

ABSTRACT
Whole mitochondrial (mt) genome analysis enables a considerable increase in analysis throughput, and improves the discriminatory power to the maximum possible phylogenetic resolution. Most established protocols on the different massively parallel sequencing (MPS) platforms, however, invariably involve the PCR amplification of large fragments, typically several kilobases in size, which may fail due to mtDNA fragmentation in the available degraded materials. We introduce a MPS tiling approach for simultaneous whole human mt genome sequencing using 161 short overlapping amplicons (average 200 bp) with the Ion Torrent Personal Genome Machine. We illustrate the performance of this new method by sequencing 20 DNA samples belonging to different worldwide mtDNA haplogroups. Additional quality control, particularly regarding the potential detection of nuclear insertions of mtDNA (NUMTs), was performed by comparative MPS analysis using the conventional long‐range amplification method. Preliminary sensitivity testing revealed that detailed haplogroup inference was feasible with 100 pg genomic input DNA. Complete mt genome coverage was achieved from DNA samples experimentally degraded down to genomic fragment sizes of about 220 bp, and up to 90% coverage from naturally degraded samples. Overall, we introduce a new approach for whole mt genome MPS analysis from degraded and nondegraded materials relevant to resolve and infer maternal genetic ancestry at complete resolution in anthropological, evolutionary, medical, and forensic applications.
Keywords: mitochondria, mtDNA, next‐generation sequencing, NGS, massively parallel sequencing, MPS
Introduction
Previous years have witnessed conspicuous progress in the establishment of the high‐copy‐number human mitochondrial DNA (mtDNA) as an imperative tool in forensic, anthropological, and medical genetics. This small, circular, double‐stranded genome has enthralled fundamental and applied geneticists with its unique features, bolstering its recognized value as a useful molecular marker when analyzing degraded or low‐copy‐number DNA samples, as often confronted at crime scenes, from old and ancient human remains, and from limited samples available in medical and anthropological applications. The higher mutation rate of mtDNA as compared with nuclear DNA, and the resulting significant sequence variability between maternally unrelated individuals, has made mtDNA an indispensable tool in various applications particularly forensics and anthropology [Wilson et al., 1995a, 1995b; Holland and Parsons, 1999; Kivisild, 2015]. Most applications employ traditional Sanger sequencing of (the hypervariable portions of) the noncoding control region of the mt genome to delineate haplotypes that enable maternal lineage identification [e.g., Gill et al., 1994; Wilson et al., 1995a; Holland and Parsons, 1999; Brandstätter et al., 2007; Palo et al., 2007]. In addition to being labor‐intensive and expensive, the control‐region Sanger sequencing approach does not always allow reliable inference of maternal haplogroups, due to the high level of homoplasy in the control region that can obscure phylogenetic signatures [Schlebusch et al., 2009; King et al., 2014] as well as the fact that many haplogroup‐defining variants are located outside the control region. Since not all the major mtDNA haplogroups can be distinguished through control‐region sequencing only, several studies [Schlebusch et al., 2009; van Oven et al., 2011; Ballantyne et al., 2012] have resorted to the use of multiplex single‐base primer extension assays for simultaneous genotyping of a limited number of mtDNA coding‐region SNPs to improve maternal haplogroup definition. However, such assays are hampered by technical limitations in terms of multiplexing capacity allowing not more than 20–40 SNPs in a single multiplex assay.
Whole mtDNA genome sequencing would augment the haplotype and haplogroup resolution to the maximum possible resolution level, thereby greatly enhancing the discriminatory power, and allowing inference of matrilineal biogeographic ancestry at a greater resolution; however, whole mtDNA genome analysis via Sanger sequencing is highly labor‐intensive given the >16 kb involved [Fendt et al., 2009; Ramos et al., 2009]. Massively parallel sequencing (MPS) or next‐generation sequencing (NGS) technology in principle provides a solution to simultaneous whole mtDNA genome analysis allowing high‐throughput analysis at reduced per‐sample costs. It had been pioneered in the field of ancient DNA analysis where degradation problems are most severe. The shotgun‐sequencing approach using MPS technology has facilitated the complete mtDNA genome analysis of extinct nonhuman species such as mammoths [Gilbert et al., 2007, 2008], cave bears, and others [Miller et al., 2009; Stiller et al., 2009; Willerslev et al., 2009; Lindqvist et al., 2010; Ho & Gilbert, 2010] as well as extinct human species such as Neanderthals [Green et al., 2008]. Recently, Davis et al. (2015) used the MPS approach to analyze the hypervariable segments (HVS)‐I and II of human mtDNA using short amplicons in tissue and bone samples keeping with the previously discussed limitations of partial mtDNA analysis. However, the most established currently available MPS protocols for whole mtDNA genome sequencing are based on PCR amplification of large mtDNA fragments, typically several kilobases in size [Sosa et al., 2012; Parson et al., 2013], which fail when applied to degraded DNA as encountered in many mtDNA applications. A very recent publication [Parson et al., 2015] described a midi‐sized amplicon approach for whole mtDNA MPS analysis using 62 PCR amplicons of 300–500 bp (average about 380 bp) in two multiplex assays, which proved useful for human hair analysis. However, many DNA samples encountered in mtDNA testing, especially for forensic and anthropological purposes are more severely degraded resulting in smaller‐sized fragments.
Recently, we introduced a MPS tool based on short amplicons (203 bp on average) using the Ion Torrent Personal Genome Machine (PGMTM) for simultaneous analysis of >530 Y‐chromosome SNPs covering the entire phylogenetic Y‐chromosome tree that allows classification of Y chromosomes into >430 worldwide Y haplogroups for ultra‐high‐resolution paternal lineage and paternal ancestry inference [Ralf et al., 2015]. As a maternal counterpart, with this study we introduce a short amplicon‐based MPS tiling approach for simultaneous whole mtDNA genome analysis using the Ion Torrent PGMTM allowing to obtain maximum‐resolution maternal lineage and maternal ancestry inference from degraded and nondegraded DNA. We tested the performance of the newly developed method using geographically diverse DNA samples with available whole mtDNA genome data based on Sanger sequencing, and using samples for which we generated whole mtDNA genome data based on an alternative MPS protocol. Additionally, we assessed the efficacy of sample pooling, the sensitivity, and the robustness of our new approach regarding experimental and natural DNA degradation.
Materials and Methods
Primer Design
Primer pairs were designed for a total of 161 partially overlapping amplicons in two separate primer pools (pool 1 with 80 pairs and pool 2 with 81 pairs). Each individual primer pool produces a battery of amplicons separated by gaps across the entire mt genome. By combining the PCR amplicons resulting from each primer pool, the gaps were complemented by amplicons from the other primer pool and the entire 16.5‐kb mt genome could be covered. Amplicons of comparable sizes were preferred (∼200 bp); however, some regions proved to be difficult to amplify because of, for example, high GC content or repetitive sequences. Also, highly polymorphic positions need to be avoided to overlap with primer annealing sites as much as possible; to overcome these issues, primer positions had to be shifted leading to more variation in amplicon size. Several rounds of test sequencing and primer redesigning were needed to get to the final protocol. All primers were synthesized by Life Technologies, a part of Thermo Fisher Scientific Inc., using their proprietary AmpliSeq™ modification, which allows multiplexing hundreds or even thousands of targets in one reaction. The primers were all received at a concentration of 614 μM. For each pool, 1 μl of each primer is combined and each pool is then brought to a volume of 3,070 μl. Each primer is now at a concentration of 200 nM. For the 20‐μl PCR, 10 μl of primer pool is used, so the final concentration in the PCR is 100 nM for each primer. Supp. Table S1 outlines the primer sequences used in this study without the proprietary modification. In the final version, and with the primer pools 1 and 2 together, the entire human mtDNA genome is covered via 161 overlapping amplicons with fragment size of 144–230 bp (average across amplicons: 200 bp).
DNA Samples
With the aim to cover the global variation of the mtDNA phylogeny [van Oven and Kayser 2009], 20 DNA samples known to belong to widely divergent haplogroups were selected from four different sources: the Centre d'Etude du Polymorphisme Humain (CEPH, Paris, France) Human Genome Diversity Project (HGDP) panel (http://www.ceph.fr/HGDP‐CEPH‐Panel), individuals from across The Netherlands [Lao et al., 2013], the commercially available Ethnic Diversity DNA panel (EDP‐1) manufactured by the European Collection of Cell Cultures (ECACC) (http://www.phe‐culturecollections.org.uk/products/dna/ethnicdna.jsp), and distributed by Sigma, and volunteers recruited at the Erasmus MC with informed consent.
Library Construction
DNA libraries were constructed with 10 ng of input DNA using the Ion Ampliseq™ Library Kit 2.0 (Life Technologies, a part of Thermo Fisher Scientific Inc., Foster City, CA) and 18 cycles with two separate primer pools, amplifying the 161 amplicons simultaneously, following the manufacturer's recommendations. The quantity of DNA was determined using the Qubit® dsDNA BR Quantification Kit and a Qubit® 2.0 Fluorometer (Invitrogen, Life Technologies, a part of Thermo Fisher Scientific Inc., Grand Island, NY). In order to test the feasibility of sequencing multiple samples simultaneously, Ion Xpress™ Barcode Adapters (Life Technologies, a part of Thermo Fisher Scientific Inc.) were used.
To evaluate the sensitivity of the tiling approach, variable amounts of DNA input ranging from 10 ng to 100 pg were run and their sequences were evaluated. Three of the 20 DNA samples, hailing from different populations—Italy (haplogroup, T2b), The Netherlands (haplogroup, V3c), and South Africa (haplogroup, L3d3a1a), were sequenced at 10 and 1 ng, 500, 250, and 100 pg of genomic DNA input. All the DNA dilutions were quantified and confirmed in duplicates with the Quantifiler® Human DNA Quantification kit (Applied Biosystems, Foster City, CA) following the manufacturer's guidelines.
The robustness of the assay to successfully type degraded materials was tested by subjecting an additional DNA sample (in this case from an Indian individual belonging to mtDNA haplogroup M35a) to DNase I treatment at different time intervals: 5, 10, 15, 20, 30, and 45 min. Three microliter of each of these DNase‐treated samples was used for the MPS. Furthermore, sample 15 (from Italy with haplogroup T2b) was subjected to two different degradation methods—exposure to ultraviolet (UV) light for 30 min using a Bio‐Link (Vilber Lourmat) at a strength of 50 J/cm2 and enzymatic shearing using the Ion Shear™ Plus Reagents Kit (Life Technologies, a part of Thermo Fisher Scientific Inc.). Three microliter of each of the degraded samples was used for the NGS.
In addition, we applied our whole mt genome MPS method to DNA extracted from six different bones and teeth samples from naturally degraded human remains found in Poland. Samples 1, 2, 4, 5, and 6 were extracted from teeth, whereas sample 3 was extracted from a femoral bone. Samples 1, 3, 4, and 5 gave a full profile with the AmpFℓSTR® NGM™ (Applied Biosystems), whereas samples 2 and 6 gave partial profiles with only 4 and 6 loci, respectively, out of the 16 loci (15 STRs plus amelogenin) of a full NGM profile. For samples 4, 5, and 6, the control region was analyzed via the Sanger sequencing (unpublished study). The remains that gave rise to sample 2 are believed to be from an archaeological stand from the XV–XVI century. Sample 5 likely comes from a recently deceased individual as decomposed soft tissue was found together with the hard tissues. The remains that gave rise to sample 1 were found in soil and the time since death was reported to be approximately 7–8 months, whereas those of sample 3 were found in a river. The samples 1–6 were PCR‐quantified using the Quantifiler® Human DNA Quantification Kit (Applied Biosystems) and their concentrations were 0.05, 0.1, 0.045, 5.4, 1.82, and 0.4 ng/μl, respectively. For MPS, we used 4 μl of each of the samples.
Furthermore, as a comparison to currently available NGS assays, five out of the 20 DNA samples were also sequenced using a different library preparation method. The entire mtDNA genome was amplified with two overlapping >8 kb fragments using the amplification primers from Fendt et al. (2009). The SequalPrep™ Long PCR Kit with dNTPs (Invitrogen) was used for the long‐range amplification following the manufacturer's guidelines. The amplified samples were quantified using the Qubit® dsDNA BR Quantification Kit and a Qubit® 2.0 Fluorometer (Invitrogen) and the amplified samples were normalized to 100 ng of input DNA. Libraries were constructed using the Ion Shear™ Plus Reagents Kit (Life Technologies, a part of Thermo Fisher Scientific Inc.) for enzymatic fragmentation of DNA, followed by barcoded adapter ligation using the Ion Xpress™ Barcode Adapters (Life Technologies, a part of Thermo Fisher Scientific Inc.) and Ion Plus Fragment Library Kit (Life Technologies, a part of Thermo Fisher Scientific Inc.) as per the manufacturer's protocol. The libraries were size selected using the E‐Gel® SizeSelect™ 2% Agarose Gel (Invitrogen, Carlsbad, CA).
Template Preparation
Using emulsion PCR, the generated libraries are attached to beads and further amplified. The concentration of each of the libraries was determined with qPCR using the Ion Library Quantitation Kit (Life Technologies, a part of Thermo Fisher Scientific Inc.), and the template dilution factors were calculated. High‐quality templated Ion Sphere™ particles, containing massively parallel clonally amplified DNA, were prepared for the 200 base‐read libraries using the Ion PGM™ Template OT2 200 Kit and the Ion OneTouch™ 2 System (Life Technologies, a part of Thermo Fisher Scientific Inc.). The template‐positive Ion Sphere™ particles were enriched with the Ion OneTouch™ Enrichment System (Life Technologies, a part of Thermo Fisher Scientific Inc.) as per the manufacturer's guidelines.
PGMTM Sequencing
Semiconductor sequencing on the Ion PGMTM detects the change in pH when the proton (H+) is released during nucleotide incorporation. The sequencing was conducted on the Ion 318™ Chip v2 using the Ion PGM™ Hi‐Q™ Sequencing Kit (six barcoded samples per chip) coupled with the PGMTM sequencer (Life Technologies, a part of Thermo Fisher Scientific Inc.). A chlorite cleaning, followed by 18 MΩ water wash (Thermo Fisher Scientific, Millipore, MA) was performed prior to the initialization of the Ion PGM™ System.
MPS Data Analysis
Commercially available NextGENe® software (v2.4.0.2) was used to analyze all the sequences generated on the Ion PGMTM sequencer. All the sequences were trimmed by 20 bases on both the 3′ and the 5′ ends and aligned to the mtDNA revised Cambridge Reference Sequence (rCRS) [Andrews et al., 1999] (GenBank NC_012920.1). The arbitrary threshold of 50 reads was considered as a minimum required to address full sequencing coverage to reliably report variants/polymorphisms. This threshold is the coverage minimum to be able to see heteroplasmies and to be able to clearly identify stochastic sequencing errors. A BED file that contains the amplicon regions was also uploaded for the analysis. The NextGENe® software allows visualization of sequence reads in addition to summarizing the polymorphic sites in a tabular column.
The NextGENe® software uses BLAST‐like Alignment Tool to align the sequence reads to the reference. Nonetheless, in some cases, manual inspection was needed to confirm certain variants and anomalies, especially regarding length heteroplasmy. It is known that the corrected version of the Cambridge Reference Sequence (the rCRS) retained the same nucleotide numbering scheme as the original reference sequence [the CRS; Anderson et al., 1981], and to correct one of the erroneous positions, an “N” was recorded at position 3,107 to represent a deletion at that position [Andrews et al., 1999]. The NextGENe® software lists it as 3,107d in any tested sample, but as this deletion does not represent a genuine sequence variant, it shall be ignored. Additionally, this software does not adhere to the nomenclature recommendations detailed by the forensic community [Carracedo et al., 2000; Bandelt and Parson 2008]. In particular, rather than reporting the common dinucleotide deletion in the dimeric repeat region between 514 and 524 as m.523A>del and m.524C>del (placing the deletion at the most 3′ position possible), NextGENe® employs a 5′ indel alignment and notes these variants as m.513G>del and m.514G>del. Furthermore, we noticed that the two sequential transitions that sometimes co‐occur at positions 151 and 152 were instead reported as a deletion at np 151 and an insertion at 152, following the recommendations by Wilson et al. (2000a, 2000b).
With the mtDNA sequence variants detected, the haplogroups were inferred using the Web‐based bioinformatics tool, MitoTool [Fan and Yao, 2011], which uses the most recent version of PhyloTree (http://www.phylotree.org; Build 16) [van Oven and Kayser, 2009].
Results and Discussion
In recent years, DNA sequencing technology has evolved from traditional Sanger sequencing of single reads to sequencing thousands of reads with high coverage in a massively parallel fashion with MPS technologies such as for analyzing human mtDNA [Sosa et al., 2012, Parson et al. 2013, 2015; Davis et al, 2015]. Contrary to the large‐fragment sequencing strategy employed mostly so far for MPS analysis of whole mt genomes [Sosa et al., 2012; Parson et al., 2013], here we introduce an MPS approach for analyzing the entire human mtDNA genome using 161 pairs of short amplicons (144–230 bp, average 200 bp) in two different primer pools. We demonstrate the whole mt genome coverage of our approach via analyzing worldwide DNA samples belonging to different mtDNA haplogroups, perform quality control via comparison with data obtained with alternative methods in the same samples and provide preliminary data on sensitivity testing, degraded DNA testing using artificially and naturally degraded DNA, and multiple DNA sample testing using barcoded adapters; the DNA variants were determined using the NextGENe® v2.4.0.2 software.
Method Application to Worldwide Samples and Quality Comparison with Alternative Methods
To test the performance of the newly designed method for simultaneous whole mt genome sequencing using the Ion Torrent PGM, we applied it to 20 carefully selected DNA samples that belong to different global mtDNA haplogroups (Table 1). For these 20 samples, the average sequencing coverage ranged from 1,302 to 5,637 reads with an average of 3,293 reads across all samples. The total number of reads per sample ranged from 211,908 to 641,486, with an average across samples of 366,327, of which 88% were successfully aligned to the rCRS (325,349 reads). Using a coverage threshold of 50 reads, 100% mt genome coverage was achieved in all the samples except three (samples 14, 17, and 20), which each miss one small piece of mtDNA sequence with our MPS approach (Table 1). In sample 17, the 9‐bp deletion at np 8,281–8,289 (which is the defining mutation of haplogroup B, among others) was not detected by the NextGENe® software in the final variant table, elucidating the current limitation in the software package used. Further improvements in the bioinformatics pipelines are needed to deal with such data. A discreet manual inspection was done to confirm the deletion. In samples 14 and 20, fragments of 66 bp (np 10,468–10,533) and 61 bp (np 10,269–10,329), respectively, were missed when using the 50‐reads threshold applied. Figure 1 illustrates the average coverage of each of the 161 amplicons across all the 20 samples. The mtDNA variants detected for all the 20 samples analyzed are detailed in Supp. Table S2.
Table 1.
Performance Summary of the 20 Geographically Diverse DNA Samples for Whole mt Genome Sequencing with the MPS Tiling Approach via 161 Short Overlapping Amplicons
| Sample ID | Total reads | Aligned reads | Percent of aligned reads | Maximum coverage | Average coverage | Percent of coverage |
|---|---|---|---|---|---|---|
| 1 | 641,486 | 535,003 | 83.40 | 67,847 | 5,593 | 100% |
| 2 | 502,725 | 427,031 | 84.94 | 54,668 | 4,852 | 100% |
| 3 | 268,055 | 224,250 | 83.66 | 68,677 | 4,494 | 100% |
| 4 | 408,522 | 343,567 | 84.10 | 44,394 | 4,285 | 100% |
| 5 | 355,691 | 305,619 | 85.92 | 50,539 | 3,804 | 100% |
| 6 | 493,881 | 485,970 | 98 | 32,082 | 3,143 | 100% |
| 7 | 289,696 | 258,720 | 89.31 | 43,687 | 2,607 | 100% |
| 8 | 264,828 | 232,310 | 87.72 | 23,412 | 1,488 | 100% |
| 9 | 264,231 | 204,023 | 77.21 | 47,124 | 3,777 | 100% |
| 10 | 352,588 | 279,159 | 79.17 | 53,959 | 3,496 | 100% |
| 11 | 476,939 | 426,050 | 89.33 | 66,262 | 5,637 | 100% |
| 12 | 268,968 | 235,011 | 87.38 | 37,458 | 2,560 | 100% |
| 13 | 249,338 | 198,790 | 79.73 | 46,631 | 1,840 | 100% |
| 14 | 433,566 | 389,219 | 89.77 | 52,840 | 3,358 | 99.59% |
| 15 | 541,295 | 532,291 | 98.34 | 44,460 | 3,488 | 100% |
| 16 | 211,908 | 194,369 | 91.72 | 25,927 | 1,302 | 100% |
| 17 | 307,441 | 278,527 | 90.60 | 30,230 | 1,865 | 100%a |
| 18 | 324,954 | 299,098 | 92.04 | 48,897 | 2,006 | 100% |
| 19 | 420,730 | 416,278 | 98.94 | 38,839 | 3,644 | 100% |
| 20 | 249,701 | 241,699 | 96.80 | 37,051 | 2,628 | 99.62% |
| Average | 366,327 | 325,349 | 88 | 45,749 | 3,293 |
The 9‐bp deletion at np 8,281–8,289 in sample 17 is the defining mutation of haplogroup B. Hence, the coverage for sample 17 was considered to be 100% despite the 9‐bp deletion.
Figure 1.
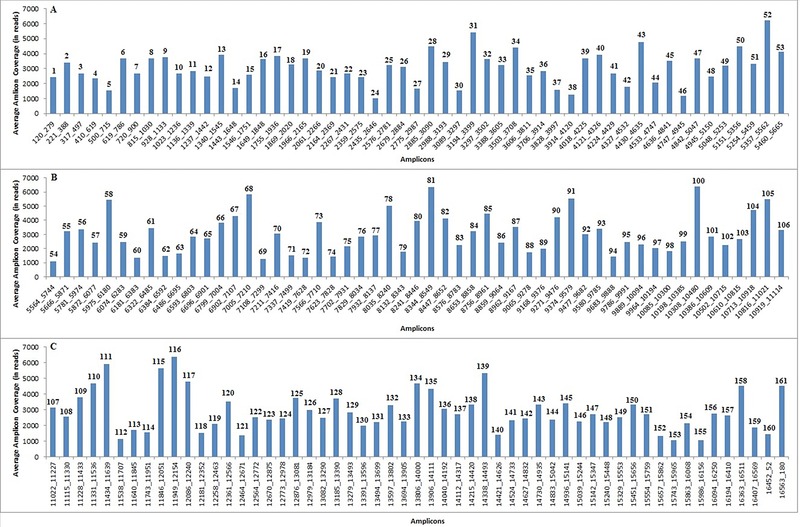
The average amplicon coverage, across all the 20 samples tested with the newly developed MPS tiling approach for whole mt genome sequencing, are presented in three plots. A: Amplicons 1–53. B: Amplicons 54–106. C: Amplicons 107–161.
Further, the whole mt genome data we generated via our MPS approach from the samples belonging to the CEPH–HGDP panel (samples 1–11) were compared with those previously reported by Hartmann et al. (2009) based on whole mt genome sequencing using the Sanger approach. The remaining nine samples (12–20) we analyzed via MPS were subjected to de novo Sanger sequencing of the control region (unpublished study, data not shown). Some of the positions in the coding region were further compared with the results previously obtained from the five SNaPshot multiplex assays established earlier (data not shown) [Chaitanya et al., 2014]. The data obtained via our PGM method were then compared with the data generated by alternative methods for quality control purposes. The 9‐bp deletion at np 8,281–8,289 in sample 17 was not tabulated in the variant table generated by NextGENe®. However, this deletion was evident from results obtained with the SNaPshot multiplex assay and also apparent in the NextGENe® viewer. Several mis‐calls by NextGENe® were revised after a meticulous manual inspection of the sequence in the NextGENe® viewer. Disparities observed between the Sanger sequencing/SNaPshot and MPS are tabulated in Supp. Table S3. Some of the discrepancies were observed in calling of the variants involving length heteroplasmy in the polycytosine stretches of the HVS‐I and HVS‐II. In samples 1, 6, 11, and 17, due to an erroneous shift in the bases, the variant was reported as m.16183A>M (an apparent heteroplasmy), instead of m.16183A>C (a homoplasmic transversion) (Supp. Fig. S1A). In the HVS‐II region, for samples 8, 13, 16, 19, and 20, only one insertion of the base C in the np 303–309 region was reported with Sanger sequencing. However, with MPS, two insertions of the base C were noted (Supp. Fig. S1B). Such differences will in practice not have any influence on haplogroup determination since the insertions at np 309, 315, 16,193, AC indels at 515–524, m.16182A>C, m.16183A>C, and m.16519T>C are usually not considered for phylogenetic reconstruction and are therefore not included in the PhyloTree [van Oven and Kayser, 2009]. The variant reported as 357M in the samples 2, 7, 14, and 15 is specious and appears due to a shift in the base position, possibly because of the polyadenine stretches as shown in Supp. Figure S2. Similarly, in samples 8, 12, and 17, the variant m.13128C>M is spurious due to the shift in the base because of the polycytosine stretches as shown in Supp. Figure S3.
Furthermore, spurious gaps were observed in the samples 8 and 12 at position 13,128 (Supp. Fig. S3 boxed region). Samples 9, 10, and 14 report an insertion 539.1C (Supp. Fig. S4), which was not evident in the Sanger sequencing data. It has been described earlier that the PGM produces a high frequency of homopolymer sequencing errors and indels [Loman et al., 2012; Seo et al., 2013]. In samples 2 and 4, a spurious deletion was noticed at position 5,824 that was not reported in the Sanger sequencing data of Hartmann et al. (2009) (Supp. Fig. S5). Further, differences to the Sanger sequencing were observed in sample 9 at position 16,209 and in sample 10 at position 204 (Supp. Table S3; Supp. Fig. S6). Spurious single‐bp deletions were observed at some sequence positions in some reads in few of the samples (Supp. Fig. S6 boxed region) but not reported in the variant table. Such gaps did not influence the final consensus sequence and therefore did not affect the final haplogroup determination.
One potential disadvantage of using a tiling approach based on small amplicons over a large fragment amplification approach is the potential detection of nuclear copies of mt sequences (NUMTs) with the tiling approach that likely are not detected with the long‐range approach simply because NUMTs typically are much smaller. To test for this, we generated comparative whole mt genome sequencing data using the previously described two overlapping 8.5‐kb amplification method [Fendt et al., 2009] in five of the samples (samples 1, 2, 3, 4, and 20) we used for PGM sequencing. It is noteworthy that the variants detected were in concordance with those detected when sequenced with the short overlapping fragments, except in sample 2. In sample 2, the polymorphisms m.10664C>Y, m.8251G>R, and m.8252C>M (Supp. Fig. S7; Table 2) were discordant with the tiling approach (m.10664C>T, m.8251G>A, and no variant at position 8,252, respectively). The erroneous calling at positions 8,251 and 8,252 could be due to the base position shifts as seen in Supp. Figure S7. The disparities observed in the HVS‐I and HVS‐II due to the length heteroplasmy were also noticed with the long‐range amplification. Hence, from the samples analyzed, we have no evidence that our tiling MPS approach picks up NUMTs.
Table 2.
Differences Between the Previously Developed Long‐Range Amplification MPS Approach and the Newly Introduced MPS Tiling Approach for Whole mt Genome Analysis Both Obtained via the PGM in Sample 2
| NGS data from NextGENe® software | |||||||
|---|---|---|---|---|---|---|---|
| %A | %C | %G | %T | %Insertions | %Deletions | ||
| Long range | m.10664C>Y | 0 | 17.46 | 0 | 78.31 | 0 | 4.23 |
| Tiling | m.10664C>T | 0 | 0.34 | 0 | 99.35 | 0 | 0.3 |
| Long range | m.8251G>R | 71.43 | 0 | 28.57 | 0 | 0 | 0 |
| m.8252C>M | 29.37 | 70.63 | 0 | 0 | 2.1 | 0 | |
| Tiling | m.8251G>A | 97.27 | 0 | 2.1 | 0 | 0 | 0.63 |
Human mitochondrial genome, rCRS (GenBank NC_012920.1).
It has been described earlier [Parson et al., 2013; Seo et al., 2015] that false deletions were observed in the mtDNA sequencing data generated using the PGM. However, no such deletions were observed when the variants reported with our tool and the long‐range PCR approach was compared. Except for the discordant calls in sample 2, no other differences in variant calling were observed. Nonetheless, this may change when more samples are tested, as in this study only a few samples have been tested.
Point heteroplasmies at 22 positions in 14 samples were reported in the NextGENe® variant table: sample 1: m.8155G>R; sample 2: m.1048C>Y, m.357A>M; sample 3: m.14423G>S; sample 4: m.4788G>R, m.14921G>R; sample 5: m.1171A>R, m.13020T>Y, m.15326A>R, m.16189T>Y; sample 6: m.7759T>Y; sample 7: m.771A>R, m.4248T>Y, m.357A>M; sample 8: m.3296T>Y, m.13128C>M; sample 9: m.10185C>Y, m.15326A>R, m.16209T>Y; sample 10: m.204T>Y, m.13928G>S, m.15326A>R, m.14831G>R; sample 12: m.13128C>M; sample 14: m.357A>M; sample 15: m.357A>M; and sample 17: m.13128C>M. Heteroplasmy threshold was set at 20% (of total coverage) [Parson et al., 2013]. Out of the 22, only seven of the point heteroplasmies (sample 1: m.8155G>R; sample 3: m.14423G>S; sample 4: m.4788G>R, m.14921G>R; sample 5: m.1171A>R, m.16189T>Y; sample 6: m.7759T>Y) were also observed in the Sanger sequencing data of Hartmann et al. (2009). A heteroplasmy at position 3,296 was not reported by Hartmann et al. (2009), contrary to the m.3296T>Y from this study. However, in a recent publication, wherein some samples from the CEPH‐HGDP panel were resequenced on the Illumina platform, a variant at 3,296 was recorded for the sample 8 as m.3296T>N [Lippold et al., 2014], suggesting that this heteroplasmy could be genuine.
In general, the availability of a viewer in the NextGENe® software proved advantageous in manually evaluating the discrepancies and solving them accordingly, especially in assessing the true heteroplasmy.
mtDNA Haplogroup Assignment and Maternal Ancestry Inference
The haplogroups for the 20 samples were inferred from the obtained whole mtDNA genome data using MitoTool [Fan and Yao, 2011] and are presented in Table 3 together with the geographic sampling origin and the previously reported geographic region of haplogroup origin indicative of maternal biogeographic ancestry. All the haplogroups determined for the 20 samples were in agreement with their biogeographic origin. It is important to assert that mtDNA construes only the matrilineal ancestry information of an individual. To achieve comprehensive biogeographic ancestry inference from DNA, in addition to the global matrilineal biogeographic ancestry assignment, information about paternal ancestry using male‐specific Y‐chromosomal DNA (in the case of males) and from biparental ancestry using ancestry‐informative autosomal DNA markers have to be considered [Kayser and de Knijff, 2011].
Table 3.
Haplogroups of the 20 DNA Samples Whole mt Genome Sequenced with the MPS Tiling Tool and Interpreted Using MitoTool
| Sample ID | Haplogroup | Broad haplogroup | Known sampling region | Main geographic region of the (broad) haplogroup origin | References |
|---|---|---|---|---|---|
| 1 | L3d3a1a | L3*(xM,N) | South Africa | Africa, West Asia | Behar et al. (2008) |
| 2 | L0d1a1a (199 missing) | L0 | South Africa | Southern Africa | Behar et al. (2008); Barbieri et al. (2013) |
| 3 | H1c | H | Russia (Caucasus) | West Eurasia, Northern Africa | Loogväli et al. (2004); Achilli et al. (2004); Roostalu et al. (2007) |
| 4 | U7a2 | U7 | Israel | West Eurasia, Central Asia, Southern Asia | Palanichamy et al. (2004); Brisighelli et al. (2009) |
| 5 | P1d1 | P | New Guinea | Oceania (Papuan, Melanesian, and Australian Aborigines) | Friedlaender et al. (2007); Hudjashov et al. (2007) |
| 6 | V | HV*(xH) | Algeria | West Eurasia, Northern Africa | Achilli et al. (2005); Álvarez‐Iglesias et al. (2009) |
| 7 | A1a1 (missing 235) | A*(xA2) | China | East Asia | Kong et al. (2006); Derenko et al. (2007) |
| 8 | J2b1a | J | Italy | West Eurasia | Pala et al. (2012) |
| 9 | M7c1a2a | M*(xM1,C,D) | China | South Asia, East Asia, Southeast Asia | Kong et al., 2006; Derenko et al. (2007) |
| 10 | F4a1a | R9 | China | East Asia, Southeast Asia | Kong et al. (2006) |
| 11 | X2b5 | X | Orkney Islands | West Eurasia, Northern Africa, Americas | Reidla et al. (2003); Achilli et al. (2008) |
| 12 | J2b1(J2b1a1: missing16278) | J | Italy | West Eurasia | Pala et al. (2012) |
| 13 | P | P | Australia (Aborigine) | Oceania (Papuan, Melanesian, and Australian Aborigines) | Friedlaender et al. (2007); Hudjashov et al. (2007) |
| 14 | M72a | M*(xM1,C,D) | Thailand | South Asia, East Asia, Southeast Asia | Tabbada et al. (2010); Peng et al. (2010) |
| 15 | T2b | T | Italy | West Eurasia | Pala et al. (2012) |
| 16 | K1a12 | U | The Netherlands | West Eurasia | Achilli et al. (2005); Behar et al. (2006) |
| 17 | B5a1a | B5 | The Netherlands | East Asia | Kong et al. (2006) |
| 18 | W5a1a | W | The Netherlands | West Eurasia | Finnilä et al. (2001); Palanichamy et al. (2004) |
| 19 | V3c | HV*(xH) | The Netherlands | West Eurasia, Northern Africa | Achilli et al. (2005); Álvarez‐Iglesias et al. (2009) |
| 20 | H23 | H | The Netherlands | West Eurasia, Northern Africa | Loogväli et al. (2004); Achilli et al. (2004); Roostalu et al. (2007) |
Preliminary Sensitivity Testing
In order to determine the limit of detection provided the 50 reads coverage threshold used and given the sensitivity of the developed PGM assay, preliminary sensitivity tests were performed with differing starting amounts of DNA at 100, 250, and 500pg, and 1 and 10 ng (measured as genomic DNA). For this, we used three of the 20 DNA samples originating from different populations with different haplogroups—South Africa with haplogroup L3d3a1a (sample 1); Italy with T2b (sample 15); and The Netherlands with V3c (sample 19). The results of the sensitivity study on the samples are summarized in Supp. Table S4. For all the sample dilutions, it was possible to detect the correct haplogroup down to merely 100 pg input genomic DNA, even though the input manufacturer's recommendation for library construction is 10 ng. Regarding whole mt genome coverage, we achieved 100% for all sample dilutions, except for sample 19 at 250 pg (99.89% at 576 reads on average), and for all three samples at 100 pg (99.93% at 2,591 average reads, 99.91% at 1,175 average reads, and 99.74% at 462 average reads for samples 1, 15, and 19, respectively). Notably, samples were further diluted to 50 pg input, but could not proceed to sequencing as the template dilution factors calculated were below one, whereas one is recommended by the manufacturer, thus implying that the amount of DNA library generated from such low quantity DNA samples was not sufficient to perform optimal emulsion PCR [Ralf et al., 2015]. Previous studies have shown that it is possible to achieve successful sequencing results with the Sanger protocol using 50 pg of input DNA, and sometimes even at 10 pg of input DNA with minimal failures [Lyons et al., 2013; Just et al., 2014]. However, to achieve full mtDNA coverage at a high resolution with the Sanger approach, several individual sequences are required, as opposed to the parallel nature of the MPS where a high volume of data is generated in relatively short time. Though it was possible to achieve correct haplogroup information at 100 pg, increasing the number of PCR cycles could result in good coverage with input below 100 pg of DNA. Hence, further testing needs to be done to show the full‐sensitivity limits of this system.
Preliminary Analysis of Experimentally and Naturally Degraded DNA Samples
The robustness of our MPS assay regarding DNA degradation was tested by sequencing experimentally and naturally degraded DNA samples. Aliquots of a sample (1 ng genomic DNA), belonging to haplogroup M35a1, was subjected to DNase treatment at different time intervals: 5, 10, 15, 20, 30, and 45 min. The effect of DNA degradation in these samples was first monitored by analyzing them with the AmpFlSTR® Identifiler® PCR Amplification Kit (Applied Biosystems) targeting 15 autosomal STRs plus the amelogenin sex typing system, which is routinely used for human identification purposes (Fig. 2). From Figure 2, it is evident that the DNA degradation‐induced STR locus dropouts are clearly correlated with PCR fragment size, as expected. Particularly, at 5 min of DNase treatment, there were complete locus dropouts at three STRs CSF1PO (allelic fragment length 330 bp), FGA (280 bp), and D2S1338 (310 bp); at 10 min, additional dropout at D7S820 (271 bp), D16S539 (280 bp), and D18S51 (270 bp); at 15 min, additional dropouts at TPOX (238 bp) and D13S317 (222 bp); at 20 min, additional dropout at D21S11 (206 bp), TH01 (172 bp), and vWA (178 bp); at 30 min, additional dropout at D5S818 (154 bp), D8S1179 (146 bp), and D19S433 (128 bp). After 45 min of DNase treatment, all of the 16 loci covering allelic fragment sizes of about 128–330 bp dropped out, except the two loci with shortest alleles, amelogenin (108 bp) and D3S1358 (116 bp). This analysis provides a rough idea on DNA fragmentation in these experimentally degraded samples as relevant for the subsequent mtDNA genome sequencing using our MPS tiling approach.
Figure 2.
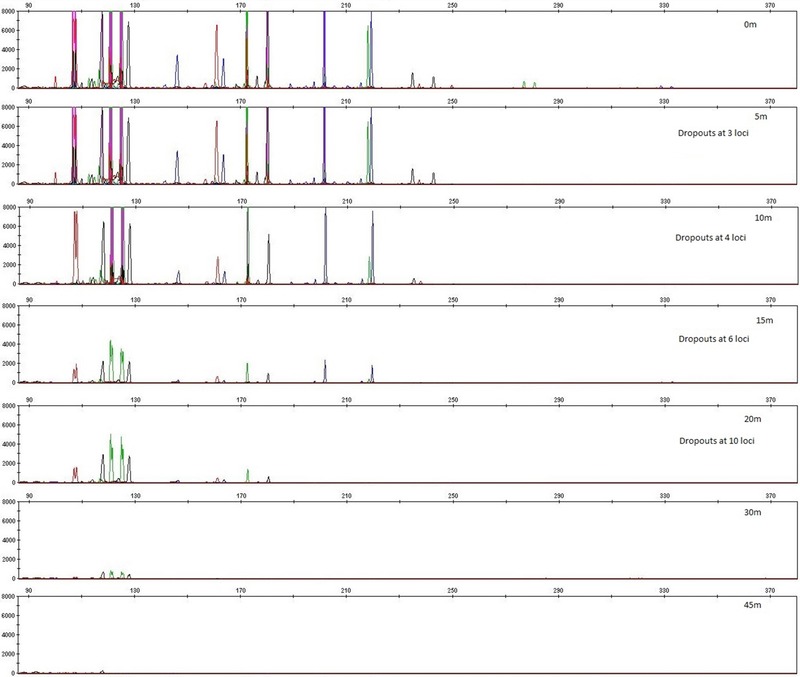
STR profiles from the AmpFlSTR® Identifiler® PCR Amplification Kit (Applied Biosystems) targeting 15 autosomal STRs plus amelogenin to illustrate the degree of DNA fragmentation for the sample treated with DNase at different time intervals: 5, 10, 15, 20, 30, and 45 min.
Table 4 enlists the performance summary of our whole mt genome tiling MPS approach in the DNase‐treated samples at different time intervals. Most notable, for the samples degraded for 5, 10, and 15 min, we obtained 100% mt genome coverage using a 50x threshold, at 6,810, 3,886, and 3,007 average reads, respectively. Prior STR analysis showed that eight loci with allelic fragment length from 222 to 330 bp already dropped out in these three degraded samples. For samples degraded for 20, 30, and 45 min, the coverage dropped down to 94.4%, 82.6%, and 19.25%, respectively, at 2,073, 1,183, and 487 average reads, respectively. Regarding the total number of mtDNA variants detected in these samples, after 5 min of DNase treatment, it was 37 as well as after 10 and 15 min where we obtained 100% mt genome coverage, whereas it dropped down to 30, 24, and 13 in the samples that received DNase treatment for 20, 30, and 45 min, respectively. However, despite the loss of reads, fragments, and thus DNA variants, it was still possible to determine the correct mtDNA haplogroup (M35a1) in all degraded DNA samples from 5 to 45 min of DNase treatment. Figure 3 elucidates the average amplicon coverage across all amplicons for the different time intervals of enzymatic degradation clearly showing the loss of sequence reads with increased degradation time, as expected. Figure 4 illustrates the amplicon coverage according to amplicon length for the different time intervals of enzymatic degradation, clearly demonstrating the effect of DNA degradation on amplicon length and number of reads. It is to be noted that successful sequencing achieving 100% mt genome coverage was obtained from degraded DNA samples with a maximal fragment size of about 220 bp (Fig. 2).
Table 4.
Performance Summary of PGM‐Based Whole mt Genome Sequencing of DNase‐Treated Sample at Different Time Intervals
| Total reads | Aligned reads | Percent of aligned reads | Maximum coverage | Average coverage | Percent of coverage | Haplogroup | ||
|---|---|---|---|---|---|---|---|---|
| DNase‐treated sample at different time intervals | 5 min | 463,854 | 452,505 | 97.55% | 51,989 | 6,810 | 100% | M35a1 |
| 10 min | 242,109 | 234,156 | 96.72% | 45,167 | 3,886 | 100% | ||
| 15 min | 163,854 | 152,505 | 93.07% | 38,249 | 3,007 | 100% | ||
| 20 min | 60,988 | 59,085 | 96.88% | 31,262 | 2,073 | 94.40% | ||
| 30 min | 83,565 | 80,367 | 96.17% | 10,731 | 1,183 | 82.60% | ||
| 45 min | 15,642 | 13,606 | 86.98% | 9,625 | 487 | 19.25% | ||
| Sample 15 | UV for 30 min | 258,179 | 255,277 | 98.88% | 21,901 | 1,362 | 88.65% | T2b |
| Enzymatic shearing | 315,030 | 278,648 | 88.45% | 5,827 | 975 | 89.90% | ||
| Ancient and degraded bone and teeth samples | Sample 1 | 542,889 | 525,140 | 96.73% | 9,572 | 4,042.52 | 87.87% | U5b2b |
| Sample 2 | 95,396 | 58,311 | 61.13% | 2,379 | 225.57 | 50.36% | H4a1 | |
| Sample 3 | 538,548 | 512,826 | 95.22% | 7,273 | 3,321.23 | 90.12% | H | |
| Sample 4 | 529,929 | 480,856 | 90.74% | 6,048 | 3,141.47 | 88.32% | T2b | |
| Sample 5 | 477,578 | 450,506 | 94.33% | 8,218 | 2,924.11 | 59.59% | U4a2 | |
| Sample 6 | 175,642 | 140,021 | 79.72% | 3,535 | 612.48 | 75.25% | T1a |
Sample 15 subjected to three different degradation methods and six different highly degraded bone and teeth samples.
Figure 3.
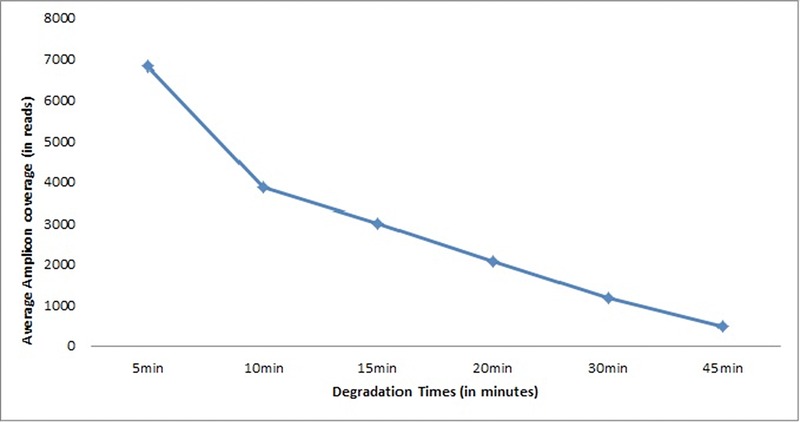
The average mt DNA amplicon coverage across all amplicons for the DNase‐treated sample at the different time intervals as obtained with the MPS tiling approach.
Figure 4.
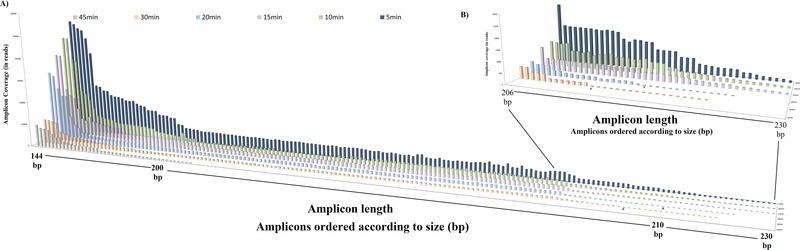
A: The amplicon coverage (number of times amplicons were observed in the MPS data) of all the 161 amplicons used to obtain complete mt genome coverage with our MPS approach, arranged according to amplicon length from the shortest amplicon used (144 bp) on the left‐hand side to the largest amplicon used (230 bp) on the right‐hand side, for the different time intervals of enzymatic DNA degradation: 5, 10, 15, 20, 30, and 45 min. All the amplicons below X represents the coverage below the 50 reads threshold. B: Additional zoomed‐in image of the longer amplicons from amplicon lengths of 206 bp (left‐hand side) to 230 bp (right‐hand side).
To test two additional experimental degradation methods, aliquots of 1 ng genomic DNA of sample 15, belonging to haplogroup T2b, were exposed to UV radiation for 30 min using a Bio‐Link (Vilber Lourmat) at a strength of 50 J/cm2 and enzymatic shearing using the Ion Shear™ Plus Reagents Kit (Life Technologies, a part of Thermo Fisher Scientific Inc.). The performance summary of the samples is depicted in Table 4. At 1 ng unexposed DNA, the total number of mtDNA variants detected was 41 at 100% mt genome coverage (1,865 reads on average); when the sample was exposed to UV for 30 min, only 38 of the 41 variants were detected (missed variants were m.7310T>C, m.8697G>A, and m.10463T>C) with a mt genome coverage of 88.65% at 1,362 reads on average. At position 13,368, the variant was reported as m.13368G>R (49.36% A and 50.61% G), whereas the true variant was m.13368G>A. In contrast, all the 41 variants were correctly detected when the sample was subjected to enzymatic shearing, even though only 89.9% mt genome coverage was obtained at 975 reads on average. The variant m.357A>M, which was explained earlier as a sequencing error probably because of a base shift position due to the poly‐A stretch, was detected in both degradation approaches. However, both degradation methods allowed our MPS approach to determine the correct haplogroup of sample 15 (i.e., T2b).
Additionally, DNA extracts from six human remains (i.e., teeth and bones) were sequenced with our MPS tiling approach to investigate naturally degraded DNA (Table 4). Notably, samples 1, 3, 4, and 5 delivered a complete 16 loci (15 STRs plus amelogenin) DNA profile with the AmpFℓSTR® NGM™ Kit (Thermo Fisher Scientific) regularly used for human identification, whereas samples 2 and 6 delivered only a partial NGM profile with 12 and 10 of the 16 loci missing, respectively. With our MPS approach, we obtained mt genome coverage of close to 90% for samples 1, 3, and 4, whereas for samples 6, 5, and 2, our method delivered 75%, 60%, and 50% of mt genome coverage (Table 4). Notably, sample 2 with the lowest observed mt genome coverage of 50% (225 reads on average) likely originates from the XV–XVI century. For samples 4, 5, and 6, mtDNA control region HVI and HVII data were obtained successfully via Sanger sequencing and compared with those of the tiling MPS approach. Sanger sequencing of these samples was performed as part of another unpublished study and hence the protocol was not described here. These data were comparable, except that the variant m.16519T>C, which was clearly evident in the tiling MPS approach, but was not reported in the Sanger sequencing data for all the three samples. However, Sanger sequencing mtDNA control region data were not available for samples 1, 2, and 3.
Although mt genome coverage was only obtained to the degree of 50%–90% from these six naturally degraded DNA samples, our MPS tiling approach allowed haplogroup assignment for all of them: U5b2b, H4a1, H, T2b, U4a2, and T1a for samples 1–6, respectively.
The AmpliSeq‐based MPS approach we developed here for complete human mt genome analysis is designed for and suited to nondegraded and mildly to considerably degraded DNA leading to DNA fragmentation down to a size range of around 200 bp and larger, as often confronted with in forensic, medical, and anthropological studies. Our approach is not suitable for more severely degraded DNA as often confronted with in ancient DNA studies. For such strongly fragmented DNA, hybridization capture methods are more suitable than AmpliSeq‐based approaches and have been developed in the field of ancient DNA research [Noonan et al., 2005; Anderung et al., 2008; Briggs et al., 2009; Templeton et al., 2013]. The AmpliSeq system we used in our approach is optimized for this kind of multiplexed amplification and the workflow requires fewer steps, which reduces the chance of contamination and mix‐up. Additionally, more targets are generated with the Ampliseq PCR system than with a hybridization capture‐based system, allowing for deeper coverage, mitigating sequencing errors.
Preliminary Testing of Sample Multiplexing via DNA Barcoding
In order to test the feasibility of sequencing multiple samples simultaneously in one sequencing run, barcode adapters were used. Six DNA samples were pooled (12 libraries, as two primer pools were needed) per each sequencing run and barcoded, using the Ion Xpress™ Barcode Adapters (Life Technologies, a part of Thermo Fisher Scientific Inc.). The ability to multiplex six samples via barcoding is a valuable asset with regard to the increase in throughput and cost‐effectiveness. Further research should involve sequencing more than six samples in a single run on one chip.
Conclusion
Our study shows that analyzing the complete human mt genome in a simultaneous way via a tiling approach targeting short amplicons is feasible. With the short overlapping fragments we employed in covering the entire mt genome, and supported by our preliminary data on experimentally and naturally degraded DNA samples, we expect our approach to be particularly useful for analyzing degraded materials. As this was a preliminary study introducing the new method, future studies with a larger sample set including more degraded samples need to be conducted to explore the complete value of this tool. However, based on the preliminary data presented here, we already expect our method to be highly useful in many mtDNA applications using degraded and nondegraded DNA where maternal lineage identification and maternal ancestry inference on the maximum possible resolution level is appreciated such as in forensic, anthropological, and medical genetics.
In the longer run, we envision this MPS tool for complete mt genome analysis allowing maximal maternal lineage determination and maternal ancestry inference being combined with MPS tools for ultra‐high‐resolution paternal lineage and paternal ancestry identification, such as the PGM tool we recently introduced for simultaneous analysis of >530 Y‐chromosomal SNPs allowing to detect >430 worldwide Y haplogroups [Ralf et al., 2015]. Since with the current whole mt genome tool using 161 short overlapping amplicons (144–230 bp, average 200 bp), the sequence capacity limits of the PGM (or alternative devices such as MiSeq) are not reached, further enlargements of combined targeted MPS tools may be expected from future work. For instance, we envision the additional addition of autosomal ancestry‐informative SNPs into combined tools together with Y and mtDNA allowing to not only detect lineages (as possible with mtDNA and Y analysis) but moreover to resolve individual genetic admixture, and to obtain an overall and more extensive estimate of an individual's biogeographic ancestry from all three ancestry components: maternal, paternal, and biparental. For some applications, such as to answer forensic and anthropological questions, the additional incorporation of phenotypic markers into comprehensive ancestry tool(s) such as SNPs predictive for human pigmentation traits or for other externally visible characteristics if available [Kayser, 2015] would further add to the investigative value available with such comprehensive targeted MPS tools.
Disclosure statement: The authors declare no conflict of interest.
Supporting information
Supplementary Material
Acknowledgments
The authors thank Thierry Jurado and Philipp Habermeier (both from Life Technologies, a part of Thermo Fisher Scientific Inc.) for their help in setting‐up and hosting this work.
Contract grant sponsors: Life Technologies (a part of Thermo Fisher Scientific Inc.); Erasmus MC University Medical Center.
Communicated by Peter J. Oefner
References
- Achilli A, Perego UA, Bravi CM, Coble MD, Kong QP, Woodward SR, Salas A, Torroni A, Bandelt HJ. 2008. The phylogeny of the four pan‐American MtDNA haplogroups: implications for evolutionary and disease studies. PLoS One 3:e1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achilli A, Rengo C, Battaglia V, Pala M, Olivieri A, Fornarino S, Magri C, Scozzari R, Babudri N, Santachiara‐Benerecetti AS, Bandelt HJ, Semino O, et al. 2005. Saami and Berbers—an unexpected mitochondrial DNA link. Am J Hum Genet 76:883–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achilli A, Rengo C, Magri C, Battaglia V, Olivieri A, Scozzari R, Cruciani F, Zeviani M, Briem E, Carelli V, Moral P, Dugoujon JM, et al. 2004. The molecular dissection of mtDNA haplogroup H confirms that the Franco‐Cantabrian glacial refuge was a major source for the European gene pool. Am J Hum Genet 75: 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez‐Iglesias V, Mosquera‐Miguel A, Cerezo M, Quintáns B, Zarrabeitia MT, Cuscó I, Lareu MV, García O, Pérez‐Jurado L, Carracedo A, Salas A. 2009. New population and phylogenetic features of the internal variation within mitochondrial DNA macro‐haplogroup R0. PLoS One 4:e5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderung C, Persson P, Bouwman A, Elburg R, Götherström A. 2008. Fishing for ancient DNA. Forensic Sci Int Genet 2:104–107. [DOI] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, et al. 1981. Sequence and organization of the human mitochondrial genome. Nature 290:457–465 [DOI] [PubMed] [Google Scholar]
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. 1999. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 23:147. [DOI] [PubMed] [Google Scholar]
- Ballantyne KN, van Oven M, Ralf A, Stoneking M, Mitchell RJ, van Oorschot RA, Kayser M. 2012. MtDNA SNP multiplexes for efficient inference of matrilineal genetic ancestry within Oceania. Forensic Sci Int Genet 6:425–436. [DOI] [PubMed] [Google Scholar]
- Bandelt HJ, Parson W. 2008. Consistent treatment of length variants in the human mtDNA control region: a reappraisal. Int J Legal Med 122:11–21. [DOI] [PubMed] [Google Scholar]
- Barbieri C, Vicente M, Rocha J, Mpoloka SW, Stoneking M, Pakendorf B. 2013. Ancient substructure in early mtDNA lineages of southern Africa. Am J Hum Genet 92:285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar DM, Metspalu E, Kivisild T, Achilli A, Hadid Y, Tzur S, Pereira L, Amorim A, Quintana‐Murci L, Majamaa K, Herrnstadt C, Howell N, et al. 2006. The matrilineal ancestry of Ashkenazi Jewry: portrait of a recent founder event. Am J Hum Genet 78:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar DM, Villems R, Soodyall H, Blue‐Smith J, Pereira L, Metspalu E, Scozzari R, Makkan H, Tzur S, Comas D, Bertranpetit J, Quintana‐Murci L, et al. 2008. The dawn of human matrilineal diversity. Am J Hum Genet 82:1130–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund EC, Kiialainen A, Syvänen AC. 2011. Next‐generation sequencing technologies and applications for human genetic history and forensics. Investig Genet 24:2–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandstätter A, Niederstätter H, Pavlic M, Grubwieser P, Parson W. 2007. Generating population data for the EMPOP database—an overview of the mtDNA sequencing and data evaluation processes considering 273 Austrian control region sequences as example. Forensic Sci Int 166:164–175. [DOI] [PubMed] [Google Scholar]
- Briggs AW, Good JM, Green RE, Krause J, Maricic T, Stenzel U, Lalueza‐Fox C, Rudan P, Brajković D, Kucan Ž, Gušić I, Schmitz R, et al. 2009. Targeted retrieval and analysis of five neandertal mtDNA genomes. Science 325:318–321. [DOI] [PubMed] [Google Scholar]
- Brisighelli F, Capelli C, Alvarez‐Iglesias V, Onofri V, Paoli G, Tofanelli S, Carracedo A, Pascali VL, Salas A. 2009. The Etruscan timeline: a recent Anatolian connection. Eur J Hum Genet 17:693–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A, Bär W, Lincoln P, Mayr W, Morling N, Olaisen B, Schneider P, Budowle B, Brinkmann B, Gill P, Holland M, Tully G, et al. 2000. DNA commission of the international society for forensic genetics: guidelines for mitochondrial DNA typing. Forensic Sci Int 110:79–85. [DOI] [PubMed] [Google Scholar]
- Chaitanya L, van Oven M, Weiler N, Harteveld J, Wirken L, Sijen T, de Knijff P, Kayser M. 2014. Developmental validation of mitochondrial DNA genotyping assays for adept matrilineal inference of biogeographic ancestry at a continental level. Forensic Sci Int Genet 11:39–51. [DOI] [PubMed] [Google Scholar]
- Davis C, Peters D, Warshauer D, King J, Budowle B. 2015. Sequencing the hypervariable regions of human mitochondrial DNA using massively parallel sequencing: enhanced data acquisition for DNA samples encountered in forensic testing. Leg Med (Tokyo) 17:123–127. [DOI] [PubMed] [Google Scholar]
- Derenko M, Malyarchuk B, Grzybowski T, Denisova G, Dambueva I, Perkova M, Dorzhu C, Luzina F, Lee HK, Vanecek T, Villems R, Zakharov I. 2007. Phylogeographic analysis of mitochondrial DNA in northern Asian populations. Am J Hum Genet 81:1025–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Yao YG. 2011. MitoTool: a web server for the analysis and retrieval of human mitochondrial DNA sequence variations. Mitochondrion 11:351–356. [DOI] [PubMed] [Google Scholar]
- Fendt L, Zimmermann B, Daniaux M, Parson W. 2009. Sequencing strategy for the whole mitochondrial genome resulting in high quality sequences. BMC Genomics 10:139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnilä S, Lehtonen MS, Majamaa K. 2001. Phylogenetic network for European mtDNA. Am J Hum Genet 68:1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlaender JS, Friedlaender FR, Hodgson JA, Stoltz M, Koki G, Horvat G, Zhadanov S, Schurr TG, Merriwether DA. 2007. Melanesian mtDNA complexity. PLoS One 2:e248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert MT, Tomsho LP, Rendulic S, Packard M, Drautz DI, Sher A, Tikhonov A, Dalén L, Kuznetsova T, Kosintsev P, Campos PF, Higham T, et al. 2007. Whole‐genome shotgun sequencing of mitochondria from ancient hair shafts. Science 317:1927–1930. [DOI] [PubMed] [Google Scholar]
- Gilbert MT, Drautz DI, Lesk AM, Ho SY, Qi J, Ratan A, Hsu CH, Sher A, Dalén L, Götherström A, Tomsho LP, Rendulic S, et al. 2008. Intraspecific phylogenetic analysis of Siberian woolly mammoths using complete mitochondrial genomes. Proc Natl Acad Sci USA 105:8327–8332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill P, Ivanov PL, Kimpton C, Piercy R, Benson N, Tully G, Evett I, Hagelberg E, Sullivan K. 1994. Identification of the remains of the Romanov family by DNA analysis. Nat Genet 6:130–135. [DOI] [PubMed] [Google Scholar]
- Green RE, Malaspinas AS, Krause J, Briggs AW, Johnson PL, Uhler C, Meyer M, Good JM, Maricic T, Stenzel U, Prüfer K, Siebauer M, et al. 2008. A complete Neandertal mitochondrial genome sequence determined by high‐throughput sequencing. Cell 134:416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann A, Thieme M, Nanduri LK, Stempfl T, Moehle C, Kivisild T, Oefner PJ. 2009. Validation of microarray‐based resequencing of 93 worldwide mitochondrial genomes. Hum Mutat 30:115–122. [DOI] [PubMed] [Google Scholar]
- Ho SYW, Gilbert MTP. 2010. Ancient mitogenomics. Mitochondrion (Kidlington) 10:1–11. [DOI] [PubMed] [Google Scholar]
- Holland MM, Parsons TJ. 1999. Mitochondrial DNA sequences analysis—validation and use for forensic casework. Forensic Sci Rev 11:21–49. [PubMed] [Google Scholar]
- Hudjashov G, Kivisild T, Underhill PA, Endicott P, Sanchez JJ, Lin AA, Shen P, Oefner P, Renfrew C, Villems R, Forster P. 2007. Revealing the prehistoric settlement of Australia by Y chromosome and mtDNA analysis. Proc Natl Acad Sci USA 10:8726–8730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just RS, Scheible MK, Fast SA, Sturk‐Andreaggi K, Higginbotham JL, Lyons EA, Bush JM, Peck MA, Ring JD, Diegoli TM, Röck AW, Huber GE, Nagl S, Strobl C, Zimmermann B, Parson W, Irwin JA. 2014. Development of forensic‐quality full mtGenome haplotypes: success rates with low template specimens. Forensic Sci Int Genet 10:73‐79. [DOI] [PubMed] [Google Scholar]
- Kayser M. 2015. Forensic DNA phenotyping: predicting human appearance from crime scene material for investigative purposes. Forensic Sci Int Genet pii:S1872‐4973(15)00032‐0. [DOI] [PubMed] [Google Scholar]
- Kayser M, de Knijff P. 2011. Improving human forensics through advances in genetics, genomics and molecular biology. Nat Rev Genet 12:179–192. [DOI] [PubMed] [Google Scholar]
- King JL, LaRue BL, Novroski NM, Stoljarova M, Seo SB, Zeng X, Warshauer DH, Davis CP, Parson W, Sajantila A, Budowle B. 2014. High‐quality and high‐throughput massively parallel sequencing of the human mitochondrial genome using the Illumina MiSeq. Forensic Sci Int Genet 12:128–135. [DOI] [PubMed] [Google Scholar]
- Kivisild T. 2015. Maternal ancestry and population history from whole mitochondrial genomes. Investig Genet 6:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong QP, Bandelt HJ, Sun C, Yao YG, Salas A, Achilli A, Wang CY, Zhong L, Zhu CL, Wu SF, Torroni A, Zhang YP. 2006. Updating the East Asian mtDNA phylogeny: a prerequisite for the identification of pathogenic mutations. Hum Mol Genet 15:2076–2086. [DOI] [PubMed] [Google Scholar]
- Lao O, Altena E, Becker C, Brauer S, Kraaijenbrink T, van Oven M, Nurnberg P, de Knijff P, Kayser M. 2013. Clinal distribution of human genomic diversity across the Netherlands despite archaeological evidence for genetic discontinuities in Dutch population history. Investig Genet 4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist C, Schuster SC, Sun YZ, Talbot SL, Qi J, Ratan A, Tomsho LP, Kasson L, Zeyl E, Aars J, Miller W, Ingólfsson Ó, et al. 2010. Complete mitochondrial genome of a Pleistocene jawbone unveils the origin of polar bear. Proc Natl Acad Sci USA 107:5053–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippold S, Xu H, Ko A, Li M, Renaud G, Butthof A, Schröder R, Stoneking M.2014. Human paternal and maternal demographic histories: insights from high‐resolution Y chromosome and mtDNA sequences. Investig Genet 5:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman NJ, Misra RV, Dallman TJ, Constantinidou C, Gharbia SE, Wain J, Pallen MJ.2012. Performance comparison of benchtop high‐throughput sequencing platforms. Nat Biotechnol 30:434–439. [DOI] [PubMed] [Google Scholar]
- Loogväli EL, Roostalu U, Malyarchuk BA, Derenko MV, Kivisild T, Metspalu E, Tambets K, Reidla M, Tolk HV, Parik J, Pennarun E, Laos S, et al. 2004. Disuniting uniformity: a pied cladistic canvas of mtDNA haplogroup H in Eurasia. Mol Biol Evol 21:2012–2021. [DOI] [PubMed] [Google Scholar]
- Lyons EA, Scheible MK, Sturk‐Andreaggi K, Irwin JA, Just RS. 2013. A high‐throughput Sanger strategy for human mitochondrial genome sequencing. BMC Genomics 16:14–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhoe JA, Holland MM, Makova KD, Su MS, Paul IM, Baker CH, Faith SA, Young B. 2014. Development and assessment of an optimized next‐generation DNA sequencing approach for the mtgenome using the Illumina MiSeq. Forensic Sci Int Genet 13:20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Kircher M, Gansauge MT, Li H, Racimo F, Mallick S, Schraiber JG, Jay F, Prüfer K, de Filippo C, Sudmant PH, Alkan C, et al. 2012. A high‐coverage genome sequence from an archaic Denisovan individual. Science 338:222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller W, Drautz DI, Janecka JE, Lesk AM, Ratan A, Tomsho LP, Packard M, Zhang Y, McClellan LR, Qi J, Zhao F, Gilbert MT, et al. 2009. The mitochondrial genome sequence of the Tasmanian tiger (Thylacinus cynocephalus). Genome Res 19:213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noonan JP, Hofreiter M, Smith D, Priest JR, Rohland N, Rabeder G, Krause J, Detter JC, Pääbo S, Rubin EM. 2005. Genomic sequencing of Pleistocene cave bears. Science 309:597–600. [DOI] [PubMed] [Google Scholar]
- Pala M, Olivieri A, Achilli A, Accetturo M, Metspalu E, Reidla M, Tamm E, Karmin M, Reisberg T, Hooshiar Kashani B, Perego UA, Carossa V, et al. 2012. Mitochondrial DNA signals of late glacial recolonization of Europe from Near Eastern refugia. Am J Hum Genet 90:915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanichamy MG, Sun C, Agrawal S, Bandelt HJ, Kong QP, Khan F, Wang CY, Chaudhuri TK, Palla V, Zhang YP. 2004. Phylogeny of mitochondrial DNA macrohaplogroup N in India, based on complete sequencing: implications for the peopling of South Asia. Am J Hum Genet 75:966–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palo JU, Hedman M, Söderholm N, Sajantila A. 2007. Repatriation and identification of the Finnish World War II soldiers. Croat Med J 48:528–535. [PMC free article] [PubMed] [Google Scholar]
- Parson W, Huber G, Moreno L, Madel MB, Brandhagen MD, Nagl S, Xavier C, Eduardoff M, Callaghan TC, Irwin JA. 2015. Massively parallel sequencing of complete mitochondrial genomes from hair shaft samples. Forensic Sci Int Genet 15:8–15. [DOI] [PubMed] [Google Scholar]
- Parson W, Strobl C, Huber G, Zimmermann B, Gomes SM, Souto L, Fendt L, Delport R, Langit R, Wootton S, Lagacé R, Irwin J. 2013. Evaluation of next generation mtGenome sequencing using the Ion Torrent Personal Genome Machine (PGM). Forensic Sci Int Genet 7:543–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng MS, Quang HH, Dang KP, Trieu AV, Wang HW, Yao YG, Kong QP, Zhang YP. 2010. Tracing the Austronesian footprint in Mainland Southeast Asia: a perspective from mitochondrial DNA. Mol Biol Evol 27:2417–2430. [DOI] [PubMed] [Google Scholar]
- Ralf A, van Oven M, Zhong K, Kayser M. 2015. Simultaneous analysis of hundreds of Y‐chromosomal SNPs for high‐resolution paternal lineage classification using targeted semiconductor sequencing. Hum Mutat 36:151–159. [DOI] [PubMed] [Google Scholar]
- Ramos A, Santos C, Alvarez L, Nogués R, Aluja MP. 2009. Human mitochondrial DNA complete amplification and sequencing: a new validated primer set that prevents nuclear DNA sequences of mitochondrial origin co‐amplification. Electrophoresis 30:1587–1593. [DOI] [PubMed] [Google Scholar]
- Rasmussen M, Li YR, Lindgreen S, Pedersen JS, Albrechtsen A, Moltke I, Metspalu M, Metspalu E, Kivisild T, Gupta R, Bertalan M, Nielsen K, et al. 2010. Ancient human genome sequence of an extinct Palaeo‐Eskimo. Nature 463:757–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidla M, Kivisild T, Metspalu E, Kaldma K, Tambets K, Tolk HV, Parik J, Loogväli EL, Derenko M, Malyarchuk B, Bermisheva M, Zhadanov S, et al. 2003. Origin and diffusion of mtDNA haplogroup X. Am J Hum Genet 73:1178–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roostalu U, Kutuev I, Loogväli EL, Metspalu E, Tambets K, Reidla M, Khusnutdinova EK, Usanga E, Kivisild T, Villems R. 2007. Origin and expansion of haplogroup H, the dominant human mitochondrial DNA lineage in West Eurasia: the Near Eastern and Caucasian perspective. Mol Biol Evol 24:436–448. [DOI] [PubMed] [Google Scholar]
- Schlebusch CM, Naidoo T, Soodyall H. 2009. SNaPshot minisequencing to resolve mitochondrial macro‐haplogroups found in Africa. Electrophoresis 30:3657–3664. [DOI] [PubMed] [Google Scholar]
- Seo SB, King JL, Warshauer DH, Davis CP, Ge J, Budowle B. 2013. Single nucleotide polymorphism typing with massively parallel sequencing for human identification. Int J Legal Med 127:1079–1086. [DOI] [PubMed] [Google Scholar]
- Seo SB, Zeng X, King JL, Larue BL, Assidi M, Al‐Qahtani MH, Sajantila A, Budowle B. 2015. Underlying Data for Sequencing the Mitochondrial Genome with the Massively Parallel Sequencing Platform Ion TorrentTM PGMTM . BMC Genomics 16(Suppl 1):S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa MX, Sivakumar IK, Maragh S, Veeramachaneni V, Hariharan R, Parulekar M, Fredrikson KM, Harkins TT, Lin J, Feldman AB, Tata P, Ehret GB, et al. 2012. Next‐generation sequencing of human mitochondrial reference genomes uncovers high heteroplasmy frequency. PLoS Comput Biol 8:e1002737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiller M, Knapp M, Stenzel U, Hofreiter M, Meyer M. 2009. Direct multiplex sequencing (DMPS)—a novel method for targeted high‐throughput sequencing of ancient and highly degraded DNA. Genome Res 19:1843–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabbada KA, Trejaut J, Loo JH, Chen YM, Lin M, Mirazón‐Lahr M, Kivisild T, De Ungria MC. 2010. Philippine mitochondrial DNA diversity: a populated viaduct between Taiwan and Indonesia? Mol Biol Evol 27:21–31. [DOI] [PubMed] [Google Scholar]
- Templeton JE, Brotherton PM, Llamas B, Soubrier J, Haak W, Cooper A, Austin JJ. 2013. DNA capture and next‐generation sequencing can recover whole mitochondrial genomes from highly degraded samples for human identification. Investig Genet 4:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oven M, Kayser M. 2009. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat 30:386–394. [DOI] [PubMed] [Google Scholar]
- van Oven M, Vermeulen M, Kayser M. 2011. Multiplex genotyping system for efficient inference of matrilineal genetic ancestry with continental resolution. Investig Genet 23:2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veras AAO, de Sã PHCG, Pinheiro KC, das Gracas DA, Baraúna RA, Schneider MPC, Azevedo V, Ramos RTJ, Silva A. 2014. Efficiency of Corynebacterium pseudotuberculosis 31 genome assembly with the Hi‐Q enzyme on an Ion Torrent PGM sequencing platform. J Proteomics Bioinform 7:374–378. [Google Scholar]
- Wilson MR, Allard MW, Monson KL, Miller KWP, Budowle B. 2002a. Recommendations for consistent treatment of length variants in the human mtDNA control region. Forensic Sci Int 129:35–42. [DOI] [PubMed] [Google Scholar]
- Wilson MR, Allard MW, Monson KL, Miller KWP, Budowle B. 2002b. Further discussion of the consistent treatment of length variants in the human mitochondrial DNA control region. Forensic Sci Commun 4:4. [DOI] [PubMed] [Google Scholar]
- Wilson MR, DiZinno JA, Polanskey D, Replogle J, Budowle B. 1995a. Validation of mitochondrial DNA sequencing for forensic casework analysis. Int J Legal Med 108:68–74 [DOI] [PubMed] [Google Scholar]
- Wilson MR, Polanskey D, Butler J, DiZinno JA, Replogle J, Budowle B. 1995b. Extraction, PCR amplification and sequencing of mitochondrial DNA from human hair shafts. Biotechniques 18:662–669. [PubMed] [Google Scholar]
- Willerslev E, Gilbert MT, Binladen J, Ho SY, Campos PF, Ratan A, Tomsho LP, da Fonseca RR, Sher A, Kuznetsova TV, Nowak‐Kemp M, Roth TL, et al. 2009. Analysis of complete mitochondrial genomes from extinct and extant rhinoceroses reveals lack of phylogenetic resolution. BMC Evol Biol 9:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaragoza MV, Fass J, Diegoli M, Lin D, Arbustini E. 2010. Mitochondrial DNA variant discovery and evaluation in human cardiomyopathies through next‐generation sequencing. PLoS One 5:e12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material