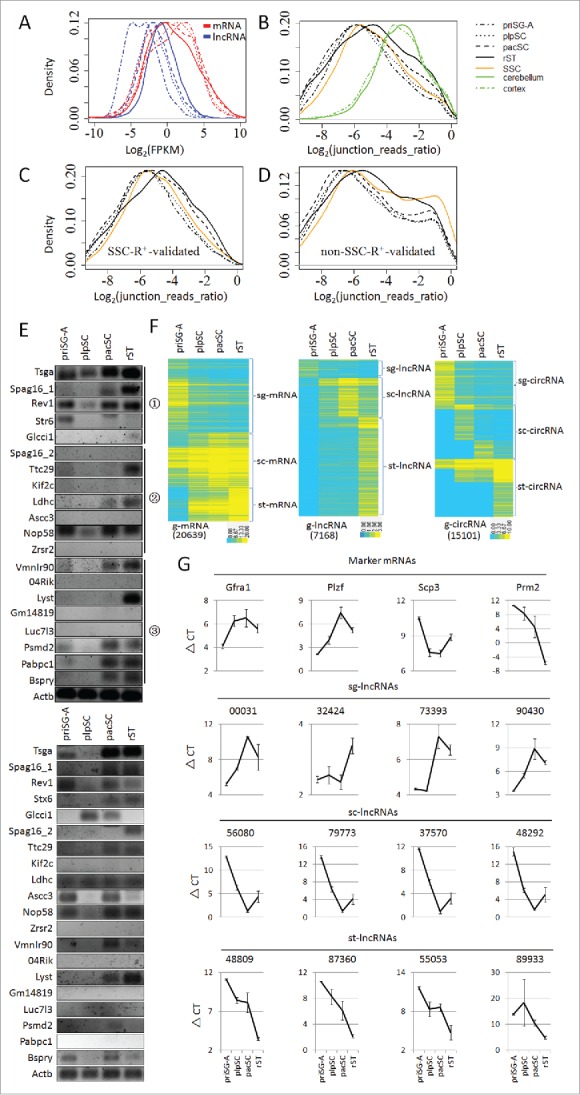
Expression of mRNAs, lncRNAs and circRNAs in primitive type A spermatogonia (priSG-A), preleptotene spermatocytes (plpSC), pachytene spermatocytes (pacSC), round spermatids (rST), collectively referred to as 4 spermatogenic cell (SPC) types. (A) Frequency distributions of the log2-transformed FPKM values of mRNAs and lncRNAs expressed in the 4 SPC types. (B) Distributions of the relative abundances of circRNAs to linear RNAs evaluated in log2-transformed ratios of junction reads representing circRNAs over the junction reads representing linear RNAs in the 4 SPC types and cultured spermatogonial stem cells (SSC) as well as in cerebellum and cortex, the RNA-seq data of which were reported by Rybak-Wolf et al. 34 (C, D) Distributions of the relative abundances of circRNAs overlapping (C) or not overlapping (D) those detected in the RNase R treated SSC sample (SSC-R+). (E) Identity validation of circRNAs by RT-PCR using 2 reverse transcriptases in 2 independent experiments (upper panel: MMLV-derived reverse transcriptase; lower panel: AMV-derived reverse transcriptase). The circRNAs were represented by the names of the corresponding genes. The circled numbers on the right side mark the subsets, from which the circRNAs were selected: 1, sets shown in (C); 2 and 3, sets corresponding to the left and right peaks of the plots in (D), respectively. (F) Clusterings of m/lnc/circRNAs expressed in the 4 SPC types, which are named g-m/lnc/circRNAs, respectively. (G) Expression validation of lncRNAs from the 3 clusters (sg-lncRNAs, sc-lncRNAs and st-lncRNAs) by qRT-PCRs. The expression levels were represented by delta-CT values. The expression of 4 well-known protein coding mRNAs, which are differentially expressed in the 4 SPC types, were included to show that these mRNAs were expressed with the expected dynamics in the isolated cells (Marker mRNAs). Results for 4 lncRNAs from each cluster were shown. Results for all lncRNAs are shown by Fig. S1.
Due to the low abundances of circRNAs, we used RNase R to treat RNA samples of cultured mouse SSCs to enrich circRNAs (Table 2). Many more circRNAs were identified in the RNase R-treated samples (SSC-R+) than in the samples without treatment (SSC). Interestingly, a large proportion of circRNAs were found to be unique to the untreated SSCs, and it is possible that they contain true positives. circRNAs from the 4 isolated SPC types are also partitioned into sets that overlap the SSC-R+ circRNAs and sets that do not overlap (Table 2). Based on the distribution curves of the relative abundances of circRNAs to linear RNAs, the average relative abundances of the circRNAs in the overlapping sets are similar to the averages of the total circRNAs. Interestingly, the non-overlapping sets show bimodal distribution indicating the existence of 2 subsets, one having a lower average expression than the overlapping sets while the other has a higher average expression (Fig. 1C and 1D). We selected a total of 20 circRNAs, 5 from the overlapping set, 7 from the non-overlapping low expression subset and another 8 from the non-overlapping high expression subset, to validate their identities by RT-PCRs. Using the MMLV-derived reverse transcriptase, 5, 3, and 5 circRNAs from these 3 sets were detected, respectively, showing that many circRNAs identified from samples without RNase R treatment are true positives (Fig. 1E, upper panel). To reduce false positive results potentially caused by self-ligation in the reverse transcription reaction, we also repeated the experiments using the AMV-derived reverse transcriptase in an independent experiment, and similar results were obtained except that the Pabpc1 circRNA was not detected this time (Fig. 1E, lower panel). Since most of circRNAs identified from the RNA-seq method were validated with RT-PCR experiments, we included all the predicted circRNAs for further analysis in order to reduce false negatives.
Clustering analysis showed that g-mRNAs, g-lncRNAs, and g-circRNAs could all be similarly partitioned into 3 big clusters based on their highest abundances in spermatogonia, spermatocytes, and spermatids, and were accordingly named sg-, sc-, and st-m/lnc/circRNAs, respectively (Fig. 1F). We selected 12, 10, and 14 g-lncRNAs from each of the 3 clusters, respectively, and validated their expression by quantitative RT-PCR (qRT-PCR) using independent samples of isolated SPCs, and the expression patterns of 5, 10, and 14 lncRNAs from the corresponding clusters were indeed confirmed to be consistent with the RNA-seq results, respectively (Fig. 1G, Fig. S1).
Spermatogenic cell-specific RNAs
Brawand et al. studied gene expression in multiple mammalian organs including the mouse testis, heart, liver, cerebellum, kidney, and brain.33 We used these data sets to identify mRNAs and lncRNAs that are specifically expressed in the mouse testis. We used the Jensen-Shannon (JS) divergence score defined by Cabili et al. to evaluate the tissue specificity of gene expression.7 The JStestis scores of 332 mRNAs and 3,281 lncRNAs are 1, and these 2 RNA sets comprise the most testis-specific sets of mRNAs (ts-mRNAs-1) and lncRNAs (ts-lncRNAs-1) (Table 1). However, we found that setting JStestis score to be 1 is too stringent to identify even the bona fide germ cell-specific transcripts such as Sycp3, the JStestis score of which is 0.53. We compiled a list of 90 testis-specific genes reported by literature and found that 87 of them have a JStestis score higher than 0.5 (Table S2). Consequently, new sets of testis-specific RNAs based on this threshold were defined for mRNAs and lncRNAs (ts-mRNAs-0.5 and ts-lncRNAs-0.5). The validity of this analysis was supported by the identification of spermatogenic cell-specific protein-coding genes such as Pou5f1, Dppa4, Dmrt1 in spermatogonia, Dmc1 in plpSC, Sycp3 and Tdrd1 in pacSC, and Rfx2, Tnp1, Prm1, Prm2 and Spz1 in rST (Table S1). More convincingly, 20 ts-mRNAs were randomly selected for examination of expression in various tissues by RT-PCR (Fig. 2A). Fourteen genes (70%) were exclusively expressed in testis, and almost all of the remaining genes were expressed in 1–3 additional tissues. These results indicate that our bioinformatics mining of ts-RNAs is highly reliable. We found that 92% of ts-mRNAs ((274+2545)/3077) and 73% of ts-lncRNAs ((2108+2786)/6707) are expressed in SPCs (Table 1). On the other hand, 14% of g-mRNAs are testis-specific ((274+2545)/20639) while 68% of the 7168 g-lncRNAs are testis-specific ((2108+2786)/7168). The percentage of g-ts-lncRNAs in all lncRNAs (15934) is 31% ((2108+2786)/ 15934). This estimation of the mouse lncRNA testis-specificity is close to the 30% human lncRNA testis-specificity estimated by Cabili et al. 7 Based on the Fisher exact test, lncRNAs are significantly more testis-specific than mRNAs (p < 2.2E-16). The expression patterns of tissue-specific m/lncRNAs across the examined 6 tissue types and their JStestis scores indicates that there are many more ts-m/lncRNAs than other types of tissue-specific m/lncRNAs (Fig. 2B).
Figure 2.
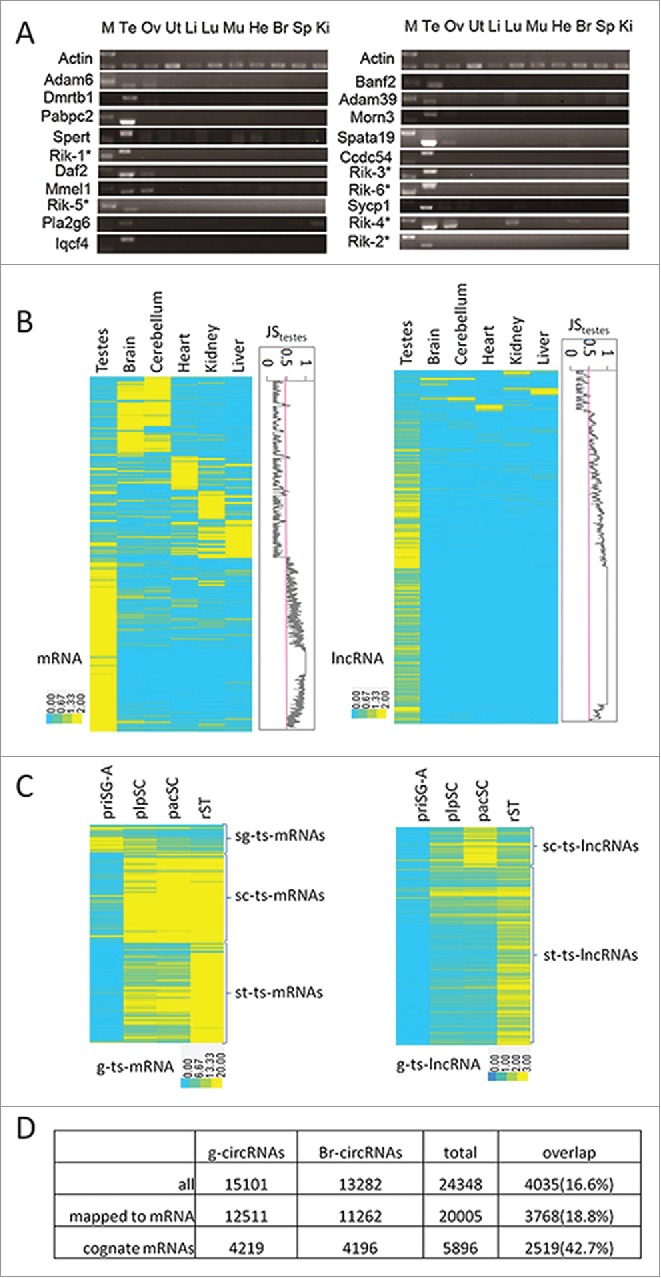
Identification of testis-specific m/lnc/circRNAs. (A) RT-PCR valuation of predicted ts-mRNAs. M, marker; Te, testis; Ov, ovary; Ut, uterus; Li, liver; Lu, lung; Mu, muscle; He, heart; Br, brain; Sp, spleen; Ki, kidney. Rik-1*:4932414N04Rik; Rik-2*:1700011F14Rik; Rik-3*:1700019N12Rik; Rik-4*: 1700001C02Rik; Rik-5*: 4930480E11Rik; Rik-6*: 4930407I10Rik. (B) Clustering of all tissue-specific m/lncRNAs (max JS score >0.5). Plots of JStestis scores were shown to the right of the heat maps. (C) Expression patterns of g-ts-m/lncRNAs in the 4 SPC types. (D) Comparisons of circRNAs and their cognate mRNAs in SPCs and the brain.
We next examined the dynamic expression patterns of g-ts-m/lncRNAs during spermatogenesis by conducting a clustering analysis (Fig. 2C). Similar to the g-m/lncRNA sets, these g-ts-m/lncRNAs are also roughly portioned into 3 clusters according to whether their highest abundances are found in SG, or SC, or ST (Table S3). From the numbers of genes in different clusters, it is interesting to see that more g-mRNAs are expressed highly in SG than in SC or ST while the majority of g-ts-mRNAs are highly expressed in SC or ST. In contrast, the majority of both g-lncRNAs and g-ts-lncRNAs are highly expressed in SC and ST. Particularly, very few g-ts-lncRNAs are highly expressed in SG and 81% of them are highly expressed in ST. Collectively, our data suggests that, g-ts-m/lncRNAs are highly expressed during or after meiosis.
Based on Gene Ontology (GO) analysis (Table S4), sg-ts-mRNAs are enriched with GO terms related to cell cycle regulation, mitosis and meiosis reflecting that SG undergo active mitosis and prepare for meiosis. sc-ts-mRNAs are enriched with terms related to cell cycle, meiosis and postmeiotic development reflecting that SC undergo meiosis and prepare for postmeiotic activities. st-ts-mRNAs are enriched with terms related to postmeiotic events and activities of sperm as ST undergo postmeiotic development to become sperm that are potent for fertilization.
Necsulea et al. identified 425 lncRNAs conserved during an evolutionary history of 300 million years.8 We found that 70 of these (16%) are in the ts-lncRNA set, which is 31% of the total lncRNAs, suggesting that ts-lncRNAs are significantly depleted of evolutionarily conserved lncRNAs (p-value = 1.2E-11 based on Fisher Exact Test).
A recent study reported the identification of 15,849 circRNAs in the mouse brain using RNA samples without RNase R treatment.34 We re-processed this raw data using the CIRI program and recovered 13,282 circRNAs and compared this data with our g-circRNA set. Interestingly, these 2 sets overlap only by a small fraction (4035/(11066+4035+9247) = 17%, Fig. 2D). We then compared the overlap of pre-mRNA-derived circRNAs between these 2 tissue types with the overlap of their cognate mRNAs and found that the percentage of circRNA overlap (18.8%) is significantly lower than that of the mRNAs (42.7%) (Fisher exact test, p < 2.2E-16). Therefore, the circRNA production is likely a highly regulated cell/tissue-type specific process.
Comparisons of expression levels of related RNAs
We next compared the dynamic changes of circRNAs and their cognate mRNAs (Fig. 3). In general, the average level of circRNAs drops slightly in pacSC compared with in priSG-A and plpSC and then peaks in rST while the abundances of mRNAs increase continually, again suggesting that the generation of circRNAs during spermatogenesis is a regulated process. When the comparisons were conducted for each of the sg/sc/st-clusters, the differences in the dynamics of circRNAs and mRNAs are more apparent (Fig. 3A). For the sg-cluster, both of the circRNA and mRNA levels drop immediately before meiosis but the circRNA level keeps dropping while the mRNA level increases during and after meiosis. For the sc-cluster, both the circRNA and mRNA levels increase immediately before meiosis but the circRNA level drops while the mRNA level continues increasing. For the st-cluster, both of the circRNA and mRNA levels increase continuously during the process of spermatogenesis except for a slight decrease of circRNA in pacSC, and this pattern is similar to that of the whole set of circRNAs. These observations suggest that the production of circRNAs during spermatogenesis is developmentally regulated; otherwise, the levels of circRNAs and their cognate mRNAs would be positively correlated.
Figure 3.
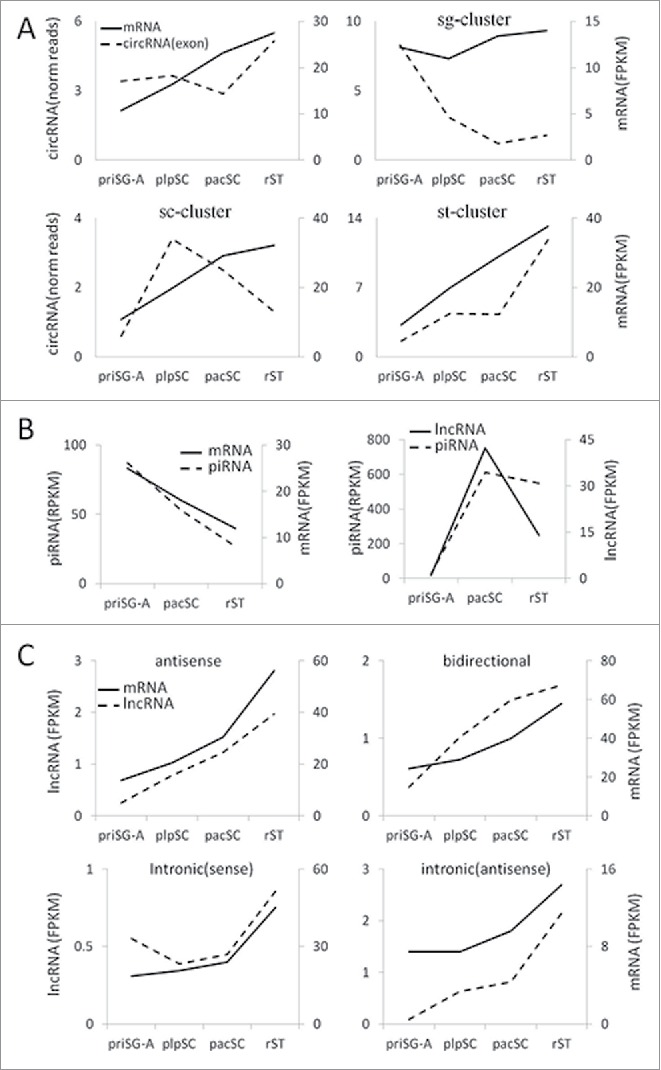
Comparisons of the expression of related RNAs. (A) Expression of different clusters of circRNAs and their cognate linear mRNAs. (B) Expression of piRNAs and their precursor m/lncRNAs. The expression of piRNAs were based on previously published RNA-seq data of small RNAs in 3 types of SPCs.28 (C) Expression of lncRNAs that map to the exons or introns or the nearby region of mRNA genes and the expression of these mRNAs.
Li et al. reported that 467 transcripts (214 genomic loci) are precursors of majority piRNAs.32 Of these loci, 114 are congruent with protein-coding genes, and the remaining 100 were thought to encode lncRNAs. We named these m/lncRNA precursors of piRNAs pi-m/lncRNAs. Eighty-five pi-lncRNA genes are intergenic, 12 are antisense to protein coding genes, 2 are sense to protein-coding genes but longer than the protein gene, and 1 is intronic. Interestingly, we found that ∼2% of piRNAs map to the 100 pi-lncRNA genes in priSG-A and ∼85% map to them in pacSC and rST while only ∼3.3% map to the pi-mRNAs. Our sequencing data of the 4 SPC types indicates that the average expression levels of both pi-mRNAs and their piRNAs dropped continuously from priSG-A to pacSC then to rST. In contrast, while the abundances of pi-lncRNAs increase dramatically from priSG-A to plpSC, and then drop quickly in rST, the abundances of piRNAs remain relatively stable when generated in large quantities since pacSC (Fig. 3B). These results suggest that the production of piRNAs from the m/lncRNA precursors is also a regulated process.
We also compared the expression of lncRNAs that map to the exons or introns or the nearby region of mRNA genes with the expression of these mRNAs (Fig. 3C). Interestingly, in all cases, expression of lncRNAs and the related mRNAs increases continuously from priSG-A to rST. This subset of mRNAs is enriched with GO terms related to protein phosphorylation.
In vitro screening of functional sg-lncRNAs
We selected 10 sg-lncRNAs and investigated their potential functions using cultured mouse SSCs. The selection was based on the following criteria: 1) FPKM in priSG-A > 1; 2) expression level in priSG-A is at least 2-fold higher than that in plpSC; 3) selected lncRNAs should be conserved between mouse and humans. Six of the selected sg-RNAs were successfully knocked down by 1 siRNA (Fig. 4A). For 4 of these 6 sg-lncRNAs, c-Kit positive cells were observed when they were knocked down (Fig. 4C, right pannel), indicating that SSCs became differentiating SG. Interestingly, the proliferation of the cells was not changed (Fig. 4B).
Figure 4.
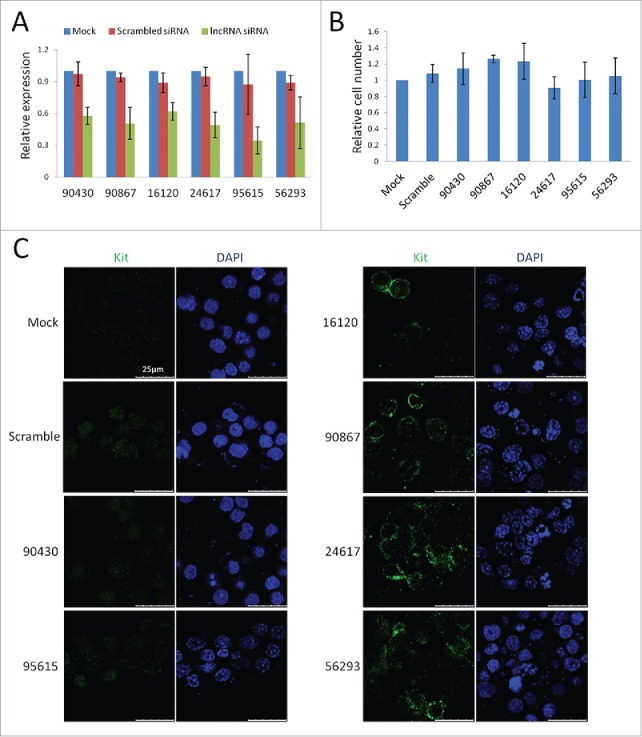
A small scale screening for functional sg-lncRNAs. (A) qRT-PCR validation of the knockdown (KD) of 6 sg-lncRNAs by siRNAs. (B) The proliferation of the KD SSCs relative to that of the normally cultured SSCs. SSCs were also transfected with siRNAs of scrambled sequence to show that the proliferation was not changed by the transfection reagents. (C) Immunofluorescent images of the KD SSCs to show the appearance of c-KIT-positive cells in some of the KO SSC cultures. The left panel shows the negative staining of c-KIT in controls and 2 lncRNA-KD samples while the right panel shows the positive staining of c-KIT in 4 lncRNA-KD samples. Note the membrane-localized typical staining pettern of c-KIT signal in contrast to the ubiquitous nonspecific background.
Identification of putative transcription factors regulating g-RNAs
To reveal how g-RNAs are regulated, we developed a tool named transcription factor binding sites (TFBS) enrichment analysis (TEA) to examine whether the proximal promoters of genes for different clusters of RNAs are enriched with TFBSs. TEA identifies TFBSs that are enriched in the evolutionarily conserved promoter regions of a group of genes. A total of 2 Kilobase (Kb) conserved region of each gene was scanned for 903 TFBSs, the position weight matrix (PWM) of which are compiled in the JASPAR 2016 database.35 A p-value of 0.05 or lower was considered to be significant. Ten TFBSs were significantly enriched in the promoters of sg-ts-mRNAs (Fig. 5A, Table S5). The most significantly enriched is for LIN54 (p = 4.3E-4), which is an essential core component of the DREAM/LINC complex that is an important regulator of cell cycle genes.36 Interestingly, 4 other enriched TFBSs are for the E2F-related TFs, which has been reported to associate with the DREAM/LINC complex.37 Among the 14 enriched TFBSs for the sc-ts-mRNAs, 10 are for the RFX family TFs, 3 are for the MYB family, and LIN54 is again identified although at the end of the list. Transcripts of Rfx1-4 are all highly expressed in haploid cells, but the expression of only Rfx2 is confined to testis and is up-regulated in meiotic cells.38 We have recently shown that RFX2 is a key regulator of the post-meiotic development of SPCs using knock-out (KO) mice and that it regulates the transcription of a large number of mRNAs, many of which are st-ts-mRNAs.39 While the mRNAs of B-MYB are detected in SPCs from gonocytes to early SC, that of A-MYB was detected in a subpopulation of SG and primary SC but not in ST.40 The ablation of A-MYB causes an arrest of meiosis at the pacSC step in KO mice.41 Notably, A-MYB also regulates the expression of RFX2.42 Moreover, B-MYB also interacts with the DREAM/LINC complex.37 Twenty-3 TFBSs were significantly enriched in st-ts-mRNAs, and they are mainly for the ATF/CREB-related TFs, the JUN/FOS-, and the RFX-related ones. The others are for the C/EBP-, SOX-, zinc finger-, and TEF-related TFs. CREM is a master regulator of gene expression in meiotic and post-meiotic SPCs and spermatogenesis arrests at the early round spermatid step when it is knocked out.43,44
Figure 5.
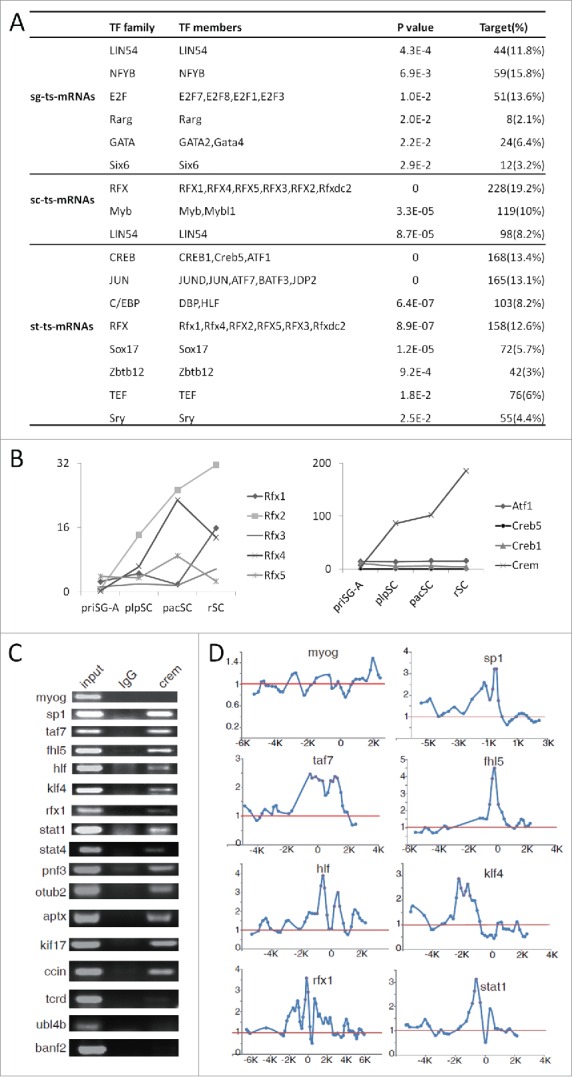
Transcription factor binding sites (TFBSs) enrichment analysis. (A) TF families and members, the TFBSs of which are enriched in the promoters of different sets of ts-mRNAs. Similar TFBSs enriched in each gene set were combined into a family and the lowest p-value of the members was used as the representative p-value of this group. (B) Expression of the CREB and RFX family TFs based on RNA-seq data. (C) ChIP-PCR validation of CREM targets supported by ChIP-chip data (see Table S12 for primer information). (D) Plots of ChIP-chip signals of some genes that were validated by ChIP-PCR in (C). Myog is a muscle specific gene and used as a negative control. The red dots label the signals of the probes on the microarray.
In light of the poor conservation of lncRNA genes, we scanned their promoter regions from −300 bp to 100 bp relative to the genomic position marked by the 5´-most lncRNA reads for enriched TFBSs using only the mouse sequences. The original p-values were multiplied by the number of the PWMs in order to generate the adjusted p-values. Using 0.05 as a cutoff for adjusted p-value, 42 enriched TFBSs were identified for sc-ts-lncRNAs, and most of them are for the ETS-related TFs (Table. S6). By comparing with the enriched TFBSs for sc-ts-mRNAs, only A-MYB was shared by both sets (adjusted p-value = 0.0064). Consistently, it was reported recently that A-MYB was a key regulator of pachytene lncRNAs.45 Twenty-one TFBSs were enriched for st-ts-lncRNAs. Interestingly, 19 of them are in the 23 TFBS set enriched for the st-ts-mRNAs. Moreover, the 2 unique ones are for CREM and SOX9, which are actually members of the enriched families of TFs identified for the st-ts-mRNAs. These data suggest that mRNAs and lncRNAs specifically expressed in postmeiotic SPCs may share a common transcription regulatory mechanism.
We next examined whether any members of the enriched TF families are indeed expressed in the corresponding stages using the RNA-seq data (Fig. 5B, S2). For all the TF families, at least one member is highly expressed at the stage corresponding to the clusters of ts-m/lncRNAs, for which the TFBSs are enriched. Therefore, both TFBSs and TFs are in place for the transcription of these ts-m/lncRNAs that are highly expressed at a certain stage of spermatogenesis.
CREM and RFX2 target validation using high throughput methods
We next used the ChIP-chip assay to screen for potential target genes of CREM in order to evaluate the performance of our TEA program. The predicted and ChIP-Chip-supported targets of CREM were named CTPs (CREM Targets by Prediction) and CTCs (CREM Targets by ChIP-chip), respectively. Cell lysates were prepared from the testes of 5 adult mice, immunoprecipited by a rabbit polyclonal antibody against the mouse CREM, followed by chip analysis using the Agilent G4490A mouse promoter microarray, which contains probes for the promoters of ∼17,000 Refseq genes (from −5.5 Kb to +2.5 Kb of the transcription start sites). Here, 1,932 CREM-bound DNA fragments (average size being 600 bp) corresponding to 1,819 genes were identified (Table S7). 57% of the DNA fragments contain the CREB/CREM/ATF family TFBSs, and this is a 5.6-fold TFBS enrichment compared with the average abundance of CREB/CREM/ATF family TFBS in the genome. If the length of the CREM-bound fragments was extended to 1000 bp, 80% of them (1,552) contain the TFBSs. To test the validity of the ChIP-chip technology, 16 CTCs and 1 non-CTC (myog) were selected to perform ChIP-PCR, the CREM TFBSs of 13 genes (81%) were confirmed (Fig. 5C, D). Of the 1,819 genes, 193 encode ts-mRNAs. On the other hand, 39 of the 168 CTPs of the st-ts-mRNAs (13.4%) are in the CTC set (1,819). This indicates that CTPs of the st-ts-mRNAs are significantly enriched with CTCs (Fisher exact test, p = 1.72E-12). Therefore, by cross-validation using CTPs and CTCs, we identified potential targets of CREM and also confirmed validity of bioinformatics predictions.
In a separate study, we generated Rfx2 KO mice and found that the males but not the females were sterile due to an arrest of spermatogenesis at the early ST step.39 Here, 822 mRNAs were found to change their expression levels based on the RNA-seq data of the KO testicular cells, and were referred to as RFX2-regulated mRNAs. In this study, 344 of these RFX2-regulated mRNAs are sc/st-ts-mRNAs, suggesting RFX2 also regulates the transcription of many other mRNAs in addition to the sc/st-ts-mRNAs although its TFBS was first observed to be enriched in the latter sets. On the other hand, 109 out of the 359 sc/st-ts-mRNAs from the predicted target genes of the RFX-related TFs are regulated by RFX2, suggesting that the predicted targets are significantly enriched with RFX2-regulated ones (Fisher exact test, p < 2.2E-16). Moreover, 498 g-lncRNAs were also found to be regulated by RFX2, and 365 of these are st-ts-lncRNAs (Table S8). These data show that RFX2 is a key regulator of both mRNAs and lncRNAs and that predicted target genes of TFs are more likely to be regulated by TFs than other genes.
Discussion
Several studies indicate that the metazoan testes possess the most complex transcriptomes as a result of the many types of cells contained in this unique organ.2,3 However, simultaneous comparisons of the expression of different types of transcripts such as m/lnc/circRNAs in SPCs have not been conducted systematically, and how these transcriptomes are established is still an open question. In the present study, we used 4 types of freshly isolated SPCs representing the mitotic, meiotic and postmeiotic phases of spermatogenesis to compare the dynamic profiles of m/lnc/circRNAs with particular interest in their testis-specific subsets. We then applied our TEA tool to identify TFBSs enriched in the promoters of different sets of ts-mRNA/lncRNA genes in order to understand how the complex transcriptomes in SPCs are stablished at the transcription level.
Historically, global analysis of transcripts in testes and/or SPCs and their testis-specific subsets have been attempted by using various high throughput methods. Based on EST data deposited in the NCBI GEO database and their own whole-testis microarray expression data, Shultz et al. first estimated in a seminal paper that ∼4.7% of the mouse transcriptome is restricted to the testis and that ∼3.8% is dedicated to expression in meiotic and postmeiotic SPCs.4 Unfortunately, only 346 testis-specific genes represented by UniGene accession numbers were identified. Using the similar microarray platforms and isolated SPCs, 2 other groups studied the dynamics of several thousands of transcripts, mostly mRNAs, during mouse spermatogenesis and identified several hundred testis-specific mRNAs, and 1 group also found that the expression patterns of many transcripts are conserved from mouse to human during evolution.46,47 A more recent microarray-based study used total testicular cells to show that several thousand m/lncRNAs are expressed dynamically during mouse spermatogenesis. The study failed, however, to identify different transcripts and their relative abundances in separate types of SPCs.48 Using the same microarray platform with isolated SPCs, a second group also identified transcripts that were differentially expressed between adjacent SPC types of mouse spermatogenesis but did not evaluate the dynamics quantitatively.49 Moreover, neither study investigated the testis specificity of m/lncRNAs, which is a prominent feature of testicular transcripts. Due to the hybridization involved in the microarray technology, expression data suffer from high noise and low sensitivity. In addition, the large numbers of transcripts represented by microarray probes are highly redundant due to the lack of complete annotations of the lncRNAs, therefore, the real numbers of lncRNAs expressed in SPCs remain elusive. In the present study, we used rRNA-depleted strand-specific RNA-seq and non-redundant references to profile the expression of m/lncRNAs as well as circRNAs, we have been able to not only compile the most complete and the least redundant lists of diverse transcripts but also to compare their dynamic changes in a quantitative manner.
Based on our expression cutoff of 0.1 FPKM, ∼80% of the mRNAs (20,639 out of 26,023) are expressed in SPCs, close to the estimate by Ramskold et al. who stated that 84% of mRNAs are expressed in the testis with a cutoff of 0.3. Using a cutoff value of 0.5 for the JStestis scores,7 which was trained by a set of 90 testis-specific genes extensively suggested in the literature, 2,819 mRNAs were found to be testis-specific, occupying ∼11% of the total mRNAs in the mouse transcriptome. This estimated percentage is higher than that suggested by Shultz et al.4 One likely cause of this discrepancy is that we used only 6 organs to evaluate the organ-specificity of the transcripts. As shown by our RT-PCR validation results, 14 of the 20 predicted ts-mRNAs are uniquely present in the testis among the 10 examined organs, and 4 are also present in the ovary, the female gonads. Therefore, the value 11% should be multiplied by a factor of 0.7 (14/20) to give a more reasonable estimate of 7.7%, which is of course still an overestimate because we have not examined all organs. In sharp contrast, both our mouse result and the human result by Cabili et al. indicate that 30% of the total lncRNAs are testis-specific.7 It is also noteworthy that a majority of these ts-m/lncRNAs are expressed in SPCs, and most likely only in SPCs but not in the somatic cells as implied by the GO terms enriched in the different clusters of ts-mRNAs.
For the first time, we identified a large number of circRNAs expressed in SPCs. Similar to what was observed by others in other organs/tissues, circRNAs are mainly spliced from exons and expressed at abundances about one order of magnitude lower than their cognate linear transcripts. circRNAs have been regarded as mis-spliced products since their discovery. However, recent deep sequencing and functional studies have revealed the existence of a very large number of circRNAs, which has challenged this idea. Our profiling data from SPCs support that the generation of circRNAs is a regulated rather than a random process with 3 lines of evidence: 1) The abundance distributions of circRNAs do not follow a standard normal distribution. This is more apparent from the distribution curves of circRNAs that do not overlap the SSC-R+ set. 2) circRNAs from SPCs overlap those from the brain by only a small proportion. 3) The correlations between the dynamic patterns of circRNAs and their cognate linear forms are poor (Fig. 3). Because both circRNAs and piRNAs are products of posttranscriptional processings and piRNAs have been shown to play important roles in degrading other RNA species and in regulating epigenetic status,50-53 we also compared the dynamic patterns of piRNAs and their precursor m/lncRNAs and found that the discrepancy between the dynamic patterns of piRNAs and their lncRNA precursors is more apparent than that between those of piRNAs and their mRNA precursors. Interestingly, lncRNA-derived piRNAs are much more abundant than mRNA-dervied ones and the former are believed to mediate RNA degradation in round spermatids while the function of the latter is unknown. Therefore, it seems that the generation of functional piRNAs is more likely to be regulated than the generation of mRNA-derived ones, the function of which remains elusive.
Our data show that SPCs express a large number of lncRNAs (7,186), most of which are expressed with higher abundances in SC and ST than in SG. Similar observations were made in a recent study, in which ∼8,600 lncRNAs were found to be expressed in mouse testes, and most of them were expressed at the meiotic and postmeiotic stages.3 The percentage of testis-specific lncRNAs was also consistent with a previous estimate.7 Some researchers proposed that the expression of a large number of intergenic lncRNAs, a majority of which are testis-specific, in addition to other intergenic elements, probably represents transcription noises due to a more open chromatin state as a result of remodeling.3 While this proposition could be true, it is also possible that lncRNAs play a role in chromatin remodeling. Our finding that the promoters of both the st-ts-mRNA and st-ts-lncRNA genes are enriched with TFBSs for the same families of TFs seems to support the second possibility. While most lncRNAs, particularly those with low expression levels, are probably non-functional, it is certainly true that there exists a subset of functional ones.54 Impressively, the knockdown of 4 of the 6 sg-lncRNAs selected for functional screening based on high levels of expression in SG and conservation during evolution resulted in differentiation of cultured SSCs as indicated by the expression of c-KIT, a marker of differentiating SG. Therefore, functional lncRNAs could be distinguished from non-functional ones when the initial bioinformatics selection was rational.
Cell type-specific transcripts are the major component contributing to the unique transcriptome of a specific cell type, and it is fundamental to understand whether their transcriptions share any common regulatory mechanism. We investigated this question by analyzing whether different sets of ts-m/lncRNAs are enriched with TFBSs at the promoter regions of their genes. Our analyses addressed this question in the following specific ways: 1) Different sets of ts-RNAs are enriched with typical families of TFBSs. 2) The families of TFs are related in terms of physical interactions or sequential regulatory relationships. For example, TFBSs of LIN54 and E2F-related factors are enriched for sg-ts-mRNAs, and the LNC complex with LIN54 as the core component interacts with E2F proteins and with B-MYB, the TFBS of which is enriched for sc-ts-mRNAs. Moreover, TFBSs of A-MYB and RFX2 are enriched for sc-ts-mRNAs and it has been known that A-MYB regulates the transcription of RFX2. 3) Some members of a TF family, the TFBSs which are enriched for the ts-lncRNAs of a cell type, are highly expressed in this particular cell type. These observations indicate that TFs, the TFBSs of which are enriched significantly based on bioinformatics prediction, may actually execute their regulatory functions. Further evidence arises from the result that predicted targets of CREM are significantly enriched with targets supported by ChIP-chip assays and that predicted targets of RFX2 are enriched with genes differentially expressed in RFX2-KO SPCs. CREM has been long known to be a key TF for the postmeiotic development of SPCs. Many lncRNAs and mRNAs in SPCs are regulated by RFX2 as indicated by RNA-seq data using the RFX2-KO mice, which arrest spermatogenesis at an early rST step39, supporting the idea that RFX2 is another key regulator of spermatogenesis.
Conclusions
Our data demonstrate that the mouse transcriptomes of SPCs are highly complex and dynamic by expressing a large number of m/lnc/circRNAs, many of which are specifically expressed in the testes. A majority of the testis-specific transcripts are expressed in SPCs, particularly in meiotic and postmeiotic ones. The testis-specificity of lncRNAs is significantly higher than that of mRNAs. By comparing the circRNAs from SPCs and the brain, we find that these 2 sets overlap significantly less than the cognate mRNAs. Moreover, the expression patterns of circRNAs are also different from their cognate mRNAs in SPCs. Therefore, the production of circRNAs in different tissue/cell types seems to be a regulated process although their abundances are low. Similarly, the generation of piRNAs from the precursor lncRNAs is also a regulated process. Functional screenings using cultured mouse SSCs show that a considerable fraction of lncRNAs conserved across species and expressed in SG at higher abundances than in other types of SPCs might be functional. Different sets of ts-m/lncRNAs might be regulated by different but related TFs as shown by the bioinformatics analyses and experimental validations. Particularly, ts-mRNAs and ts-lncRNAs, both highly expressed in postmeiotic SPCs might share similar transcriptional regulatory mechanisms. The list of transcripts generated from the present study will serve as a valuable resource of newly identified candidate genes playing important roles in mammalian spermatogenesis. Further studies are needed to elucidate the functions of these candidate genes, and to this end, high throughput functional screening platforms for new types of transcripts such as lncRNAs and circRNAs are needed.
Materials and methods
RNA preparation and sequencing
Animal studies followed standard procedures in accordance with regulations of the Animal Care and Use Committee of the Institute of Zoology, Chinese Academy of Sciences. F1 pups of DBA/2 and C57BL6 mice were used for cell isolation and SSC culture. priSG-A, plpSC, and pacSC were isolated from prepubertal mice undergoing the first wave of spermatogenesis while rST were from 60 dpp adult mice. The isolation and characterization of SPCs were conducted by using the STAPUT method, which was described in details previously.30,55 Mouse SSCs were cultured using our previously published protocol.56 Total RNAs were isolated using TRIzol reagent by following standard protocol. DNase I (Qiagen) treatment was carried out for total RNA to remove genomic DNA contamination. 10 μg of purified RNA was then subjected to rRNA (rRNA) depletion by Ribo-Zero Magnetic Gold Kit (Human/Mouse/Rat) (Epicentre). Then the rRNA-depleted RNA was divided into 2 groups, one (1/5 amount) is for a dUTP-based strand-specific RNA-seq library construction approach (Zhong et al., 2011), and the other (4/5 amount) is for RNase R treatment. To digest linear RNA, rRNA-depleted RNA was denatured at 65°C for 5 min, followed by adding 10 × RNase R buffer, 5 units of RNase R (Epicentre) and incubation at 37°C for 3 hours. Agencout RNAclean XP magnetic beads were used to purify the RNase R treated RNA. The purified RNA was randomly sheared by heating with magnesium and subsequent RNA-seq libraries were constructed according to the previously published protocol (Zhong et al. 2011). RNA-seq libraries with and without RNase R treatment were subjected to deep sequencing by Illumina Hiseq 2000 instrument in a 2×100 bp manner.
RNA sequence analysis
Two biological replicates of RNA samples were sequenced for each cell type. The correlation coefficients of transcriptomes from biological duplicates were all higher than 0.90, indicating that our RNA-seq data are highly reproducible. Subsequently, duplicate data were pooled for further analyses. The number of reads corresponding to each sample ranged from 35–54 million. The sequencing reads were mapped to the mouse genome (UCSC mm9) by using the TopHat package (version 2.0.6). The RNA expression level of mRNA and lncRNAs was represented by fragments per kb of exon model per million mapped fragments (FPKM) calculated using the Cufflinks package (version 2.0.2). The differentially expressed genes were identified using the Cuffdiff package. RefSeq mRNAs downloaded from UCSC and the lncRNA set (15,934) compiled by Necsulea, et al. 8 were used as the reference sets for mRNA and lncRNA analyses, respectively. An mRNA or lncRNA was regarded to be present in a sample if its FPKM was bigger than 0.1. CircRNAs were identified using the CIRI program, which was recently reported to have better performance than other similar tools in circRNA identification using RNA-seq data derived from RNA samples that were not treated with RNase R to remove linear transcripts.57 Briefly, sequencing reads were mapped to the mouse genome by using the BWA-MEM software. The Perl program of the CIRI algorithm implemented by Gao, et al. 57 was installed locally and was used to identify reads representing circRNAs and to count their numbers. The default values were used for all parameters.
RT–PCR confirmation of circ/lncRNA expression in spermatogenic cells
Information of primers used in RT-PCR identification of circRNAs were listed in Table S9. Total RNAs were isolated from independent SPC samples from those for RNA-seq experiments. 2 μg of RNAs were reverse transcribed in a 20 μl reaction using random primers by following the instructions of the Applied Biosystems High-Capacity cDNA Reverse Transcription Kits. PCRs were carried out by using the Takara PrimeStar system in a volume of 20 μl. The other parameters for PCRs were as the following: a single denaturing step (94°C, 5 min) followed by 35 cycles of amplification (denaturing at 94°C for 30 s, annealing at 60°C for 30 s and elongation at 72°C for 30 s). To evaluate the expression of lncRNAs, qPCR reactions were performed using the 384-plate format of the Roche LightCycler 480 Real-Time PCR system using the UltraSYBR Mixture from Beijing CoWin Biotech. The primers and other information were listed in Table S10. The reactions were set up in the following recipe: 0.2 μl cDNA template, 7.5 μl 2× PCR Mix, 0.4 μl forward and reverse primers each (10 μM), 6.5 μl ddH2O. PCR reactions were carried out with a denaturing step (95°C 10 min) and 45 cycles of amplification (denaturing at 95°C for 15 s followed by annealing and elongation at 60°C for 1 min). The geometric mean of Actb, Ubc and Nudcd3 expression values were used as internal control for all qPCR analysis.30
Mining of RNAs specially expressed in mouse testis
Dataset GSE30352, which contains expression data of 6 organs (testis, heart, liver, cerebellum, kidney, brain),33 was downloaded from NCBI GEO database. The Jensen-Shannon (JS) scores for each organ defined by Cabili et al. were calculated using an R script.7 The cutoff of JStestis was trained by using a list of 90 reported testis-specific genes (Table S2). Clustering analysis of the RNA expression was conducted using the Cluster 3.0 software. Clusterings of g-m/lnc/circRNAs were initially identified using the hierarchical algorithm, and re-processed using the K-Mean algorithm when a value of K is determined to be 3 by visual inspection of the heatmap of the hierarchical clustering results. Clustering of tissue-specific m/lncRNAs was performed using the hierarchical algorithm. The correlation coefficient of expression values of 2 genes across organ samples was used as the similarity measure for clustering. Clustering results were visualized using the TreeView program. Enrichment analysis of GO terms of Biological Processes was performed by using the web-based software DAVID (http://david.abcc.ncifcrf.gov/). Statistical significance was decided basen on 2 parameters: Benjamini p-value < 0.01, FDR < 0.01.
Knockdown of lncRNAs using siRNAs in cultured mouse SSCs
SiRNAs for lncRNA knockdown were designed using the BLOCK-iT RNAi Designer of Invitrogen (Table S11). For transfection, SSCs were incubated with medium containing siRNAs mixed with the Invitrogen RNAiMAX reagent at a final concentration of 100 nM for 48 hours (h). Aliquots of transfected cells were harvested and total RNAs isolated to examine the knockdown efficiency using qRT-PCRs. Four days after transfection, the remaining cells were subjected to immunostaining and flow cytometry analyses of c-KIT expression, which marks the differentiation of SG.
Transcription factor binding site enrichment analysis
The key steps of TFBS enrichment analysis (TEA) are outlined as a flowchart shown in Fig. S3. Briefly, promoter sequences of mouse and human orthologous genes were aligned, evolutionarily conserved regions (ECRs) were identified and saved in the database. After, 903 PWMs were collected from Jarspar2016 (519 JASPAR CORE Vertebrata PWMs; 208 JASPAR PBM PWMs for 104 mouse transcription factors; 176 JASPAR PBM HOMEO PWMs). ECRs of different gene sets were scanned for potential TFBSs using pre-determined match score cutoff values for each PWM. See supplemental info for more details.
CREM ChIP-chip experiment
Testicular cells from 5 adult mouse testes were fixed in 5 ml of 1% formaldehyde medium for 10 min at 37°C. The fixed germ cells were washed twice with PBS, resuspended in PBS and sonicated (power 40%, pulser 60%, ON 60s, OFF 60s, 5 cycles by Ruptor 250, Omni, USA). 200 μl of supernatant were diluted in 1800 μl ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, 167 mM NaCl and 1 mM PMSF). Diluted complexes were immunoprecipitated with anti-CREM (sc-440, Santa Cruz) and anti-goat IgG (negative control) at 4°C overnight. The immune complexes were precipitated with protein G followed by washes with low salt buffer, high salt buffer, LiCl buffer and TE. The complexes were eluted with elution buffer and cross-linking of DNA and protein was reverted in 5 M NaCl at 65°C overnight. The solution was digested with proteinase K and the DNA were purified by PheOH/CHCl3 extractions and EtOH precipitations. Purified DNA was first used in PCRs to check the amplification of some reported target genes and then subjected to Agilent mouse promoter microarray (G4490A) for signal detection.
ChIP-chip peak analysis and motif analysis
CREM ChIP-chip raw data were normalized by using the LOESS method.58 We implemented an algorithm similar to ChIPOTle59 using a perl program. Briefly, log-transformed ratios of signals of the antibody and control IgG immunoprecipitated samples detected by the probes within a sliding window were averaged over the numbers of probes. Then it was determined whether the average value of this window was significantly different from the average of all probes over the genome. The size of the sliding window was set to 500 bp consistent with the average size of the sonicated DNA fragments. The p-value cutoff of statistical significance was set to be 0.05 after Bonferroni correction. Windows with enriched signals were identified and adjacent ones were merged as TF bound regions (peaks).
Supplementary Material
Disclosure of potential conflicts of interest
The authors declare that they have no competing interests.
Funding
This work was supported by grants from the Ministry of Science and Technology of China [2015CB943000, 2012CB966702, 2013CB945001], National Natural Science Foundation of China [31271379, 31471349].
Authors' contributions
WB, TN, and CH conceived the project and designed the experiments. XL, MH, ZZ, TN and CH conducted the analyses. LC, JC, TS, MW and ZZ performed experiments. CH, XL and JC wrote the manuscript. All authors edited and approved the final manuscript.
References
- 1.Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet 2014; 15:423-37; PMID:24776770; http://dx.doi.org/ 10.1038/nrg3722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramskold D, Wang ET, Burge CB, Sandberg R. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Computational Biol 2009; 5:e1000598; PMID:20011106; http://dx.doi.org/ 10.1371/journal.pcbi.1000598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soumillon M, Necsulea A, Weier M, Brawand D, Zhang X, Gu H, Barthes P, Kokkinaki M, Nef S, Gnirke A, et al.. Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep 2013; 3:2179-90; PMID:23791531; http://dx.doi.org/ 10.1016/j.celrep.2013.05.031 [DOI] [PubMed] [Google Scholar]
- 4.Schultz N, Hamra FK, Garbers DL. A multitude of genes expressed solely in meiotic or postmeiotic spermatogenic cells offers a myriad of contraceptive targets. Proc Natl Acad Sci U S A 2003; 100:12201-6; PMID:14526100; http://dx.doi.org/ 10.1073/pnas.1635054100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gardini A, Shiekhattar R. The many faces of long noncoding RNAs. FEBS J 2015; 282:1647-57; PMID:25303371; http://dx.doi.org/ 10.1111/febs.13101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li L, Chang HY. Physiological roles of long noncoding RNAs: insight from knockout mice. Trends Cell Biol 2014; 24:594-602; PMID:25022466; http://dx.doi.org/ 10.1016/j.tcb.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Gen Dev 2011; 25:1915-27; PMID:21890647; http://dx.doi.org/ 10.1101/gad.17446611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, Baker JC, Grutzner F, Kaessmann H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 2014; 505:635-40; PMID:24463510; http://dx.doi.org/ 10.1038/nature12943 [DOI] [PubMed] [Google Scholar]
- 9.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature 2008; 456:470-6; PMID:18978772; http://dx.doi.org/ 10.1038/nature07509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al.. Landscape of transcription in human cells. Nature 2012; 489:101-8; PMID:22955620; http://dx.doi.org/ 10.1038/nature11233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al.. The evolutionary landscape of alternative splicing in vertebrate species. Science 2012; 338:1587-93; PMID:23258890; http://dx.doi.org/ 10.1126/science.1230612 [DOI] [PubMed] [Google Scholar]
- 12.Shen T, Han M, Wei G, Ni T. An intriguing RNA species-perspectives of circularized RNA. Protein Cell 2015; 6:871-80; PMID:26349458; http://dx.doi.org/ 10.1007/s13238-015-0202-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol 2014; 32:453-61; PMID:24811520; http://dx.doi.org/ 10.1038/nbt.2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu MT, Coca-Prados M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature 1979; 280:339-40; PMID:460409; http://dx.doi.org/ 10.1038/280339a0 [DOI] [PubMed] [Google Scholar]
- 15.Nigro JM, Cho KR, Fearon ER, Kern SE, Ruppert JM, Oliner JD, Kinzler KW, Vogelstein B. Scrambled exons. Cell 1991; 64:607-13; PMID:1991322; http://dx.doi.org/ 10.1016/0092-8674(91)90244-S [DOI] [PubMed] [Google Scholar]
- 16.Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013; 19:141-57; PMID:23249747; http://dx.doi.org/ 10.1261/rna.035667.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PloS one 2012; 7:e30733; PMID:22319583; http://dx.doi.org/ 10.1371/journal.pone.0030733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell-type specific features of circular RNA expression. PLoS Genet 2013; 9:e1003777; PMID:24039610; http://dx.doi.org/ 10.1371/journal.pgen.1003777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al.. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013; 495:333-8; PMID:23446348; http://dx.doi.org/ 10.1038/nature11928 [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, Zhu S, Yang L, Chen LL. Circular intronic long noncoding RNAs. Mol Cell 2013; 51:792-806; PMID:24035497; http://dx.doi.org/ 10.1016/j.molcel.2013.08.017 [DOI] [PubMed] [Google Scholar]
- 21.Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature 2013; 495:384-8; PMID:23446346; http://dx.doi.org/ 10.1038/nature11993 [DOI] [PubMed] [Google Scholar]
- 22.Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, Zhong G, Yu B, Hu W, Dai L, et al.. Exon-intron circular RNAs regulate transcription in the nucleus. Nature structural & molecular biology 2015; 22:256-64; PMID:25664725; http://dx.doi.org/ 10.1038/nsmb.2959 [DOI] [PubMed] [Google Scholar]
- 23.Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell 2014; 56:55-66; PMID:25242144; http://dx.doi.org/ 10.1016/j.molcel.2014.08.019 [DOI] [PubMed] [Google Scholar]
- 24.Zhang XO, Wang HB, Zhang Y, Lu X, Chen LL, Yang L. Complementary sequence-mediated exon circularization. Cell 2014; 159:134-47; PMID:25242744; http://dx.doi.org/ 10.1016/j.cell.2014.09.001 [DOI] [PubMed] [Google Scholar]
- 25.Lasda E, Parker R. Circular RNAs: diversity of form and function. Rna 2014; 20:1829-42; PMID:25404635; http://dx.doi.org/ 10.1261/rna.047126.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, Wang Z. Efficient backsplicing produces translatable circular mRNAs. Rna 2015; 21:172-9; PMID:25449546; http://dx.doi.org/ 10.1261/rna.048272.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo JU, Agarwal V, Guo H, Bartel DP. Expanded identification and characterization of mammalian circular RNAs. Genome Biol 2014; 15:409; PMID:25070500; http://dx.doi.org/ 10.1186/s13059-014-0409-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gan H, Lin X, Zhang Z, Zhang W, Liao S, Wang L, Han C. piRNA profiling during specific stages of mouse spermatogenesis. RNA 2011; 17:1191-203; PMID:21602304; http://dx.doi.org/ 10.1261/rna.2648411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gan H, Cai T, Lin X, Wu Y, Wang X, Yang F, Han C. Integrative Proteomic and Transcriptomic Analyses Reveal Multiple Post-transcriptional Regulatory Mechanisms of Mouse Spermatogenesis. Mol Cell Proteomics 2013; 12:1144-57; PMID:23325766; http://dx.doi.org/ 10.1074/mcp.M112.020123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gan H, Wen L, Liao S, Lin X, Ma T, Liu J, Song CX, Wang M, He C, Han C, et al.. Dynamics of 5-hydroxymethylcytosine during mouse spermatogenesis. Nat Commun 2013; 4:1995; PMID:23759713; http://dx.doi.org/ 10.1038/ncomms2995 [DOI] [PubMed] [Google Scholar]
- 31.Thomson T, Lin H. The biogenesis and function of PIWI proteins and piRNAs: progress and prospect. Annu Rev Cell Dev Biol 2009; 25:355-76; PMID:19575643; http://dx.doi.org/ 10.1146/annurev.cellbio.24.110707.175327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li XZ, Roy CK, Dong X, Bolcun-Filas E, Wang J, Han BW, Xu J, Moore MJ, Schimenti JC, Weng Z, et al.. An ancient transcription factor initiates the burst of piRNA production during early meiosis in mouse testes. Mol Cell 2013; 50:67-81; PMID:23523368; http://dx.doi.org/ 10.1016/j.molcel.2013.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brawand D, Soumillon M, Necsulea A, Julien P, Csardi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, et al.. The evolution of gene expression levels in mammalian organs. Nature 2011; 478:343-8; PMID:22012392; http://dx.doi.org/ 10.1038/nature10532 [DOI] [PubMed] [Google Scholar]
- 34.Rybak-Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S, Hanan M, Behm M, Bartok O, Ashwal-Fluss R, et al.. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol Cell 2015; 58:870-85; PMID:25921068; http://dx.doi.org/ 10.1016/j.molcel.2015.03.027 [DOI] [PubMed] [Google Scholar]
- 35.Mathelier A, Fornes O, Arenillas DJ, Chen CY, Denay G, Lee J, Shi W, Shyr C, Tan G, Worsley-Hunt R, et al.. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 2016; 44:D110-5; PMID:26531826; http://dx.doi.org/ 10.1093/nar/gkv1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmit F, Cremer S, Gaubatz S. LIN54 is an essential core subunit of the DREAM/LINC complex that binds to the cdc2 promoter in a sequence-specific manner. FEBS J 2009; 276:5703-16; PMID:19725879; http://dx.doi.org/ 10.1111/j.1742-4658.2009.07261.x [DOI] [PubMed] [Google Scholar]
- 37.Sadasivam S, DeCaprio JA. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer 2013; 13:585-95; PMID:23842645; http://dx.doi.org/ 10.1038/nrc3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kistler WS, Horvath GC, Dasgupta A, Kistler MK. Differential expression of Rfx1-4 during mouse spermatogenesis. Gene Expr Patterns 2009; 9:515-9; PMID:19596083; http://dx.doi.org/ 10.1016/j.gep.2009.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu Y, Hu X, Li Z, Wang M, Li S, Wang X, Lin X, Liao S, Zhang Z, Feng X, et al.. Transcription Factor RFX2 Is a Key Regulator of Mouse Spermiogenesis. Sci Rep 2016; 6:20435; PMID:26853561; http://dx.doi.org/ 10.1038/srep20435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Latham KE, Litvin J, Orth JM, Patel B, Mettus R, Reddy EP. Temporal patterns of A-myb and B-myb gene expression during testis development. Oncogene 1996; 13:1161-8; PMID:8808690 [PubMed] [Google Scholar]
- 41.Bolcun-Filas E, Bannister LA, Barash A, Schimenti KJ, Hartford SA, Eppig JJ, Handel MA, Shen L, Schimenti JC. A-MYB (MYBL1) transcription factor is a master regulator of male meiosis. Development 2011; 138:3319-30; PMID:21750041; http://dx.doi.org/ 10.1242/dev.067645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horvath GC, Kistler MK, Kistler WS. RFX2 is a candidate downstream amplifier of A-MYB regulation in mouse spermatogenesis. BMC Dev Biol 2009; 9:63; PMID:20003220; http://dx.doi.org/ 10.1186/1471-213X-9-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blendy JA, Kaestner KH, Weinbauer GF, Nieschlag E, Schutz G. Severe impairment of spermatogenesis in mice lacking the CREM gene. Nature 1996; 380:162-5; PMID:8600391; http://dx.doi.org/ 10.1038/380162a0 [DOI] [PubMed] [Google Scholar]
- 44.De Cesare D, Fimia GM, Sassone-Corsi P. CREM, a master-switch of the transcriptional cascade in male germ cells. J Endocrinol Invest 2000; 23:592-6; PMID:11079454; http://dx.doi.org/ 10.1007/BF03343781 [DOI] [PubMed] [Google Scholar]
- 45.Li XZ, Roy CK, Moore MJ, Zamore PD. Defining piRNA primary transcripts. Cell Cycle 2013; 12:1657-8; PMID:23673320; http://dx.doi.org/ 10.4161/cc.24989 [DOI] [PubMed] [Google Scholar]
- 46.Shima JE, McLean DJ, McCarrey JR, Griswold MD. The murine testicular transcriptome: characterizing gene expression in the testis during the progression of spermatogenesis. Biol Reprod 2004; 71:319-30; PMID:15028632; http://dx.doi.org/ 10.1095/biolreprod.103.026880 [DOI] [PubMed] [Google Scholar]
- 47.Chalmel F, Rolland AD, Niederhauser-Wiederkehr C, Chung SS, Demougin P, Gattiker A, Moore J, Patard JJ, Wolgemuth DJ, Jegou B, et al.. The conserved transcriptome in human and rodent male gametogenesis. Proc Natl Acad Sci U S A 2007; 104:8346-51; PMID:17483452; http://dx.doi.org/ 10.1073/pnas.0701883104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bao J, Wu J, Schuster AS, Hennig GW, Yan W. Expression profiling reveals developmentally regulated lncRNA repertoire in the mouse male germline. Biol Reprod 2013; 89:107; PMID:24048575; http://dx.doi.org/ 10.1095/biolreprod.113.113308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang M, Li W, Tian H, Hu T, Wang L, Lin Y, Li Y, Huang H, Sun F. Sequential expression of long noncoding RNA as mRNA gene expression in specific stages of mouse spermatogenesis. Sci Rep 2014; 4:5966; PMID:25097017; http://dx.doi.org/ 10.1038/srep05966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goh WS, Falciatori I, Tam OH, Burgess R, Meikar O, Kotaja N, Hammell M, Hannon GJ. piRNA-directed cleavage of meiotic transcripts regulates spermatogenesis. Gen Dev 2015; 29:1032-44; PMID:25995188; http://dx.doi.org/ 10.1101/gad.260455.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gou LT, Dai P, Yang JH, Xue Y, Hu YP, Zhou Y, Kang JY, Wang X, Li H, Hua MM, et al.. Pachytene piRNAs instruct massive mRNA elimination during late spermiogenesis. Cell Res 2014; 24:680-700; PMID:24787618; http://dx.doi.org/ 10.1038/cr.2014.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang P, Kang JY, Gou LT, Wang J, Xue Y, Skogerboe G, Dai P, Huang DW, Chen R, Fu XD, et al.. MIWI and piRNA-mediated cleavage of messenger RNAs in mouse testes. Cell Res 2015; 25:193-207; PMID:25582079; http://dx.doi.org/ 10.1038/cr.2015.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watanabe T, Cheng EC, Zhong M, Lin H. Retrotransposons and pseudogenes regulate mRNAs and lncRNAs via the piRNA pathway in the germline. Genome Res 2015; 25:368-80; PMID:25480952; http://dx.doi.org/ 10.1101/gr.180802.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Palazzo AF, Lee ES. Non-coding RNA: what is functional and what is junk? Front Genet 2015; 6:2; PMID:25674102; http://dx.doi.org/ 10.3389/fgene.2015.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bellve AR, Cavicchia JC, Millette CF, O'Brien DA, Bhatnagar YM, Dym M. Spermatogenic cells of the prepuberal mouse. Isolation and morphological characterization. J Cell Biol 1977; 74:68-85; PMID:874003; http://dx.doi.org/ 10.1083/jcb.74.1.68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang S, Wang X, Wu Y, Han C. IGF-1R Signaling Is Essential for the Proliferation of Cultured Mouse Spermatogonial Stem Cells by Promoting the G2/M Progression of the Cell Cycle. Stem Cells Dev 2015; 24:471-83; PMID:25356638; http://dx.doi.org/ 10.1089/scd.2014.0376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao Y, Wang J, Zhao F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol 2015; 16:4; PMID:25583365; http://dx.doi.org/ 10.1186/s13059-014-0571-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smyth GK, Speed T. Normalization of cDNA microarray data. Methods 2003; 31:265-73; PMID:14597310; http://dx.doi.org/ 10.1016/S1046-2023(03)00155-5 [DOI] [PubMed] [Google Scholar]
- 59.Buck MJ, Nobel AB, Lieb JD. ChIPOTle: a user-friendly tool for the analysis of ChIP-chip data. Genome Biol 2005; 6:R97; PMID:16277752; http://dx.doi.org/ 10.1186/gb-2005-6-11-r97 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.