

Abstract
No independent cross-validation of success rate for studies utilizing small interfering RNA (siRNA) for gene silencing has been completed before. To assess the influence of experimental parameters like cell line, transfection technique, validation method, and type of control, we have to validate these in a large set of studies. We utilized gene chip data published for siRNA experiments to assess success rate and to compare methods used in these experiments. We searched NCBI GEO for samples with whole transcriptome analysis before and after gene silencing and evaluated the efficiency for the target and off-target genes using the array-based expression data. Wilcoxon signed-rank test was used to assess silencing efficacy and Kruskal–Wallis tests and Spearman rank correlation were used to evaluate study parameters. All together 1,643 samples representing 429 experiments published in 207 studies were evaluated. The fold change (FC) of down-regulation of the target gene was above 0.7 in 18.5% and was above 0.5 in 38.7% of experiments. Silencing efficiency was lowest in MCF7 and highest in SW480 cells (FC = 0.59 and FC = 0.30, respectively, P = 9.3E−06). Studies utilizing Western blot for validation performed better than those with quantitative polymerase chain reaction (qPCR) or microarray (FC = 0.43, FC = 0.47, and FC = 0.55, respectively, P = 2.8E−04). There was no correlation between type of control, transfection method, publication year, and silencing efficiency. Although gene silencing is a robust feature successfully cross-validated in the majority of experiments, efficiency remained insufficient in a significant proportion of studies. Selection of cell line model and validation method had the highest influence on silencing proficiency.
Keywords: cell line, cancer, gene silencing; microarrays, RNA interference, transfection
Introduction
The discovery of RNA interference (RNAi) enabled targeted functional screens to investigate new gene functions and pathways. In RNAi, a double-stranded RNA molecule is introduced into the cells, which then triggers suppression of gene expression at the mRNA level. Techniques used to mediate RNAi effects include small interfering RNA (siRNA), short hairpin RNA, and microRNA—all these modulators of gene expression are among the noncoding RNAs constituting 98% of the entire transcriptome.1
Endogenous encoded microRNAs are ~22 nucleotides long and regulate more than two-third of genome by cleavage or transcriptional repression2 as well as destabilization of the target mRNAs.3 MicroRNA precursors are processed by the Drosha RNase III endonuclease and then transported from the nucleus to the cytoplasm where they are further processed by Dicer-mediated ribonuclease III and by the RNA-induced silencing complex.4 Finally, the sense strand of the duplex is removed, while the antisense strand directs the recognition of the target mRNAs via complementary base-pairing interactions. siRNAs are short, 19–21 bp double stranded RNAs with two-nucleotide overhangs at both 3′ ends generated from double-stranded RNAs by Dicer-mediated cleavage.5 Similarly to the microRNA-induced silencing process, the duplex associates with RNA-induced silencing complex, which in turn catalyzes the specific degradation of homologous mRNAs.
In the last 15 years, experimental RNAi has become a widespread tool for evaluating loss-of-function phenotypes. However, some technological difficulties stalled its universal introduction. The most important of these include a large variation of silencing efficiency of siRNAs synthesized against different sites on the same target mRNA6 and the occurrence of unintended off-target effects leading to high false positive rate.7 In a genome-wide analysis of the efficacy and specificity of silencing of two genes the expression profile was found to be siRNA-specific rather than target-specific.8 To improve efficiency and reduce off-target effects, a number of empirical rules,9,10 advanced design tools,11,12 and chemical modifications13 have been reported, all of these help to increase the power of specific gene silencing. However, despite the guidelines and technological advances in RNAi, it is a matter of serious concern that the issues of specificity, noise, and heterogeneity remain unaddressed. Pharmaceutical companies have now abridged interest in pursuing RNAi specific treatment strategies.14 Methodological and technical problems also lead to a significant number of publications with nonreproducible results in the last few years even in major journals.15,16,17,18
Gene chips are capable to simultaneously measure the expression of almost all human genes. One can use microarrays to identify the gene to be silenced19 or to evaluate outcome of the silencing. In this, by combining a measurement before and after RNAi treatment we can identify both target and off-target effects at the same time. To date, numerous studies utilizing multiple cell lines and various experimental technologies employed gene arrays to measure the effect of an intended silencing on a transcriptomic scale.
Since many of these experiments have published all the raw experimental data, we can mine the gene expression pairs to investigate factors affecting RNAi gene silencing efficiency. Here, we aimed to process available microarray datasets to investigate RNAi silencing efficiency across a large number of independent studies and to identify factors empowering a successful experiment.
Results
Database setup
Using the GEO search, all together 145,693 samples were downloaded. CEL files were available for 134,289 samples and of these 28,853 represent a treatment-control pair (Figure 1a). Reduction to RNAi experiments delivered 1,643 samples. The total number of pairs (n = 3,160) is higher than the actual number of samples as many studies performed repetitions for the silencing and for the control samples as well. In the statistical analysis we used each possible pair within one dataset. The complete database including all GSM IDs; the normalized gene expression table for all JetSet genes20; and a table containing descriptive characteristics of the original studies including dataset, platform, cell line name, as well as treatment options with corresponding control samples are available upon request from the authors.
Figure 1.

Overview of the study and the studies evaluated. Flowchart of the database setup starting with a GEO search and ending up having 3,631 siRNA treatment-control pairs (a). The utilized statistical tests (b) start using these pairs. Characteristics of the utilized models and techniques across all studies (c).
We have evaluated the characteristics of the 207 studies included in the analysis (Figure 1b). Only five cell lines were utilized in at least 10 independent analyses, these are MCF7, A375, HELA, MDA231, and SW480. These five constitute 47% of all used cell lines, with MCF7 taking the lion's share by 25%. When checking the transfection method, employment of a transfection reagent was leading with 71.7% over a smaller proportion of infection and electroporation, but we have to note that a quarter of the studies did not disclosed this information. The vast majority used nontargeting RNA oligos as negative control treatment. Empty vector (in case of infection based transfection), mock (in case of reagent based transfection) were both below 5% of the studies while utilization of completely untreated cell occurred in only 16 studies.
In respect to our study goals, the most important characteristic is the used method for validation of silencing efficiency. Unfortunately, 13 publications did not disclose the used method and 78 studies lacked a relevant publication. Among the other studies, the gene-chip and Western-blot based techniques were equally popular with one third of all studies, and a smaller proportion of 11.4% of all studies used quantitative polymerase chain reaction . We have to note that some studies measured both mRNA and protein level for the same gene—these studies were added to the Western blot cohort. Characteristics of the investigated studies are summarized in Figure 1c.
Proportion of significant silencing
We evaluated all investigated genes in each study. This approach enables to compare the specificity of the silencing against the background noise of nontarget genes. A heat map demonstrating the efficacy of gene silencing across all genes is presented in Figure 2 and in Supplementary Figure S1.
Figure 2.

Heat map demonstrating gene silencing efficacy across all experiments. Columns include the silenced genes and the rows depict the expression for each gene (also ranked according to the order of the columns). Genes and experiments are ranked so that the diagonal green line resembles the silencing efficiency of the targeted gene across all comparisons—fluctuations in the remaining area correspond to noise and off-target effects of the silencing. Green equals lower expression in the siRNA treated sample compared with control. The line is not straight as some genes were silenced in multiple experiments and some experiment contained multiple genes.
Overall, 429 RNAi silencing experiments from the 207 studies were evaluated (see Supplementary Table S1). The down-regulation was below 0.7 in 341 experiments (81.5%) and below 0.5 in 257 experiments (61.3%). When evaluating those with at least two pairs enabling computation of a Wilcoxon test, the down-regulation was significant in 131 experiments. A single pair of measurements was available for 60 experiments. The highest significance was achieved in two studies using 10 repeated experiments (HNF1B in GSE37290: P = 3.9E−18 and CREB1 in GSE12056: P = 3.8E−17). The average number of silenced samples in the nonsignificant cohort with multiple pairs was 1.61 only.
An opposite up-regulation with a fold change (FC) over one was observed in 27 experiments, the highest increase was 7.85. Ranked FC for all genes is presented in Figure 3.
Figure 3.

Silencing effectiveness across all genes. A ranked order of all silenced genes based on fold change—values below 0 correspond to down-regulation, and values over 0 to up-regulation. All together 81.5% of measurements displayed a down-regulation below 0.7 and 30.5% were significant (red columns). Down-regulation was insufficient in 18.5% of studies, 6.3% even delivered an up-regulation instead of lower expression.
Effect of used methods on silencing efficacy
Since nearly half the samples originated in five cell lines, we evaluated the silencing efficiency across all genes within these cell lines. Remarkably, the most widely used MCF7 breast cancer cell line demonstrated the worst response to down regulation (mean FC = 0.59 ± 0.06). Best results were obtained in the SW480 epithelial colon cancer cell line (FC = 0.30 ± 0.16), followed by the MDAMB231 breast cancer cell line (FC = 0.35 ± 0.20). We must note that these results were delivered in only 10 and 12 silencing experiments, respectively. The overall average FC in all other cell lines was 0.48 ± 0.06 (see Figure 4a).
Figure 4.

Correlation between study methods and silencing power. Selection of the cell line model had a marked effect on silencing efficiency (a), while utilization of different transfection methods or controls was not significant (b,c). There was no difference between cancer and noncancer studies (d). Protocols validating the silencing by Western-blot reached the highest reduction in the gene chip data as well (e). When comparing silencing for the same gene within independent experiments (n = 62 genes), only the cell line selection of had a significant effect (f). In each figure, lower fold change values correspond to better silencing efficiency.
There was no significant effect on silencing efficiency of the used transfection method and of the type of the applied control sample (Figure 4b,c). When comparing studies working on cancer cell lines and those working with other cell lines (keratinocytes, airway epithelial, HEK, stem cells, etc., all together n = 45 experiments) there was no significant difference in the achieved silencing power (Figure 4d).
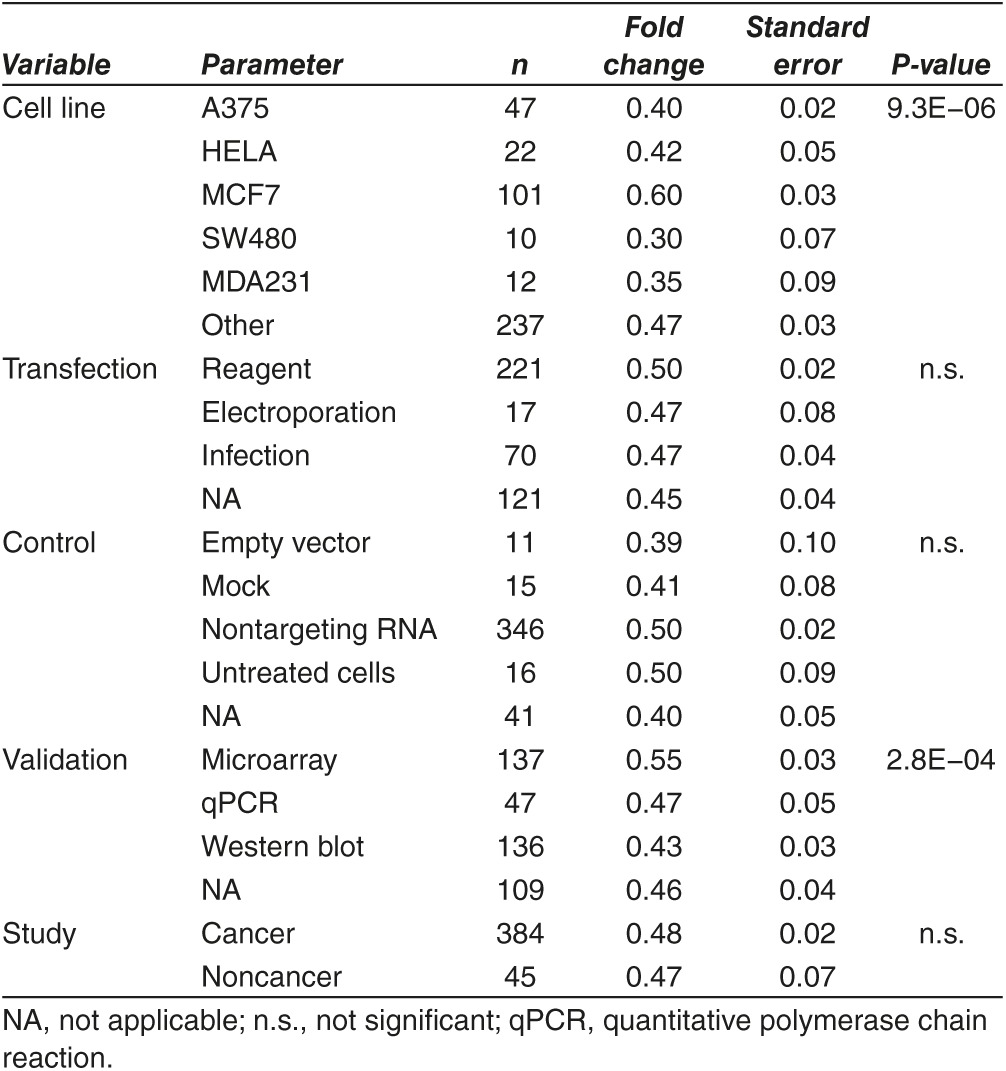
When comparing the methods used to validate silencing, Western-blot reached the lowest mean FC (0.43 ± 0.06), followed by quantitative polymerase chain reaction methods (0.47 ± 0.10) and microarray based validation was performing worst (0.55 ± 0.06). The differences between the methods was significant by Kruskal–Wallis test (P = 2.8E−04, see Figure 4e). A summary of the silencing achievements across all studies for the investigated parameters is presented in Table 1.
Table 1. A summary of achieved silencing across all experiments for the investigated variables.
Fifty genes were silenced in two independent studies, eight genes (ALK, CDK2, CDK4, SRC1, STAT1, MAPK1, HIF1A, and EZH2) in three studies, two (ESR1 and MYC) in four, and two (TP53 and CTNNB1) in six studies. To compare the effect on the same target gene under the different conditions, we extracted the achieved FC from the mean FC for these genes using the repeated experiments only. In this analysis, solely the used cell line had a significant effect (Figure 4f). Transfection method, used control and expression validation methods were not significant.
Silencing efficacy and publication
To evaluate the effect of journal prestige on experimental success, we compared the achieved P-values to the journal impact factors (IF) which was available for 297 experiments. The Spearman rank correlation between P-value and IF was −0.43 (P = 2.9E−15); and between FC and IF it was −0.15 (P = 0.003). As our analysis included datasets spanning a 10-year release range, we evaluated a possible improved performance depending on time. When comparing year of publication and achieved P-value or FC, the Spearman correlation was not significant (P = 0.16 and P = 0.47, respectively).
Noise and off-target effects
The overall reliability and reproducibility of microarrays has been extensively studied previously.21 Here, to evaluate variations in unaffected genes, we computed the FCs across all genes and across all experiments. The median FC in each experiment across all genes was 1.002 (range = 0.73–1.28) and the median FC in each gene across all experiments was 1.001 (range = 0.69–1.19). The close position of the median values to 1 indicates that there was no systematic distortion of the results and that the small variation (also seen in Figure 2) is mostly due to noise in the experiments.
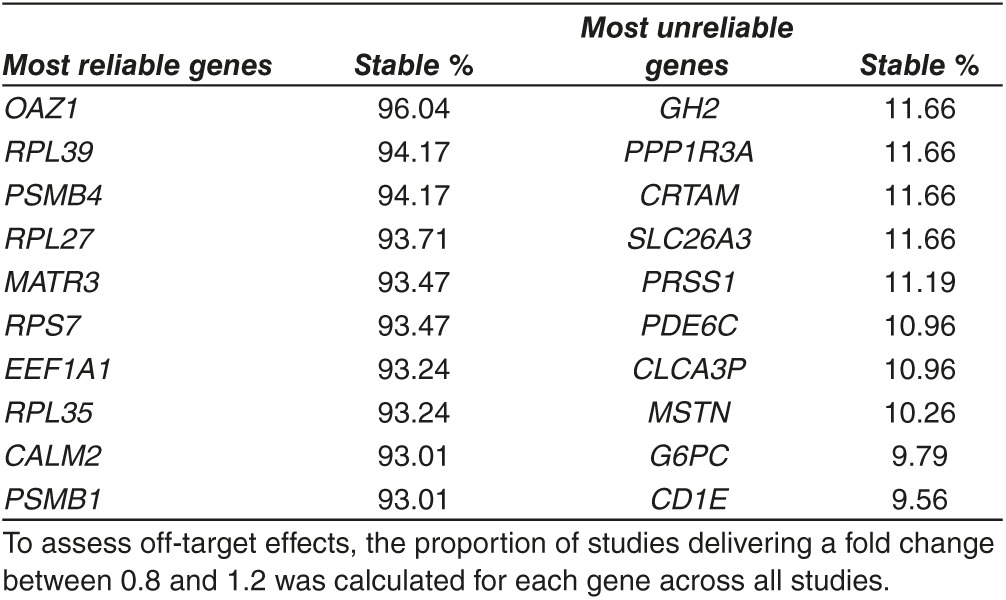
In our analysis we normalized the genes across all probe sets present in the entire gene chips. In several settings, like in case of polymerase chain reaction based validation normalization, a reliable housekeeping gene is required, thus selection of a gene minimally affected by the off-target effects of RNA silencing is of upmost importance. To identify the most reliable housekeeping genes, we computed expression rate across all 12,209 JetSet best genes present on the arrays.20 The proportion of the 419 experiments showing an absolute FC below 1.2 are listed for each gene in Supplementary Table S2. The most stable genes are on the top of this list while the most unstable genes are at the bottom of the list—the top and bottom 10 genes are presented in Table 2.
Table 2. Top 10 most stable genes (highest proportion unchanged) and top 10 most unreliable genes (lowest proportion unchanged) are listed.
Discussion
Here, we performed a large-scale validation of the efficiency of sequence-specific experimental gene silencing initiated by small RNAs homologous to the silenced gene. A major advantage of our study is the exclusive utilization of actual experimental results (in other words computationally inferred effects were left out in the statistical computations). By evaluating all together 429 independent experiments with published raw gene array data, the overall success rate was 81.5% while 6% of the studies delivered an opposite outcome (e.g., increased expression instead of silencing). Multiple issues influencing silencing power including transfection method, the utilized cell line, the type of control, and the validation method were also assessed.
To effectively activate RNAi and induce gene silencing, siRNAs must be delivered to the cytoplasm. However, the cellular membrane is relatively impermeable to siRNAs because of their negative charge and size. Multiple different delivery methods are at hand including the use of a cationic lipid transfection reagent, increasing cell-membrane permeability with an electrical field in electroporation and the utilization of a lentiviral agent to supply the oligos to the target cell. Previously, a broad and significant change was observed in the transcriptome in response to the transcription reagent.22 In our study, there was no significant difference between the available techniques, even though the lipid-based transfection reagents reached the numerically lowest efficiency. Interestingly, comparing the deviations across all measurements show that the reagent based transfection had the smallest confidence interval range compared with the other methods. We have to note that in about one-quarter of the studies the transfection method was not available at all.
A second critical issue is the use of different cell line models. Cell lines used in screening and in the validation studies do not process the hairpins at the same efficiency. Previously, variable hairpin activity in terms of silencing results was described in two independent cell lines.23 Only five cell lines were used in at least 10 experiments, but these gave almost half of all studies—of these, silencing was least significant in the MCF7 breast cancer cell line, which accounted for 25% of all experiments and most significant in the SW480 colon adenocarcinoma cell line. The marked differences between the cell lines emphasize the need for multiple model systems in order to exclude the possibility of a cell line specific false negative result.
Once the experiment is done, one has to verify the reduction of gene expression. This can be done on the protein level by Western blot, on the mRNA level by quantitative polymerase chain reaction for single genes and on the mRNA level for all genes together by gene arrays. There are other techniques available as well like Northern-blot, sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE), enzyme-linked immunosorbent assay (ELISA) or RNA-seq—however, only very few studies used these other methods and therefore, we could not evaluate their success rate. Of the three most common methods, studies using Western blot for validation achieved the strongest silencing. Our results support the current practice in multiple journals requiring Western blot validation for studies including siRNA gene silencing.
One could expect more reliable measurement in prestigious journals. To evaluate such an effect, we compared the achieved P-values to the IF. In this analysis, we found an inverse correlation between P-value and IF and between FC and IF. These results support the notion that higher ranking journals deliver more reliable outcomes. However, we have to note that we only included studies having not only multiple level of data but also whole transcriptome analysis. To this end, the average IF for the journals publishing these more complex experiments was 8.51, which is strikingly higher than the average IF across all life science journals.
At the same time, there was no correlation between publication date and silencing efficiency suggesting that current techniques are not superior to methods available a decade ago.
Many genes are affected by an RNAi experiment and only those siRNAs which have decreased off-target effect will enable the identification of essential genes in cancer.24 To assess off-target effect across all genes, we focused on spotting the most stable genes, e.g., those having the lowest proportion of studies with a FC out of the 0.8–1.2 range. Conventional housekeeping genes were at the top of this list (RPLP0 at #100, GAPDH at #137, and ACTB at #197). However, some other genes like PSMB4 or RPLP1 have reached an even higher rank. When looking at the average expression we observed higher variability for genes with a lower average expression. Our results can guide the selection of highly expressed control genes most infallible for off-target effects.
To date, RNAi treatments are not yet established in clinical setting. The first successful phase 1 clinical trial utilizing siRNA has been completed for macular degeneration a decade ago.25 However, use of siRNA without a targeted delivery vector is only possible for organs with direct access like the respiratory system, skin, and the eyes because systemic administration results in swift RNase degradation and elimination of the oligos.26 The inability to deliver RNA molecules to cell populations in vivo roots in their instability, in the limited cell uptake and short circulation half-life. Today, a wide variety of delivery systems are under development/optimization to enable site specific delivery of siRNA with some already in clinical investigation.27,28 Validation of these approaches in a similar study will only be possible once we will have access to transcriptomic data generated after in vivo siRNA treatments.
In summary, here we took advantage of the significant proportion of studies with available transcriptomic profiles and performed a large-scale validation of RNAi gene silencing efficiency. Altogether, we can endorse RNAi as a robust feature successfully cross-validated in most of the experiments. However, silencing efficiency was insufficient in nearly one-fifth of all experiments. Thus, careful study design is necessary including the cautious selection of cell line model and validation method as these had the most significant influence on silencing proficiency.
Materials and methods
Constructing a database of RNAi experiments. A GEO search was made on 1 August 2015 and results were saved into an SQL database. NCBI GEO (www.ncbi.nlm.nih.gov/geo/) is the largest microarray repository available and the only one providing free download of all data. The analysis was performed in the R statistical environment (http://www.r-project.org). The search was set to include all studies published in the last 10 complete years—starting on the 1st of January 2005. Then, the GEOsql package was used to perform a text based search in the database. In this, the search terms were the cell line names in the field's title, characteristics, source name, description of the samples, and title, summary, and overall design of the datasets. Cell line names were collected from ATCC (http://www.atcc.org), COSMIC (http://cancer.sanger.ac.uk/cosmic) and the Cancer Cell Line Encyclopedia (http://atlasgeneticsoncology.org/cell_lines.html). The search was performed using the “like” command to enable identification of dissimilar nomenclature (e.g., “MCF7” or “MCF-7” or “MCF 7”).
In the second step, each of the identified experiments was assessed manually to identify those with an RNAi study. To be included in the final database, the study design had to fulfill two main criteria: (i) there is at least one control sample for each experiment, (ii) only one treatment is administered in the treatment-control pair.
We have excluded studies focusing solely on drug treatment, hyper- or hypothermia, radiation, viral, and bacterial infection. Also, studies performing the experiments at different passage number were excluded. In this sense, studies with long-term treatment (e.g., over 2 weeks), studies in Matrigel or polyamide gels, utilization of cell lines with an evolutional selection were included in the study as long as the requirement for an untreated control-treated pair was fulfilled. A study was also included in case the silencing was performed against a mutated form of the gene.
Description of the methods was not unambiguous in the overwhelming majority of the studies. To enable a reliable analysis, each sample in entire database was processed by three independent team members (Z.P. or N.S., and B.B.) and the final database was set up by assessing the concordance of these. In case there was discordance in the interpretation, the original study was also read by a third team member (G.M.) and the final designation was set up by selecting the concordant interpretation of at least three investigators.
Processing of gene microarrays. First, all gene chips were downloaded and normalized using MAS5 in the R-statistical environment. We have selected MAS5, because it performed among the best normalization methods when compared with reverse-transcription polymerase chain reaction validated gene expression results in our previous study.29 Then, the quality of each gene chip was assessed to check all parameters suggested in the Affymetrix white paper (http://www.affymetrix.com/support/technical/whitepapers.affx). In this, we have evaluated the following parameters: percentage present calls, background, noise, spike-in controls, and RNA degradation as described previously.30
Processing of RNAi experiments. In each experiment, the silenced and the corresponding control experiments were paired. Within each study, the repetitions were treated as sovereign pairs, and every treatment replica was compared with all available controls. To assess the magnitude of the silencing, we computed mean FC across all possible pairs for each gene in each experiment. Finally, Wilcoxon signed-rank test was performed to statistically validate the silencing effect. In case there were no repetitions available, then the FC of expression alteration was computed only.
Statistical analyses. Dichotomous variables were compared with the RNAi silencing efficiency using Wilcoxon test. Multiple groups including method of validation, utilized cell line, transfection method, and type of control samples were compared using Kruskal–Wallis test. Continuous variables including impact factor and year of publication were compared using Spearman rank correlation. Statistical significance was set at P < 0.05.
Off-target effects. To identify genes most infallible for off-target regulation (e.g., housekeeping genes) and genes most fallible to show altered expression after RNAi treatments, we calculated the FCs and the false discovery rates across all genes in all experiments. Genes which are expected to change their expression in relation to the given RNAi treatment identified by using the Pathway Commons database31 were excluded from this analysis. Finally, the proportion of all experiments showing an absolute FC between 0.8 and 1.2 and a false discovery rate <5% was computed.
SUPPLEMENTARY MATERIAL Figure S1. The heat map of Figure 2 at full resolution. Table S1. Results of the Wilcoxon test for each gene in each study separately. Table S2. Gene ranking for stability against off-target effects for all genes.
Acknowledgments
The study was supported by the OTKA K108655 grant.
Supplementary Material
References
- Mattick, JS (2001). Non-coding RNAs: the architects of eukaryotic complexity. EMBO Rep 2: 986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, RC, Farh, KK, Burge, CB and Bartel, DP (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagga, S, Bracht, J, Hunter, S, Massirer, K, Holtz, J, Eachus, R et al. (2005). Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 122: 553–563. [DOI] [PubMed] [Google Scholar]
- Munkácsy, G, Tulassay, Z and Gyorffy, B (2007). [RNA interference and its clinical applications]. Orv Hetil 148: 2235–2240. [DOI] [PubMed] [Google Scholar]
- Zamore, PD, Tuschl, T, Sharp, PA and Bartel, DP (2000). RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 101: 25–33. [DOI] [PubMed] [Google Scholar]
- Holen, T, Amarzguioui, M, Wiiger, MT, Babaie, E and Prydz, H (2002). Positional effects of short interfering RNAs targeting the human coagulation trigger Tissue Factor. Nucleic Acids Res 30: 1757–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, AL and Linsley, PS (2010). Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov 9: 57–67. [DOI] [PubMed] [Google Scholar]
- Jackson, AL, Bartz, SR, Schelter, J, Kobayashi, SV, Burchard, J, Mao, M et al. (2003). Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol 21: 635–637. [DOI] [PubMed] [Google Scholar]
- Patzel, V, Rutz, S, Dietrich, I, Köberle, C, Scheffold, A and Kaufmann, SH (2005). Design of siRNAs producing unstructured guide-RNAs results in improved RNA interference efficiency. Nat Biotechnol 23: 1440–1444. [DOI] [PubMed] [Google Scholar]
- Naito, Y and Ui-Tei, K (2013). Designing functional siRNA with reduced off-target effects. Methods Mol Biol 942: 57–68. [DOI] [PubMed] [Google Scholar]
- Boudreau, RL, Spengler, RM, Hylock, RH, Kusenda, BJ, Davis, HA, Eichmann, DA et al. (2013). siSPOTR: a tool for designing highly specific and potent siRNAs for human and mouse. Nucleic Acids Res 41: e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito, Y, Yoshimura, J, Morishita, S and Ui-Tei, K (2009). siDirect 2.0: updated software for designing functional siRNA with reduced seed-dependent off-target effect. BMC Bioinformatics 10: 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, AL, Burchard, J, Leake, D, Reynolds, A, Schelter, J, Guo, J et al. (2006). Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA 12: 1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledford, H (2010). Drug giants turn their backs on RNA interference. Nature 468: 487. [DOI] [PubMed] [Google Scholar]
- [Retraction] Interleukin-11 induces the expression of matrix metalloproteinase 13 in gastric cancer SCH cells partly via the PI3K-AKT and JAK-STAT3 pathways. Molecular Medicine Reports 12: 5601 (2015). [DOI] [PubMed] [Google Scholar]
- Retraction. American journal of physiology. Lung cellular and molecular physiology 302: L976 (2012). [DOI] [PubMed] [Google Scholar]
- Roos, E (2011). Retraction. Effect of the chemokine receptor CXCR7 on proliferation of carcinoma cells in vitro and in vivo. Br J Cancer 104: 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipardi, C and Paterson, BM (2011). Retraction for Lipardi and Paterson, “Identification of an RNA-dependent RNA polymerase in Drosophila involved in RNAi and transposon suppression”. Proc Natl Acad Sci USA 108: 15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkácsy, G, Abdul-Ghani, R, Mihály, Z, Tegze, B, Tchernitsa, O, Surowiak, P et al. (2010). PSMB7 is associated with anthracycline resistance and is a prognostic biomarker in breast cancer. Br J Cancer 102: 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q, Birkbak, NJ, Gyorffy, B, Szallasi, Z and Eklund, AC (2011). Jetset: selecting the optimal microarray probe set to represent a gene. BMC Bioinformatics 12: 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L. Shi et al., The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat Biotechnol 24, 1151–1161 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raof, NA, Rajamani, D, Chu, HC, Gurav, A, Johnson, JM, LoGerfo, FW et al. (2016). The effects of transfection reagent polyethyleneimine (PEI) and non-targeting control siRNAs on global gene expression in human aortic smooth muscle cells. BMC Genomics 17: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettcher, M and Hoheisel, JD (2010). Pooled RNAi screens–technical and biological aspects. Curr Genomics 11: 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C, Liu, Z, Yang, F, Liu, W, Wang, D, Dong, E et al. (2015). siRNAs with decreased off-target effect facilitate the identification of essential genes in cancer cells. Oncotarget 6: 21603–21613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, J, Samul, R, Silva, RL, Akiyama, H, Liu, H, Saishin, Y et al. (2006). Suppression of ocular neovascularization with siRNA targeting VEGF receptor 1. Gene Ther 13: 225–234. [DOI] [PubMed] [Google Scholar]
- Vaishnaw, AK, Gollob, J, Gamba-Vitalo, C, Hutabarat, R, Sah, D, Meyers, R et al. (2010). A status report on RNAi therapeutics. Silence 1: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, ME, Zuckerman, JE, Choi, CH, Seligson, D, Tolcher, A, Alabi, CA et al. (2010). Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 464: 1067–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabernero, J, Shapiro, GI, LoRusso, PM, Cervantes, A, Schwartz, GK, Weiss, GJ et al. (2013). First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov 3: 406–417. [DOI] [PubMed] [Google Scholar]
- Gyorffy, B, Molnar, B, Lage, H, Szallasi, Z and Eklund, AC (2009). Evaluation of microarray preprocessing algorithms based on concordance with RT-PCR in clinical samples. PLoS One 4: e5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Győrffy, B, Benke, Z, Lánczky, A, Balázs, B, Szállási, Z, Timár, J et al. (2012). Recurrence Online: an online analysis tool to determine breast cancer recurrence and hormone receptor status using microarray data. Breast Cancer Res Treat 132: 1025–1034. [DOI] [PubMed] [Google Scholar]
- Cerami, EG, Gross, BE, Demir, E, Rodchenkov, I, Babur, O, Anwar, N et al. (2011). Pathway commons, a web resource for biological pathway data. Nucleic Acids Res 39 (Database issue): D685–D690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.