

Abstract
In recent years, accumulating evidence has demonstrated the important role of inflammation in cerebrovascular diseases. The inflammation can last for a long period after the initial insult. Hence, modulation of the inflammation in a wider therapeutic window is a practical approach to treat these cerebrovascular diseases. Despite the acute upregulation of many growth factors after the injury, it is not sufficient to protect and to regenerate the brain. In this mini review, we discuss major growth factors and their beneficial properties to combat the inflammation in cerebrovascular diseases. Emerging biotechnologies that facilitate the therapeutic effects of growth factors are also discussed in an effort to provide insights into the future combination therapies incorporating both central and peripheral abrogation of inflammation. Strategies designed to robustly maintain upregulation of growth factors in the injured brain and the circulation may prove as a potent regenerative approach in sequestering neuroinflammation associated with cerebrovascular diseases.
Keywords: cerebral ischemia, trauma, growth factors, inflammation, neuroprotection, biomedical engineering, stem cells
1. Introduction
Cerebrovascular diseases, as defined by the American Association of Neurological Surgeons, cover all of the disorders in which an area of the brain is temporarily or permanently affected by ischemia or bleeding and one or more of the cerebral blood vessels are involved in the pathological process [1]. The reduction in blood flow may occur from intracranial stenosis, aneurysms, and vascular malformations [1]. In this review we focus on stroke and traumatic brain injury (TBI), which are characterized by cerebrovascular pathologic symptoms accounting for a large patient population with limited therapeutic options and have huge medical costs associated to them. A common feature among cerebrovascular diseases is the occurrence of neuroinflammation as a key neurodegenerative cell death pathway during the progression of the disease. As the need for effective therapeutic treatments for cerebrovascular diseases increases, a relatively new line of research investigation has focused on modulating growth factors in sequestering the inflammatory response following the onset of these diseases. Growth factors have been extensively studied in experimental models of many diseases because of the feasibility to control experimental groups and the direct medical application to the current unmet clinical need. The regulatory functions of these molecules in the brain after such injury include neurogenesis, angiogenesis, anti-apoptosis, anti-inflammation, and among many others [2,3]. There are many growth factors including, but not limited to, nerve growth factor (NGF), fibroblast growth factor (FGF), hepatocyte growth factor (HGF), glial cell line derived neurotrophic factor (GDNF), vascular endothelial growth factor (VEGF), stromal derived factor 1 (SDF-1), stem cell factor (SCF), and brain-derived neurotrophic factor (BDNF). In this review, we describe the relationship between inflammation and cerebrovascular diseases with a focus on stroke and TBI; then we discuss specific growth factors, namely GDNF, VEGF, SDF-1, SCF and BDNF, summarizing relevant experimental studies that support their applications in cerebrovascular diseases with emphasis on stroke and TBI. We have focused on only these five growth factors, which have generated much preclinical evidence to date. Finally, we present the challenges facing growth factor therapy and the approaches taken to enhance their functional outcomes in experimental models.
A stroke is defined as loss of blood flow to one or multiple parts of the brain either by obstruction of blood vessels which leads to ischemic stroke or rupture of blood vessels which results in hemorrhagic stroke [4]. While ischemic stroke is the more common type of stroke, hemorrhagic stroke has higher mortality rate [4]. Currently stroke is ranked the 4th leading cause of death in the United States [4]. In 2012, the U.S. spends $71.6 billion in total stroke-related cost and it is expected to triple in 2030 [4]. Despite the improvement in awareness and care for stroke, the only FDA approved treatment for stroke is limited to tissue-type plasminogen activator (tPA) with small therapeutic window and serious complications [4]. In recent years, several novel drugs have been shown effective in experimental models of stroke, but have failed in the clinic. Among these drugs, Cilostazol has generated some interest as a stroke therapeutic, by acting as a phosphodiesterase (PDE) inhibitor of cyclic adenosine monophosphate (cAMP) [5]. Cilostazol inhibits the platelet aggregation induced by collagen and adenosine diphosphate (ADP), reduces thrombus formation through inhibition of monocyte chemoattractant protein-1 (MPC-1), and may prevent the progression of symptomatic intracranial artery stenosis [5]. Nonetheless, there are side effects associated with Cilostazol treatment, such as headache, gastrointestinal disturbance, and dizziness, incidences of bleeding, requiring much more rigorous preclinical research on its safety and efficacy as a stroke drug. Hence, there is still an urgent need to develop a more effective treatment for stroke. Among the many pathological features implicated in stroke such as necrosis, apoptosis, Ca2+ toxicity, glutamate excitotoxicity, compromised blood brain barrier (BBB) and downregulation of specific growth factors, the topic of inflammation has received favorable attention when contemplating growth factor therapy [6,7]. In the ischemic core, the cells are generally considered irreversibly succumbed to cell death due to energy failure, characterized by an influx of Na+ and Ca2+, causing swelling and breaking down of the plasma membrane. On the other hand, the cells in the penumbra, the area surrounding the ischemic core, which are stressed due to the hypoxic environment may be resuscitated. These cells secrete pro-inflammation cytokines, including interleukin 1beta (IL-1β), IL-6, and tumor necrosis factor – alpha (TNF-α), which activate immune cells such as microglia from within the brain and leukocytes from the vascular, and peripheral systems. The pro-inflammatory cytokines trigger a cascade of events that eventually leads to secondary cell death and the expansion of the ischemic core if no intervention is initiated.
The susceptibility of the cerebrovasculature to mechanical impact or blast injury has defined the complex pathology of TBI. Depending on the severity of the TBI either macrovessels or microvessels will be sheared [8]. Each year, 1.7 million TBIs occur in the U.S which result in approximately 52,000 deaths and 275,000 hospitalizations [9]. The treatment for TBI is limited to managing the symptoms and rehabilitation [9]. Therefore, there is a significant unmet need to develop a comprehensive treatment for TBI. TBI has two distinct phases, acute and chronic, with the latter accompanied by dementia and chronic traumatic encephalopathy (CTE) [8]. The secondary cell death arising from the primary insult in TBI include impaired metabolism, deficient mitochondrial function, BBB breakdown, diffuse axonal injury, and reduced growth factors [10,11]. Similar to the delayed phase of stroke, pro-inflammatory cytokines plague the progression of secondary cell death in TBI and expands the damage in the peri-impact area, the equivalent of the penumbra in stroke. Sequestration of neuroinflammation via growth factor therapy may retard and even halt this secondary cell death after TBI.
2. Relationship between Inflammation and Cerebrovascular Diseases
The role of inflammation as a key component of pathological processes has been implicated in cell death in many cerebrovascular diseases [12-14]. Figure 1 summarizes the role of inflammation in the pathological progress of cerebrovascular diseases. The inflammation in the brain not only can be originated from brain tissues, but also from the vasculature through the infiltration of immune cells in diseases such as atherosclerosis, stroke and TBI. Atherosclerosis has been reported to be linked with plaque-filled blood vessels that have high levels of cholesterol and inflammation cytokines [15-18]. These plaques can reduce and/or block blood flow to the brain can result in stroke [18], or exacerbate brain insults such as TBI [19-21]. Similarly, aneurysms, abnormal blood vessels, can increase the deposition of cholesterol and promote inflammation responses [22]. Rupture of aneurysms in the brain leads to hemorrhagic stroke, a less common but more lethal form of stroke [4]. Once stroke or TBI ensues, the primary insult triggers a cascade of secondary cell death events with inflammation around the core damaged tissue, a key degenerative process of the neurovascular unit [13,23,24]. Hence, maintenance of the blood vessel integrity, including the preserving BBB permeability, towards sequestration of the inflammation-induced secondary cell death is a crucial step in retarding the progressive pathology of cerebrovascular diseases. Of note, growth factors and their receptors tend to be highly expressed in blood vessels immediately after injury which implicate to the therapeutic effects of these factors in rescuing the neurovascular unit [25]. Cerebrovascular diseases have compromised neurovasculature at varying levels during the secondary cell death [26,27], further suggesting that repairing and protecting these leaky blood vessels seem to be a logical treatment approach for diseases such as stroke or TBI. In addition to reducing the damage to the blood vessels, these growth factors can reduce inflammation and eventually secondary cell death in the penumbra or peri-impact area in stroke and TBI, respectively [10,14,28,29]. Accordingly, finding effective therapeutic regimens that advance the use growth factors in preserving the vasculature and dampening the inflammation may lead to better outcome for patients with cerebrovascular diseases. Along this line of using growth factors to abrogate inflammation, growth factors such as GDNF, VEGF, SDF-1α, SCF, and BDNF have been shown as candidate molecules with anti-inflammatory effects that can preserve blood vessel integrity and its function in cerebrovascular diseases. Table 1 summarizes the functions of the five growth factors discussed in this paper. The subsequent sections will focus on each of these therapeutic molecules to gauge their safety and efficacy, and mechanism of action against neuroinflammation.
Figure 1.
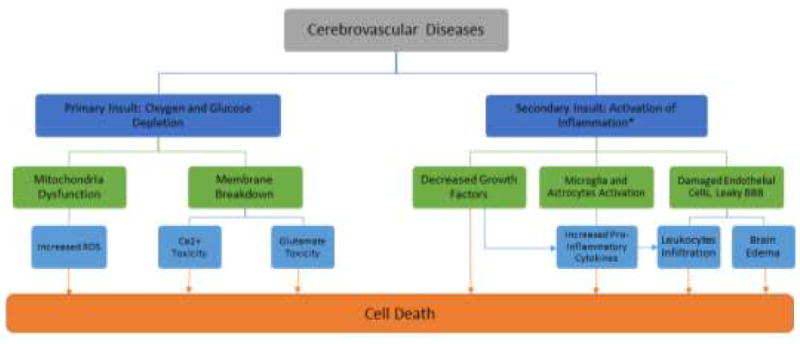
Relationship between inflammation and cerebrovascular disease. Inflammation closely accompanies the secondary cell death process which initiates within hours and may persist over days, weeks, months, and even years after the initial insult. Sequestration of inflammation can help to reduce the expansion of ischemic core and to rescue the penumbra/peri-impact. *We recognize that there are multiple secondary cell death pathways, but we focus on the inflammation pathway because of its wide therapeutic window, which is the main of this paper.
Table 1.
Summary of growth factors and their neuroprotective functions.
| Growth Factor | Function |
|---|---|
| GDNF | Neuroprotection, regulate inflammation, Differentiation, proliferation, migration, neurite outgrowth and synaptic plasticity |
| VEGF | Angiogenesis and neuroprotection |
| SDF-1 | Cell signaling, anti-inflammation, anti-apoptosis, migration, and chemotaxis |
| SCF | Differentiation, neurogenesis, neuroprotection, chemotaxis, and Migration |
| BDNF | Neuronal survival, synaptic function, anti-inflammation, and anti-apoptosis |
3. GDNF
After its milestone discovery of successfully protecting embryonic nigral neurons in vitro, GDNF has been an attractive neurotrophic factor to study in various neurological disorders. Subsequent studies have shown that GDNF also promotes the survival of dopaminergic midbrain neurons, cranial and spinal cord motor neurons, brain stem noradrenergic neurons, basal forebrain cholinergic neurons, Purkinje cells, and specific groups of dorsal ganglia and sympathetic neurons, all demonstrated in in vitro and in vivo experimental studies [30]. In the rat central nervous system (CNS), GDNF transcripts have been identified in the striatum, hippocampus, cortex, cerebellum, and spinal cord [30]. Similarly, in the human CNS, it has also been detected in the striatum, hippocampus, cortex, and spinal cord [30]. GDNF signaling is mediated through rearranged during transfection (RET) receptor tyrosine kinase, which is only activated if the ligand is bound to the GDNF-family receptor-α1 (GFRα1), a class of membrane glycosyl phosphatidylinositol anchored receptors [31,32]. GFRα1 preferentially binds to GDNF and forms a complex with the transmembrane protein RET to initiate essential intracellular signaling cascades through the Ras-MAPK, phosphoinositol 3-kinase, and Jun N-terminal kinase- and PLCγ-dependent pathways [30]. As these pathways regulate inflammation, the therapeutic effects seen with GDNF treatment include cell survival, differentiation, proliferation, migration, chemotaxis, branching morphogenesis, neurite outgrowth and synaptic plasticity [31,33-35]. To date, four GFRα receptors have been elucidated with extensive data showing the beneficial effects of GDNF in neurological disorders [31].
The therapeutic effect of GDNF is evident in the nigrostriatal pathway against inflammation-driven lypopolysaccharides (LPS) rodent model of Parkinson’s disease (PD) [36]. The unilateral intranigral infusion of LPS into the substantia nigra of male rats induced contralateral motor impairments, local microgliosis, and nigrostriatal neurodegeneration in which GDNF-transduced mesenchymal stem cells (MSCs) were effective in affording protection to the nigrostriatal terminals in its immediate vicinity [36]. In light of other studies demonstrating that higher levels of GDNF are secreted from MSCs transduced with a retrovirus to overexpress the GDNF transgene compared to their intrinsic level, this research group sought to determine the potential of GDNF in the LPS-induced inflammation model of PD [36]. Indeed, tyrosine hydroxylase (TH)-positive staining near the cell transplantation site was observed in animals that received GDNF transplants but not in control animals, indicating that the protection and regeneration of the striatal dopaminergic terminals was a result of the secreted GDNF rather than the inherent protective properties of MSCs [36].
Because GDNF is a large molecule that does not cross the intact BBB, technologies geared towards facilitating GDNF access to the brain have been explored. Indeed, GDNF has been re-engineered for BBB transport along with an anti-inflammatory agent as IgG fusion proteins fused to the heavy chain of a chimeric monoclonal antibody (MAb) against the mouse transferrin receptor (TfR) to determine the therapeutic effects of GDNF in a reversible one-hour middle cerebral artery occlusion (rMCAO) model of focal cerebral ischemia in mice [37]. Mice were treated intravenously with saline, GDNF alone, the cTfRMAb GDNF fusion protein alone, or the combined cTfRMAb–GDNF and cTfRMAb–TNFR (tumor necrosis factor receptor) fusion proteins [37]. The cTfRMAb–GDNF fusion protein alone caused a significant 25% and 30% reduction in hemispheric and cortical stroke volumes, respectively, while the combined treatment with the cTfRMAb–GDNF and cTfRMAb–TNFR fusion proteins caused a significant 54%, 69% and 30% reduction in hemispheric, cortical and subcortical stroke volumes, respectively [37]. However, intravenous GDNF had no therapeutic effect, which was attributed to the intact BBB that was not permissive for GDNF access into the brain [37]. This study also demonstrated that combination intravenous therapy may be more beneficial than stand-alone therapy in that the reduction in stroke volume with the combined use of the cTfRMAb–GDNF fusion protein and the cTfRMAb–TNFR fusion protein reduced more significantly the stroke volume than the single therapy with the cTfRMAb–GDNF fusion protein [37].
Another combination therapy examined the benefits of GDNF and another neuroprotective molecule in TBI experimental models. The administration of adenoviral vector into the forelimb sensorimotor cortex of rats (FL-SMC) containing human GDNF (AdGDNF) after a controlled cortical impact injury (CCI) and the intravenous administration of L-arginine after 30 minutes of the CCI demonstrate a significantly smaller contusion than injured rats that did not receive any treatment, or injured rats treated with either AdGDNF or l-arginine alone [38]. This implies that AdGDNF alone following a CCI was not therapeutic and although combining it with l-arginine decreased contusion size, it did not enhance behavioral recovery [38]. Similarly, the unilateral injection of an adenoviral vector harboring AdGDNF or green fluorescent protein (AdGFP) into the FL-SMC of rats one week prior to a unilateral CCI and at 2 weeks post-injury resulted in rats injected with AdGDNF displaying significantly smaller contusions, more surviving neurons, and less neurodegeneration than AdGFP injected and uninjected injured animals [39].
These encouraging preclinical studies on GDNF, either as stand-alone or in combination with other therapeutic agents, in stroke and TBI will merit from long-term investigations of safety and efficacy monitoring in order to avoid the deleterious adverse effects seen in the halted clinical trials of GDNF use in PD. While much attention was generated in assessing the safety of GDNF as a therapeutic agent in PD human patients, GDNF has not yet been evaluated for cerebrovascular diseases in clinical trials [40,41]. Of interest, the current literature has attributed the therapeutic benefits of stem cell therapy in cerebral diseases to the cocktail of neurotrophic factors that these cells pump to the extracellular milieu, often implicating GDNF and the other factors discussed in this review [42,43].
4. VEGF
Multiple forms of VEGF are secreted by endothelial cells [44]. VEGF is secreted into circulation as a cysteine-linked dimer [45]. VEGF signaling is mediated on the cell surface of endothelial cells, where the cysteine-linked dimer binds to the specific receptor tyrosine kinases – FLT1/fms, FLK1/KDR, and FLT4 [45]. Following binding of the VEGF molecule, the intracellular domains of VEGF receptors 2 (VEGFR2) are phosphorylated, which leads to activation of pro-angiogenesis signaling cascades [45]. Recent data suggest there is evidence of the neuroprotective effect in delayed VEGF treatment on neonate rodent stroke models following MCAO [46,47]. A single intracerebroventricular injection of human recombinant VEGF in this stroke model demonstrated a significant increase in endogenous angiogenesis in the peri-infarct area of the caudate [46]. Furthermore, delayed VEGF treatment revealed a notable reduction in Iba1-postive microglial cells and an increase in myelin basic protein (MBP) in the area of the ischemic caudate [46]. Therefore, cerebrovascular injury such as stroke can be reduced with delayed treatment of VEGF [46,47].
VEGF also plays a key role in providing protection following CNS trauma – specifically TBI [48]. To date, there is evidence that upregulation of VEGF-A has protective benefits in both early and late treatment of TBI [48]. A virus delivery system was coupled with an engineered zinc-finger protein transcription factor transactivator in order to monitor the upregulation of VEGF [48]. Evidence demonstrates that using VEGF zinc finger protein (VEGF-ZFP) increased neovascularization, enhanced cell survival, and increased functional recovery in both acute and delayed treatment of TBI [48].
In contrast, anti-VEGF therapy can prove as an effective treatment strategy of inflammatory CNS diseases, such as multiple sclerosis (MS). Lesions formed by inflammatory CNS diseases such as MS often compromise the BBB and cause increased permeability which can lead to entry of inflammatory processes into the brain [49]. There is evidence that suggests that VEGF-A is expressed by astrocytes and increases the permeability of the BBB in mice models [49]. To date, data show that limiting astrocytic expression of VEGF-A aids in maintaining the integrity of the BBB, thereby reducing lymphocyte infiltration, inflammatory factors, and reducing paralysis in MS mouse models [49]. This set of data supports the concept of anti-VEGF therapy against inflammatory CNS diseases by limiting BBB breakdown [49].
As mentioned above, there are limited options in the clinical treatment of ischemic stroke. Treatment with tPA activates matrix metalloproteinases (MMPs), which increase the permeability of the BBB in the earlier stages of stroke via degradation and breakdown of the extracellular matrix (ECM) [50]. Hence, the combination of tPA and VEGF in the early stage of stroke might further exacerbate the permeability of BBB and unfavorable outcomes. However, these activated MMPs are also thought to aid in neurovascular repair in the later stages of stroke through activation of the VEGF signaling cascade [50]. Although the use of VEGF therapy has shown potentially beneficial effects following cerebrovascular disease, it is important to note the possible complications with VEGF treatment in brain ischemia. A study has found an inverse relationship between angiogenesis and neuroprotection following VEGF monotherapy [51]. The complexity of VEGF mechanisms warrants further studies to elucidate the timing and dose of VEGF to treat cerebrovascular disease without inducing adverse effects.
5. SDF-1 α
Stromal derived factor 1 (SDF-1), also known as CXCL12 (Cystine-X-Cystine motif ligand 12), is a chemokine involved in many cellular processes, specifically cell motility during embryo development and cell signaling, locomotion, inflammation, chemotaxis, and adhesion after birth [52-57]. SDF-1 has two isoforms, alpha and beta [58]. SDF-1α binds to the specific CXC receptor 4 (CXCR4), causes a dimerization of the receptor, which then associates with the Gαi protein and subsequently becomes phosphorylated by the JAK2 and JAK3 kinase [55,57,59-63]. The CXCR-SDF-1α-Gαi associated complex also binds with Src homology region 2 domain-containing phosphatase-1 (Shp1) and is associated with regulating JAK2 and JAK3 activation [52,64]. This leads to the tyrosine phosphorylation of STAT proteins which lead to transcription regulations [52]. The activation of the βγ subunits of the Gαi protein leads to the activation of protein kinase C (PKC) and modulates the NF-kB, the MEK/MAPK pathway and local adhesion molecules which promote cell survival through the functions mentioned above [65,66].
SDF-1α is constituently active in the brain mainly in astrocytes and other glial cells and is found in almost every brain region in embryonic, fetal, and adult brain tissue [67]. SDF-1α is responsible for neuronal migration during development and cell proliferation, specifically in astrocytes and other glial cells, maintenance of plasticity and synaptic transmission in development. SDF-1α and CXCR4 knock-out mice have shown to not be viable and are marked with brains that are incredibly underdeveloped [53]. SDF-1α was first isolated in stromal cell lines from mice and was described as a growth factor for B cells [68]. It has also been found to be released from bone marrow stem cells (BMSC) and from astrocytes [69,70].
SDF-1α also contributes to inflammation by attracting leukocytes and aggregating platelets. Platelets function by creating hemostasis and by attracting leukocytes and modulating inflammation. SDF-1α aids in the aggregation of platelets and its subsequent effects. Because SDF-1α is expressed in the endothelial cells lining the neurovasculature, during an insult to blood vessels they release SDF-1α and through co-activators such as ADP and thrombin cause platelets to aggregate, thereafter initiating hemostasis and inflammatory processes [71]. SDF-1α acts in a classic chemokine role by attracting T cells and monocytes that are CXCR4+ to the sites of inflammation and aiding in adhesion [70]. It is also critical for the migration of neuroblasts to damaged areas in the brain, and of hematopoetic stem cells to a desired location in the body [72].
There have been a few studies that examined the levels of SDF-1α in stroke patients. However these results have not been consistent. One group reports that high SDF-1α were associated with acute strokes and that low levels of SDF-1α are associated with large diffuse strokes, the latter being the worst case [73]. Another study determined that elevated SDF-1α at the time of admission of a stroke were strongly associated with a recurrence in another ischemic stroke after a one year in Chinese patients; the study had adjusted for other variables [74]. More research needs to be conducted in order to understand the relationship between SDF-1α and stroke. In experiments where mice were subjected to stroke via MCAO, both endothelial and neuronal SDF-1α showed downregulation, while CXCR4 exhibited upregulation in non-lesioned cortex [23]. SDF-1α elevation in response to stroke leads to an increase in the infiltration and migration of T cells that express the CXCR4 receptor, which in turn increases inflammation and further tissue damage [75]. This particular research group used an antagonist, AMD3100, to ameliorate some of the harmful effects of prolonged exposure to SDF-1α. They found that AMD3100 increased sensorimotor function after stroke and reduced the amount of activated T cells in the brain without negating the beneficial effects that SDF-1α has regarding migration of progenitor cells to the site of insult [75,76].
Targeting SDF-1α’s major actions such as motility of cells that express CXCR4, adhesion, chemotaxis, secretion of matrix metalloproteases, and angiopoietic factors [77] may similarly render therapeutic benefits in stroke. In particular, SDF-1α participation with brain remodeling in post-ischemic insult has been associated with migration of progenitor cells to sites of injury [78]. One activator of SDF-1α is the hypoxia-inducible factor 1 (HIF-1) that is released during hypoxic conditions by cells. Activation of SDF-1α and CXCR4 through HIF-1 leads to adhesion and migration of progenitor cells in the wake of cerebral ischemia [78]. Immunohistochemical analyses of brains from MCAO-stroke mice was able to determine locations of SDF-1α and CXCR4 concentration, and progenitor migration over a course of 30 days [23]. SDF-1α expression was increased in MCAO-stroke 24 hours after MCAO and maintained at an elevated level for 7 days. SDF-1α was also found in the perifocal area of the insult but not in the contralaterally. SDF-1α also initiated migration of endothelial progenitor cells which are important for vascular regeneration following a cerebral insult [79].
Following a TBI event, CXCR4 is upregulated in the cortex surrounding the site of injury for 3 to 7 days [23]. Regarding angiogenesis, SDF-α plays a key role in the migration of neural stem cells (NSCs) and CD34+ cells to the site of injury. The mechanism of action remains unclear [80]. In addition to attracting CD34+ cells, SDF-1α attracts mesenchymal stem cells, monocytes, stem and endothelial progenitors, and neuroblasts through the CXCR4 receptor [73,79,81-84]. The apoptotic events that occur post TBI can be ameliorated by administering SDF-1α. SDF-1α inhibits the activation of caspase 3 a key protein in the apoptotic pathway and increases the ratio of Bcl-2/Bax an anti-apoptotic protein and pro-apoptotic pathway respectively [23]. SDF-1α also plays a role in anti-inflammation. After a TBI event SDF-1α is shown to reduce cerebral edema, protect against neuronal degeneration, reduces the production of NO, and ameliorates the TBI induced lesion through the ERK and NF-κB pathway [23]. Currently, there are no clinical trials assessing the efficacy of SDF-1α in the treatment of TBI or stroke. Thus, while SDF-1α may serve multiple therapeutic applications in stroke and TBI, ranging from acting as a biomarker to facilitating migration of progenitor cells from the bone marrow to the injured brain area, advancing SDF-1α therapy for neurological disorders will require extensive preclinical research in order to unlock its true potential.
6. SCF
Stem cell factor (SCF), also known as steel factor or KIT-ligand, has been found to have the primary role of facilitating the migration and differentiation of hematopoietic stem cells (HSCs) as well as an emerging role in neurogenesis [85,86]. SCF is expressed by perivascular stromal cells in bone marrow and hepatic cells in the liver [87,88]. In the brain, SCF has been found to be produced primarily by neurons [89] and exists mainly in its soluble form [90]. Found as a glycosylated membrane-bound cytokine and as a soluble protein, SCF has two different human isoforms which differ by the presence of a cleavage site [91]. The mechanism of action for the SCF cytokine is its binding of c-Kit (also known as CD117 or SCF receptor), which is a receptor tyrosine kinase expressed on the surface of stem cells including hematopoietic stem cells [92]. The binding of SCF induces non-covalent dimerization of c-Kit and the autophosphorylation of tyrosine residues [92]. The ligand binding of SCF triggers multiple secondary signal cascades which include the Ras/Erk, PI3k, Src, and JAK/STAT pathways [93,94],95]. Treatment with SCF promotes growth of neurites and dendritic spines, while SCF mutations are correlated with a decrease in long-term potentiation and memory [96,97,86] Administration of SCF and G-CSF was linked with increased NeuN+ (a neuronal cell marker) cells derived from bone marrow in non-ischemic murine brains, further supporting its role in facilitating neurogenesis [98,99].
After stroke, SCF has also been observed to upregulate anti-inflammatory cytokines such as IL-10 [100]. In the inflammatory process, SCF induces the chemotaxis and activation of mast cells, as well as the migration of HSCs toward areas of higher SCF concentration [101]. Injection of anti-SCF antibodies inhibits the infiltration of bone marrow mast cells into tumor tissues with high expression of SCF near the tumor [102]. As a hematopoietic cytokine, SCF must act in concert with other stimulating factors such as granulocyte colony-stimulating factor (G-CSF) to facilitate the proliferation and differentiation of stem cells.The discovery of cytokine receptors such as c-Kit and GCSFR on NSCs in the subventricular zones of adult rat brains has spurred interest in the effects of SCF and other growth factors as possible therapies for cerebrovascular disease [25]. The in vivo administration of recombinant SCF has been found to increase proliferation of immature neurons in areas of hypoxia induced by occlusion of the middle cerebral artery as measured by the incorporation of bromodeoxyuridine (BrdU), a synthetic nucleoside analog of thymidine [99]. In parallel, subtractive cDNA suppression hybridization has demonstrated the upregulation of SCF as a highly expressed cytokine in sites of brain injury [103]. Western blot in the same study has shown increased SCF protein levels compared to the brain tissue of uninjured mice. SCF immunohistochemical staining revealed that the majority of SCF was being expressed not in microglia or monocytes but in neurons. A Boyden chamber-based migration assay also demonstrated the in vitro chemotactic effect of SCF with the increased migration of NSCs to sites of injury. Administration of SCF in conjunction with G-CSF reduced the infarct area and increased the numbers of bone marrow-derived neurons and NSC-derived neurons which correlated with improved functionality [25]. Significant transition of bone marrow-derived stem cells to a reduced peri-infarct area was detected after induced ischemia [104]. SCF and G-CSF were also found to increase endothelial cells derived from bone marrow in murine brains following cerebral ischemia [105]. The combination of SCF and G-CSF has also exhibited an angiogenesis effect by reducing the apoptosis of vascular smooth muscle and increasing cerebral vascular density in a mouse model of leukoencephalopathy [106]. NF-κB has been shown to be necessary for the vascular remodeling effect of SCF and G-CSF therapy as blockage of NF-κB activation was found to reduce post-stroke functional restoration [107]. These results suggest a repair mechanism involving angiogenesis and enhanced differentiation of bone marrow stem cells into neurons and endothelial cells. Such neuroprotective and regenerative effects were notably achieved only with the combined administration of SCF and G-CSF [104]. SCF has also afforded neuroprotective effects in TBI. A stab wound model of TBI in the cerebral cortex found upregulation of c-Kit expression by activated microglia as revealed by the monoclonal anti-c-Kit antibody ACK2 [108]. Microglia activated by TBI produce several neurotoxic cytokines and chemokines that contribute to the injury although their phagocytic function may actually reduce the spread of apoptotic signals to neighboring cells [109]. SCF has been observed in cultures to inhibit the proliferation of microglia stimulated by CSF-1, which may suggest that SCF normally keeps microglia in a quiescent state [110]. Intravenous transplantation of adipose-derived stem cells was observed to reduce loss of hippocampal neurons after TBI with SCF as one of the secreted factors [111]. Such transplants of stem cells represent a combination approach of cell regeneration and the delivery of growth factors [112]. Ongoing research on SCF in combination with stem cell therapy may enhance the functional outcome of stand-alone growth factor therapy for cerebrovascular diseases, including stroke and TBI.
7. BDNF
BDNF is a secreted neurotrophin which regulates neuronal survival, development and synaptic function. BDNF has been studied extensively in many neuropsychiatry disorders such as depression, substance-related disorders, schizophrenia, anxiety, and mood disorders [113-115]. In addition, recent studies have demonstrated the neuroprotective effects of BDNF after neurovascular diseases [116-119]. BDNF is synthesized as a precursor, pro-BDNF, about 32-kDa protein with glycosylated and glycosulphated N-terminal [120,121]. Pro-BDNF is secreted and cleaved into mature BDNF by extracellular proteases [120,122]. BDNF binds to tropomyosin receptor kinase B (TrkB) and p75 receptor collectively to modulate synaptic strength and promotes survival of neurons [121,123]. Pro-BDNF has high affinity to p75 receptor whereas mature BDNF has high affinity to TrkB and low affinity to p75 receptor [121-123]. Binding of pro-BDNF to p75, member of the tumor necrosis factor receptor superfamily, activates neuronal apoptosis [121,123]. In contrast, BDNF binds with high affinity to TrkB and triggers autophosphorylation of tyrosine residues which in turn promotes dimerization and activation of downstream intracellular pathways [121].
BDNF has anti-inflammation, anti-apoptosis, and other neuroprotective effects against neurovascular diseases. Recent studies have shown that BDNF exerts anti-inflammation properties in ischemic stroke models. Anti-inflammatory effects of BDNF were characterized by decreased expression of Bcl-2 through TrkB/ERK1/2 pathway [124]. BDNF (Val66Met) polymorphism decreases angiogenesis and increases deficits in stroke rats [125]. Coincident with the anti-inflammatory effects, BDNF also carries anti-apoptotic properties [124,126]. In vivo, intravenous injection of BDNF has been shown to protect neurons against apoptosis through downregulating Bax, a pro-apoptotic protein, and upregulating Bcl-2, an anti-apoptotic protein [126].
The recognition of neuroprotective effects of BDNF has prompted studies investigating the relationship between BDNF and TBI. BDNF has been shown to upregulate after various potential treatment regimens and to improve functional outcomes [127-129]. Transplantation of human mesenchymal stem cells (hMSCs) increased expression of BDNF which resulted in functional recovery in a rat model of TBI [127]. Similar to the observed enhanced therapeutic effects when using combination therapy with other growth factors, treatment of simvastatin upregulates BDNF and VEGF and increases neurogenesis which are associated with better outcomes in TBI [130].
As we mentioned above, recent studies have demonstrated that one of the underlying pathology in both stroke and TBI is the downregulation of growth factors, specifically BDNF. To date, BDNF has been studied extensively in the context of many neuropsychiatric disorders in both basic and clinical research [113-115]. Because of mixed results in the clinical phases of BDNF, there is no current clinical trial for cerebrovascular diseases. In order to further assess the safety and efficacy of this growth factor as a therapeutic, the preclinical research on BDNF treatment for stroke and TBI may benefit from increased sample size and testing in multiple coordinated laboratories.
8. Challenges with Growth Factor Therapy
Although we have discussed research studies demonstrating the beneficial effects of growth factor therapy attributed to the dampening of inflammation through ligand-receptor mechanisms, it is essential to consider the adverse side effects of this type of therapy. For example, BDNF overexpression in the hippocampus may influence neurogenesis but it is also associated with subsequent development of epilepsy [131,132]. Like any treatment, the effective regimen of growth factor therapy will require cautious consideration in order to avoid the deleterious enhancement of brain damage after any insult. For this reason, future studies should focus on optimizing the treatment regimen of growth factor therapy in cerebrovascular diseases, as the relationship between its therapeutic properties and pathological effects remains poorly understood. The timing, dose, and route of delivery of growth factors will likely play important gating items in optimizing this regimen.
Of particular interest, the restrictive permeability of the BBB poses a challenge to new therapies that need to reach the brain. For this reason, many studies have focused on addressing this issue to facilitate the delivery of growth factors to the brain. Approaches such as encapsulated cell therapy, viral vectors, and genetically engineered molecular Trojan horses have been widely used to investigate the efficacy of growth factor therapy in experimental models [133-135]. Engineered proteins, such as the TAT-linked proteins, have been altered with polyethylene glycol (PEG) and functional peptides to increase the delivery efficiency [136]. The HIV TAT peptide, for example, has been linked to therapeutic proteins to allow efficient translocation across the BBB for ischemic stroke therapy [137]. Intravenously infused PEGylated-hemoglobin (PEG-hemoglobin), for example, has been evaluated in stroke and TBI animal models in which several advantages have been observed including increased stability and circulation time of drugs and increased diffusion to target sites [136,138]. Trojan horses, such as the human insulin receptor monoclonal antibody (HIRMab), which bind to receptors may facilitate transport of therapeutic proteins across the BBB [136]. Nanoparticles (NPs) assembled with poly-DL-lactide-coglycolide (PLGA), microparticles, and hydrogels have also been studied as drug delivery carriers [136,139,140]. Lastly, viral vectors, such as AAV, adenoviral vector, HSV, lentiviral vector, and non-viral vectors, such as liposomes and polymers, have also been extensively used as BBB transporters [133,141]. Non-viral vectors have some advantages over other vectors in that they have low immunogenicity and toxicity, and can be chemically modified easily to target brain cells [136]. However, since therapeutic genes are anti-apoptotic or growth factors, their long-term expressions may induce deleterious effects such as tumor growth [136]. Similarly, although hydrogels seem advantageous, local injection may cause brain damage [136]. Of note, a leaky BBB allows for an easier drug delivery to the brain in some cases. In the event of a stroke, for example, the BBB is severely compromised and incapable of re-establishing regular function for as long as 7 days post-trauma [142]. This time frame where the BBB remains open can be used to transport NPs to the brain, and should be taken into account when designing future therapies for CNS disorders [142]. By circumventing these potential adverse effects, such combination therapy of growth factor delivery and bioengineering hold promise for targeting neuroinflammation in cerebrovascular diseases.
There is also increasing evidence demonstrating the therapeutic effects of secreted RNAs or exosomes of growth factor after injury such as stroke or TBI. The concept of exosomes and their ability to abrogate inflammation is relatively new. Secreted RNAs not only modulate peripheral inflammation but can also cross the BBB, which is an important aspect in designing treatments for stroke and TBI. This novel line of investigation originated from the discovery of secretion of long noncoding RNA or microRNA in tumor cells in CNS [111]. The exosomes containing several microRNAs such as metastasis-associated lung adenocarcinoma transcript1 (MALAT1) and nuclear enriched abundant transcript1 (NEAT1) have also been found to be secreted by transplanted stem cells [111]. A recent in vivo study has demonstrated that the expressions of MALAT1 and NEAT1 are increased after intravenous transplantation of human adipose stem cells which result in beneficial outcomes after TBI [111]. Interestingly, these exosomes primarily abrogate the inflammation in the peripheral organs, notably in the spleen. This suggests that some of these growth factors and theirs exosomes may not need to enter the brain; but they can sequester the inflammation peripherally and still provide the neuroprotection effects against cerebrovascular diseases. This innovative concept opens new venues in targeting neuroinflammation in the traditionally considered “brain-based” cerebrovascular diseases. In the end, treatment with growth factors may need to consider both central and peripheral inflammation in combating this rampant secondary cell death associated with stroke, TBI, and other related brain disorders.
9. Conclusion
Over the last two decades, many growth factors have been discovered providing an exciting scientific basis for their use in treating many debilitating disorders including cerebrovascular diseases. This review paper focuses on the five major growth factors: VEGF, GDNF, BDNF, SDF-1a, and SCF and their ability to provide neuroprotection through various mechanisms especially in abrogating inflammation as a major strategy to retard and even halt secondary cell death. Various biomedical engineering tools may help to effectively deliver the growth factors to infiltrate the BBB to enter the brain or to circumvent the BBB by modulating the peripheral organs with the end goal of reducing the inflammation and protecting the injured brain after cerebrovascular insults. The involvement of the spleen in exacerbation of neuroinflammation also offers a new treatment approach for growth factors to peripherally sequester this secondary cell death. Optimization of growth factor therapy, its associated exosomes, and bioengineered devices will likely improve its beneficial outcome in patients with cerebrovascular diseases.
10. Expert Commentary
A unique pathologic feature of cerebrovascular diseases is neuroinflammation associated with altered CNS levels of growth factors, as seen in stroke and TBI. The current treatment for stroke and TBI are very limited, creating an enormous burden to the economy. Hence it is morally and economically important to develop a better treatment regimen for the diseases.
Over the past decades, our understanding of the neuroinflammatory response closely accompanying the progression of cerebrovascular diseases has prompted investigations into the potential benefit of growth factor therapy. Multiple studies have demonstrated the therapeutic benefits of growth factors in experimental models of stroke and TBI. Due to the complexity of the pathology, it is unlikely that one growth factor can be used as the “magic bullet” to treat cerebrovascular diseases. Combination of growth factors and anti-inflammatory modulators is more likely to provide improved outcomes in cerebrovascular diseases. On this note, transplantation of stem cells may serve as multi-pronged therapeutic agent satisfying both growth factor and anti-inflammatory agent properties, in addition to the cells’ ability to differentiate into different neural types. In other to advance the findings of growth factors from laboratories to the clinical trials, larger preclinical studies and multiple laboratory collaborations are needed to demonstrate safety and efficacy of stand-alone treatments or combination therapies in clinically relevant animal models of stroke and TBI.
11. Five-year View
As the population grows older, the incidence of cerebrovascular diseases is expected to rise for the next 5 to 10 years. Cerebrovascular diseases are significant unmet need in the clinic, thereby posing as a major health and economic burden to the society. While education and prevention are important, it is unlikely that it will be sufficient to decrease the incidence, mortality, and morbidity of these diseases.
Rigorous laboratory studies designed to model cerebrovascular diseases will be able to improve our understanding of the pathology and treatment of these diseases. In the next 5 years, large and multiple laboratory testing of growth factor therapy, either as a stand-alone or combination therapy (with stem cells or exosomes) in clinically relevant animal models should provide guidance on the efficacy and safety of this therapy for cerebrovascular diseases. New bioengineering technologies may improve bioavailability of growth factors not only in the CNS, but also in peripheral sources of inflammation such as the spleen. Coalescing research efforts towards the effective and safe sequestration of inflammation, both centrally and peripherally, will advance the translational application of growth factors from the laboratory to the clinic for stroke and TBI patients.
Key Issues.
Stroke and traumatic brain injury (TBI) are major cause of deaths, disability, and economic burden in the United States.
Inflammation plays an important pathological role in the progression of cerebrovascular diseases.
Accumulating laboratory evidence demonstrates the therapeutic effects of GDNF, VEGF, SDF-1, SCF, and BDNF in sequestering inflammation in animal models of stroke and TBI. Treatments with stem cell and exosomes may resemble the therapeutic benefits of growth factors in stroke and TBI, indicating their use as adjunctive therapies.
Optimization of dosage, timing, and delivery route remains a key lab-to-clinic issue in stand-alone or combination treatment of growth factors for cerebrovascular disorders.
Although traditionally considered as “brain diseases”, abrogating the inflammation at the peripheral level of the spleen have been shown to afford neuroprotection.
Bioengineering technologies directed at improving growth factor delivery to the brain and spleen may enhance sequestration of inflammation and therapeutic outcomes.
Acknowledgments
CV Borlongan is supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke 1R01NS071956, 1R01NS090962, and 1R21NS089851, Department of Defense W81XWH-11-1-0634, James and Esther King Foundation for Biomedical Research Program, SanBio Inc, KM Pharmaceuticals, NeuralStem Inc, and Karyopharm Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the sponsors.
Footnotes
Declaration of Interests
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Reference annotations
-
*
Of interest
-
**
Of considerable interest
- 1.Cerebrovascular disease. American Association of Neurological Surgeons; Rolling Meadows IL: 2005. [3 February 2016]. Available at: http://www.aans.org/patient%20information/conditions%20and%20treatments/cerebrovascular%20disease.aspx. [Google Scholar]
- 2.Acosta SA, Tajiri N, Shinozuka K, et al. Combination therapy of human umbilical cord blood cells and granulocyte colony stimulating factor reduces histopathological and motor impairments in an experimental model of chronic traumatic brain injury. PLoS One. 2014;9:e90953. doi: 10.1371/journal.pone.0090953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jickling GC, Liu D, Stamova B, et al. Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab. 2014;34:185–99. doi: 10.1038/jcbfm.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics--2015 update: A report from the american heart association. Circulation. 2015;131:e29–322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 5.Goto S. Cilostazol: Potential mechanism of action for antithrombotic effects accompanied by a low rate of bleeding. Atheroscler Suppl. 2005;6:3–11. doi: 10.1016/j.atherosclerosissup.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Uchida H, Morita T, Niizuma K, et al. Transplantation of unique subpopulation of fibroblasts, muse cells, ameliorates experimental stroke possibly via robust neuronal differentiation. Stem Cells. 2016;34:160–73. doi: 10.1002/stem.2206. [DOI] [PubMed] [Google Scholar]
- 7.Acosta SA, Tajiri N, Hoover J, et al. Intravenous bone marrow stem cell grafts preferentially migrate to spleen and abrogate chronic inflammation in stroke. Stroke. 2015;46:2616–27. doi: 10.1161/STROKEAHA.115.009854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blennow K, Hardy J, Zetterberg H. The neuropathology and neurobiology of traumatic brain injury. Neuron. 2012;76:886–99. doi: 10.1016/j.neuron.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 9.Traumatic Brain Injury In the United States: Epidemiology and Rehabilitation. Atlanta, GA: Centers for Disease Control and Prevention; 2015. Available at: http://www.cdc.gov/traumaticbraininjury/pdf/TBI_Report_to_Congress_Epi_and_Rehab-a.pdf. [Google Scholar]
- 10.Shlosberg D, Benifla M, Kaufer D, et al. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6:393–403. doi: 10.1038/nrneurol.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–42. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 13.Acosta SA, Tajiri N, Shinozuka K, et al. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS One. 2013;8:e53376. doi: 10.1371/journal.pone.0053376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iadecola C, Anrather J. The immunology of stroke: From mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–51. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nahrendorf M, Swirski FK. Immunology. Neutrophil-macrophage communication in inflammation and atherosclerosis. Science. 2015;349:237–8. doi: 10.1126/science.aac7801. [DOI] [PubMed] [Google Scholar]
- 17.Sun X, He S, Wara AK, et al. Systemic delivery of microRNA-181b inhibits nuclear factor-kappab activation, vascular inflammation, and atherosclerosis in apolipoprotein e-deficient mice. Circ Res. 2014;114:32–40. doi: 10.1161/CIRCRESAHA.113.302089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–6. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teasdale GM, Nicoll JA, Murray G, et al. Association of apolipoprotein e polymorphism with outcome after head injury. Lancet. 1997;350:1069–71. doi: 10.1016/S0140-6736(97)04318-3. [DOI] [PubMed] [Google Scholar]
- 20.Rom S, Dykstra H, Zuluaga-Ramirez V, et al. Mir-98 and let-7g* protect the blood-brain barrier under neuroinflammatory conditions. J Cereb Blood Flow Metab. 2015;35:1957–65. doi: 10.1038/jcbfm.2015.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedman G, Froom P, Sazbon L, et al. Apolipoprotein E-epsilon4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology. 1999;52:244–8. doi: 10.1212/wnl.52.2.244. [DOI] [PubMed] [Google Scholar]
- 22.Lai CH, Wang KC, Lee FT, et al. Toll-like receptor 4 is essential in the development of abdominal aortic aneurysm. PLoS One. 2016;11:e0146565. doi: 10.1371/journal.pone.0146565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gyoneva S, Ransohoff RM. Inflammatory reaction after traumatic brain injury: Therapeutic potential of targeting cell-cell communication by chemokines. Trends Pharmacol Sci. 2015;36:471–80. doi: 10.1016/j.tips.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dela Pena I, Sanberg PR, Acosta S, et al. Stem cells and G-CSF for treating neuroinflammation in traumatic brain injury: Aging as a comorbidity factor. J Neurosurg Sci. 2014;58:145–9. [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao LR, Singhal S, Duan WM, et al. Brain repair by hematopoietic growth factors in a rat model of stroke. Stroke. 2007;38:2584–91. doi: 10.1161/STROKEAHA.106.476457. [DOI] [PubMed] [Google Scholar]
- 26.Yang Y, Rosenberg GA. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011;42:3323–8. doi: 10.1161/STROKEAHA.110.608257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doll DN, Hu H, Sun J, et al. Mitochondrial crisis in cerebrovascular endothelial cells opens the blood-brain barrier. Stroke. 2015;46:1681–9. doi: 10.1161/STROKEAHA.115.009099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimotake J, Derugin N, Wendland M, et al. Vascular endothelial growth factor receptor-2 inhibition promotes cell death and limits endothelial cell proliferation in a neonatal rodent model of stroke. Stroke. 2010;41:343–9. doi: 10.1161/STROKEAHA.109.564229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thau-Zuchman O, Shohami E, Alexandrovich AG, et al. Vascular endothelial growth factor increases neurogenesis after traumatic brain injury. J Cereb Blood Flow Metab. 2010;30:1008–16. doi: 10.1038/jcbfm.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hof PR, Mobbs CV. Handbook of the Neuroscience of Aging. Burlington: AP; 2010. [Google Scholar]
- 31.Konishi Y, Yang LB, He P, et al. Deficiency of GDNF receptor GFRalpha1 in alzheimer’s neurons results in neuronal death. J Neurosci. 2014;34:13127–38. doi: 10.1523/JNEUROSCI.2582-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Airaksinen MS, Saarma M. The gdnf family: signalling, biological functions and therapeutic value. Nat Rev Neurosci. 2002;3:383–94. doi: 10.1038/nrn812. [DOI] [PubMed] [Google Scholar]
- 33.Jensen P, Ducray AD, Widmer HR, et al. Effects of forskolin on trefoil factor 1 expression in cultured ventral mesencephalic dopaminergic neurons. Neuroscience. 2015;310:699–708. doi: 10.1016/j.neuroscience.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y, Zhang Q, Xi J, et al. Neuroprotective effect of fasudil on inflammation through PI3K/AKT and Wnt/beta-catenin dependent pathways in a mice model of parkinson’s disease. Int J Clin Exp Pathol. 2015;8:2354–64. [PMC free article] [PubMed] [Google Scholar]
- 35.Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 36.Hoban DB, Howard L, Dowd E. GDNF-secreting mesenchymal stem cells provide localized neuroprotection in an inflammation-driven rat model of parkinson’s disease. Neuroscience. 2015;303:402–11. doi: 10.1016/j.neuroscience.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 37.Sumbria RK, Boado RJ, Pardridge WM. Combination stroke therapy in the mouse with blood-brain barrier penetrating IgG-GDNF and IgG-TNF decoy receptor fusion proteins. Brain Res. 2013;1507:91–6. doi: 10.1016/j.brainres.2013.02.022.. * Discusses the therapeutic benefit of combination therapy with both a neuroprotective neurotrophin and an anti-inflammatory agent in stroke mouse model.
- 38.Degeorge ML, Marlowe D, Werner E, et al. Combining glial cell line-derived neurotrophic factor gene delivery (AdGDNF) with l-arginine decreases contusion size but not behavioral deficits after traumatic brain injury. Brain Res. 2011;1403:45–56. doi: 10.1016/j.brainres.2011.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minnich JE, Mann SL, Stock M, et al. Glial cell line-derived neurotrophic factor (GDNF) gene delivery protects cortical neurons from dying following a traumatic brain injury. Restor Neurol Neurosci. 2010;28:293–309. doi: 10.3233/RNN-2010-0528. [DOI] [PubMed] [Google Scholar]
- 40.US National Institutes of Health. Bethesda, MD: U.S. National Institutes of Health; 2015. [16 March 2016]. AAV2-GDNF for Advanced Parkinson’s Disease. Available at: https://clinicaltrials.gov/ct2/show/NCT01621581?term=gdnf&rank=1. [Google Scholar]
- 41.US National Institutes of Health. Bethesda, MD: U.S. National Institutes of Health; 2005. [16 March 2016]. Continuously Infused Intracerebral (IC) Recombinant-Methionyl Human Glial Cell Line-Derived Neurotrophic Factor (r-metHuGDNF) for the Treatment of Idiopathic Parkinson’s Disease. Available at: https://clinicaltrials.gov/ct2/show/NCT00006488?term=gdnf&rank=5. [Google Scholar]
- 42.Wang S, Cheng H, Dai G, et al. Umbilical cord mesenchymal stem cell transplantation significantly improves neurological function in patients with sequelae of traumatic brain injury. Brain Res. 2013;1532:76–84. doi: 10.1016/j.brainres.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 43.Reyes S, Tajiri N, Borlongan CV. Developments in intracerebral stem cell grafts. Expert Rev Neurother. 2015;15:381–93. doi: 10.1586/14737175.2015.1021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neufeld G, Cohen T, Gengrinovitch S, et al. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 45.Cydzik M, Abdul-Wahid A, Park S, et al. Slow binding kinetics of secreted protein, acidic, rich in cysteine-VEGF interaction limit VEGF activation of VEGF receptor 2 and attenuate angiogenesis. FASEB J. 2015;29:3493–505. doi: 10.1096/fj.15-271775. [DOI] [PubMed] [Google Scholar]
- 46.Larpthaveesarp A, Ferriero DM, Gonzalez FF. Growth factors for the treatment of ischemic brain injury (growth factor treatment) Brain Sci. 2015;5:165–77. doi: 10.3390/brainsci5020165.. * This review notes the critical importance of the timing of VEGF treatment in treating cerebrovascular disease.
- 47.Dzietko M, Derugin N, Wendland MF, et al. Delayed vegf treatment enhances angiogenesis and recovery after neonatal focal rodent stroke. Transl Stroke Res. 2013;4:189–200. doi: 10.1007/s12975-012-0221-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siddiq I, Park E, Liu E, et al. Treatment of traumatic brain injury using zinc-finger protein gene therapy targeting VEGF-A. J Neurotrauma. 2012;29:2647–59. doi: 10.1089/neu.2012.2444. [DOI] [PubMed] [Google Scholar]
- 49.Argaw AT, Asp L, Zhang J, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in cns inflammatory disease. J Clin Invest. 2012;122:2454–68. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adibhatla RM, Hatcher JF. Tissue plasminogen activator (tPA) and matrix metalloproteinases in the pathogenesis of stroke: Therapeutic strategies. CNS Neurol Disord Drug Targets. 2008;7:243–53. doi: 10.2174/187152708784936608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Manoonkitiwongsa PS, Schultz RL, Whitter EF, et al. Contraindications of VEGF -based therapeutic angiogenesis: Effects on macrophage density and histology of normal and ischemic brains. Vascul Pharmacol. 2006;44:316–25. doi: 10.1016/j.vph.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 52.Vila-Coro AJ, Rodriguez-Frade JM, Martin De Ana A, et al. The chemokine SDF -1alpha triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. 1999;13:1699–710. [PubMed] [Google Scholar]
- 53.Ma Q, Jones D, Borghesani PR, et al. Impaired b-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci U S A. 1998;95:9448–53. doi: 10.1073/pnas.95.16.9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zou YR, Kottmann AH, Kuroda M, et al. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595–9. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 55.Nagasawa T, Hirota S, Tachibana K, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the cxc chemokine PBSF/SDF-1. Nature. 1996;382:635–8. doi: 10.1038/382635a0. [DOI] [PubMed] [Google Scholar]
- 56.Lazarini F, Tham TN, Casanova P, et al. Role of the alpha-chemokine stromal cell-derived factor (SDF-1) in the developing and mature central nervous system. Glia. 2003;42:139–48. doi: 10.1002/glia.10139. [DOI] [PubMed] [Google Scholar]
- 57.Ratajczak MZ, Zuba-Surma E, Kucia M, et al. The pleiotropic effects of the SDF-1- CXCR4 axis in organogenesis, regeneration and tumorigenesis. Leukemia. 2006;20:1915–24. doi: 10.1038/sj.leu.2404357. [DOI] [PubMed] [Google Scholar]
- 58.De La Luz Sierra M, Yang F, Narazaki M, et al. Differential processing of stromal-derived factor-1alpha and stromal-derived factor-1beta explains functional diversity. Blood. 2004;103:2452–9. doi: 10.1182/blood-2003-08-2857. [DOI] [PubMed] [Google Scholar]
- 59.Horuk R. Chemokine receptors. Cytokine Growth Factor Rev. 2001;12:313–35. doi: 10.1016/s1359-6101(01)00014-4. [DOI] [PubMed] [Google Scholar]
- 60.Hill WD, Hess DC, Martin-Studdard A, et al. SDF-1 (CXCL12) is upregulated in the ischemic penumbra following stroke: Association with bone marrow cell homing to injury. J Neuropathol Exp Neurol. 2004;63:84–96. doi: 10.1093/jnen/63.1.84. [DOI] [PubMed] [Google Scholar]
- 61.Strieter RM, Burdick MD, Mestas J, et al. Cancer CXC chemokine networks and tumour angiogenesis. Eur J Cancer. 2006;42:768–78. doi: 10.1016/j.ejca.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 62.Bagri A, Gurney T, He X, et al. The chemokine SDF1 regulates migration of dentate granule cells. Development. 2002;129:4249–60. doi: 10.1242/dev.129.18.4249. [DOI] [PubMed] [Google Scholar]
- 63.Schier AF. Chemokine signaling: Rules of attraction. Curr Biol. 2003;13:R192–4. doi: 10.1016/s0960-9822(03)00122-2. [DOI] [PubMed] [Google Scholar]
- 64.Man S, Tucky B, Cotleur A, et al. CXCL12-induced monocyte-endothelial interactions promote lymphocyte transmigration across an in vitro blood-brain barrier. Sci Transl Med. 2012;4:119ra14. doi: 10.1126/scitranslmed.3003197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cartier L, Hartley O, Dubois-Dauphin M, et al. Chemokine receptors in the central nervous system: Role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev. 2005;48:16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 66.Chernock RD, Cherla RP, Ganju RK. Shp2 and cbl participate in alpha-chemokine receptor CXCR4-mediated signaling pathways. Blood. 2001;97:608–15. doi: 10.1182/blood.v97.3.608. [DOI] [PubMed] [Google Scholar]
- 67.Mandler M, Valera E, Rockenstein E, et al. Next-generation active immunization approach for synucleinopathies: Implications for parkinson’s disease clinical trials. Acta Neuropathol. 2014;127:861–79. doi: 10.1007/s00401-014-1256-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng X, Lian YJ, Ma YQ, et al. Elevated serum levels of CXC chemokine ligand-12 are associated with unfavorable functional outcome and mortality at 6-month follow-up in chinese patients with acute ischemic stroke. Mol Neurobiol. 2016 doi: 10.1007/s12035-015-9645-9. [DOI] [PubMed] [Google Scholar]
- 69.Schajnovitz A, Itkin T, D’Uva G, et al. CXCL12 secretion by bone marrow stromal cells is dependent on cell contact and mediated by connexin-43 and connexin-45 gap junctions. Nat Immunol. 2011;12:391–8. doi: 10.1038/ni.2017. [DOI] [PubMed] [Google Scholar]
- 70.Timotijevic G, Mostarica Stojkovic M, Miljkovic D. CXCL12: Role in neuroinflammation. Int J Biochem Cell Biol. 2012;44:838–41. doi: 10.1016/j.biocel.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 71.Gear AR, Suttitanamongkol S, Viisoreanu D, et al. Adenosine diphosphate strongly potentiates the ability of the chemokines MDC, TARC, and SDF-1 to stimulate platelet function. Blood. 2001;97:937–45. doi: 10.1182/blood.v97.4.937. [DOI] [PubMed] [Google Scholar]
- 72.Liu X, Zhou C, Li Y, et al. SDF-1 promotes endochondral bone repair during fracture healing at the traumatic brain injury condition. PLoS One. 2013;8:e54077. doi: 10.1371/journal.pone.0054077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bogoslovsky T, Spatz M, Chaudhry A, et al. Stromal-derived factor-1[alpha] correlates with circulating endothelial progenitor cells and with acute lesion volume in stroke patients. Stroke. 2011;42:618–25. doi: 10.1161/STROKEAHA.110.596007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gu XL, Liu L, Lu XD, et al. Serum CXCL12 levels as a novel predictor of future stroke recurrence in patients with acute ischemic stroke. Mol Neurobiol. 2015 doi: 10.1007/s12035-015-9151-0. [DOI] [PubMed] [Google Scholar]
- 75.Ruscher K, Kuric E, Liu Y, et al. Inhibition of CXCL12 signaling attenuates the postischemic immune response and improves functional recovery after stroke. J Cereb Blood Flow Metab. 2013;33:1225–34. doi: 10.1038/jcbfm.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Takami S, Minami M, Nagata I, et al. Chemokine receptor antagonist peptide, viral MIP-II, protects the brain against focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2001;21:1430–5. doi: 10.1097/00004647-200112000-00007. [DOI] [PubMed] [Google Scholar]
- 77.Peled A, Kollet O, Ponomaryov T, et al. The chemokine sdf-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: Role in transendothelial/stromal migration and engraftment of nod/scid mice. Blood. 2000;95:3289–96. [PubMed] [Google Scholar]
- 78.Ceradini DJ, Kulkarni AR, Callaghan MJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through hif-1 induction of SDF-1. Nat Med. 2004;10:858–64. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 79.Fan Y, Shen F, Frenzel T, et al. Endothelial progenitor cell transplantation improves long-term stroke outcome in mice. Ann Neurol. 2010;67:488–97. doi: 10.1002/ana.21919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jaerve A, Schira J, Muller HW. Concise review: The potential of stromal cell-derived factor 1 and its receptors to promote stem cell functions in spinal cord repair. Stem Cells Transl Med. 2012;1:732–9. doi: 10.5966/sctm.2012-0068.. * This review discusses the crucial effects of SDF-1 in attracting stem cells in order to remyelinate neurons, aid in neurovascularization, and modulate the immune response.
- 81.Cui X, Chen J, Zacharek A, et al. Nitric oxide donor upregulation of stromal cell-derived factor-1/chemokine (CXC motif) receptor 4 enhances bone marrow stromal cell migration into ischemic brain after stroke. Stem Cells. 2007;25:2777–85. doi: 10.1634/stemcells.2007-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Imitola J, Raddassi K, Park KI, et al. Directed migration of neural stem cells to sites of cns injury by the stromal cell-derived factor 1alpha/CXC chemokine receptor 4 pathway. Proc Natl Acad Sci U S A. 2004;101:18117–22. doi: 10.1073/pnas.0408258102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Robin AM, Zhang ZG, Wang L, et al. Stromal cell-derived factor 1alpha mediates neural progenitor cell motility after focal cerebral ischemia. J Cereb Blood Flow Metab. 2006;26:125–34. doi: 10.1038/sj.jcbfm.9600172. [DOI] [PubMed] [Google Scholar]
- 84.Borlongan CV. Bone marrow stem cell mobilization in stroke: A ’bonehead’ may be good after all! Leukemia. 2011;25:1674–86. doi: 10.1038/leu.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kent D, Copley M, Benz C, et al. Regulation of hematopoietic stem cells by the steel factor/kit signaling pathway. Clin Cancer Res. 2008;14:1926–30. doi: 10.1158/1078-0432.CCR-07-5134. [DOI] [PubMed] [Google Scholar]
- 86.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–34. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med. 2014;20:833–46. doi: 10.1038/nm.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Meng F, Francis H, Glaser S, et al. Role of stem cell factor and granulocyte colony-stimulating factor in remodeling during liver regeneration. Hepatology. 2012;55:209–21. doi: 10.1002/hep.24673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Motro B, van der Kooy D, Rossant J, et al. Contiguous patterns of c-kit and steel expression: Analysis of mutations at the w and sl loci. Development. 1991;113:1207–21. doi: 10.1242/dev.113.4.1207. [DOI] [PubMed] [Google Scholar]
- 90.Anderson DM, Lyman SD, Baird A, et al. Molecular cloning of mast cell growth factor, a hematopoietin that is active in both membrane bound and soluble forms. Cell. 1990;63:235–43. doi: 10.1016/0092-8674(90)90304-w. [DOI] [PubMed] [Google Scholar]
- 91.Lennartsson J, Ronnstrand L. Stem cell factor receptor/c-kit: From basic science to clinical implications. Physiol Rev. 2012;92:1619–49. doi: 10.1152/physrev.00046.2011. [DOI] [PubMed] [Google Scholar]
- 92.Ronnstrand L. Signal transduction via the stem cell factor receptor/c-kit. Cell Mol Life Sci. 2004;61:2535–48. doi: 10.1007/s00018-004-4189-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang Z, Ruan HB, Xian L, et al. The stem cell factor/kit signalling pathway regulates mitochondrial function and energy expenditure. Nat Commun. 2014;5:4282. doi: 10.1038/ncomms5282. [DOI] [PubMed] [Google Scholar]
- 94.Kitamura Y, Hirotab S. Kit as a human oncogenic tyrosine kinase. Cell Mol Life Sci. 2004;61:2924–31. doi: 10.1007/s00018-004-4273-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zeng S, Xu Z, Lipkowitz S, et al. Regulation of stem cell factor receptor signaling by cbl family proteins (Cbl-b/c-Cbl) Blood. 2005;105:226–32. doi: 10.1182/blood-2004-05-1768. [DOI] [PubMed] [Google Scholar]
- 96.Hirata T, Morii E, Morimoto M, et al. Stem cell factor induces outgrowth of c-kit-positive neurites and supports the survival of c-kit-positive neurons in dorsal root ganglia of mouse embryos. Development. 1993;119:649–56. doi: 10.1242/dev.119.1.49. [DOI] [PubMed] [Google Scholar]
- 97.Park M, Kim WK, Song M, et al. Protein kinase c-delta-mediated recycling of active kit in colon cancer. Clin Cancer Res. 2013;19:4961–71. doi: 10.1158/1078-0432.CCR-13-0131. [DOI] [PubMed] [Google Scholar]
- 98.Corti S, Locatelli F, Strazzer S, et al. Modulated generation of neuronal cells from bone marrow by expansion and mobilization of circulating stem cells with in vivo cytokine treatment. Exp Neurol. 2002;177:443–52. doi: 10.1006/exnr.2002.8004. [DOI] [PubMed] [Google Scholar]
- 99.Jin K, Mao XO, Sun Y, et al. Stem cell factor stimulates neurogenesis in vitro and in vivo. J Clin Invest. 2002;110:311–9. doi: 10.1172/JCI15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lanfranconi S, Locatelli F, Corti S, et al. Growth factors in ischemic stroke. J Cell Mol Med. 2011;15:1645–87. doi: 10.1111/j.1582-4934.2009.00987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nervi B, Link DC, DiPersio JF. Cytokines and hematopoietic stem cell mobilization. J Cell Biochem. 2006;99:690–705. doi: 10.1002/jcb.21043. [DOI] [PubMed] [Google Scholar]
- 102.Huang B, Lei Z, Zhang GM, et al. SCF-mediated mast cell infiltration and activation exacerbate the inflammation and immunosuppression in tumor microenvironment. Blood. 2008;112:1269–79. doi: 10.1182/blood-2008-03-147033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sun L, Lee J, Fine HA. Neuronally expressed stem cell factor induces neural stem cell migration to areas of brain injury. J Clin Invest. 2004;113:1364–74. doi: 10.1172/JCI20001.. * The study shows recombinant SCF was observed to induce migration of neural stem cells to damaged CNS sites which also showed higher levels of endogenous SCF mRNA and protein.
- 104.Kawada H, Takizawa S, Takanashi T, et al. Administration of hematopoietic cytokines in the subacute phase after cerebral infarction is effective for functional recovery facilitating proliferation of intrinsic neural stem/progenitor cells and transition of bone marrow-derived neuronal cells. Circulation. 2006;113:701–10. doi: 10.1161/CIRCULATIONAHA.105.563668. [DOI] [PubMed] [Google Scholar]
- 105.Toth ZE, Leker RR, Shahar T, et al. The combination of granulocyte colony-stimulating factor and stem cell factor significantly increases the number of bone marrow-derived endothelial cells in brains of mice following cerebral ischemia. Blood. 2008;111:5544–52. doi: 10.1182/blood-2007-10-119073.. * The study demonstrates the induction of angiogenesis and reduction of infarct volume from the combined administration of SCF and G-CSF.
- 106.Liu XY, Gonzalez-Toledo ME, Fagan A, et al. Stem cell factor and granulocyte colony-stimulating factor exhibit therapeutic effects in a mouse model of cadasil. Neurobiol Dis. 2015;73:189–203. doi: 10.1016/j.nbd.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 107.Cui L, Duchamp NS, Boston DJ, et al. NF-kappaB is involved in brain repair by stem cell factor and granulocyte-colony stimulating factor in chronic stroke. Exp Neurol. 2015;263:17–27. doi: 10.1016/j.expneurol.2014.08.026. [DOI] [PubMed] [Google Scholar]
- 108.Zhang SC, Fedoroff S. Expression of stem cell factor and c-kit receptor in neural cells after brain injury. Acta Neuropathol. 1999;97:393–8. doi: 10.1007/s004010051003. [DOI] [PubMed] [Google Scholar]
- 109.Hernandez-Ontiveros DG, Tajiri N, Acosta S, et al. Microglia activation as a biomarker for traumatic brain injury. Front Neurol. 2013;4:30. doi: 10.3389/fneur.2013.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang SC, Fedoroff S. Modulation of microglia by stem cell factor. J Neurosci Res. 1998;53:29–37. doi: 10.1002/(SICI)1097-4547(19980701)53:1<29::AID-JNR4>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 111.Tajiri N, Acosta SA, Shahaduzzaman M, et al. Intravenous transplants of human adipose-derived stem cell protect the brain from traumatic brain injury-induced neurodegeneration and motor and cognitive impairments: Cell graft biodistribution and soluble factors in young and aged rats. J Neurosci. 2014;34:313–26. doi: 10.1523/JNEUROSCI.2425-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lozano D, Gonzales-Portillo GS, Acosta S, et al. Neuroinflammatory responses to traumatic brain injury: Etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat. 2015;11:97–106. doi: 10.2147/NDT.S65815.. • The study provides evidence of neuroprotective effects of stem cells through secretion of growth factors.
- 113.Ghitza UE, Zhai H, Wu P, et al. Role of BDNF and GDNF in drug reward and relapse: A review. Neurosci Biobehav Rev. 2010;35:157–71. doi: 10.1016/j.neubiorev.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wu JQ, Chen da C, Tan YL, et al. Altered BDNF is correlated to cognition impairment in schizophrenia patients with tardive dyskinesia. Psychopharmacology (Berl) 2015;232:223–32. doi: 10.1007/s00213-014-3660-9. [DOI] [PubMed] [Google Scholar]
- 115.Angelucci F, Brene S, Mathe AA. BDNF in schizophrenia, depression and corresponding animal models. Mol Psychiatry. 2005;10:345–52. doi: 10.1038/sj.mp.4001637. [DOI] [PubMed] [Google Scholar]
- 116.Lin D, De La Pena I, Lin L, et al. The neuroprotective role of acupuncture and activation of the bdnf signaling pathway. Int J Mol Sci. 2014;15:3234–52. doi: 10.3390/ijms15023234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Qin L, Jing D, Parauda S, et al. An adaptive role for BDNF Val66Met polymorphism in motor recovery in chronic stroke. J Neurosci. 2014;34:2493–502. doi: 10.1523/JNEUROSCI.4140-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chan A, Yan J, Csurhes P, et al. Circulating brain derived neurotrophic factor (BDNF) and frequency of bdnf positive T cells in peripheral blood in human ischemic stroke: Effect on outcome. J Neuroimmunol. 2015;286:42–7. doi: 10.1016/j.jneuroim.2015.06.013. [DOI] [PubMed] [Google Scholar]
- 119.Failla MD, Kumar RG, Peitzman AB, et al. Variation in the BDNF gene interacts with age to predict mortality in a prospective, longitudinal cohort with severe tbi. Neurorehabil Neural Repair. 2015;29:234–46. doi: 10.1177/1545968314542617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mowla SJ, Farhadi HF, Pareek S, et al. Biosynthesis and post-translational processing of the precursor to brain-derived neurotrophic factor. J Biol Chem. 2001;276:12660–6. doi: 10.1074/jbc.M008104200. [DOI] [PubMed] [Google Scholar]
- 121.Fayard B, Loeffler S, Weis J, et al. The secreted brain-derived neurotrophic factor precursor pro-BDNF binds to TrkB and p75NTR but not to TrkA or TrkC. J Neurosci Res. 2005;80:18–28. doi: 10.1002/jnr.20432. [DOI] [PubMed] [Google Scholar]
- 122.Notaras M, Hill R, van den Buuse M. A role for the BNF gene Val66Met polymorphism in schizophrenia? A comprehensive review. Neurosci Biobehav Rev. 2015;51:15–30. doi: 10.1016/j.neubiorev.2014.12.016. [DOI] [PubMed] [Google Scholar]
- 123.Teng HK, Teng KK, Lee R, et al. Pro-BDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci. 2005;25:5455–63. doi: 10.1523/JNEUROSCI.5123-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Atif F, Yousuf S, Sayeed I, et al. Combination treatment with progesterone and vitamin d hormone is more effective than monotherapy in ischemic stroke: The role of BDNF/TrkB/Erk1/2 signaling in neuroprotection. Neuropharmacology. 2013;67:78–87. doi: 10.1016/j.neuropharm.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Qin L, Kim E, Ratan R, et al. Genetic variant of BDNF (Val66Met) polymorphism attenuates stroke-induced angiogenic responses by enhancing anti-angiogenic mediator cd36 expression. J Neurosci. 2011;31:775–83. doi: 10.1523/JNEUROSCI.4547-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schabitz WR, Sommer C, Zoder W, et al. Intravenous brain-derived neurotrophic factor reduces infarct size and counterregulates Bax and Bcl-2 expression after temporary focal cerebral ischemia. Stroke. 2000;31:2212–7. doi: 10.1161/01.str.31.9.2212.. ** The study shows intravenous injection of BDNF reduces inflammation through modulation of Bax and Bcl-2.
- 127.Kim HJ, Lee JH, Kim SH. Therapeutic effects of human mesenchymal stem cells on traumatic brain injury in rats: Secretion of neurotrophic factors and inhibition of apoptosis. J Neurotrauma. 2010;27:131–8. doi: 10.1089/neu.2008.0818. [DOI] [PubMed] [Google Scholar]
- 128.Yenari MA, Han HS. Neuroprotective mechanisms of hypothermia in brain ischaemia. Nat Rev Neurosci. 2012;13:267–78. doi: 10.1038/nrn3174. [DOI] [PubMed] [Google Scholar]
- 129.Umschweif G, Shabashov D, Alexandrovich AG, et al. Neuroprotection after traumatic brain injury in heat-acclimated mice involves induced neurogenesis and activation of angiotensin receptor type 2 signaling. J Cereb Blood Flow Metab. 2014;34:1381–90. doi: 10.1038/jcbfm.2014.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wu H, Lu D, Jiang H, et al. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/AKT pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J Neurotrauma. 2008;25:130–9. doi: 10.1089/neu.2007.0369. [DOI] [PubMed] [Google Scholar]
- 131.Kim J, Yang M, Kim J, et al. Developmental and degenerative modulation of brain-derived neurotrophic factor transcript variants in the mouse hippocampus. Int J Dev Neurosci. 2014;38:68–73. doi: 10.1016/j.ijdevneu.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 132.Isgor C, Pare C, McDole B, et al. Expansion of the dentate mossy fiber-ca3 projection in the brain-derived neurotrophic factor-enriched mouse hippocampus. Neuroscience. 2015;288:10–23. doi: 10.1016/j.neuroscience.2014.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shen F, Kuo R, Milon-Camus M, et al. Intravenous delivery of adeno-associated viral vector serotype 9 mediates effective gene expression in ischemic stroke lesion and brain angiogenic foci. Stroke. 2013;44:252–4. doi: 10.1161/STROKEAHA.112.662965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sumbria RK, Boado RJ, Pardridge WM. Brain protection from stroke with intravenous TNFalpha decoy receptor-trojan horse fusion protein. J Cereb Blood Flow Metab. 2012;32:1933–8. doi: 10.1038/jcbfm.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Emerich DF, Orive G, Thanos C, et al. Encapsulated cell therapy for neurodegenerative diseases: From promise to product. Adv Drug Deliv Rev. 2014;67-68:131–41. doi: 10.1016/j.addr.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 136.Rhim T, Lee DY, Lee M. Drug delivery systems for the treatment of ischemic stroke. Pharm Res. 2013;30:2429–44. doi: 10.1007/s11095-012-0959-2. [DOI] [PubMed] [Google Scholar]
- 137.Zhu Y, Bu Q, Liu X, et al. Neuroprotective effect of TAT-14-3-3epsilon fusion protein against cerebral ischemia/reperfusion injury in rats. PLoS One. 2014;9:e93334. doi: 10.1371/journal.pone.0093334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Brockman EC, Bayir H, Blasiole B, et al. Polynitroxylated-pegylated hemoglobin attenuates fluid requirements and brain edema in combined traumatic brain injury plus hemorrhagic shock in mice. J Cereb Blood Flow Metab. 2013;33:1457–64. doi: 10.1038/jcbfm.2013.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yemisci M, Caban S, Gursoy-Ozdemir Y, et al. Systemically administered brain-targeted nanoparticles transport peptides across the blood-brain barrier and provide neuroprotection. J Cereb Blood Flow Metab. 2015;35:469–75. doi: 10.1038/jcbfm.2014.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gauberti M, Montagne A, Marcos-Contreras OA, et al. Ultra-sensitive molecular mri of vascular cell adhesion molecule-1 reveals a dynamic inflammatory penumbra after strokes. Stroke. 2013;44:1988–96. doi: 10.1161/STROKEAHA.111.000544.. ** This article highlights the evolution of the cerebrovascular inflammation that occurs after an ischemic and hemorrhagic stroke using an ultra-sensitive MRI and the effects of various anti-inflammatory agents.
- 141.Liu DZ, Jickling GC, Ander BP, et al. Elevating microRNA-122 in blood improves outcomes after temporary middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab. 2015 doi: 10.1177/0271678X15610786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Panagiotou S, Saha S. Therapeutic benefits of nanoparticles in stroke. Front Neurosci. 2015;9:182. doi: 10.3389/fnins.2015.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]