

Abstract
Hereditary factors are thought to play a role in at least one third of patients with colorectal cancer (CRC) but only a limited proportion of these have mutations in known high‐penetrant genes. In a relatively large part of patients with a few or multiple colorectal polyps the underlying genetic cause of the disease is still unknown. Using exome sequencing in combination with linkage analyses together with detection of copy‐number variations (CNV), we have identified a duplication in the regulatory region of the GREM1 gene in a family with an attenuated/atypical polyposis syndrome. In addition, 107 patients with colorectal cancer and/or polyposis were analyzed for mutations in the candidate genes identified. We also performed screening of the exonuclease domain of the POLE gene in a subset of these patients. The duplication of 16 kb in the regulatory region of GREM1 was found to be disease‐causing in the family. Functional analyses revealed a higher expression of the GREM1 gene in colorectal tissue in duplication carriers. Screening of the exonuclease domain of POLE in additional CRC patients identified a probable causative novel variant c.1274A>G, p.Lys425Arg. In conclusion a high penetrant duplication in the regulatory region of GREM1, predisposing to CRC, was identified in a family with attenuated/atypical polyposis. A POLE variant was identified in a patient with early onset CRC and a microsatellite stable (MSS) tumor. Mutations leading to increased expression of genes can constitute disease‐causing mutations in hereditary CRC syndromes. © 2015 The Authors. Genes, Chromosomes & Cancer Published by Wiley Periodicals, Inc.
INTRODUCTION
One third of the variance in CRC risk is presumed to be due to genetic factors, but only around 6% of the patients have mutations in known high‐penetrant genes. Definitions of hereditary CRC syndromes are based on phenotypical features like histology, location and number of polyps, extra‐intestinal manifestations, cancer risk and inheritance mode (Aaltonen et al., 2007). Syndromes characterized by polyposis include familial adenomatous polyposis (FAP), MUTYH‐associated polyposis (MAP), juvenile polyposis (JPS), Cowden syndrome, Peutz–Jeghers syndrome, serrated polyposis (SPS), hereditary mixed polyposis (HMPS), and the recently described polymerase proofreading‐associated polyposis (PPAP). This syndrome is caused by germ‐line mutations in the exonuclease domain of POLE and POLD1 (Palles et al., 2013) and is recognized by multiple or very large adenomas and early onset CRC with microsatellite stable tumors (MSS). Exome studies have also recently identified high penetrant mutations in new CRC predisposing genes e.g. the NTHL1 gene with a recessive inheritance of the phenotype, predisposing to BER associated adenomatous polyposis and CRC (Weren et al., 2015). However, in the attenuated and mixed polyposis syndromes only a fraction of the disease‐causing mutations can still be identified (Fodde et al., 1992; Miyoshi et al., 1992; Lynch et al., 1995). The low mutation‐detection rate implicates the probable presence of additional unknown disease‐causing genes and possibly also recognition of new mutation mechanisms.
The HMPS is characterized by an autosomal dominant inheritance of multiple types of colorectal polyps (including serrated, hamartomatous, and conventional adenomas) and CRC. The genetic cause of this syndrome is still to a large extent unknown but a duplication of 40 kb including the 3′ region of the SCG5 gene and upstream region of GREM1 has been described as the disease‐causing mutation in families of Ashkenazi descent (Jaeger et al., 2012).
The aim of this study was to identify a causative germ‐line mutation predisposing to CRC in a family with AFAP/atypical polyposis. Combinations of techniques were used including, exome sequencing, linkage analysis, MLPA (multiplex ligation‐dependent probe amplification) and array analyses for SNP and CNV detection. In addition, 107 patients with or without a family history of polyposis and/or CRC were analyzed for mutations in the candidate genes identified. In a subset of these patients also the exonuclease domain of the POLE gene was screened.
MATERIAL
The material consists of one large atypical AFAP family for which the clinical characteristics are described below and of 107 index patients. The 107 patients either constitute simplex cases without any known inheritance for the disease or the index patient in a family where also other members are affected. The patients are described in Supporting Information Table S1 and each index patient is numbered in bold. Based on the initial referral the patients are divided into seven groups which are the following: (I) CRC familial or unknown inheritance not polyposis without identified mutation, (II) FAP familial without identified mutation, (III) FAP or Gardner syndrome (no inheritance) without identified mutation, (IV) AFAP familial without identified mutation, (V) AFAP (no inheritance) without identified mutation, (VI) Atypical FAP/mixed polyposis/serrated polyposis, familial or unknown inheritance without identified mutation, (VII) Polymerase Proofreading Associated Polyposis (PPAP), familial or unknown inheritance. Unfortunately we did not have enough DNA from the index patients so that we could include all of them in all of the mutation and genotype analyzes.
The AFAP/Atypical Polyposis Family
This family is shown in Figure 1 with the clinical diagnoses indicated. In general the syndrome in this family resembles an AFAP phenotype but some indications of polyp morphology similar to a juvenile and a metaplastic type have also been reported. Family member IV:2 had follow‐ups since the age of 21 and due to polyps a prophylactic IRA (ileo‐rectal anastomosis) was performed at age 28. The histological examination showed one polyp in the rectum and six in the transverse colon and basophilic epithelium. Family member V:2 had follow‐ups since the age of 11. At the age of 25 coloscopy showed tubular and tubulovillous adenomas in the ascending and transverse colon. One year later a few polyps in transverse colon and left colic (splenic) flexure were removed. At the age of 27 a polyp in the transverse colon was found and the year after a polyp in the right colic (hepatic) flexure and a rectal polyp (tubular adenoma or perhaps juvenile polyp) were observed.
Figure 1.
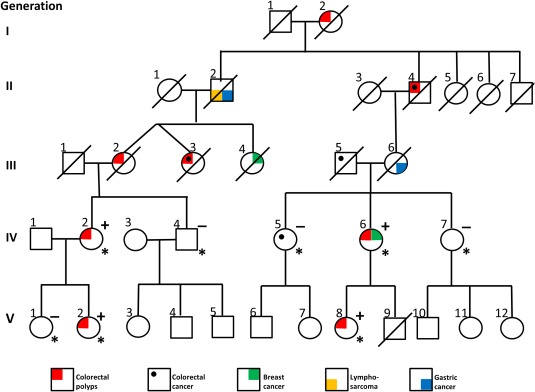
Pedigree of the AFAP/atypical polyposis family. Individuals with clinical diagnosis indicated, patients marked with * were sequenced by whole exome sequencing and linkage analysis was also performed on these patients. The duplication was present in heterozygote form in patients indicated with a (+) and family members which did not carry the mutation are indicated with a (−). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Family member III:2 had a colectomy at the age of 52 where histological examinations detected five polyps in the colon and a 4 cm polyp 12 cm from the valvula bauhinia (ileoceacal valve). Metaplastic polyps in the rectum were also found on multiple follow‐ups. Family member III:3 was operated for cancer in the transverse colon at the age of 33 with a colon resection, 18 years later polyps in the left flexure and in the rectum were found and she had CRC at age 51. A total colectomy was performed. The sister of the twin sisters (III:4) was afflicted with breast cancer, their father (II:2) with gastric cancer and lymphosarcoma, and a cousin (III:6) with gastric cancer. A grandchild (V:8) had polyps (colectomy at 26 years of age) as had her mother (IV:6) (colectomy at 47 years of age) who also developed breast cancer at 61 years of age. Her sister (IV:5) was afflicted with CRC at age 66 without polyps. She has been followed with regular colonoscopies since 20 years of age without any findings of polyps why CRC in this patient probably can be considered as sporadic. Genomic DNA from peripheral blood from four affected (IV:2, V:2, IV:6, V:8) and four unaffected (IV:4, V:1, IV:5, IV:7) family members were available for the exome and linkage analyses. In addition, RNA purified from samples from normal colon mucosa were available from individual IV:2 and V:2 for expression analyses of GREM1.
Ethical Statement
Ethical approval for the study was obtained from the regional ethics committee in Gothenburg (administration number 227‐10) and includes the family with AFAP/atypical polyposis (both affected and healthy individuals) and patient no. 106 (Supporting Information Table S1), all provided written informed consent at the time of collection of their samples, no samples were from minors. The consent also includes access to medical record data. All other samples in the study were anonymized. All samples used in this study were blood samples if not otherwise stated.
The sequence data is deposit in European Nucleotide Archive (ENA), accession number PRJEB7926.
METHODS
Sanger Sequencing, TaqMan Analyses and MLPA
Genomic DNA was extracted using the BioRobot EZ1 (Qiagen, Hilden, Germany) with the EZ1 DNA Blood 350 μl kit (Qiagen). Amplification, purification and sequencing of GREM 1 (NM_013372.6), TPRM1 (NM_001252020.1), and the exonuclease domain, exon 9‐14, of the POLE gene (NM_006231) were carried out as described previously (Kanter‐Smoler et al., 2008). Primers used for direct sequencing were identical to those used in the amplification reactions. All primer information is available in Supporting Information Table S2.
Custom TaqMan Assay Design Tool was used to design the probes for variants in GREM1 and TRPM1 and Genotyping TaqMan SNP assays (Life Technologies, Carlsbad, CA) was used for variant analysis. The analyses were performed on the Applied Biosystems® 7500 Fast Real‐Time PCR System (Life Technologies) in 96‐well plates according to the manual instructions. The results were analyzed by using the settings for the SNP assay with subsequent determination of genotypes. Since limited amounts of DNA were available subsets of patients were analyzed for the variants.
Copy‐number detection by MLPA was performed using the SALSA MLPA kits: P043 APC (version B1), P003 MLH1/MSH2 (version B2), P072 MSH6 (version C1), P008 PMS2 (version C1), and P378 MUTYH (version B1) (MRC‐Holland, Amsterdam, The Netherlands) according to the protocol provided by the supplier. The MLPA data were analyzed using GeneMapper 4.0 genotyping software (Life Technologies, and SeqPilot version 3.3.2 (JSI medical systems, GmBH, Kippenheim, Germany).
Exome Capture
DNA samples were quantified using the Qubit system (Life Technologies). Three µg of DNA was fragmented using the Covaris S2 Ultrasonicator (Covaris, Woburn, MA). The samples were then analyzed on the Bioanalyzer (Agilent Technology, Santa Clara, CA) for correct fragment sizes. Libraries were constructed using the “Agilent SureSelect XT Human All Exon 50Mb library kit” or “Agilent SureSelect_XT_Human_All_Exon_V5” for Illumina Paired End Sequencing (v3 protocol version 1.4.1 or G7530 90000_SureSelect_IlluminaXTMultiplexed_1_5) (Agilent Technologies) according to instructions. The concentration of each library was determined by use of the Qubit and the Bioanalyzer. Whole exome sequencing was performed on the Illumina HiScanSQ with 2x76 bp paired‐end reads using True Seq SBS HS‐v3 clustering and sequencing chemistry (Illumina, San Diego, CA).
Analyses of Exome Sequencing Data and Filtering of Variants
Quality assessment of the sequence reads was performed by generating QC statistics with FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc). Read alignment to the reference human genome (hg19, UCSC assembly, February 2009) was done using Burrow Wheelers Aligner (BWA) (Li and Durbin, 2009) with default parameters. A summary of the sequencing data is shown in (Supporting Information Table S3). After removal of PCR duplicates (Picard tools, http://picard.sourceforge.net) and file conversion (Samtools (Li et al., 2009)) quality score recalibration, indel realignment, and variant calling were performed with the Genome Analysis Tool Kit (GATK) package (McKenna et al., 2010). Variants were annotated with Annovar (Wang et al., 2010) using a wide range of databases such as dbSNP build 135 (Sherry et al., 2001), dbNSFP (Liu et al., 2011), KEGG (Kanehisa et al., 2012), the Gene Ontology project (Ashburner et al., 2000), MITOMAP (Ruiz‐Pesini et al., 2007) and tracks from the UCSC (Fujita et al., 2010), and also the National Heart, Lung and Blood Institute (NHLBI) Exome Sequencing Project (ESP6500) (http://evs.gs.washington.edu/EVS/).
Four affected and four unaffected individuals were available (Fig. 1). All common variants among the four affected individuals (not present among the unaffected) were filtered for technical artifacts and common polymorphic variants against an in‐house control data set (generated from exome data from 38 individuals) provided by Genomics Core facility (Gothenburg, Sweden). Variants were then compared with the following public databases; the Single Nucleotide Polymorphism database (dbSNP) together with 1000 Genomes (Abecasis et al., 2012), the National Heart, Lung and Blood Institute (NHLBI) Exome Sequencing Project (ESP6500) (http://evs.gs.washington.edu/EVS/), and the Exome Aggregation Consortium (ExAC), Cambridge, MA (URL:http://exac.broadinstitute.org). Selected variants with a MAF <1% were also compared with cbioportal (http://www.cbioportal.org/) (Cerami et al., 2012; Gao et al., 2013) and the COSMIC database (Forbes et al., 2015). To distinguish variants from local polymorphisms between 200 and 248 anonymized in‐house normal control blood samples were sequenced over the positions of interest. Variant were interpreted for their deleteriousness by using SIFT (Sorting Intolerant From Tolerant), PolyPhen‐2, (Polymorphism Phenotyping version 2), Mutation Taster, Condel (CONsensus DELeteriousness score of missense SNVs), and PON‐P (Pathogenic‐or‐Not–Pipeline) (Sunyaev et al., 2001; Kumar et al., 2009; Schwarz et al., 2010; Gonzalez‐Perez and Lopez‐Bigas, 2011; Olatubosun et al., 2012). Splice‐site prediction of variants was performed with SpliceSiteFinder‐like, MaxEntScan, NNSPLICE, GeneSplicer and Human Splicefinder (Reese et al., 1997; Zhang, 1998; Pertea et al., 2001; Fairbrother et al., 2002; Cartegni et al., 2003; Yeo and Burge, 2004; Desmet et al., 2009; Houdayer et al., 2012).
Detection of copy‐number alterations in our exome analysis, was done by applying FREEC v6.7 (Boeva et al., 2011) with default settings on reads that were uniquely mapped to the reference genome.
Detection of Copy Number Variations (CNVs) and Linkage Analyses Using Affymetrix Genome‐Wide Human SNP Array 6.0 and Cytoscan®HD Array
Samples were prepared according to standard conditions (Affymetrix Inc, Santa Clara, CA). Purification of PCR products was performed using Magnetic Beads (Agencourt Bioscience Corporation, Beverly, MA). Hybridization, washing and staining of arrays was performed as described by the supplier (Affymetrix). Affymetrix Genotyping Console 3.0.1 (GTC) and/or Chromosome Analysis Suite (ChAS) 1.0.1. software (Affymetrix) was used for genotyping and copy number analyses on both of the array platforms. The Genotyping Console Contrast Quality Control metric was used to filter out low‐quality samples using the default threshold of cQC <0.4. The birdseed analysis software package included in GTC was used to generate the genotypes. For the CN analysis the Median Absolute Pairwise Difference (MAPD) value threshold was set to MAPD <0.3. For the cytoscan HD array these values where QC >15, MAPD <0.25 and waviness Sd <0.12. To exclude regions that represent normal CNVs we compared all detected CNVs with those present in the Database of Genomics Variants (DGV) (MacDonald et al., 2014) and an in‐house data set of 44 blood controls. CNVs included in the in‐house dataset or at least twice in DGV were considered normal. In the family common CNVs among affected were manually inspected.
Parametric linkage analyses based on SNP data from both of the array platforms was done with Allegro 2.0 (Gudbjartsson et al., 2000) using a dominant model with a rare disease allele, full penetrance, and no phenocopies. The set of SNPs were pruned to 26,000 high quality SNPs with limited LD before running the analysis.
RNA Preparation and Expression Analyses by Droplet Digital PCR (ddPCR)
RNA from four tissue samples (fresh frozen normal colon mucosa) two each from patients, IV:2 and V:2 (from different locations in the colon), were extracted using RNeasy Mini Kit (Qiagen) according to the manufactureŕs instructions. RNA extracted from normal colon mucosa from six patients with sporadic CRC was included as controls. The samples were diluted to 1 ng/µl and run in duplicates, except for V:2 which was run in triplicates. Nontemplate controls (NTC) were also included in duplicates. The master mix included One‐Step RT‐ddPCR supermix (Bio‐Rad, Hercule, CA) and the TaqMan assay GREM1 (Assay ID HS000171951 (Life Technologies, Carlsbad, CA)), a total reaction volume of 20 µl was used. Five microliters RNA (1 ng/µl) was added to all reactions. Droplets were formed by adding 70 µl oil and 20 µl sample to each cartridge which was then placed in the QX100 Droplet Generator (Bio‐Rad). The instrument generates about 20,000 droplets in a mixture of sample and oil. Forty microliters droplets (the total volume generated) were carefully transferred to the PCR plate. Reverse transcription and PCR were performed in one step on T100 Thermal Cycler instrument (Bio‐Rad) with the program (reverse transcription 60°C 30 min, enzyme activation 95°C 5 min, denaturation 30 sec 94°C, annealing/extension 60°C 60 sec with a ramp rate of 2.5°C/sec for 40 cycles and finally enzyme deactivation for 10 min at 98°C). The PCR plate was placed in the QX100 Droplet Reader (Bio‐Rad), and 14,010‐18,795 droplets for each sample were analyzed by fluorescence measurement. Manual settings were used to establish a cut‐off threshold (1,212) for negative versus positive droplets (partitions). Statistical significance was performed by using Student's t‐test. The expression analyses were performed at the TATAA Biocenter AB (Gothenburg, Sweden).
Methylation Analysis of the GREM1 Region
A region of 2,166 bp (chr15:33,009,531‐33,011,696) was identified as the GREM1 CpG island region including 336 unique CpG sites. Assays were designed targeting CpG sites in the specified region of interest (ROI) using primers created with Rosefinch, Zymo Research's proprietary sodium bisulfite converted DNA‐specific primer design tool (Zymo research, Irvine, CA). Following primer validation, samples were bisulfite converted using the EZ DNA Methylation Lightning™ Kit (Zymo Research) according to the manufacturer's instructions. Multiplex amplification of all samples using ROI specific primer pairs and the Fluidigm Access Array™ System (Fluidigm, South San Francisco, CA) was performed according to the manufacturer's instructions. Targeted region was amplified in 16 amplicons. The resulting amplicons were pooled for harvesting and subsequent barcoding according to the Fluidigm instrument's guidelines. After barcoding, samples were purified and then prepared for massively parallel sequencing using a MiSeq V2 300 bp Reagent Kit (Illumina) and paired‐end sequencing according to the manufacturer's guidelines was carried out. Sequence reads were identified using standard Illumina base‐calling software and then analyzed using a Zymo Research proprietary analysis pipeline. Low quality nucleotides and adapter sequences were trimmed off during analysis QC. Sequence reads were aligned back to the reference genome using Bismark (http://www.bioinformatics.babraham.ac.uk/projects/bismark/), an aligner optimized for bisulfite sequence data and methylation calling (Krueger and Andrews, 2011). Paired‐end alignment was used as default thus requiring both read 1 and read 2 to be aligned within a certain distance, otherwise both read 1 and read 2 were discarded. Index files were constructed using the bismark_genomer_preparation command and the entire reference genome. The non‐directional parameter was applied while running Bismark. All other parameters were set to default. Nucleotides in primers were trimmed off from amplicons during methylation calling. The methylation level of each sampled cytosine was estimated as the number of reads reporting a C, divided by the total number of reads reporting a C or T. All analyses were carried out at Zymo Research.
RESULTS
In order to find the disease‐causing mutation in the AFAP/atypical polyposis family we performed two exome analyses. In the first only exome regions were targeted while in the second also 5′‐ and 3′‐UTR gene regions were included. In the initial analysis we sequenced four affected and four unaffected individuals from the family using Agilent version “All exome 50MB capture kit”. Linkage data was used to restrict our search to shared regions in the genome. We had previously determined linkage to five regions with LOD >1.5 on chromosome 3 (LOD = 2.94), 7 (LOD = 2.97), 8 (LOD = 1.58), 15 (LOD = 2.18), and X (LOD = 1.80). These regions were the only regions that fit a fully penetrant dominant model, by necessity or by random co‐segregation. The differences in max LOD is due to varying information content. Analyses for variants segregating with disease in coding sequences in these regions were performed, only one variant shared between affected but not present in unaffected individuals was found. This variant was TRPM1 c.2200G>C, p.Ala734Pro on chromosome 15. This was the only shared variant of any kind in the coding regions among the affected individuals (entire exome analyzed) after the previous filtering steps had been applied. The TRPM1 variant has an MAF of 0.63% (ExAC), and 0.12% (ESP6500). The variant was also present in 2 of 248 (0.8%) in‐house blood controls from the western part of Sweden. The G in position c.2200 is highly conserved with a phyloP value of 5.86. The amino acid substitution is predicted to be deleterious according to SIFT, probably damaging according to Polyphen‐2, deleterious according to Condel but neutral according to PON‐P. Sixty‐six index patients were analyzed for the presence of this variant (Supporting Information Table S4, Column 2). Patient 99 was also positive for the variant. As the TRPM1 variant is a known rare polymorphism and has not been associated with CRC previously it was unlikely that this variant was the disease causing mutation and therefore we performed a second exome sequencing with an updated Agilent exome kit, version 5, in which also UTŔs are included for further investigations. Two affected (IV:2, IV:6) and two unaffected (IV:4, IV:7) (Fig. 1, Supporting Information Table S3) individuals, one from each branch of the family were sequenced. In this analysis a GREM1 variant (c.−76C>G (g.33,010,288) in exon 1 (5′UTR) was found. This variant was located in one of the regions which showed linkage to the disease allele on chromosome 15.
Sanger sequencing of all affected and unaffected individuals confirmed that this variant segregated with the disease. The variant was not found to be present in ExAC, ESP6500, or in dbSNP, nor in 213 in‐house blood controls. This variant was not found in any of the 84 index patients, for which DNA were available in our material (Supporting Information Table S4, Column 3). The disease haplotype is 3.7 Mb and is located between rs12912234 and rs2928022. Several genes are located in this region, among them the SCG5 gene, in which a duplication leading to a higher expression of the GREM1 gene has been found in a large HMPS family (Jaeger et al., 2012). Analysis of CNVs in the SCG5 and GREM1 gene regions by MLPA on the same individuals as those analyzed by whole exome sequencing revealed a duplication of approximately 20 kb in affected family members. Duplication carriers were estimated to have three copies relative to controls and the duplicated region included two MLPA probes, the first probe located 42 bp downstream of the last exon in SCG5 and the second further downstream the SCG5 gene but still upstream (8,423 bp) of exon 1 in the GREM1 gene. The GREM1 gene is located downstream of SCG5. PCR amplification across the duplication breakpoints mapped it to chr.15:32,986,220‐33,002,449 (16,229 bp) and it was found to be a tandem repeat, located approximately 7.7 kb upstream of the GREM1 gene as illustrated in Figure 2.
Figure 2.
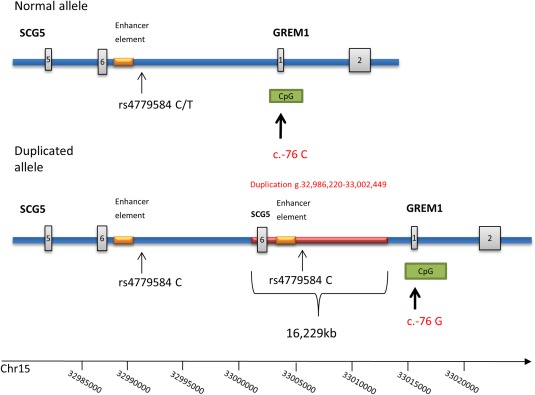
Schematic view of the duplicated region on chromosome 15. The tandem repeat of 16,229 bp (chr15:32,986,220‐33,002,449 (GRCh37/hg19) including the 3′part of the SCG5 gene. The rs4779584 C allele in the duplicated region is indicated with an arrow as is the c.−76 C/G SNP in the CpG island upstream of the GREM1 gene. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
In order to investigate if the c.−76C>G variant had any effect on the methylation among the affected individuals the whole CpG island of 2,166 bp upstream of GREM1 was sequenced in all available family members (blood), in three normal blood controls and a normal colon mucosa control. In 9 to 23 (depending on the sample analyzed) of the CpG‐sites some methylation was detected but it was not observed in more than approximately 20% of the reads and the sites were not found to be significantly methylated in any of the samples analyzed. No difference between patients and control were found and position c.−76 was not methylated to any significant degree in any of the samples analyzed and this variant could not be considered the disease‐causing mutation either (Fig. 3 and Supporting Information Fig. S1) However, the duplication, segregated with the disease in all the affected family members but was not present in any of the unaffected subjects (Fig. 1). Normal colon mucosa was available from two patients (IV:2 and V:2) and we analyzed for elevated expression of GREM1 in these samples. The two samples from different locations in the colon were analyzed with droplet digital PCR (ddPCR) using the described TaqMan expression assay for the GREM1 locus. The expression levels (copies/µl) for patients (IV:2 and V:2) compared with controls are presented in Figure 4. The controls included normal colon mucosa from sporadic colon cancer patients and a purchased control (normal colon mucosa). A significantly higher expression of the GREM1 gene was found in the patient samples compared with controls (P = 0.013). The SNP rs4779584 (C/T) located in the duplicated region have been associated with increased risk of CRC in several GWAS studies (Yang et al., 2014). We genotyped the family for this SNP and found the duplicated allele to be associated with the C allele (Fig. 5).
Figure 3.
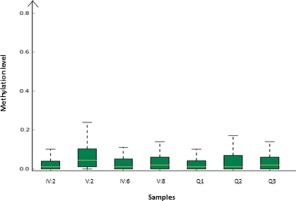
CpG‐methylation level boxplot showing the overall methylation levels of each sample compared side by side. The methylation level on the Y‐axis is the fraction of methylated reads/total number of reads. Sample IV:2, V:2, IV:6, and V:8 are from affected members and Q1, Q2, Q3 are blood controls. The overall degree of methylation detected is low and there is no significant difference between the patients and the control. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 4.

The absolute expression analysis of the GREM1 gene in patients and controls. The number of copies/µl is shown in this absolute expression analysis of GREM1. Six controls of normal colon mucosa and normal colon mucosa from two samples each from patient IV:2 and V:2 were analyzed. The GREM1 gene is significantly higher expressed in samples from colon mucosa in the affected family members compared with controls (P = 0.013). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Figure 5.
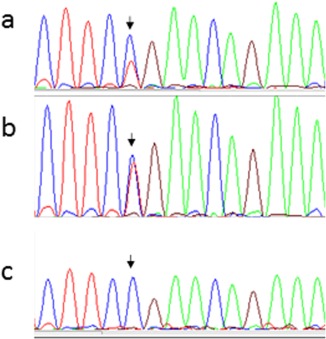
Sanger sequencing over rs4779584. (a) Family member V:2 (affected), presents with three alleles C,C,T (where C is blue and T is red). The duplication of C on the affected allele is clearly seen compared with the normal control in b. (b) Normal control (alleles C,T). (c) Family member IV:2 (affected) with three C alleles, C,C,C. The duplicated C allele cannot be distinguished from the other normal C allele in this case. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
To find out if possibly other variants in TRPM1 or GREM1 were present in other patients included in the study the entire coding regions of TRPM1 (27 exons) and GREM1 (two exons including the 5′UTR) were sequenced in 40 patients for which DNA was available (Supporting Information Table S4, Column 4). Variants with a MAF < 1% in these genes were investigated further. In the TRPM1 gene the variant c.602G>A, p.Arg201Gln (MAF: dbSNP 0.07%, ESP6500 0.06%, and ExAC 0.10%) rs188852505 was found in patient I:6. This variant was considered as a possible disease‐causing variant and was predicted deleterious by Condel and SIFT, probably damaging by Polyphen‐2 and unclassified by PON‐P. The amino acid was evolutionary conserved between species down to frog. Splice site predication tools also predicted the base change to possibly introduce a new donor site. No variants with a MAF <1% was found in the GREM1 gene To find out if a duplication in the same region upstream of GREM1 was present in any of 42 index patients for which DNA was available (Supporting Information Table S4, Column 5) MLPA analysis was performed on all of these. No duplications or deletions were found in this region in any of the patients. We also performed CNV analyses on array and exome data to exclude the presence of other large deletions or duplications in the samples from the family. All regions harboring deletions or duplications were manually inspected. No gene‐containing region that was shared between affected individuals could be found. The duplicated GREM1 region in the family was not detected with the array method.
Sanger Sequencing of the POLE Exonuclease Domain
Mutations in the DNA polymerase ɛ gene (POLE) were recently reported as predisposing for inherited colorectal cancer (Palles et al., 2013). In a previous study we present a mutation in the proof‐reading exonuclease domain of this protein in a large Swedish family with CRC (Rohlin et al., 2014), Supporting Information Table S1. This finding prompted us to search for mutations in the whole proof‐reading exonuclease domain of the POLE gene (exons 3–14) in 76 index patients for which DNA was available (Supporting Information Table S4, Column 1). A missense mutation c.1274A>G, p.Lys425Arg in the POLE gene was found in one individual VII:107 (Supporting Information Table S1). This individual was a single case with only sigmoid CRC and we had not access to any information about inheritance of the disease in the family. The tumor was MSI negative. The variant is present in the ExAC database at a frequency of 0.003%. In an attempt to try to understand if the amino‐acid substitution could confer a functional effect on the protein, in silico prediction based on the yeast DNA polymerase, polE, structure (4M8O.pdb), was performed (Hogg et al., 2014). The region of interest exhibits high amino‐acid identity. The positively charged basic amino acids, arginine and lysine, are mostly exposed to protein surface, and may take part in electrostatic interactions. In particular, the guanidinium group of arginine allows interactions in three possible directions, which enables arginine to form a higher number of electrostatic interactions compared with lysine. Considering the close proximity of the Lys425 to the negatively charged phosphate group of the DNA in the exo‐site, it is conceivable that the lysine to arginine mutation affects the proof‐reading activity of POLE (Fig. 6). The sequence Leu424, Lys425, Ala426 is also completely conserved in eukaryote polE polymerases (Supporting Information Fig. S2). Also theoretical predictions of the effect of the amino‐acid substitution suggest the variant to be deleterious (SIFT: Deleterious, Polyphen‐2: Probably Damaging, Mutation Taster: Disease Causing, Condel: Deleterious and PON‐P: Pathogenic).
Figure 6.
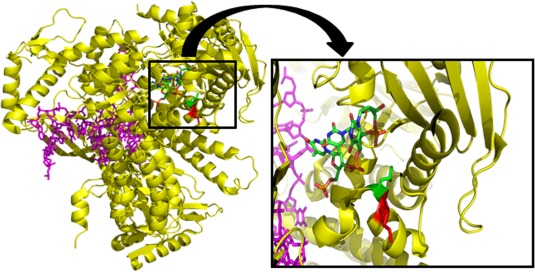
Superposition of yeast DNA polymerase and ssDNA substrate from Escherichia coli DNA polymerase Klenow fragment. Yeast DNA polymerase epsilon (yellow) and bound dsDNA (magenta) with ssDNA substrate (red, green, blue, orange) from E. coli DNA polymerase Klenow fragment superimposed onto the exonuclease domain of the yeast DNA polymerase. The magnification shows a slice through the polE structure (4M8O.pdb) superimposed with the ssDNA (red, green, blue, orange) in the exo‐site from the structure of the Klenow fragment (18Y.pdb). The side chains of the Lys425 and the Leu424 residues are shown in red and green, respectively. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
DISCUSSION
Exome resequencing is a powerful tool to find novel disease‐causing genes and mutations, although a major challenge is to be able to identify disease alleles among a large background of nonpathogenic variants. In families or simplex cases with hereditary cancer syndromes identification of new tumor suppressor genes or inactivating mutations in existing genes is most often the case (Gylfe et al., 2013; Smith et al., 2013; Gala et al., 2014; Nieminen et al., 2014; Segui et al., 2015; Weren et al., 2015). In our study an activating 16 kb duplication was found upstream of GREM1. A duplication of approximately 40 kb of an upstream region containing parts of the SCG5 gene and also a region between, but not the GREM1 gene itself, has recently been shown to over express GREM1 in colorectal epithelium (Jaeger et al., 2012; Davis et al., 2015). The GREM1 gene encodes the secreted BMP antagonist Gremlin. Compared with other tumorigenic pathways in CRC, the BMP signaling pathway is not well known. Ligands binding to BMP receptors are involved in the driving process of this pathway. Increased levels of GREM1 are expected to reduce BMP ligand levels and it is suggested that the cell favors a more stem cell‐like phenotype (Jaeger et al., 2012). The duplication which segregates with the disease phenotype in our family is about half the size (16.2 kb) of the duplication reported by Jaeger et al. (2012), but includes the same enhancer element that cause the increased GREM1 expression in this larger duplication (Fig. 2). In concordance with the results of their study an increased expression of the GREM1 transcript in normal colon epithelium from duplication carriers was expected. This was in true also found in colon mucosa from the two available duplication carriers analyzed, which confirms the disease mechanism. Interestingly, a reduction in BMP signaling is also responsible for initiation of JPS tumors. JPS can be caused by mutations in the BMPR1A gene (encoding the type 1A BMP receptor) or in SMAD4, which is a downstream effector of the BMP pathway. In fact the polyps in the family display some interesting histology resembling juvenile polyps and metaplastic polyps, which could define it as a family with mixed polyposis. In the HMPS family identified by Jaeger et al. (2012) a mixed phenotype was also suggested. To our knowledge this family has no Ashkenazi ancestry in contrast to the family by Jaeger et al. (2012). We did not find any other mutation of this kind in our cohort of FAP/AFAP or atypical FAP patients, which does point to the fact that these duplications are rare. A whole gene duplication of the GREM1 gene has been reported (Venkatachalam et al., 2011) and this duplication also contained a part (exons 3 to 6) of the SCG5 gene in a patient with early onset sigmoid colon cancer. A variant in a CpG site (g.33,010,288) in the GREM1 CpG island was also found to segregate with the disease in the family and we analyzed for methylation differences between affected, unaffected and controls. As no significant methylation was found in any of the samples, we propose that this variant does not cause the phenotype in the family. However, a SNP close to GREM1 has been found to be associated with an increased susceptibility to CRC in the general population (Lewis et al., 2014).
Separate from the exome analysis we sequenced the exonuclease domain of the POLE gene in a subset of the patients. The reason for this additional sequencing was that heterozygote mutations in the DNA polymerase genes, POLD1 and POLE, were recently identified in patients with multiple adenoma and/or CRC. The mutations were restricted to the proofreading exonuclease domain of the proteins (Palles et al., 2013). We have recently reported a family with a mutation in the exonuclease domain of POLE in Sweden. The mutation predisposes carriers to a multitumor phenotype (Rohlin et al., 2014). In our current study we identified another POLE mutation in the exonuclease domain of the POLE protein. The mutation was identified in a single case with limited information of the phenotype and inheritance (Supporting Information Table S1).The effect of this variant was analyzed using theoretical structure/function predictions and a possible deleterious effect is proposed for the Lys425Arg substitutions. This effect is supported by theoretical predictions of the consequence of the substitution as well as conservation of the amino acid between species. The close proximity to the Leu424Val mutation which has previously been shown to be the cause of CRC (Palles et al., 2013), further strengthens a pathogenic effect of the Lys425Arg substitution.
Our study demonstrates the possibilities whole‐exome sequencing provides in the demanding task to reveal new genes and mutations conferring deleterious effects caused by activation or inactivation of genes and gene products in CRC predisposing syndromes. Although exome sequencing is a powerful tool to identify new disease‐causing mutations, the GREM1 duplication in this study was not identified with this method as its location is outside the exonic region. In fact, the localization of the duplication‐site was indicated by a combination of linkage analysis and exome sequencing. The base substitution in the CpG island of the GREM1 gene that was not previously reported, was at first identified by exome‐sequencing and as the linkage analyses supported the possibility of a variant segregating with disease in this chromosomal region we focused on this gene. Finally the duplication was found with MLPA analysis. A limitation with exome sequencing is clearly illustrated with this case. Whole‐genome sequencing that includes regulatory regions outside the coding part of the gene is probably better suited to find the whole spectra of diverse mutation mechanisms in CRC and also other cancer syndromes. Interestingly the duplicated GREM1 region in the family was not detected with the array method even by careful manual inspection without any cut‐off settings, even though probes are located in the region. The region only contains a few probes and the regulatory region is quite GC‐rich which might affect hybridization, which could be one reason why this duplication was not found. In the exome data this region was not covered with any probes and therefore the duplication was not detected in this analysis either.
In this study we found one high penetrant mutation in GREM1 and also a probable causative POLE variant. In both of these genes disease‐causing alleles have recently been found in hereditary CRC families. Interestingly, candidate CRC predisposing genes in the BMP signaling pathway have been identified in GWAS and exome studies. The contribution of variants to CRC susceptibility in genes in this pathway needs further investigation. The GREM1 duplication also confirms that alterations of regions giving rise to amplifications and up‐regulation of expression can have an effect on tumor initiation also in germ‐line cells. The genotype, phenotype, and also the increased expression levels of GREM1 in our family give further support to the suggested role of activation of GREM1 as a cause of initiation and development of colorectal tumors (Jaeger et al., 2012; Davis et al., 2015).
Supporting information
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Table 1.
Supporting Information Table 2.
Supporting Information Table 3.
Supporting Information Table 4.
ACKNOWLEDGMENT
We are grateful for the contribution and support from the family members. This study was supported by grants from The Swedish Cancer Society, The Swedish state under the LUA/ALF agreement concerning research and education of doctors (no. 76310), The Health and Medical Care Committee of the Regional Executive Board, Region Västra Götaland, Project grant from Laboratory division Sahlgrenska University Hospital, The Nilsson‐Ehle Fondation, The Assar Gabrielsson Foundation, The Wilhelm and Martina Lundgren Research Foundation and The Sahlgrenska University Hospital Foundation. The exome analyses were carried out at the Genomics Core Facility at the University of Gothenburg.
REFERENCES
- Aaltonen L, Johns L, Jarvinen H, Mecklin JP, Houlston R. 2007. Explaining the familial colorectal cancer risk associated with mismatch repair (MMR)‐deficient and MMR‐stable tumors. Clin Cancer Res 13:356–361. [DOI] [PubMed] [Google Scholar]
- Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel‐Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeva V, Zinovyev A, Bleakley K, Vert J‐P, Janoueix‐Lerosey I, Delattre O, Barillot E. 2011. Control‐free calling of copy number alterations in deep‐sequencing data using GC‐content normalization. Bioinformatics (Oxford, England) 27:268–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. 2003. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res 31:3568–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. 2012. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis H, Irshad S, Bansal M. 2015. Aberrant epithelial GREM1 expression initiates colonic tumorigenesis from cells outside the stem cell niche. Nat Med 21:62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet FO, Hamroun D, Lalande M, Collod‐Beroud G, Claustres M, Beroud C. 2009. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother WG, Yeh RF, Sharp PA, Burge CB. 2002. Predictive identification of exonic splicing enhancers in human genes. Science 297:1007–1013. [DOI] [PubMed] [Google Scholar]
- Fodde R, van der Luijt R, Wijnen J, Tops C, van der Klift H, van Leeuwen‐Cornelisse I, Griffioen G, Vasen H, Khan PM. 1992. Eight novel inactivating germ line mutations at the APC gene identified by denaturing gradient gel electrophoresis. Genomics 13:1162–1168. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, Teague JW, Stratton MR, McDermott U, Campbell PJ. 2014. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015;43:D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, Diekhans M, Dreszer TR, Giardine BM, Harte RA, Hillman‐Jackson J, Hsu F, Kirkup V, Kuhn RM, Learned K, Li CH, Meyer LR, Pohl A, Raney BJ, Rosenbloom KR, Smith KE, Haussler D, Kent WJ. 2010. The UCSC Genome Browser database: Update 2011. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gala MK, Mizukami Y, Le LP, Moriichi K, Austin T, Yamamoto M, Lauwers GY, Bardeesy N, Chung DC. 2014. Germline mutations in oncogene‐induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology 146:520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Perez A, Lopez‐Bigas N. 2011. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet 88:440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, Kong A. 2000. Allegro, a new computer program for multipoint linkage analysis. Nat Genet 25:12–13. [DOI] [PubMed] [Google Scholar]
- Gylfe AE, Katainen R, Kondelin J, Tanskanen T, Cajuso T, Hanninen U, Taipale J, Taipale M, Renkonen‐Sinisalo L, Jarvinen H, Mecklin JP, Kilpivaara O, Pitkanen E, Vahteristo P, Tuupanen S, Karhu A, Aaltonen LA. 2013. Eleven candidate susceptibility genes for common familial colorectal cancer. PLoS Genet 9:e1003876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg M, Osterman P, Bylund GO, Ganai RA, Lundstrom EB, Sauer‐Eriksson AE, Johansson E. 2014. Structural basis for processive DNA synthesis by yeast DNA polymerase varepsilon. Nat Struct Mol Biol 21:49–55. [DOI] [PubMed] [Google Scholar]
- Houdayer C, Caux‐Moncoutier V, Krieger S, Barrois M, Bonnet F, Bourdon V, Bronner M, Buisson M, Coulet F, Gaildrat P, Lefol C, Leone M, Mazoyer S, Muller D, Remenieras A, Revillion F, Rouleau E, Sokolowska J, Vert JP, Lidereau R, Soubrier F, Sobol H, Sevenet N, Bressac‐de Paillerets B, Hardouin A, Tosi M, Sinilnikova OM, Stoppa‐Lyonnet D. 2012. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum Mutat 33:1228–1238. [DOI] [PubMed] [Google Scholar]
- Jaeger E, Leedham S, Lewis A, Segditsas S, Becker M, Cuadrado PR, Davis H, Kaur K, Heinimann K, Howarth K, East J, Taylor J, Thomas H, Tomlinson I. 2012. Hereditary mixed polyposis syndrome is caused by a 40‐kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1. Nat Genet 44:699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. 2012. KEGG for integration and interpretation of large‐scale molecular data sets. Nucleic Acids Res 40:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanter‐Smoler G, Fritzell K, Rohlin A, Engwall Y, Hallberg B, Bergman A, Meuller J, Gronberg H, Karlsson P, Bjork J, Nordling M. 2008. Clinical characterization and the mutation spectrum in Swedish adenomatous polyposis families. BMC Med 6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: A flexible aligner and methylation caller for Bisulfite‐Seq applications. Bioinformatics 27:1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. 2009. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073–1081. [DOI] [PubMed] [Google Scholar]
- Lewis A, Freeman‐Mills L, de la Calle‐Mustienes E, Giraldez‐Perez RM, Davis H, Jaeger E, Becker M, Hubner NC, Nguyen LN, Zeron‐Medina J, Bond G, Stunnenberg HG, Carvajal JJ, Gomez‐Skarmeta JL, Leedham S, Tomlinson I. 2014. A polymorphic enhancer near GREM1 influences bowel cancer risk through differential CDX2 and TCF7L2 binding. Cell Rep 8:983–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jian X, Boerwinkle E. 2011. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat 32:894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch HT, Smyrk T, McGinn T, Lanspa S, Cavalieri J, Lynch J, Slominski‐Castor S, Cayouette MC, Priluck I, Luce MC. 1995. Attenuated familial adenomatous polyposis (AFAP). A phenotypically and genotypically distinctive variant of FAP. Cancer 76:2427–2433. [DOI] [PubMed] [Google Scholar]
- MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. 2014. The Database of Genomic Variants: A curated collection of structural variation in the human genome. Nucleic Acids Res 42:D986–D992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi Y, Ando H, Nagase H, Nishisho I, Horii A, Miki Y, Mori T, Utsunomiya J, Baba S, Petersen G, Hamilton SR, Kinzler1 KW, Vogelstein B, Nakamura Y. 1992. Germ‐line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc Natl Acad Sci USA 89:4452–4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieminen TT, O'Donohue MF, Wu Y, Lohi H, Scherer SW, Paterson AD, Ellonen P, Abdel‐Rahman WM, Valo S, Mecklin JP, Jarvinen HJ, Gleizes PE, Peltomaki P. 2014. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology 147:595.e595–598.e595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olatubosun A, Valiaho J, Harkonen J, Thusberg J, Vihinen M. 2012. PON‐P: Integrated predictor for pathogenicity of missense variants. Hum Mutat 33:1166–1174. [DOI] [PubMed] [Google Scholar]
- Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, Sherborne A, Chubb D, Carvajal‐Carmona LG, Ma Y, Kaur K, Dobbins S, Barclay E, Gorman M, Martin L, Kovac MB, Humphray S, Lucassen A, Holmes CC, Bentley D, Donnelly P, Taylor J, Petridis C, Roylance R, Sawyer EJ, Kerr DJ, Clark S, Grimes J, Kearsey SE, Thomas HJ, McVean G, Houlston RS, Tomlinson I. 2013. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 45:136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Lin X, Salzberg SL. 2001. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res 29:1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Eeckman FH, Kulp D, Haussler D. 1997. Improved splice site detection in Genie. J Comput Biol 4:311–323. [DOI] [PubMed] [Google Scholar]
- Rohlin A, Zagoras T, Nilsson S, Lundstam U, Wahlstrom J, Hulten L, Martinsson T, Karlsson GB, Nordling M. 2014. A mutation in POLE predisposing to a multi‐tumour phenotype. Int J Oncol 45:77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz‐Pesini E, Lott MT, Procaccio V, Poole JC, Brandon MC, Mishmar D, Yi C, Kreuziger J, Baldi P, Wallace DC. 2007. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res 35:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. 2010. MutationTaster evaluates disease‐causing potential of sequence alterations. Nat Methods 7:575–576. [DOI] [PubMed] [Google Scholar]
- Segui N, Mina LB, Lazaro C, Sanz‐Pamplona R, Pons T, Navarro M, Bellido F, Lopez‐Doriga A, Valdes‐Mas R, Pineda M, Guino E, Vidal A, Soto JL, Caldes T, Duran M, Urioste M, Rueda D, Brunet J, Balbin M, Blay P, Iglesias S, Garre P, Lastra E, Sanchez‐Heras AB, Valencia A, Moreno V, Pujana MA, Villanueva A, Blanco I, Capella G, Surralles J, Puente XS, Valle L. 2015. Germline mutations in FAN1 cause hereditary colorectal cancer by impairing DNA repair. Gastroenterology 149:563–566. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CG, Naven M, Harris R, Colley J, West H, Li N, Liu Y, Adams R, Maughan TS, Nichols L, Kaplan R, Wagner MJ, McLeod HL, Cheadle JP. 2013. Exome resequencing identifies potential tumor‐supressor genes that predispose to colorectal cancer. Hum Mutat 34:1026–1034. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Koch I, 3rd Lathe W, , Kondrashov AS Bork P. 2001. Prediction of deleterious human alleles. Hum Mol Genet 10:591–597. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: Functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam R, Verwiel ET, Kamping EJ, Hoenselaar E, Gorgens H, Schackert HK, van Krieken JH, Ligtenberg MJ, Hoogerbrugge N, van Kessel AG, Kuiper RP. 2011. Identification of candidate predisposing copy number variants in familial and early‐onset colorectal cancer patients. Int J Cancer 129:1635–1642. [DOI] [PubMed] [Google Scholar]
- Weren RD, Ligtenberg MJ, Kets CM, de Voer RM, Verwiel ET, Spruijt L, van Zelst‐Stams WA, Jongmans MC, Gilissen C. 2015. A germline homozygous mutation in the base‐excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet 47:668–671. [DOI] [PubMed] [Google Scholar]
- Yang H, Gao Y, Feng T, Jin TB, Kang LL, Chen C. 2014. Meta‐analysis of the rs4779584 polymorphism and colorectal cancer risk. PLoS One 9:e89736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Burge CB. 2004. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11:377–394. [DOI] [PubMed] [Google Scholar]
- Zhang MQ. 1998. Statistical features of human exons and their flanking regions. Hum Mol Genet 7:919–932. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1.
Supporting Information Figure 2.
Supporting Information Table 1.
Supporting Information Table 2.
Supporting Information Table 3.
Supporting Information Table 4.