

Abstract
Background
Cerebral hypoxia/ischemia (H/I) is an important stress factor involved in the disruption of the blood–brain barrier (BBB) following stroke injury, yet the cellular and molecular mechanisms on how the human BBB responds to such injury remains unclear. In this study, we investigated the cellular response of the human BBB to chemical and environmental H/I in vitro.
Methods
In this study, we used immortalized hCMEC/D3 and IMR90 stem-cell derived human brain microvascular endothelial cell lines (IMR90-derived BMECs). Hypoxic stress was achieved by exposure to cobalt chloride (CoCl2) or by exposure to 1 % hypoxia and oxygen/glucose deprivation (OGD) was used to model ischemic injury. We assessed barrier function using both transendothelial electrical resistance (TEER) and sodium fluorescein permeability. Changes in cell junction integrity were assessed by immunocytochemistry and cell viability was assessed by trypan-blue exclusion and by MTS assays. Statistical analysis was performed using one-way analysis of variance (ANOVA).
Results
CoCl2 selectively disrupted the barrier function in IMR90-derived BMECs but not in hCMEC/D3 monolayers and cytotoxic effects did not drive such disruption. In addition, hypoxia/OGD stress significantly disrupted the barrier function by selectively disrupting tight junctions (TJs) complexes. In addition, we noted an uncoupling between cell metabolic activity and barrier integrity.
Conclusions
In this study, we demonstrated the ability of IMR90-derived BMECs to respond to hypoxic/ischemic injury triggered by both chemical and environmental stress by showing a disruption of the barrier function. Such disruption was selectively targeting TJ complexes and was not driven by cellular apoptosis. In conclusion, this study suggests the suitability of stem cell-derived human BMECs monolayers as a model of cerebral hypoxia/ischemia in vitro.
Electronic supplementary material
The online version of this article (doi:10.1186/s12987-016-0042-1) contains supplementary material, which is available to authorized users.
Background
The blood–brain barrier (BBB), a component of the neurovascular unit, constitutes a crucial biological barrier in the maintenance of brain homeostasis by restricting the diffusion of solutes and toxins to brain parenchyma. The presence of such barrier is supported by brain microvascular endothelial cells (BMECs), which are present in the cerebral microvasculature. BMECs provide both a physical (tight junctions complexes) and a chemical barrier (drug and nutrient transporters), which tightly regulate the diffusion of small molecules between the blood and brain. However, the integrity of the BBB is compromised in several neurological diseases including multiple sclerosis [1], neurodegenerative diseases [2–5], and stroke [1, 6, 7].
Stroke constitutes the fifth leading cause of death in industrialized countries and is a leading cause of disability [8]. The majority of stroke events are classified as an ischemic, marked by an abrupt decreased perfusion in a defined brain region, resulting in an impairment of both oxygen and nutrient supply. This ultimately leads to the onset of a cerebral hypoxic/ischemic (H/I) injury.
BMECs are the first cell type of the neurovascular to sense hypoxia and respond to such injury by disrupting barrier function. Such disruption will eventually lead to a vascular leakage. The mechanisms by which H/I impacts barrier function have been extensively studied in rodents and non-human primates [9–17], yet the literature showing similar outcomes at the human BBB remains unclear.
In this study, we investigated the effect of H/I on a stem cell-derived model of the human BBB using the IMR90-c4 induced pluripotent stem cell line [18–21] and compared their response to hCMEC/D3, an immortalized human BMEC line commonly used in the literature [22].
Methods
Cell culture
hCMEC/D3 cell line [22] was purchased from Millipore (EMD Millipore, Billerica, MA, USA) and maintained following established manufacturer protocol. IMR90-c4 induced pluripotent stem cell (iPSC) cell line [18] was purchased from WiCell (WiCell, Madison, WI). IMR90-c4 iPS cell line was maintained in mTeSR1 (Stem Cell Technologies, Vancouver, BC, USA) and grown on hPSC-qualified Matrigel (Corning Inc., Corning, NY, USA). The IMR90 cell line was differentiated into BMECs (iPSC-BMECs) following the differentiation protocol established by Lippmann and colleagues [20, 21] and summarized in Additional file 1: Figure S1. In brief, cells were seeded at 20,000 cells/cm2 5 days before differentiation and maintained in mTeSR medium. Five days after seeding, BMECs differentiation was set using unconditioned maturation medium (UMM) following the same composition as previously described: DMEM/F12 with 15 mM HEPES supplemented with 20 % KO serum replacement, 1 % MEM non-essential aminoacids, and 0.5 % Glutamax I, (ThermoFisher, Waltham, MA, USA) and 0.1 mM β-mercaptoethanol (Sigma-Aldrich, St Louis, MO, USA) for 6 days. After such differentiation, IMR90-derived BMECs were incubated in presence EC differentiation medium (EC serum free medium (ThermoFisher), supplemented with 1 % platelet-poor derived plasma serum (ThermoFisher), 20 µg/mL human basic fibroblast growth factor (R&D Systems) and 10 µM all-trans retinoic acid (Sigma-Aldrich) for 2 days. After 8 days of differentiation, cells were enzymatically dissociated and seeded at a density of 106 cells/cm2 on 12-well Transwell polyester cell culture inserts (0.4 µm pore size) coated with collagen from human placenta (Sigma-Aldrich) and fibronectin from bovine plasma (Sigma-Aldrich) at concentrations of 80 and 20 µg/cm2. After 24 h (day 9), BMECs were maintained in EC differentiation medium containing only 1 % platelet-poor plasma-derived serum. All experiments were carried out 48 h after seeding.
Barrier function
Cells were grown on 12-well Transwell polyester membranes (Corning Inc.) coated with 30 µg/cm2 collagen I (Sigma-Aldrich, St Louis, MO, USA) or with 80 µg/cm2 collagen supplemented with 20 µg/cm2 fibronectin (Sigma-Aldrich) mixture to allow hCMEC/D3 and IMR90-derived BMECs to attach respectively. Monolayer tightness was assessed by measuring transendothelial electrical resistance (TEER) using an EVOM STX2 chopstick electrode (World Precision Instruments, Sarasota, FL, USA). Paracellular permeability was assessed in monolayers by measuring the diffusion profile of 1 µM of sodium fluorescein (Sigma-Aldrich) using the clearance method described by Perriere and colleagues [23]. Fluorescence was assessed using a Synergy MX2 ELISA plate reader (Bio-Tek Instruments, Burlington, VT, USA). Average TEER and permeability values of both monolayers can be found in Table 1. TEER and permeability values from untreated monolayers were used to normalize experimental values expressed as % of control. Untreated group experimental values were arbitrarily set to 100 % control.
Table 1.
Biological values of hCMEC/D3 and IMR90-derived BMECs under resting conditions
| Cell type | hCMEC/D3 | IMR90-derived BMECs |
|---|---|---|
| Cell density (× 103 cells/cm2) | 128 ± 21 | 98 ± 19 |
| TEER (Ω cm2) | 40 ± 14 | 392 ± 113 |
| Pe fluorescein (10−4 cm/min) | 19 ± 8 | 2.44 ± 1.12 |
| Metabolic activity (OD 490 nm) | 1.26 ± 0.45 | 0.89 ± 0.34 |
Average cell density, TEER, fluorescein permeability (Pe) and metabolic activity (MTS absorbance at 490 nm wavelength). Mean ± SD, n = 18 for TEER and Pe values
Cobalt chloride and hypoxic treatment
Cobalt chloride (CoCl2, MP Biomedicals, Santa Ana, CA, USA) was freshly dissolved in complete cell medium at a stock concentration of 100 mM and further dissolved to achieve the concentrations of 10, 30 and 100 μM. Cells were incubated in presence of CoCl2 for 24 h. For the hypoxic experiments, cell medium was replaced with fresh medium and cells were incubated in a normobaric hypoxic C-chamber (Biospherix, Laconia, NY, USA) set at 1 % O2, 5 % CO2 and maintained at 37 °C for 6 or 24 h. In experiments involving oxygen deprivation (OD), cell medium was replaced with Dulbecco’s Modified Eagle Medium (DMEM) containing 1 g/L d-glucose and 1 % platelet-poor derived serum (Alfa Aesar, Ward Hill, MA, USA). In GD and OGD experiments, glucose-free and pyruvate-free DMEM were used instead of DMEM containing 1 g/L d-glucose.
Immunocytochemistry
Cells were grown to confluence on 48-well coated plates and quickly washed with ice-cold phosphate buffered saline (PBS) solution and fixed with either 4 % paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) or cold methanol (ThermoFisher) and processed as previously described [20, 21].
Cell density and viability assay
Cell density was assessed on monolayers by Trypan-blue exclusion based assay. In brief, monolayers were briefly washed with ice-cold PBS and incubated in presence of accutase (corning) or 0.25 % trypsin–EDTA (ThermoFisher) for 5 min followed by a centrifugation at 1000 rpms for 5 min. Cells were resuspended in 0.5 mL PBS and counted using a Countess II automated cell counter (ThermoFisher). Cell viability was assessed by MTS-based CellTiter 96® Aqueous (Promega, Madison, WI, USA). After treatment, cells grown on 96-well plates were incubated in presence of MTS reagent for 1 h. Absorbance was measured at 490 nm and corrected against unconditioned medium containing the same amount of MTS. Absorbance values from all samples were normalized against their respective control samples.
Statistics
All experiments were performed using cells coming from at least three independent experiments (distinct cell passages, each experiment was performed with two technical replicates). Statistical analysis was performed using one-way ANOVA followed by a post hoc analysis using Dunnett’s test with the control group as the reference group. P values lesser or equal to 0.05 were considered statistically significant.
Results
CoCl2 induces barrier disruption in IMR90-derived BMECs but not in hCMEC/D3 monolayers
In order to demonstrate the suitability of IMR90-derived BMECs (iPSC-BMECs) as an in vitro model of cerebral hypoxia/ischemia, it is important to show that such cells can respond to known hypoxic stimuli. Thus, we first assessed the cellular response of iPSC-BMEC monolayers to chemical hypoxia (Fig. 1) by exposing them to different concentrations of cobalt chloride (CoCl2), a common hypoxia-mimetic chemical used in various cell types [9, 24–29]. We compared iPSC-BMECs response to hCMEC/D3 cell monolayers, a human brain microvascular endothelial cell line commonly used in the literature [22, 30–32]. It should be noted that these cell monolayers have lower baseline TEERs and higher sodium fluorescein permeabilities that the iPSC-BMEC monolayers (Table 1).
Fig. 1.
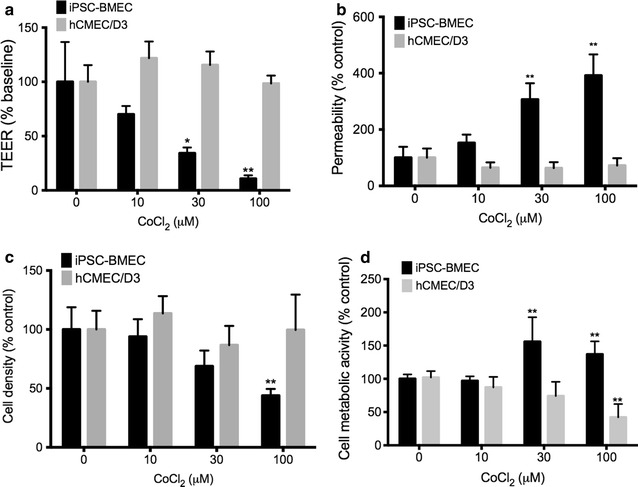
Cobalt chloride induces barrier disruption in iPSC-derived BMECs but not in hCMEC/D3 monolayers. Cells were treated with cobalt chloride (CoCl2) for 24 h and were compared to untreated cell monolayers. Changes in the monolayer integrity were assessed by TEER (a) and by sodium fluorescein (b). Note the decrease in barrier function in IMR90-derived BMECs (iPSC-BMEC) versus the hCMEC/D3 monolayers. c Cobalt chloride induced decrease in adherent cell density in iPSC-BMEC at high concentrations (100 µM). * and ** denotes P < 0.05 and P < 0.01 versus control (untreated) groups. d Metabolic activity following treatment with CoCl2 did not induce cell toxicity in IMR90-derived BMECs, as we noted no decrease versus control group. Mean ± SD, n = 3 for each group, * and ** denotes P < 0.05 and P < 0.01 versus control (untreated) groups
Upon exposure of 24 h to CoCl2, there was a decrease in the iPSC-BMECs barrier function as noted by a significant decrease in TEER (Fig. 1a) in cells treated with CoCl2 at 30 and 100 µM. However, hCMEC/D3 failed to display any changes in barrier function even when exposed at 100 µM (a dose known to trigger a barrier breakdown in rat brain endothelial cell monolayers [9]). To further confirm this observation, we investigated changes in sodium fluorescein permeability in both iPSC-BMECs monolayers and hCMEC/D3 following CoCl2 treatment (Fig. 1b). As observed in our previous experiment, CoCl2 treatment affected the barrier integrity in iPSC-BMECs at 30 and 100 µM, with a 3- to 4-fold increase in paracellular permeability to sodium fluorescein compared to untreated cells.
CoCl2 treatment differentially affects cell density and metabolism in hCMEC/D3 and IMR90-derived BMEC monolayers
Because CoCl2 treatment can induce cell apoptosis in various mammalian cell lines [33–36], we investigated changes in cell viability following CoCl2 treatment using MTS and Trypan-blue exclusion assays (Fig. 1c, d). After 24 h of CoCl2 treatment, we did not note a significant decrease in hCMEC/D3 cell density (Fig. 1c). However, we noted a 50 % decrease in cell density only in iPSC-BMEC group exposed to 100 µM, suggesting a detrimental effect of CoCl2 on the monolayer integrity. However, no change in hCMEC/D3 cell monolayer density was observed, even at the highest CoCl2 concentration.
Attempts to use trypan blue exclusion assay only provided information on adherent cells remaining after CoCl2 treatment. Furthermore, the additional mechanical stress applied to assess cell viability in non-adherent cells using such method may also interfere with the method accuracy. Therefore, we consider such method not suited for cytotoxic assays. Notably, tetrazolium-based cytotoxic assays directly assess changes in mitochondrial activity and can be directly applied to adherent and non-adherent cells. Therefore, we investigated changes in cell viability following CoCl2 treatment using an MTS-based assay (Fig. 1d), an assay commonly used to assess cytotoxicity by measuring changes in cell metabolic activity [37]. Notably, we did not notice changes in cell viability in iPSC-BMEC monolayers following exposure to CoCl2 compared to control. We even noted an increase in cell viability following 30 µM and 100 µM treatment. In contrast, we noted a dose-dependent decrease in cell viability in hCMEC/D3 monolayer with a 50 % decrease in cell metabolism.
To further understand how CoCl2 impacted barrier function in iPSC-BMECs, we investigated changes in cell junction integrity by immunocytochemistry (Fig. 2). At 30 µM, we observed alterations in the distribution of tight junction complexes (claudin-5 and occludin) with a loss of immunostaining at the cell borders and a relocalization into cell cytoplasm. Surprisingly, such relocalization was not observed in adherens junction complexes (PECAM-1 and β-catenin). Taken together, these observations are consistent with observations in RBE4 monolayers [9] and suggest the ability of IMR90-derived BMECs to respond to hypoxic stimulus in similar fashion to rodent-based in vitro models.
Fig. 2.
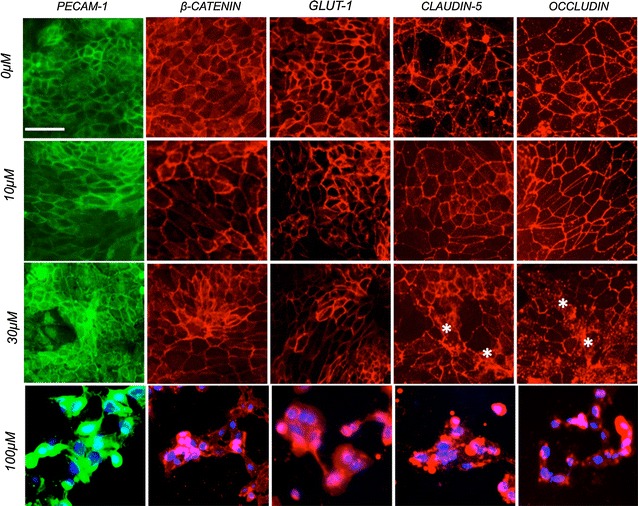
Cobalt-chloride treatment impact monolayer integrity in iPSC-derived BMECs. Representative immunocytochemistry micrograph pictures of IMR90-derived BMECs monolayers in presence of CoCl2. Cells were treated with different CoCl2 concentrations. Note the presence of irregular patterning in TJ proteins (claudin-5 and occludin) as marked by asterisks at 30 µM treatment and a complete disruption at 100 µM. Scale bar = 20 µm
Hypoxia induces barrier disruption in both IMR90-derived BMECs and hCMEC/D3 cell monolayers
Hypoxia-induced BBB disruption is a well-established phenomenon reported in both in vitro and in vivo models [9, 38–44]. Therefore, we assessed the ability of both IMR90-derived BMECs and hCMEC/D3 to respond to hypoxic stress by exposing our monolayers to 1 % O2 for 6 or 24 h (Fig. 3).
Fig. 3.
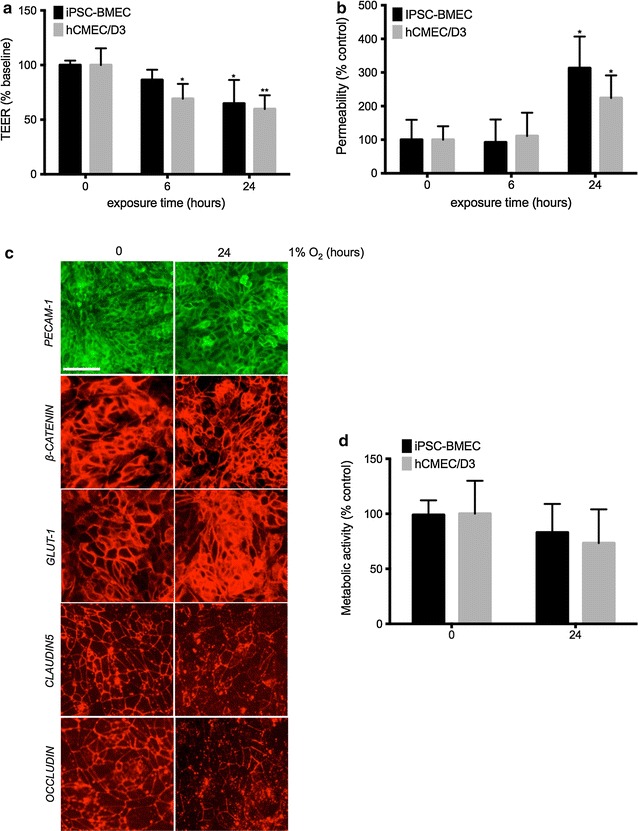
Prolonged Hypoxia impairs the barrier function in both cell monolayers. TEER (a) and sodium fluorescein (b) permeability values in both iPSC-BMECs and hCMEC/D3 cell monolayers following exposure to hypoxia for 6 and 24 h. Note the early onset of hypoxia-induced barrier disruption in hCMEC/D3 at 6 h as indicated by TEER compared to IMR90-derived BMECs, with a significant decrease at 24 h. Hypoxia-induced paracellular permeability however was only observed after 24 h in both groups. c Immunocytochemistry profile of IMR90-derived BMECs following exposure to hypoxia (1 % O2) for 24 h. Note the decreased immunoreactivity of claudin-5 and occludin following hypoxia, whereas no changes where noted in other cell junction proteins (β-catenin and PECAM-1). Scale bar = 20 µm. d Cell metabolic activity as measured by MTS assay. Following hypoxic incubation, MTS was added to the cell conditioned medium and incubated for 1 h before readout. Mean ± SD, n = 3 for each group. * and ** denotes P < 0.05 and P < 0.01 to controls (labeled as 0 h) respectively
Acute (6 h) hypoxic stress resulted in significant decrease of over 70 % in TEER in hCMEC/D3 monolayers (Fig. 3a), whereas IMR90-derived BMECs showed only a mild decrease. However, such changes in TEER were not reflected in changes in vascular permeability, as we noted no significant increase in sodium fluorescein permeability (Fig. 3b).
In contrast, prolonged hypoxic (24 h) injury resulted in loss of barrier function in both IMR90-derived BMECs and hCMEC/D3, as we observed a 50 % decrease in TEER (Fig. 3a) and a 2.5-fold increase in fluorescein permeability (Fig. 3b) compared to normoxic monolayers. These results suggest the ability of the monolayers to respond to hypoxia similarly to the existing literature.
To further understand how hypoxia affects the barrier function, we investigated changes in cell junction complexes in IMR90-derived BMECs by immunocytochemistry (Fig. 3c). After 24 h of hypoxia, we did not observe any changes in adherens junction (PECAM-1 and β-catenin) complexes. However, we noted changes in tight junction (claudin-5 and occludin) complexes with a discontinuous staining pattern. Finally, we assessed by MTS whether this insult impacted cell viability (Fig. 3d). Following 24 h of hypoxic stress, we did not find any significant changes in cell metabolic activity, as the average metabolic activity was 80 and 70 % of the normoxic levels in IMR90-derived BMECs and hCMEC/D3 monolayers, respectively. In conclusion, our data demonstrate that IMR90-derived BMECs, as well as hCMEC/D3 cell monolayers, actively respond to hypoxia by disruption of their barrier function. Such disruption appears driven by alterations in the tight junction complexes rather than cell death.
Oxygen-glucose deprivation impairs barrier function in both IMR90-derived BMECs and hCMEC/D3 monolayers
Next, we investigated how hypoxia/aglycemia affected the barrier function in our human models of the BBB by exposing our cells to hypoxia, aglycemia or to oxygen-glucose deprivation (OGD) stress (Fig. 4). Interestingly, incubation in glucose-deprived medium (GD) for 24 h resulted in a significant decrease in TEER (~50 %) in both hCMEC/D3 and IMR90-derived BMEC monolayers (Fig. 4) but failed to show any significant increase in cell permeability to sodium fluorescein (Fig. 4b) suggesting that aglycemia alone was not sufficient to induce BBB disruption. On the other hand, hypoxia (OD) was capable of inducing BBB disruption, as we noted a significant decrease in TEER and an increase in when fluorescein permeability. However, we observed the maximum disruption cells were subjected to both oxygen and glucose deprivation (OGD), as noted by the lowest TEER values (20 % of control) and highest permeability values to fluorescein (5-fold increase).
Fig. 4.
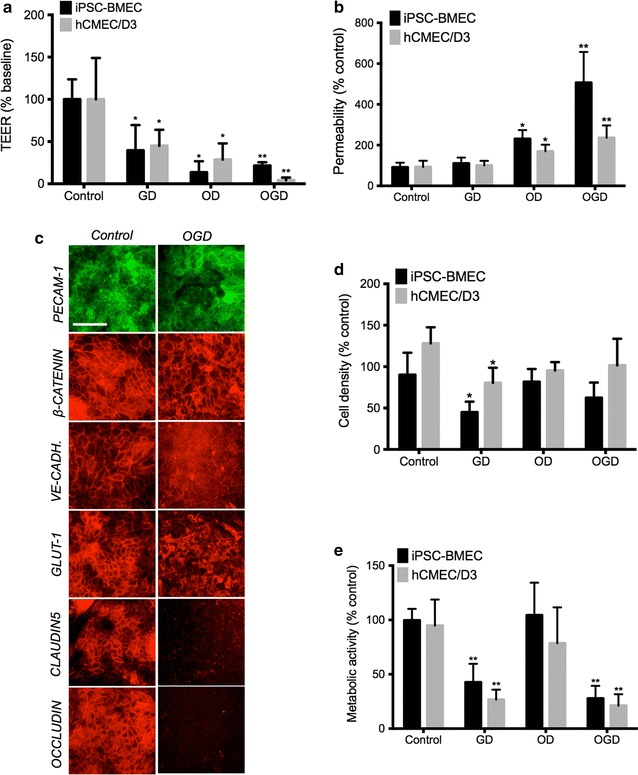
OGD induced barrier disruption in human BMECs.TEER (a) and sodium fluorescein (b) permeability values in both iPSC-BMECs and hCMEC/D3 cell monolayers following aglycemic (GD), hypoxic (OD) and oxygen-glucose deprivation (OGD) stress. Cells incubated in DMEM with glucose under normoxia served as control. Note the increase in fluorescein permeability following OD and OGD stress. c Representative immunocytochemistry pictures in iPSC-derived BMECs monolayers following 24 h of OGD stress. Note the overall degradation in cell junctions, with a quasi-disappearance in TJ complexes immunoreactivity. Scale bar = 20 µm. d IMR90-derived BMECs and hCMEC/D3 average cell density following treatment with aglycemia, hypoxia or OGD stress. Notably, aglycemia (GD) showed the lowest cell density in both cell monolayers. e Cell metabolic activity following 24 h of treatment. After 24 h of treatment, MTS reagent was added to conditioned cell medium and allowed to further incubate for 1 h. Mean ± SD, n = 3 for each group. * and ** denotes P < 0.05 and P < 0.01 to controls (DMEM with glucose, normoxia) respectively
Following such observations, we investigated changes in cell junction complexes in IMR90-derived BMECs following OGD stress by immunocytochemistry (Fig. 4c). OGD stress was capable of altering adherens junction complexes (as noted by a decrease in VE-cadherin immunoreactivity) but more importantly, we noted a complete loss of immunoreactivity for tight junction complex proteins.
Taken together, loss of tight junction complexes during OGD stress may be a contributor to the BBB disruption.
As changes in cell density and viability may impact the barrier integrity during injury, we investigated changes in cell density by trypan-blue exclusion assay (Fig. 4d). Interestingly, we noted a significant decrease in cell density after aglycemic treatment in both IMR90-derived BMECs and in hCMEC/D3 monolayers. However, we did not notice a significant decrease in cell density following hypoxia and OGD stress (two conditions shown to have an impact on the barrier function). Furthermore, we observed a similar outcome when we measured changes in cell viability by MTS assay (Fig. 4e). Aglycemia decreased metabolic activity level by 60 % compared to controls in both monolayers. However, hypoxia (OD) alone showed no differences compared to controls. Finally, cells exposed to OGD stress showed only 15–20 % of the metabolic activity observed in the control group. Taken together, our study demonstrates that both IMR90-derived BMECs and hCMEC/D3 respond to OGD stress with disrupted barrier function. However, we noted an uncoupling between BBB barrier function and cell density/cell metabolic activity following hypoxic or aglycemic treatment.
Discussion
Stroke constitutes the fifth leading cause of death and it is a major cause of disability in the United States. Although important efforts have been made to identify therapeutics capable of improving outcome in stroke patients, the ability to translate findings from animal models to patients has had little success.
Human Induced pluripotent stem cells (iPSCs) based in vitro models of the neurovascular unit (integrating BMECs, astrocytes and neurons) may help provide a screening platform to improve such translation. However, pluripotent stem cells are also highly hypoxic tolerant [45], therefore questioning the suitability of stem-cell based models for understanding the effects of hypoxic/ischemic injury in vitro.
In this study, we investigated the ability of stem cell derived BMECs to respond to hypoxic/ischemic injury in vitro by comparing their response to hCMEC/D3, an immortalized adult human BMEC line [22]. We first investigated changes in the barrier function following CoCl2 treatment, a chemical commonly used to simulate a hypoxic response in mammalian cells [25, 26, 28, 29]. We noted a significant decrease in barrier function in IMR90-derived BMECs and a loss of monolayer integrity. Such results are in agreement with a previous study by Engelhardt and colleagues [9]. In that study, the authors observed that CoCl2 was capable of significantly decreasing barrier function in RBE4 cells monolayers as early as 6 h. In contrast to their findings, we observed a significant decrease in the barrier function only after 24 h in IMR90-derived BMECs. Surprisingly, we did not observe a response in hCMEC/D3 monolayers following CoCl2 exposure, with no changes in barrier function compared to untreated groups.
A difference in response to CoCl2 between the two monolayers might be explained by differences in oxygen sensing, as CoCl2 exerts its activity through the activation of the hypoxia-induced factor (HIF)-1 pathway. Such differences maybe also inherent to the nature of the hCMEC/D3 cells, as Patak and colleagues reported an absence of up-regulation in ABCB1 and ABCC1 expression following hypoxic stress [46]. We are currently investigating such dimorphism in oxygen sensing between these two cells by comparing HIF-1α expression at protein levels and VEGF production.
Following these observations, we investigated the cellular response to environmental hypoxia by incubating cells in the presence of 1 % O2, a gas phase concentration commonly used in the literature. In our study, both IMR90-derived BMECs and hCMEC/D3 cells responded to prolonged hypoxic injury in vitro with decreased barrier function. Furthermore, IMR90-derived BMECs displayed alterations in tight junction complex distribution similar to previous reports [9]. However, oxygen-glucose deprivation (OGD) stress was necessary to achieve a major barrier disruption in both IMR90-derived BMECs and hCMEC/D3 cells. Such disruption was marked by the greatest decrease in TEER and highest increase in fluorescein permeability.
This dramatic decrease in barrier function was likely due to a major disruption in tight junction complex integrity, as we noted a significant decrease in immunoreactivity of both claudin-5 and occludin in IMR90-derived BMECs. We speculate such decreased immunoreactivity may be indicative of a cleavage of such proteins by a select number of matrix metalloproteinases (MMPs), in particular by MMP-2 and MMP-9 [47–49].
In addition to changes in barrier function due to changes in MMP activity, we also speculate that impaired energy production during hypoxic/ischemic injury may contribute to barrier disruption. A recent study by Vandekeere and colleagues suggests ATP production in brain endothelial cell metabolism is driven by glycolysis under aerobic conditions [50]. MTS-based assays are designed to measure changes in mitochondrial hydrogenase activity [51]. Thus, we were expecting little change in MTS activity following hypoxic/OGD stress in our model. Surprisingly, we noted no changes in MTS activity in hypoxic cells. Such results were consistent with a recent study published by Ogunshola and colleagues [10], in which no significant changes in cell metabolism were observed following prolonged hypoxia.
Interestingly, we noted changes in cell density following CoCl2 treatment as well as during glucose deprivation. Although a decrease in cell density was accompanied by an increase in permeability following CoCl2 treatment, we did not observe such phenomenon in glucose-deprived cells.
Finally, we observed a conundrum between metabolic activity and barrier function in BMECs, in particular between hypoxic and aglycemic treatments. Decreased cell metabolism in aglycemic cells was not accompanied by decreased barrier function, whereas we noted a loss of barrier function was not accompanied by a decrease in cell metabolism. Such data suggests that tight junction disruption following hypoxia may not be due to energy deficiency, but also suggests that IMR90-derived BMECs may utilize other nutrients to maintain mitochondrial activity through some anaploretic reactions (in particular glutamine). Thus, we are currently investigating how hypoxia/OGD stress influences energy production in BMECs; in particular we are investigating how such stress influences changes in glycolysis and oxidative phosphorylation mechanism.
Conclusions
In summary, this study demonstrates the suitability of stem cell-derived in vitro model of the human BBB as an in vitro model for H/I. This model may effectively complement existing in vivo models and help improve the identification of novel therapeutics capable of fighting H/I-induced BBB disruption following stroke.
Authors’ contributions
AA has designed and contributed in the experimental design, performed some of the experiments and analyzed the results and edited the manuscript and figures. SP and AM have contributed in the experimental design and equally contributed in the experiments. SP has contributed in the analysis and the redaction of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
None.
Competing interests
The authors declare that they have no competing interests.
Availability of data and supporting materials
Relevant raw data will be provided upon request.
Ethics approval and consent to participate
Not applicable (human cell lines purchased from repositories).
Funding
This research funding was supported by institutional support to AA.
Abbreviations
- BBB
blood-brain barrier
- BMECs
brain microvascular endothelial cells
- DMEM
Dulbecco’s modified eagle medium
- GD
glucose deprivation
- H/I
hypoxia-ischemia
- OD
oxygen deprivation
- OGD
oxygen-glucose deprivation
- PBS
phosphate-buffered saline solution
- TEER
transendothelial electrical resistance
- TJ
tight junctions
Additional file
10.1186/s12987-016-0042-1 Representative diagram of the iPSC-derived BMEC differentiation protocol.
Contributor Information
Shyanne Page, Email: shyanne.page@ttuhsc.edu.
Alli Munsell, Email: alli.munsell@ttuhsc.edu.
Abraham J. Al-Ahmad, Phone: +1 (806) 414-9369, Email: abraham.al-ahmad@ttuhsc.edu
References
- 1.Lopes Pinheiro MA, Kooij G, Mizee MR, Kamermans A, Enzmann G, Lyck R, et al. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim Biophys Acta. 2015;1862(3):461–471. doi: 10.1016/j.bbadis.2015.10.018. [DOI] [PubMed] [Google Scholar]
- 2.Duits FH, Hernandez-Guillamon M, Montaner J, Goos JD, Montanola A, Wattjes MP, et al. Matrix metalloproteinases in Alzheimer’s disease and concurrent cerebral microbleeds. J Alzheimers Dis. 2015;48(3):711–720. doi: 10.3233/JAD-143186. [DOI] [PubMed] [Google Scholar]
- 3.Drouin-Ouellet J, Sawiak SJ, Cisbani G, Lagace M, Kuan WL, Saint-Pierre M, et al. Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: potential implications for its pathophysiology. Ann Neurol. 2015;78(2):160–177. doi: 10.1002/ana.24406. [DOI] [PubMed] [Google Scholar]
- 4.Qosa H, Abuasal BS, Romero IA, Weksler B, Couraud PO, Keller JN, et al. Differences in amyloid-beta clearance across mouse and human blood-brain barrier models: kinetic analysis and mechanistic modeling. Neuropharmacology. 2014;79:668–678. doi: 10.1016/j.neuropharm.2014.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garbuzova-Davis S, Sanberg PR. Blood-CNS Barrier Impairment in ALS patients versus an animal model. Front Cell Neurosci. 2014;8:21. doi: 10.3389/fncel.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah K, Abbruscato T. The role of blood-brain barrier transporters in pathophysiology and pharmacotherapy of stroke. Curr Pharm Des. 2014;20(10):1510–1522. doi: 10.2174/13816128113199990465. [DOI] [PubMed] [Google Scholar]
- 7.Jickling GC, Liu D, Stamova B, Ander BP, Zhan X, Lu A, et al. Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab. 2014;34:185–199. doi: 10.1038/jcbfm.2013.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics–2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelhardt S, Al-Ahmad AJ, Gassmann M, Ogunshola OO. Hypoxia selectively disrupts brain microvascular endothelial tight junction complexes through a hypoxia-inducible factor-1 (HIF-1) dependent mechanism. J Cell Physiol. 2014;229(8):1096–1105. doi: 10.1002/jcp.24544. [DOI] [PubMed] [Google Scholar]
- 10.Engelhardt S, Huang SF, Patkar S, Gassmann M, Ogunshola OO. Differential responses of blood-brain barrier associated cells to hypoxia and ischemia: a comparative study. Fluids Barriers CNS. 2015;12:4. doi: 10.1186/2045-8118-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al Ahmad A, Gassmann M, Ogunshola OO. Involvement of oxidative stress in hypoxia-induced blood-brain barrier breakdown. Microvasc Res. 2012;84(2):222–225. doi: 10.1016/j.mvr.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 12.Lee B, Clarke D, Al Ahmad A, Kahle M, Parham C, Auckland L, et al. Perlecan domain V is neuroprotective and proangiogenic following ischemic stroke in rodents. J Clin Investig. 2011;121(8):3005–3023. doi: 10.1172/JCI46358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al Ahmad A, Gassmann M, Ogunshola OO. Maintaining blood-brain barrier integrity: pericytes perform better than astrocytes during prolonged oxygen deprivation. J Cell Physiol. 2009;218(3):612–622. doi: 10.1002/jcp.21638. [DOI] [PubMed] [Google Scholar]
- 14.Abbruscato TJ, Davis TP. Combination of hypoxia/aglycemia compromises in vitro blood-brain barrier integrity. J Pharmacol Exp Ther. 1999;289:668–675. [PubMed] [Google Scholar]
- 15.Yamagata K, Tagami M, Takenaga F, Yamori Y, Itoh S. Hypoxia-induced changes in tight junction permeability of brain capillary endothelial cells are associated with IL-1beta and nitric oxide. Neurobiol Dis. 2004;17:491–499. doi: 10.1016/j.nbd.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 16.Brown RC, Davis TP. Hypoxia/aglycemia alters expression of occludin and actin in brain endothelial cells. Biochem Biophys Res Commun. 2005;327:1114–1123. doi: 10.1016/j.bbrc.2004.12.123. [DOI] [PubMed] [Google Scholar]
- 17.Culot M, Mysiorek C, Renftel M, Roussel BD, Hommet Y, Vivien D, et al. Cerebrovascular protection as a possible mechanism for the protective effects of NXY-059 in preclinical models: an in vitro study. Brain Res. 2009;1294:144–152. doi: 10.1016/j.brainres.2009.07.035. [DOI] [PubMed] [Google Scholar]
- 18.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 19.Wilson HK, Canfield SG, Hjortness MK, Palecek SP, Shusta EV. Exploring the effects of cell seeding density on the differentiation of human pluripotent stem cells to brain microvascular endothelial cells. Fluids Barriers CNS. 2015;12:13. doi: 10.1186/s12987-015-0007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lippmann ES, Al-Ahmad A, Azarin SM, Palecek SP, Shusta EV. A retinoic acid-enhanced, multicellular human blood-brain barrier model derived from stem cell sources. Sci Rep. 2014;4:4160. doi: 10.1038/srep04160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lippmann ES, Azarin SM, Kay JE, Nessler RA, Wilson HK, Al-Ahmad A, et al. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat Biotechnol. 2012;30(8):783–791. doi: 10.1038/nbt.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weksler BB, Subileau EA, Perriere N, Charneau P, Holloway K, Leveque M, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005;19:1872–1874. doi: 10.1096/fj.04-3458fje. [DOI] [PubMed] [Google Scholar]
- 23.Perriere N, Demeuse P, Garcia E, Regina A, Debray M, Andreux JP, et al. Puromycin-based purification of rat brain capillary endothelial cell cultures. Effect on the expression of blood-brain barrier-specific properties. J Neurochem. 2005;93(2):279–289. doi: 10.1111/j.1471-4159.2004.03020.x. [DOI] [PubMed] [Google Scholar]
- 24.Guo L, Lan J, Lin Y, Guo P, Nie Q, Mao Q, et al. Hypoxia/ischemia up-regulates Id2 expression in neuronal cells in vivo and in vitro. Neurosci Lett. 2013;554:88–93. doi: 10.1016/j.neulet.2013.08.044. [DOI] [PubMed] [Google Scholar]
- 25.Gotoh M, Sano-Maeda K, Murofushi H, Murakami-Murofushi K. Protection of neuroblastoma Neuro2A cells from hypoxia-induced apoptosis by cyclic phosphatidic acid (cPA) PLoS ONE. 2012;7(12):e51093. doi: 10.1371/journal.pone.0051093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang B, Zou Y, Yuan ZL, Xiao JG. Genistein suppressed upregulation of vascular endothelial growth factor expression by cobalt chloride and hypoxia in rabbit retinal pigment epithelium cells. J Ocul Pharmacol Ther. 2003;19(5):457–464. doi: 10.1089/108076803322473015. [DOI] [PubMed] [Google Scholar]
- 27.Bernaudin M, Bellail A, Marti HH, Yvon A, Vivien D, Duchatelle I, et al. Neurons and astrocytes express EPO mRNA: oxygen-sensing mechanisms that involve the redox-state of the brain. Glia. 2000;30:271–278. doi: 10.1002/(SICI)1098-1136(200005)30:3<271::AID-GLIA6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 28.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minchenko A, Bauer T, Salceda S, Caro J. Hypoxic stimulation of vascular endothelial growth factor expression in vitro and in vivo. Lab Invest. 1994;71:374–379. [PubMed] [Google Scholar]
- 30.Naik P, Cucullo L. In vitro blood-brain barrier models: current and perspective technologies. J Pharm Sci. 2012;101:1337–1354. doi: 10.1002/jps.23022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poller B, Gutmann H, Krahenbuhl S, Weksler B, Romero I, Couraud PO, et al. The human brain endothelial cell line hCMEC/D3 as a human blood-brain barrier model for drug transport studies. J Neurochem. 2008;107:1358–1368. doi: 10.1111/j.1471-4159.2008.05730.x. [DOI] [PubMed] [Google Scholar]
- 32.Cucullo L, Couraud PO, Weksler B, Romero IA, Hossain M, Rapp E, et al. Immortalized human brain endothelial cells and flow-based vascular modeling: a marriage of convenience for rational neurovascular studies. J Cereb Blood Flow Metab. 2008;28(2):312–328. doi: 10.1038/sj.jcbfm.9600525. [DOI] [PubMed] [Google Scholar]
- 33.Hartwig K, Fackler V, Jaksch-Bogensperger H, Winter S, Furtner T, Couillard-Despres S, et al. Cerebrolysin protects PC12 cells from CoCl2-induced hypoxia employing GSK3 beta signaling. Int J Dev Neurosci. 2014;38:52–58. doi: 10.1016/j.ijdevneu.2014.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Guan D, Su Y, Li Y, Wu C, Meng Y, Peng X, et al. Tetramethylpyrazine inhibits CoCl2 -induced neurotoxicity through enhancement of Nrf2/GCLc/GSH and suppression of HIF1alpha/NOX2/ROS pathways. J Neurochem. 2015;134(3):551–565. doi: 10.1111/jnc.13161. [DOI] [PubMed] [Google Scholar]
- 35.Huang CY, Hsieh YL, Ju DT, Lin CC, Kuo CH, Liou YF, et al. Attenuation of magnesium sulfate on CoCl(2)-induced cell death by activating ERK1/2/MAPK and inhibiting HIF-1alpha via mitochondrial apoptotic signaling suppression in a neuronal cell line. Chin J Physiol. 2015;58(4):244–253. doi: 10.4077/CJP.2015.BAD296. [DOI] [PubMed] [Google Scholar]
- 36.Mo SJ, Hong J, Chen X, Han F, Ni Y, Zheng Y, et al. VEGF-mediated NF-kappaB activation protects PC12 cells from damage induced by hypoxia. Neurosci Lett. 2016;610:54–59. doi: 10.1016/j.neulet.2015.10.051. [DOI] [PubMed] [Google Scholar]
- 37.Sumantran VN. Cellular chemosensitivity assays: an overview. Methods Mol Biol. 2011;731:219–236. doi: 10.1007/978-1-61779-080-5_19. [DOI] [PubMed] [Google Scholar]
- 38.Ogunshola OO, Al Ahmad A. HIF-1 at the blood-brain barrier: a mediator of permeability? Special topic: the many faces of hypoxia HIF-1 at the blood-brain barrier. High Alt Med Biol. 2012;13:153–161. doi: 10.1089/ham.2012.1052. [DOI] [PubMed] [Google Scholar]
- 39.Zhu H, Wang Z, Xing Y, Gao Y, Ma T, Lou L, et al. Baicalin reduces the permeability of the blood-brain barrier during hypoxia in vitro by increasing the expression of tight junction proteins in brain microvascular endothelial cells. J Ethnopharmacol. 2011;141:714–720. doi: 10.1016/j.jep.2011.08.063. [DOI] [PubMed] [Google Scholar]
- 40.Yan J, Zhou B, Taheri S, Shi H. Differential effects of HIF-1 inhibition by YC-1 on the overall outcome and blood-brain barrier damage in a rat model of ischemic stroke. PLoS ONE. 2011;6:e27798. doi: 10.1371/journal.pone.0027798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kimura K, Teranishi S, Kawamoto K, Nishida T. Protective effect of dexamethasone against hypoxia-induced disruption of barrier function in human corneal epithelial cells. Exp Eye Res. 2011;92:388–393. doi: 10.1016/j.exer.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 42.Lochhead JJ, McCaffrey G, Quigley CE, Finch J, DeMarco KM, Nametz N, et al. Oxidative stress increases blood-brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab. 2010;30:1625–1636. doi: 10.1038/jcbfm.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Natah SS, Srinivasan S, Pittman QJ, Zhao Z, Dunn JF. Effects of acute hypoxia and hyperthermia on the permeability of the blood brain barrier in adult rats. J Appl Physiol. 2009;107(4):1348–1356. doi: 10.1152/japplphysiol.91484.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCaffrey G, Willis CL, Staatz WD, Nametz N, Quigley CA, Hom S, et al. Occludin oligomeric assemblies at tight junctions of the blood-brain barrier are altered by hypoxia and reoxygenation stress. J Neurochem. 2009;110:58–71. doi: 10.1111/j.1471-4159.2009.06113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Semenza GL. Dynamic regulation of stem cell specification and maintenance by hypoxia-inducible factors. Mol Aspects Med. 2015;47:15–23. doi: 10.1016/j.mam.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 46.Patak P, Jin F, Schafer ST, Metzen E, Hermann DM. The ATP-binding cassette transporters ABCB1 and ABCC1 are not regulated by hypoxia in immortalised human brain microvascular endothelial cells. Exp Transl Stroke Med. 2011;3:12. doi: 10.1186/2040-7378-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. 2005;50:329–339. doi: 10.1002/glia.20169. [DOI] [PubMed] [Google Scholar]
- 48.Yang T, Roder KE, Abbruscato TJ. Evaluation of bEnd5 cell line as an in vitro model for the blood-brain barrier under normal and hypoxic/aglycemic conditions. J Pharm Sci. 2007;96:3196–3213. doi: 10.1002/jps.21002. [DOI] [PubMed] [Google Scholar]
- 49.Yang Y, Rosenberg GA. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011;42:3323–3328. doi: 10.1161/STROKEAHA.110.608257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vandekeere S, Dewerchin M, Carmeliet P. Angiogenesis revisited: an overlooked role of endothelial cell metabolism in vessel sprouting. Microcirculation. 2015;22(7):509–517. doi: 10.1111/micc.12229. [DOI] [PubMed] [Google Scholar]
- 51.Berg K, Hansen MB, Nielsen SE. A new sensitive bioassay for precise quantification of interferon activity as measured via the mitochondrial dehydrogenase function in cells (MTT-method) APMIS. 1990;98(2):156–162. doi: 10.1111/j.1699-0463.1990.tb01016.x. [DOI] [PubMed] [Google Scholar]