

Abstract
Post-translational epigenetic modification of histones is controlled by a number of histone-modifying enzymes. Such modification regulates the accessibility of DNA and the subsequent expression or silencing of a gene. Human histone methyltransferases (HMTs)constitute a large family that includes histone lysine methyltransferases (HKMTs) and histone/protein arginine methyltransferases (PRMTs). There is increasing evidence showing a correlation between HKMTs and cancer pathogenesis. Here, we present an overview of representative HKMTs, including their biological and biochemical properties as well as the profiles of small molecule inhibitors for a comprehensive understanding of HKMTs in drug discovery.
Keywords: histone methyltransferases, adenosylmethionine, small molecule inhibitors, anti-cancer drugs, epigenetic modification, drug discovery
Introduction
Post-translational epigenetic modifications of histones are controlled by histone-modifying enzymes, including histone methyltransferases (HMTs), histone demethylases, histone acetyltransferases, histone deacetylases (HDACs), ubiquitin ligases, and other specific kinases phosphorylating serine residues on histones. Such modifications regulate the accessibility of DNA and the subsequent expression or silencing of a gene1,2. Histone methyltransferases include a large family of dozens of histone lysine methyltransferases (HKMTs) and histone/protein arginine methyltransferases (PRMTs), many of which have been found to play critical roles in cell differentiation, gene regulation, DNA recombination and damage repair3,4,5,6.
The methylation of histone lysine residues was first discovered in the 1960s7,8 and was believed to be irreversible in the following decades until the discovery of a lysine demethylase in 20049. Recently, increasing evidence indicating a correlation between HKMTs and cancer pathogenesis has emerged3,4,5,6. Therefore, a number of small molecule inhibitors of histone lysine methyltransferases have been identified and exhibited selective cancer cell killing activities in vitro and in vivo, suggesting the possibility of using HKMTs as targets for cancer therapy. Here, we discuss individual histone lysine methylation with the currently available small molecule inhibitors and their applications in cancer treatment.
HKMTs-catalyzed methylation
Histone lysine methyltransferases (HKMTs) transfer the methyl group from the cofactor adenosylmethionine (AdoMet), which contains a highly reactive methylthiol group, to the tailed nitrogen of the substrate lysine residue, producing mono-, di-, or tri-methylated products and its analogue adenosylhomocysteine (AdoHcy) (Figure 1)10,11. AdoHcy is structurally similar to AdoMet, binding to methyltransferases in the same orientation with a similar Kd value, but it lacks the reactive methyl group and acts as a competitive inhibitor with respect to AdoMet12. Additionally, it cannot be used as a probe in cell biology or in vivo because of its quick degradation to adenosine and homocysteine by SAH hydrolase13.
Figure 1.

Methylation of lysine residues by histone methyltransferases utilizing adenosylmethionine (AdoMet) as a cofactor.
Catalytic active sites in HKMTs
Two pockets are involved in the catalytic active sites: a SAM-binding pocket and a lysine acceptor channel, which are on opposite sides of the protein surface facilitating the entrance of two substrates to the active sites from opposite sides of the enzyme surface14. The structures of cofactor binding sites are conserved in different SAM-dependent methyltransferase classes and are represented by three motifs located in the P loop, G loop and part of strand β415. The co-crystallized structures of SET domain-containing HKMTs16 (except DOT1L) with SAM or SAH show that the cofactor adopts a 'U-shaped' configuration in contrast to DOT1L, which adopts an extended configuration17 similar to that of PRMTs. In the 'U-shaped' configuration, the methylsulfonium cation of SAM forms a juxtaposition to the ϵ-amino group of the acceptor lysine residue within the narrow lysine channel, which was induced by an H-bond function between the conserved Asp or Glu residue and the hydroxyl groups of ribose as well as a salt bridge between a positively charged lysine and the carboxylate group of SAM. A 'tyrosine (Y)-phenylalanine (F) switch', a key determinant for mono-, di-, and tri-methylation of lysine residue in the SET domain-containing HKMTs, also exists, which is demonstrated by the mutants of DIM5F281Y and SETD7Y305F 18. The location of substrate also varies among different HKMTs. For instance, the substrates of SET domain-containing HKMTs are located in the tail of unordered histone, while H3K79, the substrate of DOT1L, is located at the core structure of ordered histone octamer19,20.
Representative HKMTs and their inhibitors
DOT1L
DOT1L, the only HKMT belonging to class I methyltransferases, mono-, di-, and tri-methylates H3K79 and plays a role in embryonic development and hematopoiesis, etc20,21,22. Compared with many other SET-domain HKMTs, DOT1L is a distributive enzyme20, which releases SAH, and mono-, di- and tri-methylated products at the same time after one catalytic cycle. Several studies19,20,21,22 indicated DOT1L as a target for acute leukemia with MLL (mixed-lineage leukemia) gene translocations. MLL rearrangements result in the loss of the SET-domain H3K4 methyltransferase on the carboxyl terminal, and the remaining amino-terminal MLL protein is fused to one of more than 60 different partner genes (predominantly, AF4, AF9, AF10, and ENL), which is found to recruit DOT1L as a part of transcription protein complexes and subsequently methylates H3K79 instead of H3K4. Such an aberrant epigenetic modification leads to the overexpression of MLL-target genes such as HOXA9 and MEIS1 and causes leukemia. Therefore, DOT1L is proposed as a target for the therapeutic intervention of MLL-rearranged leukemia. Several potent small molecules have thus been developed including SAH-like scaffold21, iodoethyl-containing scaffold21, urea or benzoimidazole-containing scaffold22,23. Among them, two inhibitors (EPZ00477722 and EPZ567623) disclosed by Epizyme bearing urea or benzoimidazole group substituted on the side chain of the aminonucleoside were identified as SAM-competitive DOT1L selective inhibitors with high potencies (IC50=0.4 nmol/L for EPZ004777 and Ki=0.08 nmol/L for EPZ5676, respectively) (Table 1). They dose-dependently inhibited global H3K79 levels in MLL-rearranged leukemia cells, transcript levels of MLL fusion target genes such as HOXA9 and MEIS1, and the proliferation of MLL-rearranged leukemia cells, with little or no effect on non-MLL-rearranged leukemia cells. However, the poor pharmacokinetic properties (short half-life, etc) of these two compounds limited their conventional dosing in vivo, and they are used with a subcutaneously implanted mini-osmotic pump instead.
Table 1. The profiles of small molecule inhibitors of histone lysine methyltransferases (HKMTs).
| Compound | Target HKMTs | Mechanism | Biochemical potency (IC50) | Cellular potency (IC50) | In vivo activity | Fold selectivity | Status | Reference |
|---|---|---|---|---|---|---|---|---|
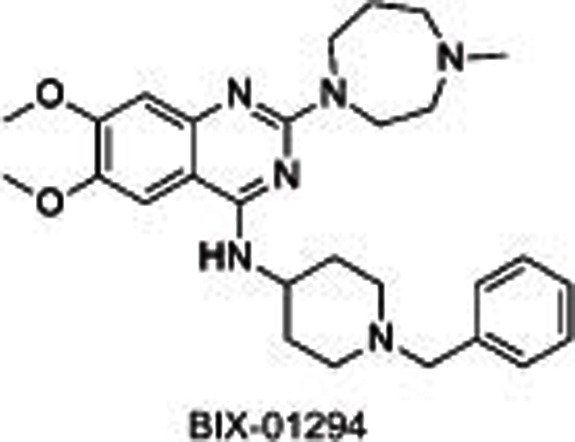 |
EHMTs | Substrate competitive | 1.9±0.1 μmol/L (EHMT2) 0.7±0.1 μmol/L (EHMT1) | 500±43 nmol/L (MDA-MB-231) | No | No inhibition observed (other HMTs) | Preclinical | 31,32,33 |
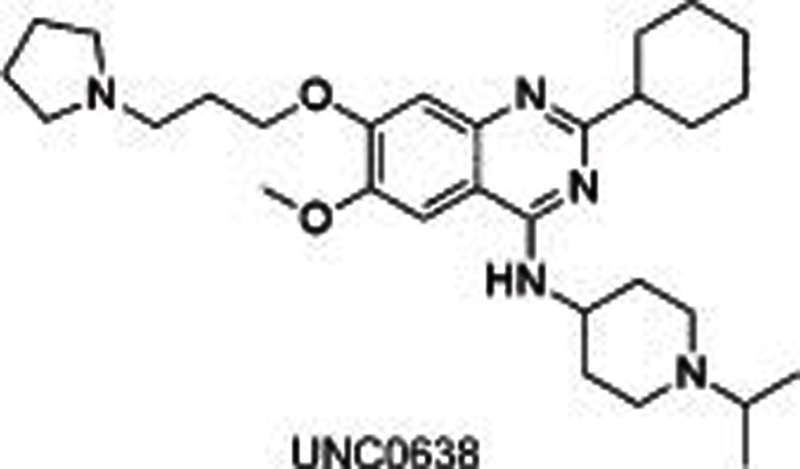 |
EHMTs | Substrate competitive | <15 nmol/L (EHMT2) 19±1 nmol/L (EHMT1) | 81±9 nmol/L (MDA-MB-231) | No | ∼500 (other HMTs) | Biological testing | 30,33 |
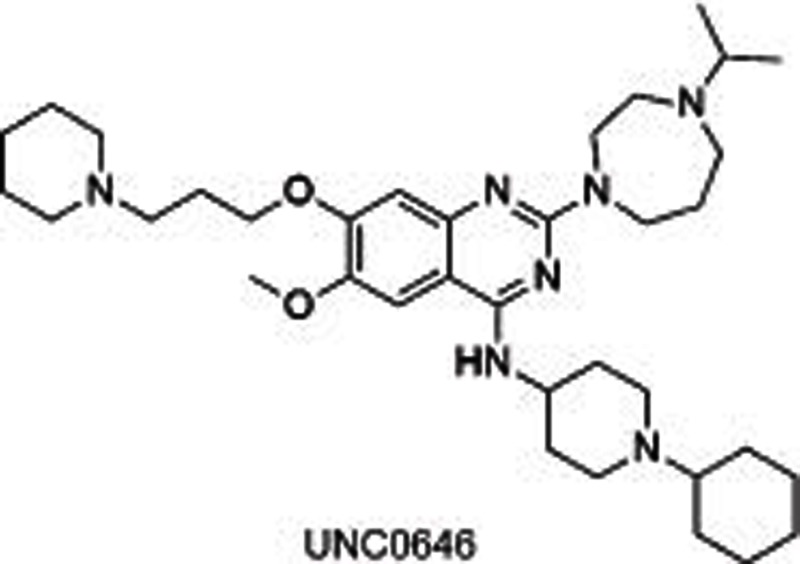 |
EHMTs | Substrate competitive | 0.006 μmol/L (EHMT2) | 0.026 μmol/L (MDA-MB-231) | No | No report | Biological testing | 34 |
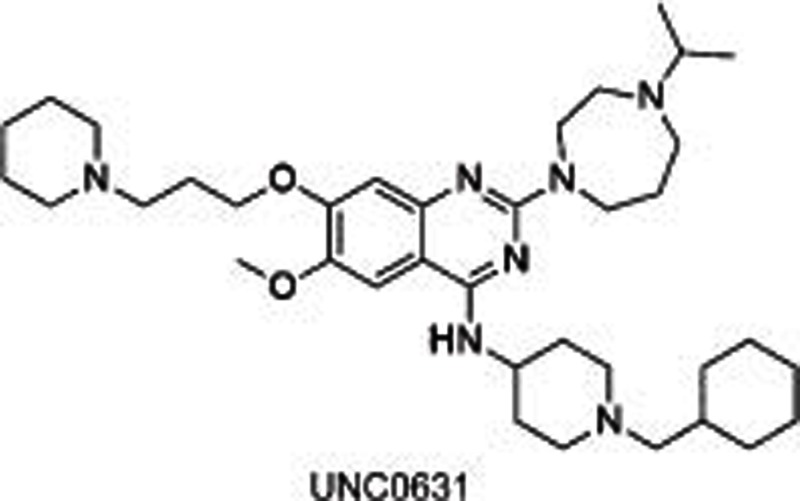 |
EHMTs | Substrate competitive | 0.004 μmol/L (EHMT2) | 0.025 μmol/L (MDA-MB-231) | No | No report | Biological testing | 34 |
 |
EHMTs | Unknown | 1.6 μmol/L (EHMT2) 1.5 μmol/L (EHMT1) | No | No | ∼100 (other 3 HMTs) | Biological testing | 35 |
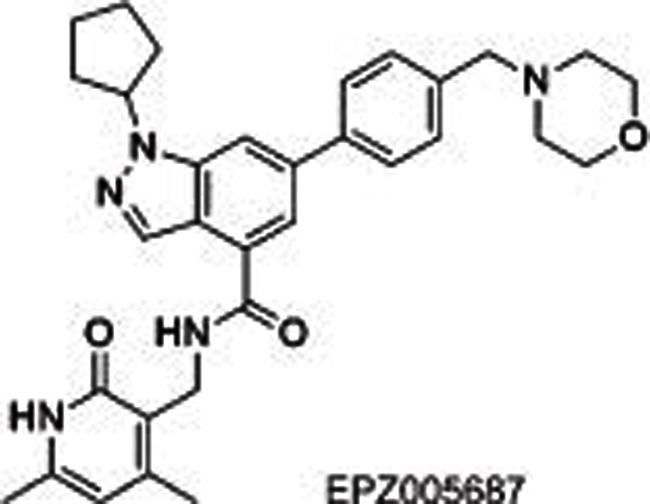 |
EZH2 | SAM competitive | 54±5 nmol/L (EZH2-PRC2) | 80±30 nmol/L (OCI-LY19 EZH2WT) | No | ∼50 (EZH1) >500 (other 15 HMTs) | Preclinical | 40 |
 |
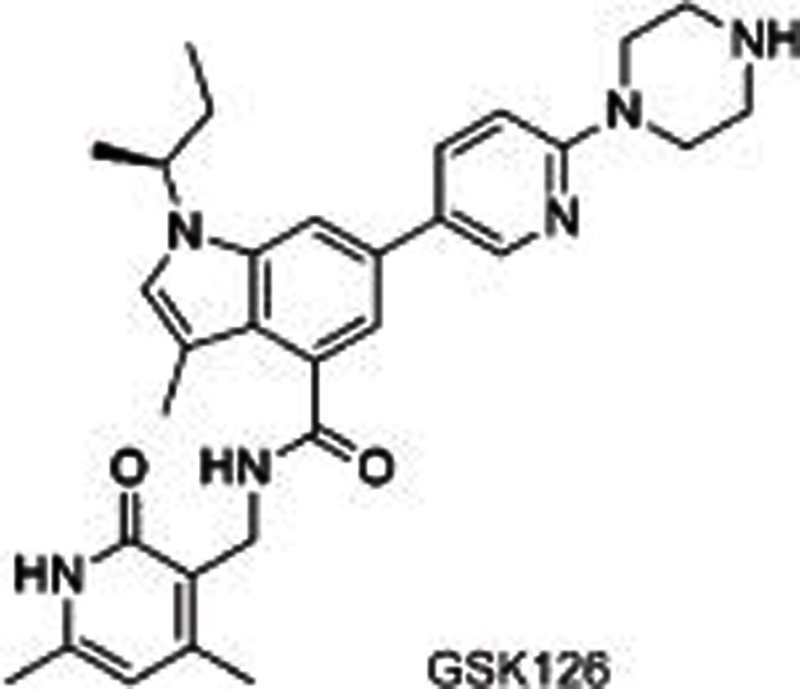
EZH2 | SAM competitive | 11±5 nmol/L (EZH2) 2–38 nmol/L (EZH2 mutants) | 8 nmol/L (OCI-LY19, EZH2WT), 2–90 nmol/L (EZH2 mutants) | Yes | ∼35 (EZH1) >4500 (other HMTs) | Phase II | 51 |
 |
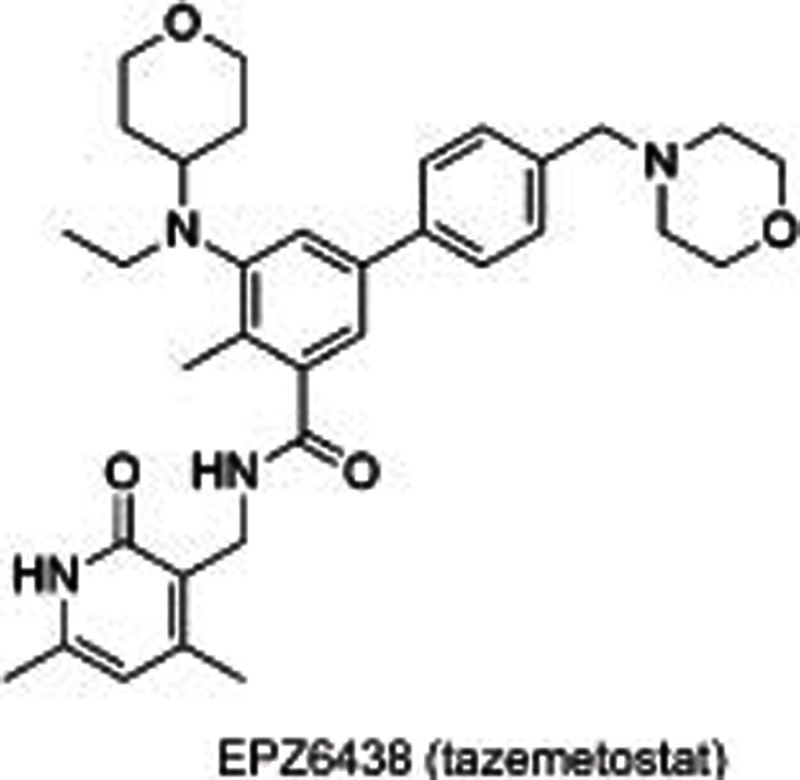
EZH2 | SAM competitive | 9.9 nmol/L (EZH2) | 28–861 nmol/L (DLBCL, EZH2 mutants) | Yes | ∼150 (EZH1) ∼1000 (other 20 HMTs) | Phase I | 49 |
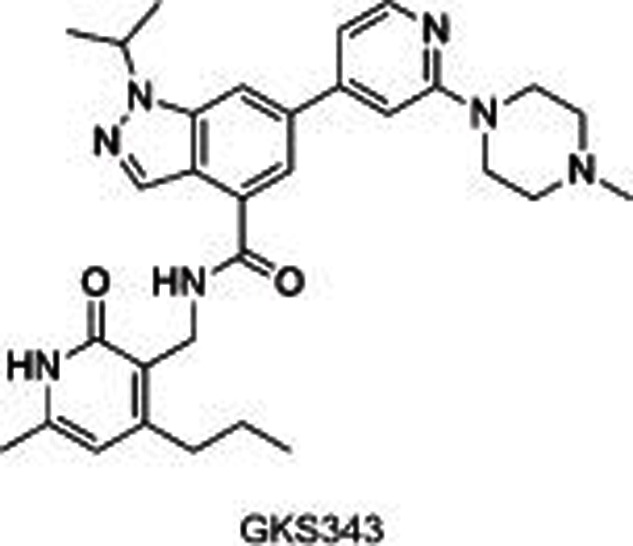 |
EZH2 | SAM competitive | 4 nmol/L (EZH2) | 174±84 nmol/L (HCC1806) | No | ∼60 (EZH1) ∼1000 (other HMTs) | Preclinical | 48 |
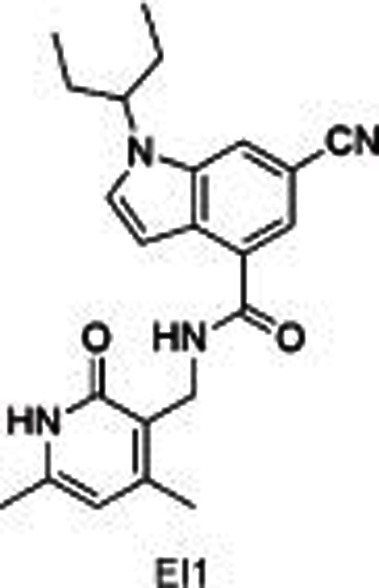 |
EZH2 | SAM competitive | 15±2 nmol/L (EZH2WT) 13±3 nmol/L (EZH2Y641F) | ∼370 nmol/L (SU-DHL6, EZH2Y641N) | No | ∼90 (EZH1) >10 000 (other HMTs) | Biological testing | 50 |
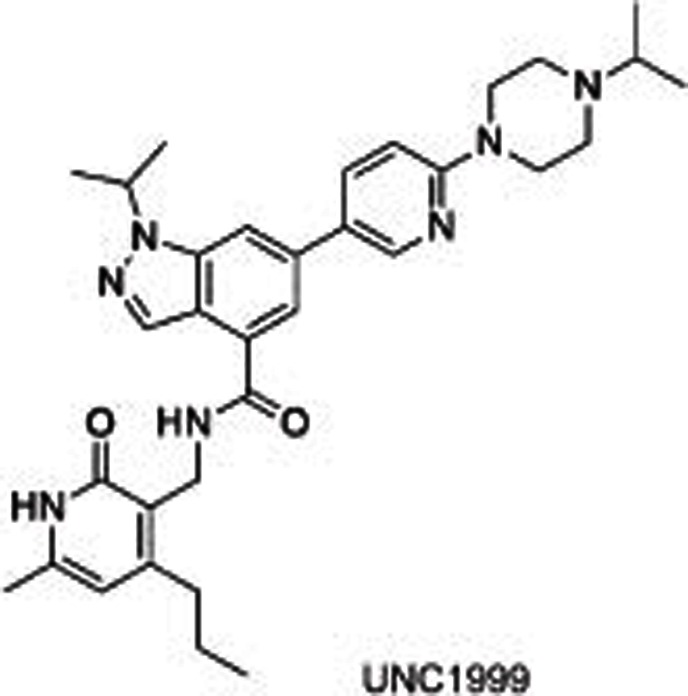 |
EZH2 | SAM competitive | <10 nmol/L (EZH2) | 124±11 nmol/L (MCF10A, EZH2WT) | Yes | ∼10 (EZH1) ∼10000 (other HMTs) | Preclinical | 47 |
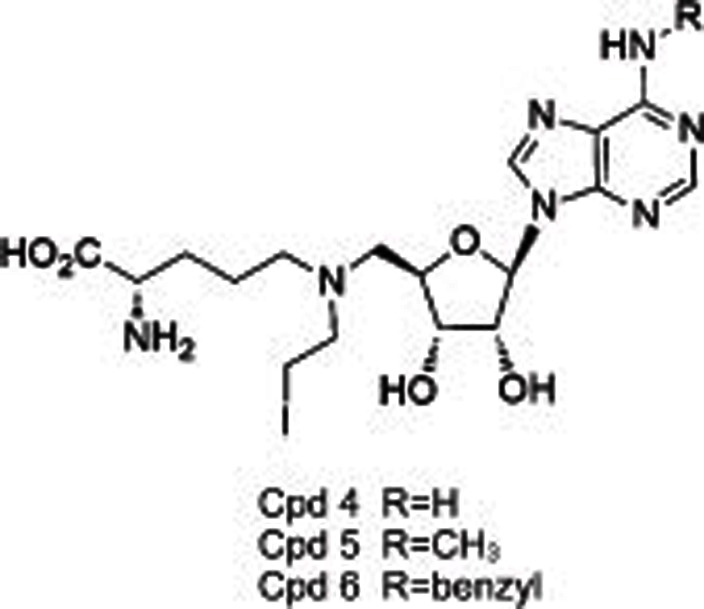 |
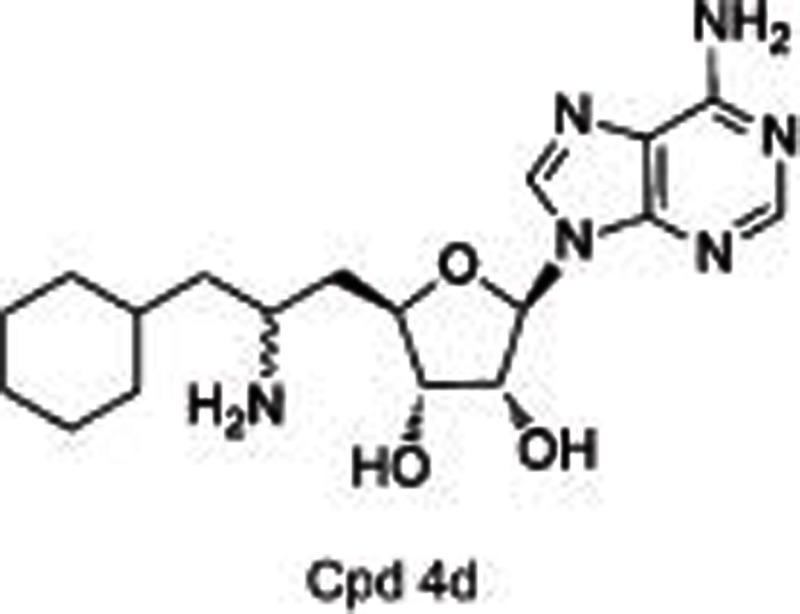
DOT1L | SAM competitive | 38 nmol/L (Cpd 4) 120 nmol/L (Cpd 5) 110 nmol/L (Cpd 6) | No | No | >29 (other 4 HMTs) | Biological testing | 21 |
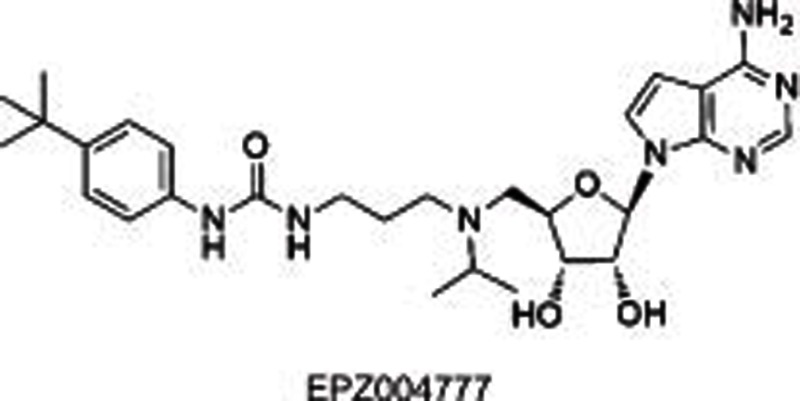 |
DOT1L | SAM competitive | 0.4±0.1 nmol/L | 0.17–6.47 μmol/L (MLL-rearranged leukemia cell lines) | Yes | >1000 (other HMTs) | Preclinical | 22 |
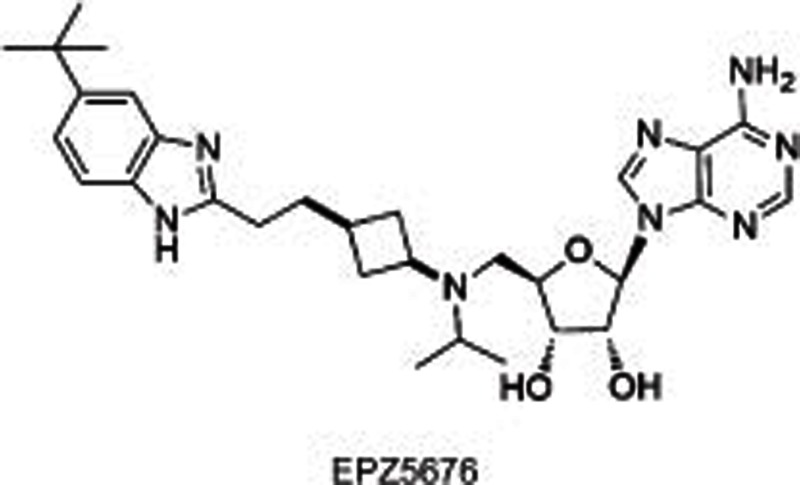 |
DOT1L | SAM competitive | Ki=0.08±0.03 nmol/L | 3.5±0.7 nmol/L (MV4-11) | Yes | >37 000 (other 15 HMTs) | Phase I | 23 |
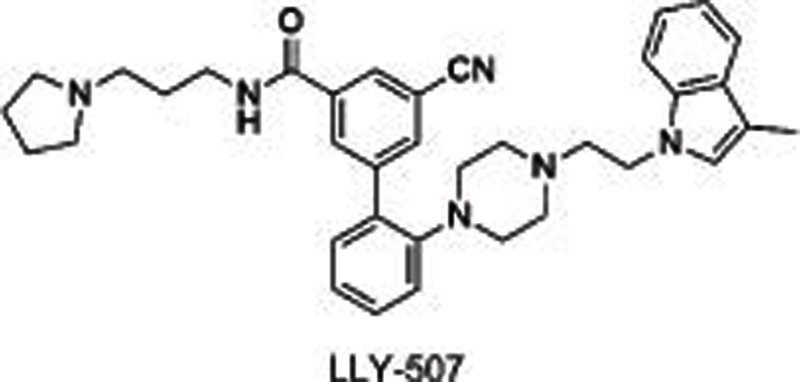 |
SMYD2 | Substrate competitive | <15 nmol/L (p53) 31 nmol/L (H4) | 0.6 μmol/L (U2OS) 0.6 μmol/L (KYSE-150) | No | >100 (other 25 HMTs and non-HMTs) | Biological testing | 56 |
 |
SMYD2 | Substrate competitive | 0.12 μmol/L | No | No | >700 (6 other HMTs) | Biological testing | 59 |
 |
SMYD2 | Substrate competitive | 2.8 nmol/L | 42% reduction of p53K370me1 (A549) | No | No significant inhibition (>50%) at 1 μmol/L for other 30 HMTs | Biological testing | 60 |
EHMT1/2
Euchromatin histone methyltransferases (EHMTs), including G9a encoded by EHMT2 and GLP (G9a-like protein) encoded by EHMT1, are an evolutionarily conserved protein family, which mono- or di-methylate H3K9 as well as other non-histone substrates such as K373 of the tumor suppressor p5324,25. EHMTs regulate transcriptional repression and activation in the form of a heterodimeric complex in the process of germ cell formation, embryogenesis and cardiac morphogenesis26,27. G9a is highly expressed in a variety of human cancers with a poor prognosis such as leukemia, prostate cancer, lung cancer, and hepatocellular carcinoma28. Reduction of G9a expression levels inhibited lung and breast cancer cell proliferation, migration and invasion in vitro and suppressed primary tumor growth and metastasis in vivo29. Renneville30 et al demonstrated in 2015 that knockdown of either EHMT1 or EHMT2 could significantly increase the expression of γ-globin genes, and the percentage of cells expressing hemoglobin F (HbF). This discovery may lead to a new therapy for sickle cell disease (SCD) because induction of HbF is a well-validated strategy, and novel epigenetic regulators such as HbF inducers are attractive targets for SCD treatment.
BIX01294, identified through a high-throughput screening, was the first reported31,32 selective small molecule inhibitor of G9a and GLP, showing an IC50 of 1.9 μmol/L for G9a and 0.7 μmol/L for GLP, separately. The crystal structure of GLP with SAH and BIX01294 demonstrated that BIX01294 did not bind to the SAM binding pocket but was in a substrate groove that was usually occupied by residues H3K4-H3R8 at the N-terminal of the target H3K9. The structure of BIX01294 consists of three parts: quinazoline, piperidine and diazepane subunits. Further structure-activity relationship (SAR) studies were conducted, including the extension of 7-methoxy into the target lysine binding channel and replacement of the benzyl group on the piperidine, represented by UNC063830,33, UNC064634 and UNC063134. All three compounds showed high potency both in a fluorescence-based SAH-coupled assay (IC50: 4–19 nmol/L) and in the H3K9me2 In-Cell Western (ICW) assay (IC50: 25–81 nmol/L), with an excellent toxicity to function ratio range of 110–140 in MDA-MB-231 (breast carcinoma) cells compared with BIX01294 (<6). UNC0638 could also dose-dependently increase human γ-globin expression, HbF expression, and mouse embryonic γ-globin gene expression without altering the cellular morphology at a concentration up to 0.25 μmol/L and without affecting cell proliferation and viability up to 0.1 μmol/L in primary adult human erythroid cells ex vivo. A combination of UNC0638 with entinostat (HDAC inhibitor) or decitabine (inhibitor of DNA methyltransferase) additively increased the gene expression of γ-globin, suggesting a different γ-globin inducing mechanism of EHMT1/2 inhibition in comparison to entinostat or decitabine30. Our group35 has also reported a group of sinefungin analogues which dose-dependently inhibited EHMT1/2 with an IC50 of 1.5–1.6 μmol/L. However, it was not competitive with SAM or substrate, which is indicative of other binding sites in EHMT1/2.
EZH2 and PRC2
The polycomb repressive complex 2 (PRC2), which is responsible for mono-, di-, and tri-methylation of H3K27, is composed of EED (embryonic ectoderm development), SUZ12, RbAp48 and an enzymatic subunit of either EZH2 (enhancer of zeste homologue 2, also known as KMT6 or KMT6A) or its homolog EZH1 (enhancer of zeste homologue 1, also known as KMT6B), which shares 96% sequence identity in the SET domains with EZH236. EZH2 overexpression is implicated in several tumor types correlated with poor prognosis such as prostate cancer, breast cancer, myeloma, hepatocellular carcinoma and gastric cancer37. Mutations of the Y641 residue within the catalytic SET domain of EZH2 occur in 22% of diffuse large B-cell lymphomas (DLBCLs) and follicular lymphomas (FL)38,39, and 8%–24% of non-Hodgkin lymphomas (NHL)40. Homology modeling41,42 and the solved apo-structure of the EZH2 SET domain43,44 have disclosed Y641 as a key amino acid for substrate specificity. Mutations of Y641 into other smaller amino acids, such as phenylalanine (F), serine (S), histidine (H) and cysteine (C) resulted in changes in pocket dimensions, as well as hydrogen bonding, which allowed the free rotation of H3K27me2 and subsequent tri-methylation to H3K27me3 (a transcriptionally repressive mark) and led to repression of key tumor suppressor genes45,46. Therefore, EZH2 and PRC2 have been proposed as attractive drug targets for cancer therapy, which has inspired several pharmaceutical companies to launch high-throughput screening campaigns to discover inhibitors of EZH2.
Several selective EZH2/1 inhibitors have been reported, which could be divided into 3 series according to their different structures: pyridone-indazole scaffold (EPZ005687, UNC1999, and GSK343), pyridone-indole scaffold (GSK126 and EI1), and pyridone-phenyl scaffold (EPZ006088 and EPZ6438). EPZ005687 was the first small molecule reported by Epizyme40 as a selective inhibitor of EZH2, with approximately 50-fold potent selectivity for EZH1 and more than 150-fold selectivity for 15 other HMTs. Docking results for EPZ005687 from the homology model of EZH2 built using GLP as a template indicated key hydrogen bonds (the central amide and N688, pyridone and H689) and hydrophobic interactions of the pyridone-indazole core with EZH2, excluding the morpholinomethyl group exposed to the solvent. Further structural modifications surrounding the morpholinomethyl moiety led to UNC199947 (N-isopropyl piperazine instead), with a desirable physicochemical property (clogP=3.1) that increased its oral bioavailability. It is worth noting that UNC1999 is a SAM-competitive, dual inhibitor of EZH2 and EZH1 (selectivity: ∼10-fold), giving it an advantage in killing cancer cells such as MLL-rearranged acute leukemia, where H3K27 is methylated by both PRC2-EZH2 and PRC2-EZH1. GSK34348, with an n-propyl group substituted on the 4-pyridone core, selectively inhibited EZH2 with an IC50 of 4 nmol/L. However, the high clearance in rat pharmacokinetics studies limited its use in vivo.
The representatives of a pyridone-indole scaffold such as GSK126 and EI1, which were reported almost at the same time as EPZ005687, were identified as selective and SAM-competitive inhibitors of EZH2. Different from EPZ005687, GSK126 showed in vivo activity, ie, subcutaneous dosing in nude mice with xenografts of KARPAS-422 and Pfeiffer cells (mutant DLBCL cell lines) dose-dependently decreased the level of global H3K27me349. The structure of EI1 was reported to be similar to GSK126, with a cyano instead of a pyridine-piperazine group. It inhibited the proliferation of EZH2 mutant DLBCL cell lines with no effect on wild types50.
Another pyridone-phenyl scaffold represented by EPZ00608851 and EPZ643851 was reported by Epizyme in 2016. Modeling of EPZ005687 indicated that the amide group joining pyridone was out of plane owing to the forces of the indazole group, which presents a surrogate pyridone-phenyl core with a methyl group adjacent to the amide providing the possibility of steric-directing effects both on the amide and aniline. EPZ6438 (tazemetostat) was thus discovered with THP substituted on the aniline core. EPZ6438 dose-dependently inhibited H3K27Me3 levels and cell proliferation in human lymphoma cell lines including EZH2WT and EZH2 mutants (A682 and Y646). Further in vivo assays in mice showed low clearance and desirable bioavailability, which led to clinical trials in patients with lymphoma (DLBCL and FL) and INI1-deficient solid tumors51.
SMYD2
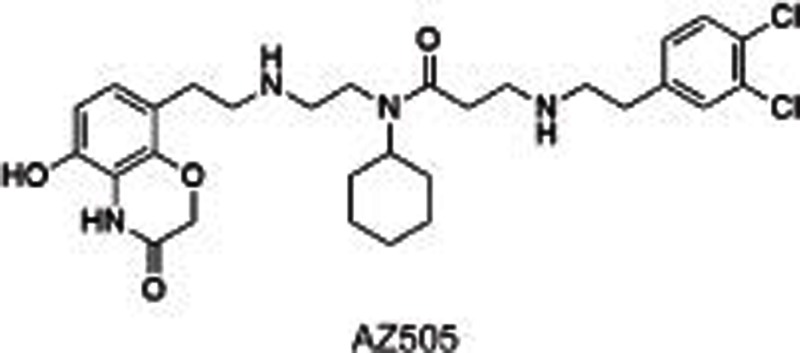
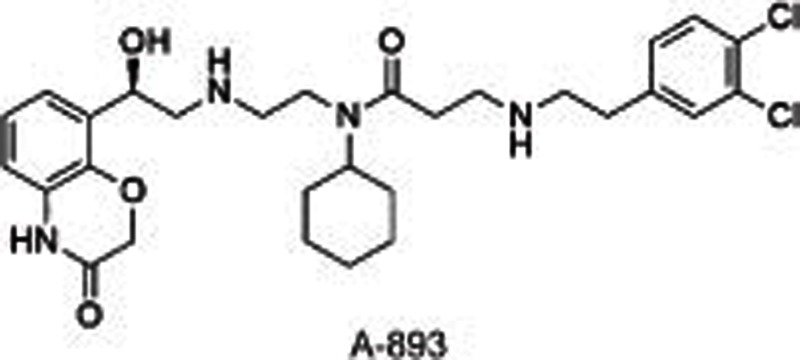
SMYD2, a SET and MYND domain containing HKMT, methylates H2B, H3, H452,53, and other non-histone substrates, including retinoblastoma tumor suppressor RbK860 and RbK81054 as well as tumor suppressors p53K37055 and HSP9056. SMYD2 is highly expressed in 75% of esophageal squamous cell carcinomas (ESCCs)57 and 95% of pediatric acute lymphoblastic leukemias58 with poor survival. All of these characteristics point to its potential as a drug target to treat cancers. Several small molecule inhibitors have been developed to investigate the roles of SMYD2 in carcinoma and other biological processes. LLY-50756 was disclosed as a selective, substrate-competitive inhibitor for SMYD2 with more than 100-fold selectivity over other HMTs and non-HMTs, including its homolog SMYD3. Cell-based assays showed that LLY-507 dose-dependently inhibited the expression of p53K370me1 in U2OS and KYSE-150 cells (ESCC cell lines) and the proliferation of ESCC, HCC and breast cancer cells. However, no obvious changes were observed in the global histone methylation levels using mass spectrometry-based proteomics, suggesting that SMYD2 is mainly present in the cytoplasm. Two other inhibitors, AZ505 and A-893, have also been reported59,60. AZ505 and A-893 have similar structures except for one hydroxy group translocating from the benzoxazinone in AZ505 to the linker in A-893. Both of these inhibitors were shown to be selective, substrate-competitive SMYD2 inhibitors. Cell-based assays indicated that these two compounds reduced the expression levels of p53K370me1 in human A549 lung carcinoma cells. The co-crystal structure of SMYD2 with AZ505/A-893 suggested that two hydrophobic pockets are present: a primary pocket occupied by the cyclohexyl group and a secondary pocket occupied by the dichlorophenyl moiety, with the benzoxazinone group occupying the lysine-binding channel. An additional hydrogen bond between the benzylic alcohol and Y240 was formed in the co-crystal structure of SMYD2 with A-893, which contributes to the increased potency of A-893 compared to AZ505 (>80-fold).
Conclusion
The human histone methyltransferases (HMTs) are a large class of enzymes (>50), many of which are associated with cell differentiation, gene regulation and DNA recombination. Overexpression of HMTs has been linked to a variety of human malignancies. Owing to their common characteristics in chemical catalytic reactions, HMTs are likely to be tractable as targets for drug discovery. At this point, several potent small molecules have been discovered and show inhibitory activities in vitro and in vivo on various cancer cells. Such inhibitors have been used either as chemical probes to study enzyme functions or as candidates for drug development. Although histone methylation has been studied extensively, investigation of non-histone methylation is still in its infancy, which calls for further systematic research.
Acknowledgments
We gratefully acknowledge the financial support from the National Health and Family Planning Commission (2012ZX09304-011, 2013ZX09401003-005, 2013ZX09507001, 2013ZX09507-002, and 2014ZX09507002-001), the National Natural Science Foundation of China (21302202), Shanghai Science and Technology Development Fund (15DZ2291600), and the Thousand Talents Program in China.
References
- Cole PA. Chemical probes for histone-modifying enzymes. Nat Chem Biol 2008; 4: 590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler BR, Archer TK. Chromatin-modifying enzymes as therapeutic targets - Part 1. Expert Opin Ther Targets 2008; 12: 1301–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128: 683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol 2009; 9: 91–105. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 2007; 8: 355–67. [DOI] [PubMed] [Google Scholar]
- Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol 2007; 14: 1008–16. [DOI] [PubMed] [Google Scholar]
- Allfrey VG, Mirsky AE. Structural modifications of histones and their possible role in the regulation of RNA synthesis. Science 1964; 144: 559. [DOI] [PubMed] [Google Scholar]
- Murray K. The occurrence of epsilon-N-methyl lysine in histones. Biochemistry 1964; 3: 10–5. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004; 119: 941–53. [DOI] [PubMed] [Google Scholar]
- Fowler B. Homocysteine: overview of biochemistry, molecular biology, and role in disease processes. Semin Vasc Med 2005; 5: 77–86. [DOI] [PubMed] [Google Scholar]
- Cantoni GL. Biological methylation: selected aspects. Annu Rev Biochem 1975; 44: 435–51. [DOI] [PubMed] [Google Scholar]
- Wu JC, Santi DV. Kinetic and catalytic mechanism of HhaI methyltransferase. J Biol Chem 1987; 262: 4778–86. [PubMed] [Google Scholar]
- Mandaviya PR, Stolk L, Heil SG. Homocysteine and DNA methylation: a review of animal and human literature. Mol Genet Metab 2014; 113: 243–52. [DOI] [PubMed] [Google Scholar]
- Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Disc 2009; 8: 724–32. [DOI] [PubMed] [Google Scholar]
- Kagan RM, Clarke S. Widespread occurrence of 3 sequence motifs in diverse S-adenosylmethionine-dependent methyltransferases suggests a common structure for these enzymes. Arch Biochem Biophys 1994; 310: 417–27. [DOI] [PubMed] [Google Scholar]
- Cheng X, Collins RE, Zhang X. Structural and sequence motifs of protein (histone) methylation enzymes. Annu Rev Biophys Biomol Struct 2005; 34: 267–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Feng Q, Li Z, Zhang Y, Xu RM. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 2003; 112: 711–23. [DOI] [PubMed] [Google Scholar]
- Trievel RC, Flynn EM, Houtz RL, Hurley JH. Mechanism of multiple lysine methylation by the SET domain enzyme Rubisco LSMT. Nat Struct Biol 2003; 10: 545–52. [DOI] [PubMed] [Google Scholar]
- Anglin JL, Deng L, Yao Y, Cai G, Liu Z, Jiang H, et al. Synthesis and structure-activity relationship investigation of adenosine-containing inhibitors of histone methyltransferase DOT1L. J Med Chem 2012; 55: 8066–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stulemeijer IJ, De Vos D, van Harten K, Joshi OK, Blomberg O, van Welsem T, et al. Dot1 histone methyltransferases share a distributive mechanism but have highly diverged catalytic properties. Sci Rep 2015; 5: 9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Chen PH, Diao JH, Cheng G, Deng L, Anglin JL, et al. Selective inhibitors of histone methyltransferase DOT1L: design, synthesis, and crystallographic studies. J Am Chem Soc 2011; 133: 16746–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011; 20: 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack-Sjodin PA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013; 122: 1017–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath SC, Marazzi I, Yap KL, Sampath SC, Krutchinsky AN, Mecklenbräuker I, et al. Methylation of a histone mimic within the histone methyltransferase G9a regulates protein complex assembly. Mol Cell 2007; 27: 596–608. [DOI] [PubMed] [Google Scholar]
- Huang J, Dorsey J, Chuikov S, Pérez-Burgos L, Zhang X, Jenuwein T, et al. G9a and GLP methylate lysine 373 in the tumor suppressor p53. J Biol Chem 2010; 285: 9636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagawa M, Nakajima K, Makino T, Ogawa S, Kojima M, Ito S, et al. Histone H3 lysine 9 methyltransferases, G9a and GLP are essential for cardiacmorphogenesis. Mech Dev 2013; 130: 519–31. [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev 2011; 25: 781–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artal-Martinez de Narvajas A, Gomez TS, Zhang JS, Mann AO, Taoda Y, Gorman JA, et al. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol Cell Biol 2013; 33: 3983–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Dorsey J, Chuikov S, Pérez-Burgos L, Zhang X, Jenuwein T, et al. G9a and GLP methylate lysine 373 in the tumor suppressor p53. J Biol Chem 2010; 285: 9636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renneville A, Van Galen P, Canver MC, McConkey M, Krill-Burger JM, Dorfman DM, et al. EHMT1 and EHMT2 inhibition induces fetal hemoglobin expression. Blood 2015; 126: 1930–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek S, O'Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell 2007; 25: 473–81. [DOI] [PubMed] [Google Scholar]
- Chang YQ, Zhang X, Horton JR, Upadhyay AK, Spannhoff A, Liu J, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat Struct Mol Biol 2009; 16: 312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol 2012; 7: 566–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Barsyte-Lovejoy D, Allali-Hassani A, He Y, Herold JM, Chen X, et al. Optimization of cellular activity of G9a inhibitors 7-aminoalkoxy-quinazolines. J Med Chem 2011; 54: 6139–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devkota K, Lohse B, Liu Q, Wang MW, Stærk D, Berthelsen J, et al. Analogues of the natural product sinefungin as inhibitors of EHMT1 and EHMT2. ACS Med Chem Lett 2014; 5: 293–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 2008; 32: 503–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res 2008; 647: 21–9. [DOI] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 2010; 42: 181–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, et al. Mutation of A677 in histonemethyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A 2012; 109: 2989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 2012; 8: 890–6. [DOI] [PubMed] [Google Scholar]
- McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, et al. Mutation of A677 in histone methyltransferase EZH2 in B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A 2012; 109: 2989–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011; 117: 2451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonysamy S, Condon B, Druzina Z, Bonanno JB, Gheyi T, Zhang F, et al. Structural context of disease-associated mutations and putative mechanism of autoinhibition revealed by X-ray crystallographic analysis of the EZH2-SET domain. PLoS One 2013; 8: e84147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Zeng H, Dong A, Li F, He H, Senisterra G, et al. Structure of the catalytic domain of EZH2 reveals conformational plasticity in cofactor and substrate binding sites and explains oncogenic mutations. PLoS One 2013; 8: e83737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz KW, Campbell JE, Keilhack H, Pollock RM, Knutson SK, Porter-Scott M, et al. The importance of being Me: magic methyls, methyltransferase inhibitors, and the discovery of Tazemetostat. J Med Chem 2016; 59: 1556–64. [DOI] [PubMed] [Google Scholar]
- Paschall AV, Yang DF, Lu C, Choi JH, Li X, Liu F, et al. H3K9 trimethylation silences Fas expression to confer colon carcinoma immune escape and 5-fluorouracil chemoresistance. J Immunol 2015; 195: 1868–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem Biol 2013; 8: 1324–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma SK, Tian XR, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, et al. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med Chem Lett 2012; 3: 1091–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012; 492: 108–12. [DOI] [PubMed] [Google Scholar]
- Qi W, Chan HM, Teng L, Li L, Chuai S, Zhang R, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A 2012; 109: 21360–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz KW, Campbell JE, Keilhack H, Pollock RM, Knutson SK, Porter-Scott M, et al. The importance of being Me: magic methyls, methyltransferase inhibitors, and the discovery of Tazemetostat. J Med Chem 2016; 59: 1556–64. [DOI] [PubMed] [Google Scholar]
- Brown MA, Sims RJ 3rd, Gottlieb PD, Tucker PW. Identification and characterization of Smyd2: a split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol Cancer 2006; 5: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Farha M, Lambert JP, Al-Madhoun AS, Elisma F, Skerjanc IS, Figeys D. The tale of two domains: proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol Cell Proteomics 2008; 7: 560–72. [DOI] [PubMed] [Google Scholar]
- Saddic LA, West LE, Aslanian A, Yates JR 3rd, Rubin SM, Gozani O, et al. Methylation of the retinoblastoma tumor suppressor by SMYD2. J Biol Chem 2010; 285: 37733–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, et al. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006; 444: 629–32. [DOI] [PubMed] [Google Scholar]
- Nguyen H, Allali-Hassani A, Antonysamy S, Chang S, Chen LH, Curtis C, et al. LLY-507, a cell-active, potent, and selective inhibitor of protein-lysine methyltransferase SMYD2. J Biol Chem 2015; 290: 13641–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu S, Imoto I, Tsuda H, Kozaki KI, Muramatsu T, Shimada Y, et al. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis 2009; 30: 1139–46. [DOI] [PubMed] [Google Scholar]
- Sakamoto LH, Andrade RV, Felipe MS, Motoyama AB, Pittella Silva F. SMYD2 is highly expressed in pediatric acute lymphoblastic leukemia and constitutes a bad prognostic factor. Leuk Res 2014; 38: 496–502. [DOI] [PubMed] [Google Scholar]
- Ferguson AD, Larsen NA, Howard T, Pollard H, Green I, Grande C, et al. Structural basis of substrate methylation and inhibition of SMYD2. Structure 2011; 19: 1262–73. [DOI] [PubMed] [Google Scholar]
- Sweis RF, Wang Z, Algire M, Arrowsmith CH, Brown PJ, Chiang GG, et al. Discovery of A-893, a new cell-active benzoxazinone inhibitor of lysine methyltransferase SMYD2. ACS Med Chem Lett 2015; 6: 695–700. [DOI] [PMC free article] [PubMed] [Google Scholar]