

Abstract
Notch receptors play a central role in skeletal development and homeostasis. Hajdu Cheney Syndrome (HCS) is a rare disease associated with mutations of NOTCH2 that lead to the translation of a truncated, presumably stable, NOTCH2 protein. As a consequence, a gain-of-NOTCH2 function is manifested. We report a subject presenting with HCS and her child, both harboring a new heterozygous mutation in Exon 34 of NOTCH2 upstream of the PEST domain. The subject presented with osteoporosis, fractures, acroosteolysis and splenomegaly but did not have neurological complications, cardiovascular defects or polycystic kidneys. Sequencing of genomic DNA revealed a previously unreported mutation at nucleotide 6667C>T leading to a Gln2223Ter protein product in the subject and her son. Preclinical studies have demonstrated that the bone loss in HCS is secondary to enhanced osteoclastogenesis and bone resorption, and the same mechanism may operate in humans. Accordingly, the case we report was treated and responded to therapy with denosumab with an increase in bone mineral density (BMD). However, acroosteolysis progressed and was not modified by denosumab. In conclusion, we report a case of HCS associated with a novel mutation in NOTCH2 and its response to denosumab on BMD.
Keywords: Notch, bone remodeling, fractures, acroosteolysis, denosumab, splenomegaly
INTRODUCTION
Hajdu Cheney Syndrome (HCS) is a rare genetic disorder characterized by acroosteolysis, severe osteoporosis with fractures and craniofacial dysmorphism[1,2]. Individuals with HCS also present with short stature, wormian bones, platybasia, neurological complications, cardiovascular septal and valvular defects and polycystic kidneys[3–5]. The disorder was originally described by Hajdu in 1948 and reported as a syndrome by Cheney in 1962[6,7]. HCS is inherited as a dominant genetic disorder, although many sporadic cases occur. HCS is associated with mutations in exon 34 of NOTCH2 upstream the proline (P), glutamic acid (E), serine (S), and threonine (T) (PEST) domain, resulting in the expression of a NOTCH2 truncated protein[8–10]. Because the PEST domain is necessary for the ubiquitination and degradation of NOTCH2, the protein is presumably stable so that the mutation causes a gain of NOTCH2 function.
Notch 1 to 4 are transmembrane proteins that determine cell fate and function, and play a critical role in embryonic development and cell renewal[11–13]. Notch receptors are activated following interactions with their ligands, and Jagged 1 and 2 and Delta-like 1, 3 and 4 are considered canonical Notch ligands. Although Notch 1 through 4 have structural similarities, their functions are not necessarily redundant. In skeletal cells, Notch activation inhibits chondrogenesis and osteoblastogenesis and causes severe osteopenia[13,14]. However, the effects of Notch are cell context dependent and when activated in osteocytes, Notch1 causes osteopetrosis[15,16]. Whereas Notch1 seems to have an inhibitory function in osteoclastogenesis, in part by inducing osteoprotegerin, Notch2 induces osteoclast maturation by interacting with Nuclear factor KappaB and inducing the transcription of nuclear factor of activated T cells 1[17,18].
Mutations in Notch receptors, their ligands or various components of the Notch signaling pathway are associated with a variety of developmental skeletal disorders[19]. Recently, a mouse model replicating the human mutation of HCS was reported[20]. A point mutation creating a STOP codon was introduced into exon 34 of Notch2 upstream sequences coding for the PEST domain by homologous recombination. The HCSNotch2 mutant mouse exhibited osteopenia secondary to increased osteoclast number and bone resorption. There was an increase in the preosteoclast cell pool as well as in the osteoclastogenetic response to Receptor activator of nuclear factor KappaB ligand (Rankl). In addition, osteoblasts and osteocytes expressed increased levels of Rankl. These observations indicated that the osteopenic phenotype in the HCSNotch2 mouse mutant was secondary to an increase in osteoclastogenesis. If these observations were to apply to the human condition, the osteoporosis of HCS could be ameliorated by the use of agents that inhibit osteoclastogenesis and bone resorption.
Because of the limited number of individuals affected by HCS, there are no controlled trials on therapeutic interventions. Occasional patients have been treated with bisphosphonates or teriparatide in an effort to lessen the osteoporosis but evidence of a beneficial response is limited[21,22].
In the present work, we describe a mother and son presenting with HCS and harboring a novel mutation in exon 34 of NOTCH2. We also report the skeletal response of the adult subject with HCS to treatment with denosumab, an agent known to block RANKL and inhibit osteoclastogenesis and bone resorption.
CASE REPORT
In October 2013, a 33-year-old female patient was seen at the Rheumatology Unit of the University of Verona (Italy) with a history of generalized bone pain, particularly in both hands, hips and back, and severe osteoporosis with fractures. She had suffered multiple vertebral fractures from 1999 to 2013 between T6 and L3 as well as atraumatic fractures of the left knee, right tibia, right fibula and 4th metatarsal bone of the right foot. From 2011 to 2013, the subject suffered two vertebral fractures (T11 and T12). The subject described progressive shortening of the middle finger of the right hand and of the 2nd and 5th finger bilaterally. The patient suffered from congenital right hip dysplasia treated with several surgical procedures and eventually with a total hip replacement. There was no history of neurological or cardiovascular complications or urinary symptoms. The subject’s son suffered a low trauma fracture of the right clavicle at 5 years of age. The report of the case was approved by the Verona Medical School Institutional Review Board (IRB) and both mother and son provided written consent and assent, respectively, for the publication of this report.
Prior to her visit to the Rheumatology Unit of the University of Verona, the subject of study had been treated with vitamin D supplementation, pamidronate a 30 mg IV infusion once in 2008, clodronate intramuscularly 100 mg twice in 2009 and with strontium ranelate 2 g daily for five months in 2010. On physical exam the patient’s height was 155 cm and weight was 63 kg. She displayed a prominent forehead, hypertelorism and micrognathia, finger deformities and hallux valgus, and her hands were short and stubby. There were no signs of local inflammation in either fingers or toes. The spleen was palpable and no abnormalities were noted on cardiovascular and neurological exams.
Complete blood count, serum electrolytes, liver and renal function tests were within the normal range. Serum, alpha-1-antitrypsin, glucose, haptoglobin, blood iron, ferritin, transferrin, growth hormone, insulin-growth factor 1, adrenocorticotropic hormone, thyroid stimulating hormone, luteinizing hormone, prolactin, tryptase, anti-nuclear antibodies, anti-extractable nuclear antigens antibodies, anti-double-strand DNA antibodies, anti-citrullinated peptide antibodies, anti-neutrophil cytoplasmic antibodies and complement were within the normal range. Serum calcium, phosphorous, parathyroid hormone (PTH), 25-hydroxy vitamin D (25OHD), osteocalcin, C-telopeptide of type I collagen (CTX), bone specific alkaline phosphatase (BSAP), and procollagen I intact N-terminal peptide (P1NP) were within the normal range (Table 1), CTX, BSAP and P1NP were measured by the IDS-ISYS Multi-Discipline Automated Analyzer (Immunodiagnostic System, Boldon, UK). Serum RANKL was elevated at 0,451 pmol/L and osteoprotegerin (OPG) was decreased at 1,244 pmol/L, when compared to values obtained in ten age matched female controls. RANKL and OPG were measured by enzyme-linked immunosorbent assay (Biomedica Medizinprodukte GmbH & Co KG, Wien, Austria). All laboratory tests were conducted in the clinical laboratories of the University of Verona Hospital. Studies in normal volunteers were approved by the Verona Medical School IRB and written consent was obtained from volunteers prior to blood drawing.
Table 1.
Biochemical Parameters on a Subject with HCS
| Baseline (8/2013) | 3 days after 1st denosumab (10/2013) | 5 days before 2nd denosumab (4/2014) | 1 day before 3rd denosumab (12/2014) | 1 day before 4th denosumab (7/2015) | 5 days after 5th denosumab (10/2015) | Last follow-up (3/2016) | |
|---|---|---|---|---|---|---|---|
| Calcium (mg/dL) (8,9–10,1) | 9.2 | 8.4 | 9.9 | 9.6 | 9.8 | 9.0 | 9.2 |
| Phosphorous (mg/dL) (2,4-4,1) | 3.1 | 2.3 | 3.0 | 3.0 | 3.4 | 2.7 | 3.0 |
| PTH (pg/mL) (10–55) | 29 | 33 | 28 | 31 | 25 | 64 | 19 |
| 25OHD (ng/mL) (>30) | 34.6 | 31.4 | 40.0 | 30.5 | 31.4 | 32.1 | 24 |
| CTX (pg/mL) (100–670) | 225 | 63 | 220 | 189 | 277 | 33 | 416 |
| BSAP (UI/L) (6–23) | 11 | 11 | 9 | 10 | 9 | 9 | 9 |
| P1NP (ng/mL) (19,3–76,3) | 20,5 | 21.1 | / | 16.0 | 20.0 | 11.2 | 34.3 |
| RANKL (pmol/L) (0,022–0.282)* | 0.451 | 0.008 | / | 0.292 | 0.269 | 0.008 | 0.328 |
| OPG (pmol/L) (4,850–6,840)* | 1.244 | 1.021 | / | 1.411 | 1.539 | 1.422 | 1.092 |
| RANKL/OPG ratio (0,004–0,041) | 0.362 | 0.007 | / | 0.206 | 0.174 | 0.005 | 0.300 |
PTH= Parathyroid hormone, 25OHD= 25 hydroxy Vitamin D, CTX= C-Telopeptide of type I collagen, BSAP= Bone specific alkaline phosphatase, P1NP= Procollagen I intact N-terminal peptide, RANKL= Receptor activator of nuclear factor Kappa-B ligand, OPG= Osteoprotegerin; Normal range values are in parenthesis.
Normal range was established by measuring serum RANKL and OPG in ten age-matched female normal volunteers.
In October 2013, prior to initiation of therapy with denosumab, dual-energy X-ray absorptiometry (DXA) performed with a QDR Hologic Delphy instrument, revealed a bone mineral density (BMD) at the lumbar spine (L1–L4) of 0,810 g/cm2 and a T-score of −3,1 (Z-score −2,9), a BMD at the left femoral neck of 0,848 g/cm2 and a T-score of −1,4 (Z-score −1.0) and at the total left hip of 0,870 g/cm2 and a T score of −1.1 (Z-score −0.8).
Chest X-ray revealed normal lungs and multiple vertebral fractures (Figure 1). Skull X-rays revealed anteroposterior diameter augmentation, paranasal sinus hypoplasia and opened lambdoid suture. Hand X-rays showed acroosteolysis involving the distal phalanx of the 2nd and 5th fingers of both hands (Figure 2). X-rays of both feet showed diffuse demineralization and severe deformities of the first metatarsophalangeal joint bilaterally (Figure 2).
Figure 1.

X-rays showing normal heart and lungs and vertebral deformities (pointed by the arrows) prior to treatment with denosumab.
Figure 2.

X-rays of hands showing acroosteolysis of distal phalanges of the 2nd and 5th finger of both hands, pointed by the arrows, and X-rays of both feet showing hallux valgus and deformities in a subject with HCS.
Spine Magnetic Resonance Imaging (MRI) demonstrated severe scoliosis, “Biconcave lens” vertebral deformities from T6 to L3, associated with angiomatous formation in the vertebral bodies of T3 and L5 (Figure 3). Abdominal ultrasound revealed an enlarged spleen of 14,5 cm in length and 8 cm in width, and no evidence of polycystic kidneys (not shown). Nailfold hand capillaroscopy, performed for the morphologic assessment of the microcirculation, showed increased capillary branching.
Figure 3.

Magnetic Resonance Imaging of the spine showing scoliosis and vertebral biconcave lens deformities in a subject with HCS. Arrows point to deformities.
To establish the diagnosis of HCS, DNA sequence analysis of the NOTCH2 coding sequence was performed on genomic DNA extracted from peripheral leucocytes at the ARC-NET Research Centre, University and Trust of Verona (Verona, Italy)[23]. A custom panel to analyze the coding sequence of NOTCH2 was designed using Ion AmpliSeq Designer software. A suitable library for samples from mother and son was obtained and analyzed on Ion Torrent PGM and loaded on chip 318v2 (Thermo Fisher, Waltham, MA). Data analysis, including alignment to the hg19 human reference genome and variant calling, was performed using the Torrent Suite Software v.5.0 (Thermo Fisher). Filtered variants were annotated using a custom pipeline based on vcflib (https://github.com/ekg/vcflib), SnpSift, the Variant Effect Predictor (VEP) software and NCBI RefSeq database[24,25]. DNA sequence analysis of the subject of this report revealed a heterozygous nonsense (NP_077719.2:p.Gln2223Ter; NM_024408.3:c.6667C>T) mutation in exon 34 of NOTCH2 not described previously (Figure 4). The 6667C>T mutation in exon 34 of NOTCH2 leads to the creation of a Stop codon and a predicted truncated protein product of 2222 amino acids.
Figure 4.

Genomic DNA from blood samples of the subject with HCS was sequenced by next generation sequencing on Ion Torrent (Thermo Fisher) for the coding sequence of the NOTCH2 gene. The representation of the reads aligned to the reference genome, as provided by the Integrative Genomics Viewer software (IGV v 2.1, Broad Institute), shows a 1:1 signal ratio of C (blue) to T (red) at nucleotide 6667 demonstrating the presence of a heterozygous 6667C>T mutation.
The subject initiated treatment with denosumab, 60 mg subcutaneously every 6 months and vitamin D3 supplementation at an oral dose of 10,000 IU weekly. Serum RANKL decreased following the administration of denosumab (Table 1). Serum CTX declined from 225 pg/ml to 63 pg/ml 3 days after denosumab was administered, but serum CTX levels were within the normal range prior to each subsequent dose of denosumab. Serum markers of bone formation, including BSAP and osteocalcin did not change with the treatment. A progressive increase in lumbar spine BMD was observed after treatment with denosumab; BMD increased by 6.8% at L1–L4 and by 2.6% at the femoral neck two years after the initiation of therapy (Figure 5). The subject did not report any non-vertebral fractures or symptoms suggestive of vertebral fractures while receiving treatment with denosumab. Absence of new vertebral fractures was verified by DXA morphometric vertebral fracture assessment (VFA) obtained following therapy with denosumab (Figure 6) and comparison with DXA VFA, vertebral XRays and MRI obtained prior to therapy.
Figure 5.

Bone mineral density (BMD) of the lumbar spine and left femoral neck obtained prior to (October 2013) and following therapy with 5 doses of denosumab, 60 mg given subcutaneously every 6 months through October 2015 in a subject with HCS. Note that scale for y-axis for lumbar and femoral neck BMD is different.
Figure 6.
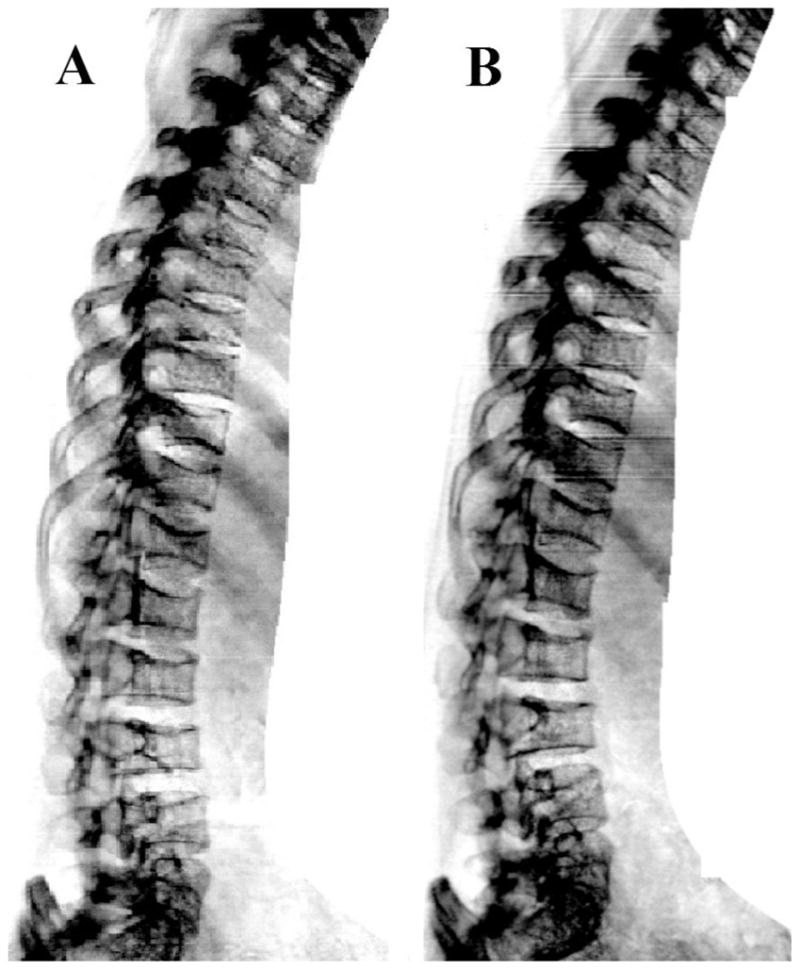
Morphometric vertebral fracture assessment by densitometry (DXA) of the spine of a subject with HCS obtained before (A) and following (B) therapy with 5 doses of denosumab, 60 mg, given subcutaneously every 6 months from October 2013 through October 2015.
During the treatment period, the patient suffered from fluctuating pain in the 3rd finger of the right hand. A radiograph of the hands was obtained after two years of treatment with denosumab, and revealed progression of acroosteolysis and newly discovered involvement of the distal phalanx of the 3rd finger of the right hand (Figure 7). Treatment with denosumab was not associated with adverse events.
Figure 7.

X-rays of hands showing acroosteolysis of the 2nd and 5th finger of both hands as well as the 3rd finger of the right hand in a subject with HCS following two years of treatment with denosumab.
The study subject’s son was assessed at 5 years of age following a low trauma fracture of the clavicle. His weight was 24 kg and his height was at the 25th percentile and, on examination, he was on stage Pubic (P)1 and Genitals (G)1 of Tanner pre-puberal scale. On physical examination, he presented micrognathia, but no other signs of HCS. DXA revealed a lumbar spine BMD of 0,633 g/cm2 (Z score −0,1) and total left hip BMD of 0,832 g/cm2 (Z score +1,7), left femoral neck BMD of 0,808 g/cm2 (Z score +1,3). X-rays of the hands did not reveal any abnormalities. Sequence analysis of genomic DNA obtained from peripheral blood revealed a 6667C>T (Gln2223Ter) mutation in exon 34 of NOTCH2.
DISCUSSION
We report a mother and her child affected by HCS associated with a novel 6667C>T (Gln2223Ter) NOTCH2 heterozygous mutation. The subject presented with a variety of skeletal abnormalities including osteoporosis with fractures, scoliosis and acroosteolysis. Polycystic kidneys and cardiovascular developmental anomalies, including patent ductus arteriosus, atrial and ventricular septal defects and valve defects can be clinical manifestations of HCS[4,26,27]. The subject of this report did not present with any cardiovascular or renal defects, but had splenomegaly, a possible consequence of increased NOTCH2 activity in the marginal zone of the spleen and not previously reported in HCS[28]. Work conducted in murine models of Notch2 global and conditional inactivation in cells of the hematopoietic or the B-cell lineage has demonstrated that Notch2 is indispensable for marginal zone B-cell development[29,30]. Moreover, the constitutive activation of Notch2 in the B-cell lineage results in an increase in marginal zone B-cells of the spleen[28]. HCS is an autosomal dominant inherited disorder, although sporadic cases may occur. DNA sequencing information was not available and could not be obtained from the subject’s parents since the subject had lost contact with them. Consequently, we could not document the type of inheritance in the subject of this report with certainty. DNA sequence analysis indicated a heterozygous mutation which is in accordance with previously reported cases of HCS[8–10]. It is possible that homozygous mutations result in embryonic lethality.
Previous reports have demonstrated the presence of point mutations or short deletions in exon 34 of NOTCH2, upstream the PEST domain in families affected by HCS[8,9]. The mutations create a stop codon and the premature termination of the protein product leading to the accumulation of a stable protein and presumably persistent NOTCH2 signaling[8,9]. The individual we report had a novel mutation 6667C>T in NOTCH2 creating Gln2223Ter at the amino acid level. In accordance with previously described subjects afflicted by HCS, the mutation was in exon 34 upstream the PEST domain. Recently, we conducted studies in a mouse model carrying a 6955C>T Notch2 corresponding to a 6949C>T mutation in NOTCH2 found in a subject affected by HCS[20]. The Notch2 mutation in this mouse model is in exon 34 upstream the PEST domain, and leads to the translation of a truncated protein of 2318 amino acids and enhanced Notch2 signaling in skeletal cells[20]. There was higher expression of Notch target genes (Hes, Hey) in osteoblasts and osteoclasts as well as in bone extracts from mice carrying the HCSNotch2 mutation. Mice harboring HCSNotch2 mutations exhibit cancellous and cortical bone osteopenia, and bone histomorphometric analysis showed increased osteoclast number and bone resorption[20]. In vitro studies demonstrated that osteoclastogenesis and the bone resorptive capacity of osteoclasts were enhanced. These observations were attributed to a direct effect of Notch2 on osteoclast precursors as well as to an induction of Rankl in osteoblasts and osteocytes[18,20]. In line with these observations, the individual we report had increased serum levels of RANKL and a favorable BMD response to denosumab, an agent known to inhibit osteoclastogenesis. There was a progressive increase in BMD and the subject was free of fractures during the two years of therapy as demonstrated by DXA VFA, whereas she had suffered multiple fractures in the preceding years. Although the patient presented with multiple vertebral fractures and these might have affected the DXA values, the degree of deformity in the L1–L4 region was relatively modest so that it is reasonable to believe that the changes in vertebral BMD represent a response to the therapeutic intervention. Moreover, an increase in BMD, although modest, also was observed in the femoral neck.
Clinical studies have offered limited information on the pathogenesis of the acroosteolysis of HCS. It is accompanied by neovascularization, inflammation and fibrosis, but the underlying mechanism is not clear and appears to be unrelated to the one responsible for the bone loss and fractures[31,32]. In accordance with this hypothesis, the acroosteolysis of the individual we report progressed while the osteoporosis improved with denosumab.
It is of interest that somatic NOTCH2 mutations causing loss of the PEST domain exhibit enhanced NOTCH activation and have been identified in B-cell lymphoma and lymphomas of the marginal zone of the spleen[33–35]. There is no apparent increase in the incidence of B-cell lymphoma in HCS. However, the individual we report had splenomegaly, a manifestation of the disease not reported previously. Splenomegaly can be explained by the fact that Notch2 is required for the formation of the marginal zone of the spleen, and overexpression of Notch2 in B-cells results in increased cellularity of the marginal zone[34,35]. It is not known, however, whether Notch activation in the marginal zone plays any role in the skeletal disease of subjects affected by HCS.
In conclusion, we report a novel NOTCH2 mutation in an individual with HCS and her child. The individual presented with significant osteoporosis and a favorable response to treatment with denosumab.
HIGHLIGHTS.
Hajdu Cheney Syndrome (HCS) is associated with NOTCH2 mutations
A novel 6667C>T mutation in NOTCH2 is reported in a case of HCS
Subject with HCS presented with osteoporosis, acroosteolysis and splenomegaly
Bone mineral density, but not acroosteolysis, responded to denosumab
Acknowledgments
The authors express their gratitude to the subject of this report and her son for agreeing to the publication of this work. The authors thank Mary Yurczak for secretarial support and Prof. Silvano Adami who left us with the love for research and clinical dedication.
This work was supported in part by a grant from the National Institutes of Health (NIH) DK045227 (EC). The work is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Disclosure Statement: The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Canalis E, Zanotti S. Hajdu-Cheney syndrome: a review. Orphanet J Rare Dis. 2014;9:200. doi: 10.1186/s13023-014-0200-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Canalis E, Zanotti S. Hajdu-Cheney Syndrome, a Disease Associated with NOTCH2 Mutations. Curr Osteoporos Rep. 2016 doi: 10.1007/s11914-016-0311-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Descartes M, Rojnueangnit K, Cole L, Sutton A, Morgan SL, Patry L, Samuels ME. Hajdu-Cheney syndrome: phenotypical progression with de-novo NOTCH2 mutation. Clin Dysmorphol. 2014;23:88–94. doi: 10.1097/MCD.0000000000000034. [DOI] [PubMed] [Google Scholar]
- 4.Gray MJ, Kim CA, Bertola DR, Arantes PR, Stewart H, Simpson MA, Irving MD, Robertson SP. Serpentine fibula polycystic kidney syndrome is part of the phenotypic spectrum of Hajdu-Cheney syndrome. Eur J Hum Genet EJHG. 2012;20:122–124. doi: 10.1038/ejhg.2011.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isidor B, Le Merrer M, Exner GU, Pichon O, Thierry G, Guiochon-Mantel A, David A, Cormier-Daire V, Le Caignec C. Serpentine fibula-polycystic kidney syndrome caused by truncating mutations in NOTCH2. Hum Mutat. 2011;32:1239–1242. doi: 10.1002/humu.21563. [DOI] [PubMed] [Google Scholar]
- 6.Hajdu N, Kauntze R. Cranio-skeletal dysplasia. Br J Radiol. 1948;21:42–48. doi: 10.1259/0007-1285-21-241-42. [DOI] [PubMed] [Google Scholar]
- 7.Cheney WD. ACRO-OSTEOLYSIS. Am J Roentgenol Radium Ther Nucl Med. 1965;94:595–607. [PubMed] [Google Scholar]
- 8.Isidor B, Lindenbaum P, Pichon O, Bézieau S, Dina C, Jacquemont S, Martin-Coignard D, Thauvin-Robinet C, Le Merrer M, Mandel J-L, David A, Faivre L, Cormier-Daire V, Redon R, Le Caignec C. Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis. Nat Genet. 2011;43:306–308. doi: 10.1038/ng.778. [DOI] [PubMed] [Google Scholar]
- 9.Simpson MA, Irving MD, Asilmaz E, Gray MJ, Dafou D, Elmslie FV, Mansour S, Holder SE, Brain CE, Burton BK, Kim KH, Pauli RM, Aftimos S, Stewart H, Kim CA, Holder-Espinasse M, Robertson SP, Drake WM, Trembath RC. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nat Genet. 2011;43:303–305. doi: 10.1038/ng.779. [DOI] [PubMed] [Google Scholar]
- 10.Zhao W, Petit E, Gafni RI, Collins MT, Robey PG, Seton M, Miller KK, Mannstadt M. Mutations in NOTCH2 in patients with Hajdu-Cheney syndrome. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA. 2013;24:2275–2281. doi: 10.1007/s00198-013-2298-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fortini ME. Notch signaling: the core pathway and its posttranslational regulation. Dev Cell. 2009;16:633–647. doi: 10.1016/j.devcel.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 12.Mumm JS, Kopan R. Notch signaling: from the outside in. Dev Biol. 2000;228:151–165. doi: 10.1006/dbio.2000.9960. [DOI] [PubMed] [Google Scholar]
- 13.Zanotti S, Canalis E. Notch signaling in skeletal health and disease. Eur J Endocrinol Eur Fed Endocr Soc. 2013;168:R95–103. doi: 10.1530/EJE-13-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zanotti S, Canalis E. Notch suppresses nuclear factor of activated T cells (NFAT) transactivation and Nfatc1 expression in chondrocytes. Endocrinology. 2013;154:762–772. doi: 10.1210/en.2012-1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canalis E, Adams DJ, Boskey A, Parker K, Kranz L, Zanotti S. Notch signaling in osteocytes differentially regulates cancellous and cortical bone remodeling. J Biol Chem. 2013;288:25614–25625. doi: 10.1074/jbc.M113.470492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canalis E, Parker K, Feng JQ, Zanotti S. Osteoblast lineage-specific effects of notch activation in the skeleton. Endocrinology. 2013;154:623–634. doi: 10.1210/en.2012-1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bai S, Kopan R, Zou W, Hilton MJ, Ong C, Long F, Ross FP, Teitelbaum SL. NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J Biol Chem. 2008;283:6509–6518. doi: 10.1074/jbc.M707000200. [DOI] [PubMed] [Google Scholar]
- 18.Fukushima H, Nakao A, Okamoto F, Shin M, Kajiya H, Sakano S, Bigas A, Jimi E, Okabe K. The Association of Notch2 and NF-κB Accelerates RANKL-Induced Osteoclastogenesis. Mol Cell Biol. 2008;28:6402–6412. doi: 10.1128/MCB.00299-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zanotti S, Canalis E. Notch Signaling and the Skeleton. Endocr Rev. 2016;37:223–253. doi: 10.1210/er.2016-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Canalis E, Schilling L, Yee SP, Lee SK, Zanotti S. Hajdu Cheney Mouse Mutants Exhibit Osteopenia, Increased Osteoclastogenesis, and Bone Resorption. J Biol Chem. 2016;291:1538–1551. doi: 10.1074/jbc.M115.685453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galli-Tsinopoulou A, Kyrgios I, Giza S, Giannopoulou EZ, Maggana I, Laliotis N. Two-year cyclic infusion of pamidronate improves bone mass density and eliminates risk of fractures in a girl with osteoporosis due to Hajdu-Cheney syndrome. Minerva Endocrinol. 2012;37:283–289. [PubMed] [Google Scholar]
- 22.McKiernan FE. Integrated anti-remodeling and anabolic therapy for the osteoporosis of Hajdu-Cheney syndrome: 2-year follow-up. Osteoporos Int. 2007;19:379–380. doi: 10.1007/s00198-007-0461-6. [DOI] [PubMed] [Google Scholar]
- 23.Simbolo M, Gottardi M, Corbo V, Fassan M, Mafficini A, Malpeli G, Lawlor RT, Scarpa A. DNA Qualification Workflow for Next Generation Sequencing of Histopathological Samples. PLOS ONE. 2013;8:e62692. doi: 10.1371/journal.pone.0062692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cingolani P, Patel VM, Coon M, Nguyen T, Land SJ, Ruden DM, Lu X. Using Drosophila melanogaster as a Model for Genotoxic Chemical Mutational Studies with a New Program, SnpSift. Front Genet. 2012;3:35. doi: 10.3389/fgene.2012.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaler SG, Geggel RL, Sadeghi-Nejad A. Hadju-Cheney syndrome associated with severe cardiac valvular and conduction disease. Dysmorphol Clin Genet. 1990;4:43–47. [Google Scholar]
- 27.Sargin G, Cildag S, Senturk T. Hajdu Cheney syndrome with ventricular septal defect. Kaohsiung J Med Sci. 2013;29:343–344. doi: 10.1016/j.kjms.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 28.Hampel F, Ehrenberg S, Hojer C, Draeseke A, Marschall-Schröter G, Kühn R, Mack B, Gires O, Vahl CJ, Schmidt-Supprian M, Strobl LJ, Zimber-Strobl U. CD19-independent instruction of murine marginal zone B-cell development by constitutive Notch2 signaling. Blood. 2011;118:6321–6331. doi: 10.1182/blood-2010-12-325944. [DOI] [PubMed] [Google Scholar]
- 29.Witt CM, Won WJ, Hurez V, Klug CA. Notch2 Haploinsufficiency Results in Diminished B1 B Cells and a Severe Reduction in Marginal Zone B Cells. J Immunol. 2003;171:2783–2788. doi: 10.4049/jimmunol.171.6.2783. [DOI] [PubMed] [Google Scholar]
- 30.Saito T, Chiba S, Ichikawa M, Kunisato A, Asai T, Shimizu K, Yamaguchi T, Yamamoto G, Seo S, Kumano K, Nakagami-Yamaguchi E, Hamada Y, Aizawa S, Hirai H. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity. 2003;18:675–85. doi: 10.1016/s1074-7613(03)00111-0. [DOI] [PubMed] [Google Scholar]
- 31.Elias AN, Pinals RS, Anderson HC, Gould LV, Streeten DHP. Hereditary osteodysplasia with acro-osteolysis (the Hajdu-Cheney syndrome) Am J Med. 1978;65:627–636. doi: 10.1016/0002-9343(78)90851-3. [DOI] [PubMed] [Google Scholar]
- 32.Nunziata V, di Giovanni G, Ballanti P, Bonucci E. High turnover osteoporosis in acro-osteolysis (Hajdu-Cheney syndrome) J Endocrinol Invest. 2014;13:251–255. doi: 10.1007/BF03349553. [DOI] [PubMed] [Google Scholar]
- 33.Kiel MJ, Velusamy T, Betz BL, Zhao L, Weigelin HG, Chiang MY, Huebner-Chan DR, Bailey NG, Yang DT, Bhagat G, Miranda RN, Bahler DW, Medeiros LJ, Lim MS, Elenitoba-Johnson KSJ. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J Exp Med. 2012;209:1553–1565. doi: 10.1084/jem.20120910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee S, Kumano K, Nakazaki K, Sanada M, Matsumoto A, Yamamoto G, Nannya Y, Suzuki R, Ota S, Ota Y, Izutsu K, Sakata-Yanagimoto M, Hangaishi A, Yagita H, Fukayama M, Seto M, Kurokawa M, Ogawa S, Chiba S. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer Sci. 2009;100:920–926. doi: 10.1111/j.1349-7006.2009.01130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, Monti S, Vaisitti T, Arruga F, Famà R, Ciardullo C, Greco M, Cresta S, Piranda D, Holmes A, Fabbri G, Messina M, Rinaldi A, Wang J, Agostinelli C, Piccaluga PP, Lucioni M, Tabbò F, Serra R, Franceschetti S, Deambrogi C, Daniele G, Gattei V, Marasca R, Facchetti F, Arcaini L, Inghirami G, Bertoni F, Pileri SA, Deaglio S, Foà R, Dalla-Favera R, Pasqualucci L, Rabadan R, Gaidano G. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med. 2012;209:1537–1551. doi: 10.1084/jem.20120904. [DOI] [PMC free article] [PubMed] [Google Scholar]