

Abstract
Aims
We evaluated the effects of patiromer, a potassium (K+)‐binding polymer, in a pre‐specified analysis of hyperkalaemic patients with heart failure (HF) in the OPAL‐HK trial.
Methods and results
Chronic kidney disease (CKD) patients on renin–angiotensin–aldosterone system inhibitors (RAASi) with serum K+ levels ≥5.1 mEq/L to <6.5 mEq/L (n = 243) received patiromer (4.2 g or 8.4 g BID initially) for 4 weeks (initial treatment phase); the primary efficacy endpoint was mean change in serum K+ from baseline to week 4. Eligible patients (those with baseline K+ ≥5.5 mEq/L to <6.5 mEq/L and levels ≥3.8 mEq/L to <5.1 mEq/L at the end of week 4) entered an 8‐week randomized withdrawal phase and were randomly assigned to continue patiromer or switch to placebo; the primary efficacy endpoint was the between‐group difference in median change in the serum K+ over the first 4 weeks of that phase. One hundred and two patients (42%) had heart failure (HF). The mean [± standard error (SE)] change in serum K+ from baseline to week 4 was −1.06 ± 0.05 mEq/L [95% confidence interval (CI), −1.16,−0.95; P < 0.001]; 76% (95% CI, 69,84) achieved serum K+, 3.8 mEq/L to <5.1 mEq/L. In the randomized withdrawal phase, the median increase in serum K+ from baseline of that phase was greater with placebo (n = 22) than patiromer (n = 27) (P < 0.001); recurrent hyperkalaemia (serum K+, ≥5.5 mEq/L) occurred in 52% on placebo and 8% on patiromer (P < 0.001). Mild‐to‐moderate constipation was the most common adverse event (11%); hypokalaemia occurred in 3%.
Conclusion
In patients with CKD and HF who were hyperkalaemic on RAASi, patiromer was well tolerated, decreased serum K+, and, compared with placebo, reduced recurrent hyperkalaemia.
Keywords: chronic kidney disease, heart failure, hyperkalaemia, patiromer
Introduction
Renin–angiotensin–aldosterone system inhibitors (RAASi) have been shown to be effective in reducing mortality as well as hospitalizations for heart failure (HF) in patients with chronic HF and a reduced left ventricular ejection fraction (HFrEF) and have been accorded a class 1 indication in international guidelines.1, 2, 3, 4, 5, 6 In patients with chronic kidney disease (CKD) complicating their HF, renal excretion of potassium (K+) is compromised and is in part compensated by an increase in colonic excretion, which often is not sufficient to avoid hyperkalaemia.7 Patients with CKD and HFrEF are therefore at an increased risk of death but paradoxically often receive suboptimal doses of RAASi because of fear of inducing hyperkalaemia (serum K+ >5.0 mEq/L) and its consequences, including sudden cardiac death.8, 9, 10 While there are several options for the treatment of acute hyperkalaemia, options for the management of chronic hyperkalaemia, and thereby maintaining RAASi use in patients with HF, are limited. Agents that have been employed to manage persistently high K+ levels include diuretics (with or without sodium bicarbonate) and the K+‐binding resins, calcium polystyrene sulphonate (CPS) and sodium polystyrene sulphonate (SPS).11, 12 Calcium polystyrene sulphonate and SPS can lead to serious gastrointestinal (GI) adverse events (AEs), including colonic necrosis and perforation,11, 12, 13, 14 which has led to warnings in the prescribing information.11, 12, 15 In addition, because sodium is the counter exchange ion with SPS, caution is advised when treating patients who cannot tolerate even small increases in sodium load (e.g. those with severe congestive HF, severe hypertension, or marked oedema).11 Patients with chronic HF who develop hyperkalaemia therefore often have their dose of RAASi reduced or discontinued,8, 10, 16 thereby exposing them to increased cardiovascular risk.
The active moiety of patiromer is a non‐absorbed oral K+‐binding polymer. It acts primarily in the distal colon, where the concentration of free K+ is the highest, to increase fecal K+ excretion.17, 18 Patiromer consists of smooth, spherical beads approximately 100 µm in diameter that are insoluble, free‐flowing, and that do not swell appreciably when mixed with water.18 The OPAL‐HK17 study showed patiromer to be generally well tolerated and effective in reducing serum K+ levels in CKD patients with mild and moderate‐to‐severe hyperkalaemia (serum K+ ≥5.1 mEq/L to <6.5 mEq/L).17
We evaluated patiromer's efficacy and safety in a pre‐specified analysis of OPAL‐HK in the subgroup of CKD patients with HF and compared those results with CKD patients without HF.
Methods
Study population
The OPAL‐HK study has been described previously.17 In brief, eligible patients were 18–80 years of age, had stage 3 or 4 CKD [estimated glomerular filtration rate (eGFR) of 15 mL/min./1.73 m2 to <60 mL/min./1.73 m2 of body surface area], serum K+ 5.1 mEq/L to <6.5 mEq/L, indicative of hyperkalaemia, and had been receiving a stable dose of ≥1RAASi for ≥28 days. Patients who were also on anti‐hypertensive medication, loop and thiazide diuretics, or beta‐blockers were receiving them at stable doses for ≥28 days.
Patients were excluded if at screening they presented with K+ ‐related electrocardiographic changes, severe GI disorders, uncontrolled or unstable arrhythmias or clinically significant ventricular arrhythmias, type 1 diabetes, New York Heart Association (NYHA) class IV HF, acute coronary syndrome, or confirmed systolic blood pressure >180 mmHg or <110 mmHg, or diastolic blood pressure >110 mmHg or <60 mmHg. Patients were also excluded if they underwent recent cardiac surgery, kidney or heart transplantation, had a transient ischaemic attack or stroke within the previous 2 months, or received emergent treatment for type 2 diabetes or acute HF within the previous 3 months.
The study (NCT01810939) was conducted in accordance with the International Conference on Harmonisation Guideline for Good Clinical Practice and complied with the Declaration of Helsinki. The study protocols were reviewed and approved by institutional review boards and patients provided written informed consent.
Study protocol
This study was carried out in two parts: an initial treatment phase and a randomized withdrawal phase. The initial treatment phase was a 4‐week, single‐group, single‐blind assessment of patiromer in patients with CKD taking RAASi. At the beginning of this phase, patients were assigned one of two patiromer doses based on their screening serum K+ levels. Patients with mild hyperkalaemia (serum K+ 5.1 mEq/L to <5.5 mEq/L) received 4.2 g of patiromer twice daily; patients with moderate‐to‐severe hyperkalaemia (serum K+ ≥ 5.5 mEq/L to <6.5 mEq/L) received 8.4 g of patiromer twice daily. Patiromer was administered as an oral suspension in 40–120 mL of water, depending on dose, with breakfast and dinner. Subsequent doses were adjusted to reach and maintain target serum K+ levels based on a pre‐specified treatment algorithm (see the Supplementary material online, Table S1). The target serum K+ levels were conservative to avoid hypokalaemia. The RAASi dose was not adjusted to facilitate interpretation of primary study endpoints during the initial treatment phase. Patients discontinued RAASi and their participation in the study if their serum K+ was ≥6.5 mEq/L during the initial treatment phase.
Following the initial treatment phase was a randomized withdrawal phase (an 8‐week, single‐blind, placebo‐controlled, parallel group assessment of patiromer withdrawal). Patients who completed the initial treatment phase were eligible to start the randomized withdrawal phase if they had moderate‐to‐severe hyperkalaemia (serum K+ ≥ 5.5 mEq/L to < 6.5 mEq/L) at baseline of the initial treatment phase and were normokalaemic (serum K+ level within target range of ≥ 3.8 mEq/L to <5.1 mEq/L) at week 4 of the initial treatment phase (baseline of the randomized withdrawal phase). Patients were eligible for continuation to the randomized withdrawal phase if they were still taking patiromer and ≥ 1 RAASi. Eligible patients were randomized in a single‐blind manner to either continue patiromer at the daily dose they were receiving at week 4 of the initial treatment phase or to switch to placebo, in a 1:1 ratio. The randomization was performed centrally and stratified based on the presence of type 2 diabetes as well as baseline serum K+ [moderate (≥ 5.5 mEq/L to <5.8 mEq/L) vs. severe (≥5.8 mEq/L) hyperkalaemia].
Use of RAASi and recurrence of hyperkalaemia were monitored during the randomized withdrawal phase. Hyperkalaemia was defined as a serum K+ measurement ≥5.5 mEq/L during the first 4 weeks of the randomized withdrawal phase, and ≥5.1 mEq/L during the last 4 weeks of this phase. Recurrences of hyperkalaemia were managed with a pre‐specified treatment algorithm (see the Supplementary material online, Table S2) either by increasing the patiromer dose (patiromer group) or by RAASi modification (placebo group) at the time of the first occurrence of hyperkalaemia. Subsequent occurrences required discontinuation of RAASi. Neither of these interventions was to be used during the first 4 weeks of the randomized withdrawal phase (unless serum K+ reached ≥5.5 mEq/L) to facilitate the interpretation of the primary efficacy endpoint.
Serum K+ levels were measured at local and central laboratories at baseline, day 3, and weekly throughout both parts of the study. Safety data were recorded at each of these visits. During the study, site staff counselled patients to restrict foods high in K+ content (>250 mg/100 g) and to have a target K+ intake of ≤2–3 g/day (approximately 50–75 mEq/day). Up to three safety follow‐up visits occurred within 1–2 weeks after discontinuation of patiromer or placebo (including patients who withdrew from the study or did not qualify for the randomized withdrawal phase) to monitor AE occurrences and serum K+ levels.
Clinical endpoints
The primary efficacy endpoint of the treatment phase was the mean change in serum K+ level from baseline to week 4 in patients who received ≥1 dose of patiromer and had at least one K+ measurement after day 3. The secondary endpoint was the proportion of patients whose serum K+ was within target range (≥ 3.8 mEq/L to <5.1 mEq/L) at week 4. The primary efficacy endpoint of the randomized withdrawal phase was the difference between the patiromer and placebo groups in the median change in serum K+ from baseline to either week 4—if serum K+ stayed in target range—or the earliest visit when serum K+ was outside that range. The secondary endpoints were the proportions of patients with a recurrence of hyperkalaemia according to two definitions: serum K+ ≥5.1 mEq/L or ≥5.5 mEq/L. An exploratory efficacy endpoint of the randomized withdrawal phase was the proportion of patients requiring an intervention to manage a recurrence of hyperkalaemia [i.e. an increase of the patiromer dose (patiromer group) or RAASi dose reduction (placebo group) at the first occurrence of hyperkalaemia; RAASi discontinuation at subsequent occurrences (both treatment groups)]. Adverse events were monitored and recorded during both parts of the study and for up to 2 weeks after discontinuation from the study.
Statistical analysis
The mean changes in serum K+ (primary endpoint) and the associated 95% confidence interval (CI) were estimated using a longitudinal repeated measures model of the centrally measured, weekly post‐baseline serum K+ values. Consequently, only patients with at least one weekly post‐baseline measurement were included in the analyses. For the subgroups defined by presence or absence of HF, the model used one binary covariate (presence of type 2 diabetes) and one continuous covariate (baseline serum K+). Patients were included in the HF subgroup based on a history of HF (per the investigator's judgment), which included NYHA class, date of diagnosis (if known), and diagnosis of systolic or diastolic dysfunction (if determined).
To assess the primary efficacy endpoint of the randomized withdrawal phase, changes in serum K+ from baseline in placebo and patiromer groups were compared using an analysis of variance of rank‐transformed data. All randomized patients who entered the withdrawal phase were included in these analyses. The between‐group differences in median serum K+ change from baseline and the associated 95% CI were calculated using a Hodges–Lehmann estimator. The comparison of the treatment groups used rank of change carried forward to account for and include patients who discontinued study drug before week 4 of the randomized withdrawal phase. For both secondary endpoint calculations, proportions of patients in the two treatment groups were compared using a Mantel–Haenszel test with baseline strata. The analyses for the subgroups defined by the presence or absence of HF did not include any adjustment for multiplicity. A more detailed description of the statistical analysis methods is provided in the primary publication describing the overall results of the OPAL‐HK study.17
With 240 patients enrolled, this provided more than 99% power to detect a mean change from baseline in serum K+ of at least 0.3 mEq/L. This calculation was based on a 2‐sided, single‐sample, paired t‐test, a significance level of α = 0.05, and the assumption of a standard deviation (SD) of 0.55. We used SAS Version 9.2 for statistical analyses (SAS Institute Inc. 100 SAS Campus Drive Cary, NC 27513‐2414, USA).
Results
Patients
A total of 243 patients with CKD were enrolled in the initial treatment phase, 102 (42%) with HF and 141 (58%) without HF (Table 1). Of the patients with HF, 39 (38%) entered the study with mild hyperkalaemia and 63 (62%) with moderate‐to‐severe hyperkalaemia; of the patients without HF, 53 (38%) had mild hyperkalaemia and 88 (62%) had moderate‐to‐severe hyperkalaemia. All patients received at least one dose of patiromer.
Table 1.
Baseline demographics and clinical characteristics
| Characteristic | Heart failure (n = 102) | Non‐heart failure(n = 141) |
|---|---|---|
| Male, n (%) | 56 (55%) | 84 (60%) |
| Age (years), mean (SD) | 67.4 (8.6) | 61.9 (11.1) |
| White, n (%) | 102 (100%) | 137 (97%) |
| eGFR (mL/min./1.73 m2), n (%) | ||
| 60 to ≤90, Stage 2 | 9 (9%) | 13 (9%) |
| 45 to <60, Stage 3A | 20 (20%) | 29 (21%) |
| 30 to <45, Stage 3B | 28 (27%) | 35 (25%) |
| <30, Stage 4/5 | 45 (44%) | 64 (45%) |
| Serum K+ (mEq/L), mean (SD) | 5.6 (0.6) | 5.5 (0.4) |
| Type 2 diabetes, n (%) | 55 (54%) | 84 (60%) |
| Time since diagnosis of type 2 diabetes (years), mean (SD) | 12.0 (9.9) | 14.0 (8.9) |
| NYHA HF class, n (%) | ||
| I | 19 (19%) | NA |
| II | 66 (65%) | NA |
| III | 17 (17%) | NA |
| Myocardial infarction, n (%) | 33 (32%) | 27 (19%) |
| Hypertension, n (%) | 97 (95%) | 139 (99%) |
| RAASi medication, n (%) | 102 (100%) | 141 (100%) |
| ACE inhibitor | 70 (69%) | 100 (71%) |
| ARB | 37 (36%) | 55 (39%) |
| Aldosterone antagonist | 20 (20%) | 2 (1%) |
| Renin inhibitor | 2 (2%) | 0 |
| Dual RAASi blockade,* n (%) | 25 (25%) | 16 (11%) |
| On maximal RAASi dose,† n (%) | 42 (41%) | 64 (45%) |
| Other concomitant medication for HF | ||
| Beta blocker | 60 (59%) | 68 (48%) |
| Thiazide | 27 (26%) | 43 (30%) |
| Loop | 44 (43%) | 33 (23%) |
Data are number of patients and per cent.
ACE, angiotensin‐converting enzyme; ARB, angiotensin receptor blocker; eGFR, estimated glomerular filtration rate; HF, heart failure; NYHA, New York Heart Associations; RAASi, renin–angiotensin–aldosterone system inhibitor.
Any combination of two or more of the following: ACE inhibitor, ARB, aldosterone antagonist, renin inhibitor.
As judged by the investigator in accordance with local standards of care.
A total of 91 (89%) patients with HF completed the initial treatment phase. Of those, 42 patients (46%) were not eligible to continue to the randomized withdrawal phase. The most common reason for ineligibility was a centrally measured baseline serum K+ of <5.5 mEq/L (40 patients, 44%); 1 patient was ineligible solely because their serum K+ fell outside the target range at week 4. The remaining 49 patients with HF (54%) eligible for the randomized withdrawal phase were randomly assigned either to continue patiromer (27 patients) or to switch to placebo (22 patients). A total of 12 patients with HF discontinued the randomized withdrawal phase prematurely: 5 (19%) patients in the patiromer group and 7 (32%) patients in the placebo group. Most of the discontinuations resulted from an elevated serum K+ that met the pre‐specified withdrawal criteria [5 patients with HF (23%) in the placebo group and 0 patients with HF in the patiromer group]. Comprehensive disposition information for patients with and without HF can be found in the Supplementary material online, Figure S1 (initial treatment phase) and Figure S2 (randomized withdrawal phase).
At the start of the trial, the proportion of HF patients with stage 3 and stage 4/5 CKD, respectively, was 47% and 44%; in patients without HF, the corresponding proportions were 46% and 45%. In patients with and without HF, 9% had stage 2 CKD based on central laboratory eGFR measurements and were included in the study because they had met entry criteria on the basis of eGFR measurements obtained at local laboratories. The mean serum K+ (± SD) at baseline was 5.6 ± 0.6 mEq/L in patients with HF and 5.5 ± 0.4 mEq/L in patients without HF.
Efficacy
Initial treatment phase
The mean [± standard error (SE)] change in serum K+ from baseline to week 4 in patients with HF (100 patients who had at least one serum K+ measurement after day 3) was −1.06 ± 0.05 mEq/L (95% CI −1.16 to −0.95, P < 0.001, Figure 1 A). The mean (± SE) change in serum K+ from baseline to week 4 in HF patients with mild hyperkalaemia (n = 38) was −0.74 ± 0.08 mEq/L (95% CI −0.91 to −0.57), and for patients with HF with moderate‐to‐severe hyperkalaemia (n = 62) the change from baseline was −1.26 ± 0.07 mEq/L (95% CI −1.40 to −1.12). Figure 1 B shows the observed mean serum K+ over time. By the end of the 4‐week initial treatment phase, 76% of patients with HF achieved a serum K+ in the target range (≥ 3.8 mEq/L to <5.1 mEq/L) (95% CI 69–84). The primary and secondary efficacy endpoints were similar in patients without HF (Figure 1). The mean daily dose of patiromer received over the 4‐week initial treatment phase was 17.8 g for patients with HF and 18.4 g for those without HF, with a similar mean number of dose adjustments in each subgroup (0.8 and 0.9, respectively).
Figure 1.
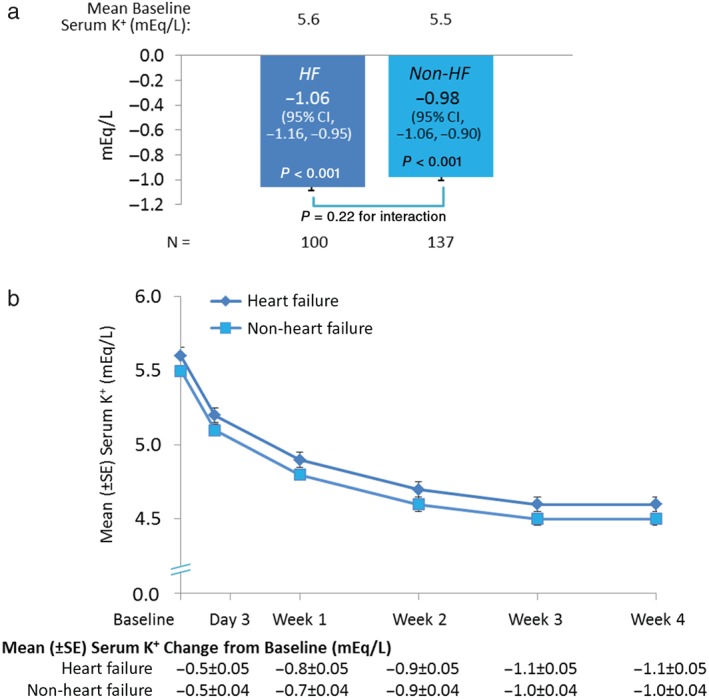
Serum K+ levels over time during the treatment phase. a, Treatment phase primary endpoint: Mean serum K+ change from baseline to week 4. b, Mean serum K+ change from baseline to week 4 over time. Secondary endpoint: 76% and 75% of patients with and without HF, respectively, had serum K+ 3.8 to < 5.1 mEq/L at week 4. HF, heart failure; CI, confidence interval; K+, potassium; SE, standard error.
Randomized withdrawal phase
At baseline of the randomized withdrawal phase (week 4 of the initial treatment phase), which included only those patients whose serum K+ was controlled during the initial treatment phase, mean serum K+ was 4.52 mEq/L in patients with HF randomized to patiromer and 4.56 mEq/L in patients with HF randomized to placebo. The estimated median change in serum K+ from baseline to week 4 of the randomized withdrawal phase was 0.74 mEq/L for patients with HF taking placebo and 0.10 mEq/L for those taking patiromer, for a between‐group difference of 0.64 mEq/L (95% CI 0.29–0.99; P < 0.001; Figure 2).
Figure 2.
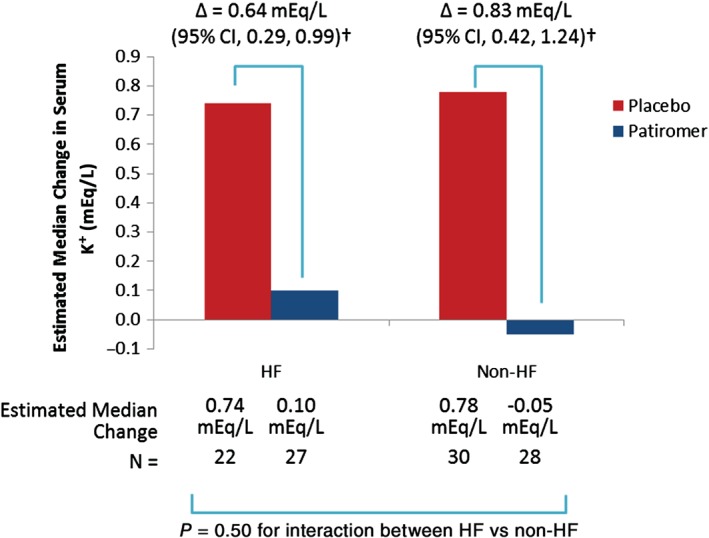
Effect of patiromer on serum K+ in patients with and without HF during the randomized withdrawal phase. †P < 0.001; CI, confidence interval; HF, heart failure; K+, potassium.
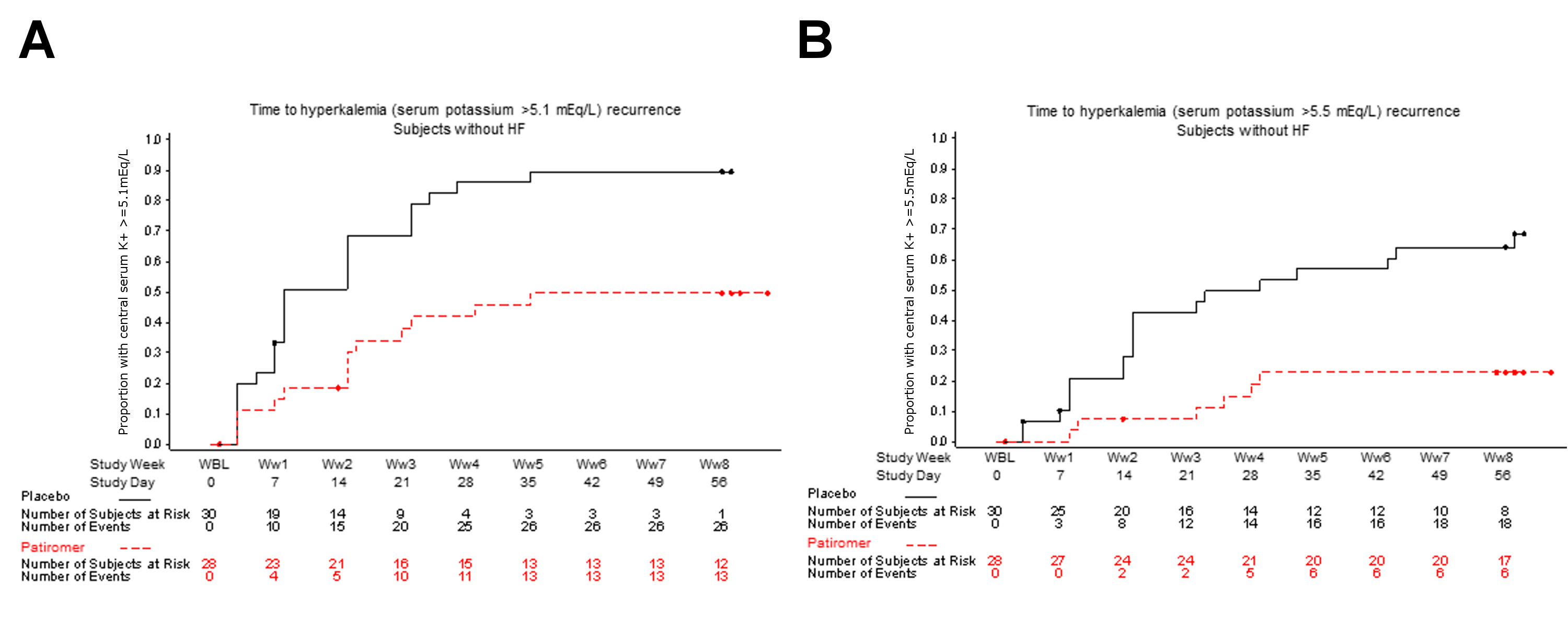
In patients with HF, 52% (95% CI 30–74) of those randomized to placebo compared with 8% (95% CI 1–25) of those randomized to patiromer had at least 1 serum K+ of ≥5.5 mEq/L during the 8‐week randomized withdrawal phase (P < 0.001 for placebo–patiromer group difference). A total of 95% (95% CI 77 to 99) of patients with HF randomized to placebo and 36% (95% CI 19–57) of those randomized to patiromer had at least 1 serum K+ of ≥5.1 mEq/L (P < 0.001). Figure 3 shows the time to hyperkalaemia (serum K+ ≥5.1 mEq/L and ≥5.5 mEq/L) recurrence in patients with HF, time to hyperkalaemia in patients without HF can be found in the Supplementary material online, Figure S3.
Figure 3.
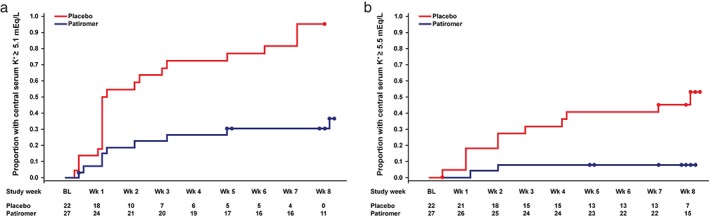
Time to first recurrence of hyperkalaemia [(a) K+ ≥5.1 mEq/L; (b) K+ ≥5.5 mEq/L] in patients with HF during the randomized withdrawal phase. Circles indicate censored observations. BL, baseline; HF, heart failure; K+, potassium; Wk, week.
In a pre‐specified exploratory analysis, 13 (59%) patients with HF taking placebo compared with 3 (11%) patients taking patiromer required an intervention to manage their hyperkalaemia recurrence; by the end of the randomized withdrawal phase, 55% of HF patients on placebo and 100% of HF patients on patiromer were still receiving RAASi. Figure 4 shows the time to the discontinuation of RAASi. Results were similar in patients without HF (Figures 2, 3, 4).
Figure 4.
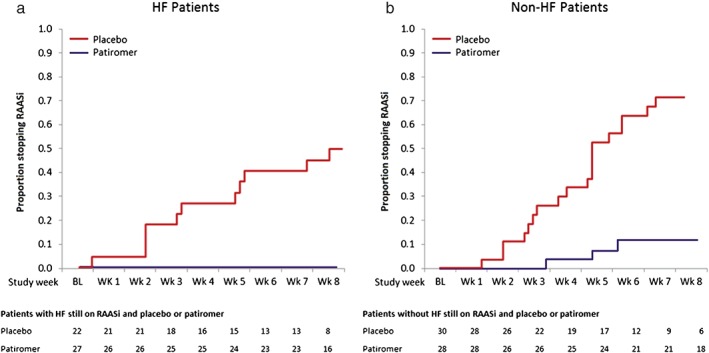
Proportion of patients discontinuing RAASi therapy during the randomized withdrawal phase. (a) HF patients; (b) non‐HF patients. BL, baseline of withdrawal; HF, heart failure; RAASi, renin‐angiotensin‐aldosterone system inhibitor; Wk, week.
Safety
During the initial treatment phase and its follow‐up period, 41% of patients with HF and 51% of patients without HF reported at least 1 AE (see the Supplementary material, Table S3A). The most common AEs that occurred during the initial treatment phase are shown in Table 2. The majority of AEs were gastrointestinal. None of the GI events that occurred in this phase were severe. Adverse events that led to discontinuation of patiromer occurred in 7 (7%) patients with HF and 8 (6%) patients without HF. Three patients (2 with HF and 1 without HF) experienced non‐fatal serious AEs during the initial treatment phase; the investigators did not consider the events to be related to patiromer.
Table 2.
Adverse events occurring in at least 3% of patients with and without HF during the initial treatment phase and through the safety follow‐up period for that phase*
| Heart failure (n = 102) | Non‐heart failure (n = 141) | |
|---|---|---|
| ≥1 Adverse event | 42 (41%) | 72 (51%) |
| Constipation | 11 (11%) | 15 (11%) |
| Diarrhoea | 4 (4%) | 4 (3%) |
| Nausea | 1 (1%) | 7 (5%) |
| Hypomagnesaemia | 3 (3%) | 5 (4%) |
| Anaemia | 4 (4%) | 3 (2%) |
| Chronic renal failure | 1 (1%) | 6 (4%) |
| Hyperkalaemia | 3 (3%) | 3 (2%) |
| Left ventricular hypertrophy | 0 | 6 (4%) |
| Dyslipidaemia | 0 | 4 (3%) |
| ≥1 Serious adverse event | 2 (2%) | 1 (1%) |
Data are number of patients and percent. HF, heart failure
The safety follow‐up period was 1–2 weeks after discontinuation of the study drug in the initial treatment phase. In the Supplementary material online, Table 3 A shows adverse events that occurred in at least 2 patients with or without HF in each dose group and Table 3 B shows all serious adverse events.
The proportion of patients with and without HF experiencing at least one AE during the 8‐week randomized withdrawal phase and its follow‐up period was similar between the placebo and patiromer groups (HF 64% and 56%, respectively; non‐HF 40% and 39%; Table 3). The most common GI AEs reported in the patiromer group during this phase of the study were diarrhoea (HF 7%; non‐HF 0%) and nausea (HF 7%; non‐HF 0%)—all mild or moderate. Cardiac disorders as a class were reported as AEs in 8% of HF and 7% of non‐HF patients during the treatment phase; all individual cardiac AEs occurred in 2 or fewer HF patients in either phase. One HF patient experienced worsening of HF in the treatment phase. In non‐HF patients, left ventricular hypertrophy was reported in 6 (4%) patients and first‐degree atrioventricular block was reported in 3 (2%) patients in the treatment phase; all other individual cardiac AEs occurred in 2 or fewer non‐HF patients in either phase. Study drug discontinuation because of AEs occurred in two HF patients [1 (5%) taking placebo and 1 (4%) taking patiromer], and in none of the patients without HF.
Table 3.
Adverse events occurring in at least 2 patients in the patiromer group regardless of HF diagnosis during the randomized withdrawal phase and through the safety follow‐up period for that phase*
| Heart failure | Non‐heart failure | |||
|---|---|---|---|---|
| Placebo (n = 22) | Patiromer (n = 27) | Placebo (n = 30) | Patiromer (n = 28) | |
| ≥1 Adverse event | 14 (64%) | 15 (56%) | 12 (40%) | 11 (39%) |
| Headache | 3 (14%) | 1 (4%) | 1 (3%) | 1 (4%) |
| Supraventricular extrasystoles | 1 (5%) | 1 (4%) | 0 | 1 (4%) |
| Diarrhoea | 0 | 2 (7%) | 0 | 0 |
| Nausea | 0 | 2 (7%) | 0 | 0 |
| Constipation | 0 | 1 (4%) | 0 | 1 (4%) |
| ≥1 Serious adverse event | 1† (5%) | 0 | 0 | 0 |
Data are number of patients and percent. HF, heart failure
The safety follow‐up period was 1–2 weeks after discontinuation of the study drug.
Mesenteric vessel thrombosis leading to death occurred in 1 patient.
Two patients with HF had at least 1 serious AE during the treatment phase. In 1 HF patient, the serious AE was atrial fibrillation leading to hospitalization. However, the patient had a previous medical history of atrial fibrillation and this serious event was deemed not to be related to patiromer (by the investigator). The other serious AEs occurring during the treatment phase, all of which occurred in the other HF patient, are listed in the Supplementary material online, Table S3B. One HF patient on placebo had a serious AE during the withdrawal phase (mesenteric vessel thrombosis leading to death). None of the serious AEs (in either phase) were considered related to patiromer by the investigator.
During the initial treatment phase and its follow‐up period, hypokalaemia (serum K+ <3.5 mEq/L) occurred in 3% of patients with and without HF. These patients had serum K+ in range of ≥ 3.2–3.4 mEq/L; hypokalaemia was most often transient after adjustment of the patiromer dose. During the randomized withdrawal phase and its follow‐up period, hypokalaemia requiring withdrawal from the study (serum K+ <3.8 mEq/L) occurred in 7% and 4% of patients with and without HF taking patiromer, respectively, in none of the patients with HF, and in 1 (3%) patient without HF taking placebo.
The mean serum magnesium level remained within normal range throughout both phases of the trial in patients with and without HF. Small mean decreases in serum magnesium were observed at the end of the initial treatment phase in patients with HF [−0.20 mg/dL (−0.16 mEq/L)] and without HF [−0.16 mg/dL (−0.13 mEq/L)], with no apparent dose effect. At the end of the randomized withdrawal phase, a small mean increase from baseline was observed in serum magnesium in placebo patients with HF (+0.19 mEq/L) and without HF (+0.05 mEq/L). There was no significant change in serum magnesium levels in patiromer patients with and without HF during the randomized withdrawal phase. A serum magnesium level of <1.4 mg/dL was observed in 4 HF patients and 3 non‐HF patients during the initial treatment phase. During the randomized withdrawal phase, no patient (with or without HF) had a serum magnesium level of <1.4 mg/dL. No patient (with or without HF) had serum magnesium levels <1.2 mg/dL during either phase of the study. Magnesium replacement therapy was prescribed in 9 patients (3 with HF and 6 without HF) taking patiromer during the initial treatment phase. No clinically relevant changes in renal function or in levels of serum calcium, fluoride, or other electrolytes (e.g. bicarbonate) were observed in either phase of the study. Two patients in the initial treatment phase (1 with HF and 1 without HF) and 1 patient without HF on patiromer in the randomized withdrawal phase had electrocardiographic changes consistent with hyperkalaemia; none had changes consistent with hypokalaemia. No clinically relevant changes in renal function were observed (see the Supplementary material online, Table S4).
Discussion
The results of the present pre‐specified analysis of the OPAL‐HK trial show that patiromer reduced mean serum K+ to within the normal range in patents with HF. Compared with placebo, patiromer also reduced the percentage of patients with recurrent hyperkalaemia and, importantly, there were no differences in patients with and without HF in regard to these effects of patiromer. Mean serum K+ level was reduced from ≥ 5.6 mEq/L to <5.0 mEq/L in patients with HF during the initial treatment phase of the study, and 76% of patients with HF achieved normokalaemia. Conversely, in the randomized withdrawal phase, only 8% of patients on patiromer developed recurrent hyperkalaemia (when defined as serum K+ ≥5.5 mEq/L) compared with 52% on placebo, allowing significantly more patients to remain on guideline‐recommended RAASi. This has important implications for patients with HF because RAASi including mineralocorticoid receptor antagonists (MRAs) have been shown to reduce mortality in patients with HFrEF.1, 2, 3, 4, 5, 6 Despite the proven efficacy of RAASi and their class 1 indication in guidelines for patients with HFrEF, many clinicians have avoided their use, especially MRAs, because of the fear of inducing hyperkalaemia.8, 10 In those patients with HF in whom MRAs are initiated, they are often discontinued during the first several months because of an increase in serum K+ or worsening renal function.9, 19, 20, 21 While many clinicians consider a serum potassium of ≥5.5 mEq/L indicative of hyperkalaemia and an increased risk for sudden cardiac death, increasing evidence indicates that in patients with HF and CKD a serum K+ of >5.0 mEq/L is associated with an increase in cardiovascular risk.22 Recurrent hyperkalaemia, when defined by serum K+ ≥5.1 mEq/L, occurred in 95% of placebo patients with HF compared with 36% of patiromer patients with HF. The present analysis also suggests that hyperkalaemia occurring in a patient with HF can be relatively easily controlled with patiromer as shown by the finding that all of the HF patients on patiromer were able to be maintained on their RAASi during the randomized withdrawal phase despite the need for patiromer dose adjustment to manage hyperkalaemia in 11% of patients. Importantly, the use of patiromer in patients with HF has the potential to allow titration of RAASi doses to target levels and to maintain these dose levels. Target doses of RAASi appear to be more effective than lower doses in patients with HF as shown by the results in the HEAAL study.23
The tolerability and safety profile of patiromer in CKD patients both with and without HF in the present analysis supports the hypothesis that patiromer can be used to maintain the use of guideline‐recommended RAASi. The proportion of patiromer patients discontinuing because of AEs was 7% while gastrointestinal intolerance, hypokalaemia, and hypomagnesaemia were easily managed. Our study did not measure intracellular magnesium levels. In the present study, no new‐onset clinically significant arrhythmias were observed; therefore, it is unlikely that tissue magnesium levels were reduced to critically low levels.
The relatively good tolerability and safety profile of patiromer in the present study, both in patients with and without HF, is supported by the findings in the recent AMETHYST‐DN study,24 in which patiromer was administered to patients with hyperkalaemia, CKD, type 2 diabetes, hypertension, and receiving RAASi over a 1‐year study period. In a study of patiromer for the prevention of hyperkalaemia (PEARL‐HF),25 normokalaemia patients with HF with a history of discontinuation of RAASi or beta adrenergic blocking agent because of hyperkalaemia were randomized to patiromer or placebo and titrated to 50 mg/day of spironolactone. In that study, significantly fewer patients on patiromer developed hyperkalaemia (serum K+ >5.5 mEq/L) compared with patients on placebo (7% vs. 25%, P = 0.015).25 In the subgroup of patients with CKD (eGFR <60 mL/min./1.73 m2) in PEARL‐HF, hyperkalaemia developed in 7% of patients randomized to patiromer compared with 39% of patients randomized to placebo (P = 0.041). While GI AEs occurred more frequently with patiromer than with placebo (21% vs. 6%) they were mostly mild‐to‐moderate in severity, and similar proportions of patients in each group discontinued owing to AEs.25
There are limitations to the study. No placebo or active control was used in the initial treatment phase of this study as it was considered unethical to allow patients with hyperkalaemia to remain untreated. Although the withdrawal phase was randomized, it was not blinded to the investigators. The sponsor provided treatment algorithms to minimize bias and to standardize interventions when hyperkalaemia or hypokalaemia occurred. However, it was possible that changes in treatment regimen implemented by the investigator in response to hyperkalaemia or hypokalaemia may have indicated treatment assignment to the patient. The higher proportion of patiromer patients still receiving RAASi at the end of the withdrawal phase may have been partly because of the treatment algorithm, which allowed investigators to increase the dose of patiromer at the first occurrence of hyperkalaemia in the patiromer group. The estimates for mean change from baseline in serum K+ during the initial treatment phase used only weekly post‐baseline measurements (i.e. 2 HF patients with post‐baseline measurements only at day 3 were excluded from the analysis). As day 3 is not included in the model, our limitations include an i nability to estimate day 3 from the repeated measures model. Instead, day 3 was estimated separately using an analysis of variance model with the presence of type 2 diabetes included as a binary covariate and baseline serum K+ as a continuous covariate. In the withdrawal phase, the comparison of the treatment groups used rank of change carried forward to account for and include patients who discontinued study drug before week 4 because of hyperkalaemia or hypokalaemia. Missing serum K+ values of patients who discontinued study drug before week 4 for other reasons were imputed using multiple imputation. Patients were included in the HF subgroup based on a history of HF per the investigator's judgment, therefore inaccuracies in assessing the differences between the groups with and without HF may have occurred because no specific instructions for HF diagnosis were provided. During the study, a low proportion of patients received concomitant aldosterone antagonist therapy, potentially because these agents are contraindicated in patients with hyperkalaemia or with reduced renal function (eGFR <30 mL/min./1.73 m2).26, 27 While this may limit the conclusions that can be drawn regarding the effect of patiromer in CKD patients with HF receiving aldosterone antagonists, the PEARL‐HF study previously demonstrated that patiromer prevented hyperkalaemia in HF patients with normal serum K+ levels receiving spironolactone.25
In conclusion, the efficacy and safety profile of patiromer demonstrated in the present analysis and in the AMETHYST‐DN and PEARL‐HF studies24, 25 suggests that patiromer may have an important role in initiating and maintaining RAASi in patients with CKD and HF, with the potential for consequent reduction in cardiovascular death in these high risk patients. Further adequately powered prospective randomized studies will be required to evaluate this hypothesis and to determine the cost‐effectiveness of this strategy.
Funding
This work was supported by Relypsa, Inc.
Conflict of interest: B.P. has received consulting fees from Pfizer, Inc.; Bayer AG; AstraZeneca plc; Stealth Biotherapeutics, Inc.; Sarfez Pharmaceuticals, Inc.; and Eli Lilly and Company. He has received consulting fees and holds stock from Relypsa, Inc.; Aura Sense Therapeutics; Tricida, Inc.; scPharmaceuticals, Inc.; and DaVinci BioSciences, LLC. He is on the data safety monitoring board for Novartis AG; Johnson & Johnson; and Oxygen Biotherapeutics. G.L.B. has received consulting fees from Relypsa, Inc.; AbbVie, Inc.; Daiichi‐Sankyo Ltd; Janssen Pharmaceuticals; Novartis; Bayer; and Medtronic plc; and grants from Takeda Pharmaceutical Company LT. M.R.W. has received consulting fees from Relypsa, Inc.; and ZS Pharma, Inc. D.A.B. has received consulting fees from Relypsa, Inc. L.B., M.R.M., D.G., and Y.S. are employees of Relypsa, Inc. H.C‐S. is an employee of the Statistics Collaborative.
Supporting information
Figure S1. Patient disposition with and without HF in the initial treatment phase. eGFR, estimated glomerular filtration rate; HF, heart failure; K+, potassium.
Figure S2. Patient disposition with and without HF in the randomized withdrawal phase. eGFR, estimated glomerular filtration rate; HF, heart failure; K+, potassium.
Figure S3. Time to hyperkalaemia recurrence in patients without HF. Circles indicate censored observations. eGFR, estimated glomerular filtration rate; HF, heart failure; K+, potassium.
{kind=link}
Table S1. Titration algorithm for initial treatment phase.
Table S2. Titration algorithm for first 4 weeks of the withdrawal phase (day 3 to week 3 visits of this phase).
Table S3. (A) Individual adverse events occurring in at least 2 patients with or without HF in each dose group; (B) All Serious adverse events in the initial treatment phase.
Table S4. Kidney function tests in patients with and without HF.
References
- 1. Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, Palensky J, Wittes J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999;341:709–717. [DOI] [PubMed] [Google Scholar]
- 2. Zannad F, McMurray JJ, Krum H, van Veldhuisen, DJ , Swedberg K, Shi H, Vincent J, Pocock SJ, Pitt B. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011;364:11–21.21073363 [Google Scholar]
- 3. Sorensen MV, Matos JE, Praetorius HA, Leipziger J. Colonic potassium handling. Pflugers Arch 2010;459:645–656. [DOI] [PubMed] [Google Scholar]
- 4. McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Böhm M, Dickstein K, Falk V, Filippatos G, Fonseca C, Gomez‐Sanchez MA, Jaarsma T, Køber L, Lip GY, Maggioni AP, Parkhomenko A, Pieske BM, Popescu BA, Rønnevik PK, Rutten FH, Schwitter J, Seferovic P, Stepinska J, Trindade PT, Voors AA, Zannad F, Zeiher A. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2012;14:803–869. [DOI] [PubMed] [Google Scholar]
- 5. Kidney Disease Outcomes Quality Initiative (K/DOQI) . K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am J Kidney Dis 2004;43:S1–S290. [PubMed] [Google Scholar]
- 6. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJV, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WHW, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013;62:e147–e239. [DOI] [PubMed] [Google Scholar]
- 7. Einhorn LM, Zhan M, Hsu VD, Walker LD, Moen MF, Seliger SL, Weir MR, Fink JC. The frequency of hyperkalemia and its significance in chronic kidney disease. Arch Intern Med 2009;169:1156–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yildirim T, Arici M, Piskinpasa S, Aybal‐Kutlugun A, Yilmaz R, Altun B, Erdem Y, Turgan C. Major barriers against renin‐angiotensin‐aldosterone system blocker use in chronic kidney disease stages 3–5 in clinical practice: a safety concern? Ren Fail 2012;1095–1099. [DOI] [PubMed] [Google Scholar]
- 9. Palmer BF. Managing hyperkalemia caused by inhibitors of the renin‐angiotensin‐aldosterone system. N Engl J Med 2004;351:585–592. [DOI] [PubMed] [Google Scholar]
- 10. Albert NM, Yancy CW, Liang L, Zhao X, Hernandez AF, Peterson ED, Cannon CP, Fonarow GC. Use of aldosterone antagonists in heart failure. JAMA 2009;302:1658–1665. [DOI] [PubMed] [Google Scholar]
- 11.Kayexalate® (Sodium Polystyrene) Package Insert. Sanofi‐Aventis US LLC. Bridgewater, NJ 08807. Revised December 2010. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/011287s023lbl.pdf (21 June 2015).
- 12.Resonium Calcium® (Calcium Polystyrene Sulfonate) Package Insert. Sanofi‐Aventis Canada Inc. Laval, Quebec H7V 0A3. Revised December 2013. Available at: http://products.sanofi.ca/en/resonium‐calcium.pdf (21 June 2015).
- 13. Minford EJ, Hand T, Jones MC. Constipation and colonic perforation complicating calcium resonium therapy. Postgrad Med J 1992;68:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sterns RH, Rojas M, Bernstein P, Chennupati S. Ion‐exchange resins for the treatment of hyperkalemia: are they safe and effective? J Am Soc Nephrol 2010;21:733–735. [DOI] [PubMed] [Google Scholar]
- 15.Food and Drug Administration. The association with colonic necrosis led the US Food and Drug Administration to issue in 2009 a warning advising against the use of SPS mixed with sorbitol. Available at: http://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm186845.htm (17 June 2015).
- 16. Pappoe LS, Winkelmayer WC. ACE inhibitor and angiotensin II type 1 receptor antagonist therapies in elderly patients with diabetes mellitus: are they underutilized? Drugs Aging 2010;27:87–94. [DOI] [PubMed] [Google Scholar]
- 17. Weir MR, Bakris GL, Bushinsky DA, Mayo MR, Garza D, Stasiv Y, Wittes J, Christ‐Schmidt H, Berman L, Pitt B, OPAL‐HK Investigators . Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N Engl J Med 2015;372:211–221. [DOI] [PubMed] [Google Scholar]
- 18. Buysse JM, Huang IZ, Pitt B. PEARL‐HF: prevention of hyperkalemia in patients with heart failure using a novel polymeric potassium binder, RLY5016. Future Cardiol 2012;8:17–28. [DOI] [PubMed] [Google Scholar]
- 19. Eschalier R, McMurray JJ, Swedberg K, van Veldhuisen DJ, Krum H, Pocock SJ, Shi H, Vincent J, Rossignol P, Zannad F, Pitt B, EMPHASIS‐HF Investigators . Safety and efficacy of eplerenone in patients at high risk for hyperkalemia and/or worsening renal function: analyses of the EMPHASIS‐HF study subgroups (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure). J Am Coll Cardiol 2013;62:1585–1593. [DOI] [PubMed] [Google Scholar]
- 20. Rossignol P, Dobre D, Gregory D, Massaro J, Kiernan M, Konstam MA, Zannad F. Incident hyperkalemia may be an independent therapeutic target in low ejection fraction heart failure patients: insights from the HEAAL study. Int J Cardiol 2014;173:380–387. [DOI] [PubMed] [Google Scholar]
- 21. Lazich I, Bakris GL. Prediction and management of hyperkalemia across the spectrum of chronic kidney disease. Semin Nephrol 2014;34:333–339. [DOI] [PubMed] [Google Scholar]
- 22. Pitt B, Collins AJ, Reaven N, Funk S, Bakris GL, Bushinsky DA. Effect of cardiovascular comorbidities on the mortality risk associated with serum potassium. Circulation 2014;130:A13320 (abstr.). [Google Scholar]
- 23. Konstam MA, Neaton JD, Dickstein K, Drexler H, Komajda M, Martinez FA, Riegger GA, Malbecq W, Smith RD, Guptha S, Poole‐Wilson PA, HEAAL Investigators . Effects of high‐dose versus low‐dose losartan on clinical outcomes in patients with heart failure (HEAAL study): a randomised, double‐blind trial. Lancet 2009;374:1840–1848. [DOI] [PubMed] [Google Scholar]
- 24. Bakris GL, Pitt B, Weir MR, Freeman MW, Mayo MR, Garza D, Stasiv Y, Zawadzki R, Berman L, Bushinsky DA, AMETHYST‐DN Investigators . Effect of patiromer on serum potassium level in patients with hyperkalemia and diabetic kidney disease: the AMETHYST‐DN Randomized Clinical Trial. JAMA 2015;314:151–161. [DOI] [PubMed] [Google Scholar]
- 25. Pitt B, Anker SD, Bushinsky DA, Kitzman DW, Zannad F, Huang IZ, PEARL‐HF Investigators . Evaluation of the efficacy and safety of RLY5016, a polymeric potassium binder, in a double‐blind, placebo‐controlled study in patients with chronic heart failure (the PEARL‐HF) trial. Eur Heart J 2011;32:820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pfizer Pharmaceuticals . Aldactone® [Full Prescribing Information]. New York, NY: Pfizer Pharmaceuticals, Inc., 2014. [Google Scholar]
- 27. Pfizer Pharmaceuticals . Inspra™ [Full Prescribing Information]. New York, NY Pfizer Pharmaceuticals, Inc., 2008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Patient disposition with and without HF in the initial treatment phase. eGFR, estimated glomerular filtration rate; HF, heart failure; K+, potassium.
Figure S2. Patient disposition with and without HF in the randomized withdrawal phase. eGFR, estimated glomerular filtration rate; HF, heart failure; K+, potassium.
Figure S3. Time to hyperkalaemia recurrence in patients without HF. Circles indicate censored observations. eGFR, estimated glomerular filtration rate; HF, heart failure; K+, potassium.
Table S1. Titration algorithm for initial treatment phase.
Table S2. Titration algorithm for first 4 weeks of the withdrawal phase (day 3 to week 3 visits of this phase).
Table S3. (A) Individual adverse events occurring in at least 2 patients with or without HF in each dose group; (B) All Serious adverse events in the initial treatment phase.
Table S4. Kidney function tests in patients with and without HF.