

Abstract
Background
Mild parkinsonian signs (MPS) are common in older people and are associated with an increased risk of different neurodegenerative diseases. This study prospectively evaluates the longitudinal course of cognitive performance in older individuals with MPS.
Methods
From the TREND study, 480 individuals neurologically healthy at baseline, aged between 50 and 80 years, with complete follow-up data for three assessments within a mean of 43.8 months, were included in this analysis. Participants underwent a detailed cognitive test battery, evaluation of prodromal markers for neurodegenerative diseases and history of vascular diseases at each study visit. In addition, plasma levels of amyloid-beta (Aβ)1–40 and Aβ1–42 were evaluated longitudinally.
Results
In 52 (11 %) of the 480 participants, MPS could be detected at baseline. These individuals had cognitive deficits significantly more often compared with controls at each time point and their cognitive performance showed a steeper decline during follow-up. In addition, their levels of plasma Aβ1–42 were significantly lower than those of controls, and declined more rapidly over time.
Conclusions
This longitudinal study shows that MPS are associated with cognitive decline and decrease in plasma Aβ1–42, possibly indicating an ongoing neurodegenerative process.
Electronic supplementary material
The online version of this article (doi:10.1186/s13195-016-0209-7) contains supplementary material, which is available to authorized users.
Keywords: Amyloid-beta, Dementia, Prospective, Longitudinal, Cohort study
Background
Symptoms of bradykinesia, rigidity and tremor occurring in isolation as well as in combination are common in older people who do not meet the criteria for Parkinson’s disease (PD) or other neurodegenerative diseases [1, 2]. These are referred to as mild parkinsonian signs (MPS). Obviously these symptoms can be caused by a variety of non-neurological conditions as well as by neurological disorders. MPS have been shown to be associated with several different diseases and adverse health outcomes, such as PD, dementia, functional disability and mortality [2–5]. Three prospective studies have so far demonstrated an association between the presence of MPS in non-demented older adults and the development of incident dementia (including Alzheimer’s disease (AD)) during follow-up. Wilson et al. [6] showed already that a slight progression of MPS doubles the risk for developing AD. Further, Louis et al. [4] showed in a study with 1028 individuals that those with MPS at baseline had an increased risk of 56 % for developing incident dementia after a mean follow up of 5.6 years. In a large prospective study including 1851 individuals, the same group found dementia in 16 % of the MPS participants after a mean follow up of 3.7 years, with AD being the most common type (86 %) [7]. Further, there are several cross-sectional analyses showing an association between MPS and mild cognitive impairment in the “prodromal phase” of AD [8–10]. Taken together, there is evidence that MPS might be a “prodromal marker” for future incident dementia. The “prodromal phase” of a neurodegenerative disease is defined as the phase in which neurodegeneration is ongoing but a clinical diagnosis cannot be made. Symptoms of the prodromal phase comprise motor and non-motor markers (“marker” here refers to any disease indicator, whether a symptom, sign or biomarker).
An opportunity to better estimate the possible role of MPS as a prodromal stage of AD is the longitudinal evaluation of clinical and biofluid markers in individuals at risk for this form of dementia.
To the best of our knowledge, no study has yet evaluated individuals with MPS with regard to cognitive symptoms and biological/biochemical markers longitudinally with biennial evaluations. To address this issue we investigated a large cohort of healthy older individuals with and without MPS, longitudinally over a study period of 3.5 years, using a cognitive test battery, self-administered questionnaires for prodromal AD markers and co-morbidities and ultrasound for the measurement of the intima-media thickness (IMT) of the carotids. Moreover we analysed biomaterial of the participants for amyloid-beta (Aβ)1–40 and Aβ1–42 and apolipoprotein (ApoE) genotypes.
Methods
Study population
The TREND study is a prospective follow-up study initiated in 2009, which included individuals aged ≥50 years at baseline and provides biennial assessments until death/autopsy. For a detailed outline of the TREND study, as well as inclusion and exclusion criteria, see Gaenslen et al. [11]. A large assessment battery with mainly quantitative, unobtrusive measurements designed to be repeated easily and objectively is being applied. To make sure that there is no bias in data acquisition, all investigators are blinded to the results of all other examinations. The TREND study initially comprised 715 participants of whom 593 individuals had three evaluations, the prerequisite for being eligible for analyses in this study. Of these 593 individuals, those with complete datasets of neurological examination, a cognitive test battery and plasma levels of Aβ1–40 and Aβ1–42 were included in this analysis. In total, 480 individuals (52 with persistent MPS and 428 without) were analysed.
Neurological examination
Each participant underwent a standardized neurological examination and the motor part of the revised Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) applied by an experienced movement disorder specialist [12]. In accordance with other large-scale studies [13, 14], MPS were defined as present when any of the following conditions was met: at least two MDS-UPDRS III items ≥1; or at least one MDS-UPDRS III item ≥2; iii) at least one MDS-UPDRS rest tremor item ≥1. Only individuals in whom MPS were present at all three time points were defined as individuals with persistent MPS. Individuals in whom the results of the neurological examination revealed PD according to the UK Brain Bank Criteria were excluded from this analysis [15].
Assessment of demographics and medical history
Each participant underwent a structured medical interview including demographics, medical history and medication. A family history questionnaire was used to obtain information on all first-degree and second-degree relatives of the participants. Vascular risk factors were assessed with a self-assembled medical questionnaire which includes questions about history of ischaemic stroke or transient ischaemic attack, heart attack, arrhythmia, angina pectoris, occlusive disease of the peripheral arteries, congestive heart failure, hypertension, hypercholesterolaemia and diabetes mellitus. This questionnaire allows one to rate the existence or absence of the afore-mentioned disturbances (“yes/no”). In addition, the intake of medication was recorded and used to verify the answers.
Assessment of prodromal markers
Impaired olfaction
Olfaction was tested using the 16 Sniffin’ sticks battery (Burghart Medizintechnik, Germany) as described by Hummel et al. [16]. According to the suggestion of Hummel and colleagues, individuals identifying less than 75 % of odours correctly were classified as having hyposmia. Some of the 480 participants had co-morbidities prohibiting the correct determination of olfaction (report of allergic rhinitis/cold or nose surgery). At baseline, olfaction could not be tested in 24 participants (21 healthy controls and three individuals with persistent MPS).
Rapid eye movement sleep behaviour disorder
Presence of rapid eye movement sleep behaviour disorder (RBD) was determined by a self-administered RBD screening questionnaire (RBDSQ). The RBDSQ is a recently developed questionnaire, comprising 10 items to describe the most prominent clinical features of RBD [17]. Presence of RBD was accepted in participants with ≥5 points in the questionnaire (sensitivity 96 % and specificity 92 %).
Depression
Depressive symptoms were assessed with the Beck Depression Inventory I (BDI-I). The BDI-I is a 21-item self-report questionnaire, ranging from 0 to 63 (no depression, 0–10; mild to moderate depression, 11–17; severe depression, 18–63) [18].
Intima-media thickness
Measurement of the right common carotid artery (CCA) was performed in the first follow-up using a 5–10 MHz linear array transducer (VF10-5; Siemens, Erlangen, Germany). Participants were examined in a supine position with their head tilted backwards. The CCA was differentiated from the internal jugular vein in a transverse plane, displayed in a longitudinal scan while the course of the CCA was followed up to the carotid bulb. IMT was measured 1 cm proximal of the carotid bulb using a fourfold magnification of the ultrasound image [19]. The IMT of the CCA was defined as the distance between the intima line and the media–adventitia border at the far wall of the CCA [20]. Measurements were documented and images were stored. IMT was defined as pathologic if the measurement was >1.0 mm [21, 22].
Cognition
Cognition was tested using an established and standardized cognitive test battery. We used the German version of the extended Consortium to Establish a Registry for Alzheimer’s Disease neuropsychological battery (CERAD-Plus) [23]. The battery contains the following subtests: verbal fluency, Boston Naming Test, Mini Mental State Examination, word list memory, word list recall, word list recognition, constructional praxis, recall of constructional praxis, phonematic fluency and the Trail-Making Test Parts A and B (TMT-A and TMT-B). The TMT consists of two parts and evaluates executive function, cognitive flexibility and working memory [24, 25]. In TMT-A, subjects have to connect randomly spread numbers from 1 to 25 in ascending order. In TMT-B, participants are asked to connect randomly spread numbers (1–13) and letters (A–L) in alternating numeric and alphabetical order (1–A–2–B–3–C–…–13–L). In the case of an error, the examiner draws the attention of the participant to the error, to allow completion of the task without errors at the expense of additional time. The maximum time allowed is 180 s for TMT-A and 300 s for TMT-B. After this time the investigator discontinues the experiment [26]. TMT performance was calculated taking the time needed to perform TMT-B minus the time needed for TMT-A. This ΔTMT value prevents possible bias due to differences in upper extremity motor speed, simple sequencing, visual scanning and psychomotor functioning [24, 25]. The subtests of the CERAD-Plus battery were grouped into four domains: executive, memory, language and visuospatial. For the analysis of these domains, demographically adjusted z scores were used. The total score of the CERAD battery, ranging from 0 to 100, has been shown to provide an effective global measure of cognitive functioning [27].
Biomaterial and analyses of biomarkers in plasma
Standard operating procedures were defined for the collection, preparation, storage and analysis of biomaterial obtained from the study participants.
Plasma Aβ1–38, Aβ1−40 and Aβ1–42 levels
EDTA plasma was centrifuged at 2000 × g, 4 °C for 10 min and stored at −80 °C within 60 min after collection. Plasma levels of Aβ1–42 and Aβ1–40 were quantified by EUROIMMUN Beta-Amyloid 1–42 Plasma ELISA and EUROIMMUN Beta-Amyloid 1–40 Plasma ELISA, respectively. Instead of a manual procedure, the automated workup on the ANALYZER-I (EUROIMMUN) was applied. Test specifications and characteristics for the methodology are shown in Additional file 1. For quality control (QC) purposes, QC samples were produced by pooling of EDTA plasma samples. Some samples were used native and others were spiked to higher concentrations with calibrator material. After aliquoting, samples were freeze dried for long-term stability. The samples were coded R2, R3, R6 and R8 and used over the three plasma amyloid assays. For test run monitoring, three QC samples were included (single testing) in each test run in parallel with the test samples. Only runs with available data points for the three samples were considered. The coloured blocks show the 95 % confidence interval (±2 SD). For details, see Additional file 2.
ApoE genotypes
Genomic DNA was extracted from EDTA blood using standardized protocols. ApoE genotypes were analysed by a multiplex SNaPshot assay (Applied Biosystems Life Technologies GmbH, Darmstadt, Germany) (PMID:15505371). All SNPs investigated were in Hardy–Weinberg equilibrium (data not shown).
Statistical analysis
Statistical analysis was performed using SPSS 22.0 software for Windows (SPSS Inc., Chicago, IL, USA). Because of the asymmetric distribution between the two groups, differences of non-categorical data were evaluated with the Wilcoxon rank-sum test. The Fisher’s exact test was used for categorical data. Descriptive statistics are given either as median (range) and mean ± standard deviation for non-categorical data or as percentages of total for categorical data. CERAD total score, TMT and plasma levels of Aβ1–40 and Aβ1–42 have been evaluated for each time point adjusting for age, gender and educational levels (only cognition). For the CERAD subdomains, demographically adjusted z scores were used. Differences were considered significant at p < 0.05.
Results
The 480 TREND participants had a mean duration of total follow-up of 43.8 ± 4.2 months (baseline (BL) to first follow-up (1FU) 19.8 ± 3.4 months; 1FU to second follow-up (2FU) 23.6 ± 2.0 months). Persistent MPS was present in 52 participants (11 %). The remaining individuals were defined as controls.
At baseline, individuals with persistent MPS compared with the control cohort were more often male (65 % vs. 43 %; p < 0.01), older (67 years vs. 62 years; p < 0.01), more often had RBD (35 % vs 19 %; p = 0.01), had fewer years of education (13 years vs. 14 years; p = 0.03) and less often reported a positive family history for dementia (23 % vs. 39 %; p = 0.03; see Table 1). Participants with persistent MPS did not significantly differ from controls in terms of prevalence of hyposmia, occurrence of lifetime depression, severity of depressive symptoms, ApoE4 status and frequency of vascular co-morbidities. Table 1 and Additional file 3 present a complete overview of all parameters. According to the grouping criteria, MDS-UPDRS III was significantly higher in individuals with MPS (4 points vs. 0 points; p < 0.001).
Table 1.
Baseline characteristics of participants with persistent MPS and controls
| Controls (n = 428) | Persistent MPS (n = 52) | p value | |
|---|---|---|---|
| Male gender (N (%)) | 182 (43) | 34 (65) | 0.002 |
| Age (years) | 62 (50–80) | 67 (51–77) | <0.001 |
| (62 ± 7) | (67 ± 6) | ||
| Education (years) | 14 (9–21) | 13 (9–21) | 0.030 |
| (15 ± 3) | (14 ± 3) | ||
| Family history PD (%) | 63 (15) | 8 (15) | 0.838 |
| Family history dementia (%) | 167 (39) | 12 (23) | 0.033 |
| MMSE (0–30) | 29 (25–30) | 29 (25–30) | 0.173 |
| (29 ± 1) | (28 ± 1) | ||
| MDS-UPDRS-III (0–132) | 0 (0–10) | 4 (2–17) | <0.001 |
| (1 ± 2) | (5 ± 3) | ||
| Lifetime depression (%) | 155 (36) | 20 (39) | 0.762 |
| Hyposmia (%) | 95 (22) | 15 (29) | 0.216 |
| RBD (%) | 81 (19) | 18 (35) | 0.011 |
| BDI-I (0–63) | 6 (0–38) | 6 (0–38) | 0.121 |
| (7 ± 6) | (9 ± 7) | ||
| Hyperechogenic substantia nigra (%) | 106 (25) | 19 (37) | 0.075 |
| ApoE4 positive (%) | 94 (22) | 12 (23) | 0.931 |
Data are presented as median (range) (mean ± standard deviation) or number (percentage of total)
ApoE4 apolipoprotein 4, BDI-I Beck Depression Inventory I, MMSE Mini Mental State Examination, MPS mild parkinsonian signs, PD Parkinson’s disease, RBD rapid eye movement sleep behaviour disorder, MDS-UPDRS-III Unified Parkinson’s Disease Rating Scale part 3
Individuals with persistent MPS showed worse cognitive performance compared with controls in the CERAD total score, TMT-A and TMT-B at all three time points. Further, they showed a non-significant trend to be worse in ΔTMT-B – TMT-A at baseline. This difference became significant within the follow-up evaluations (Table 2). During the follow-up period (BL to 2FU), controls had stable results in the longitudinally assessed CERAD total scores (p = 0.210) and TMT-A (p = 0.422) but an improvement in their performance in the TMT-B (p < 0.001). In contrast, individuals with persistent MPS showed stable results only in the TMT-A (p = 0.684) but tended (non-significant trend) to deteriorate in the TMT-B (p = 0.078) and in the CERAD total score (p = 0.062) during the follow-up period. The two groups did not differ in the memory, language or visuospatial domain of the CERAD-Plus battery but differed in the executive domain, represented by the TMT.
Table 2.
Cognitive test performance in controls and individuals with persistent MPS
| Examination | Controls | Persistent MPS | p value | |
|---|---|---|---|---|
| CERAD total score (0–100) | BL | 87 (64–100) | 84 (60–96) | 0.002a |
| (87 ± 7) | (83 ± 8) | |||
| 1FU | 86 (56–100) | 82 (44–97) | 0.002a | |
| (85 ± 8) | (80 ± 8) | |||
| 2FU | 88 (57–100) | 83 (50–98) | <0.001a | |
| (87 ± 8) | (82 ± 9) | |||
| TMT-A (s) | BL | 34 (15–100) | 39 (24–81) | 0.005a |
| (36 ± 12) | (41 ± 12) | |||
| 1FU | 34 (17–103) | 41 (21–71) | 0.016a | |
| (36 ± 14) | (43 ± 12) | |||
| 2FU | 33 (15–90) | 40 (24–110) | 0.014a | |
| (36 ± 11) | (43 ± 14) | |||
| TMT-B (s) | BL | 80 (34–300) | 96 (47–300) | 0.050a |
| (88 ± 33) | (105 ± 40) | |||
| 1FU | 76 (26–300) | 93 (55–300) | <0.001a | |
| (82 ± 34) | (107 ± 49) | |||
| 2FU | 73 (25–300) | 97 (47–300) | 0.001a | |
| (81 ± 37) | (112 ± 61) | |||
| TMT-B – TMT-A (s) | BL | 46 (11–185) | 54 (8–206) | 0.083a |
| (52 ± 30) | (64 ± 40) | |||
| 1FU | 40 (5–243) | 51 (13–229) | 0.001a | |
| (46 ± 29) | (64 ± 43) | |||
| 2FU | 39 (0–230) | 52 (19–265) | 0.004a | |
| (46 ± 33) | (70 ± 54) | |||
| Executive domain | BL | 0.406 ± 1.0 | 0.136 ± 1.0 | 0.288 |
| 1FU | 0.690 ± 1.2 | 0.215 ± 1.1 | 0.010 | |
| 2FU | 0.887 ± 1.3 | 0.368 ± 1.1 | 0.014 | |
| Memory domain | BL | 0.002 ± 0.8 | 0.028 ± 0.8 | 0.862 |
| 1FU | 0.177 ± 0.8 | 0.002 ± 0.8 | 0.157 | |
| 2FU | 0.169 ± 0.9 | 0.124 ± 0.8 | 0.463 | |
| Language domain | BL | 0.416 ± 0.7 | 0.370 ± 0.7 | 0.956 |
| 1FU | 0.004 ± 0.7 | −0.042 ± 0.7 | 0.575 | |
| 2FU | 0.343 ± 0.7 | 0.121 ± 0.7 | 0.124 | |
| Visuospatial domain | BL | 0.284 ± 0.9 | 0.221 ± 1.3 | 0.073 |
| 1FU | 0.431 ± 0.9 | 0.074 ± 1.3 | 0.697 | |
| 2FU | 0.041 ± 1.0 | 0.488 ± 1.2 | 0.115 |
Values are presented as median (range) (mean ± standard deviation) for the CERAD total score and the TMT. For the four subdomains, values are presented as mean ± standard deviation of demographically adjusted z scores
a p values were corrected for age, gender and years of education
BL baseline, CERAD Consortium to Establish a Registry for Alzheimer’s Disease, 1FU first follow-up, 2FU second follow-up, MPS mild parkinsonian signs, TMT Trail Making Test
The overall relative risk of individuals with persistent MPS to develop cognitive impairment (defined as being in the fourth quartile with more than >98 sec needed for completion of TMT-B at the 2FU) was 2.06 (95 % confidence interval: 1.45–2.92; p < 0.001).
After correction for age and gender, individuals with persistent MPS had significantly lower Aβ1–42 values than controls at all time points (BL, p = 0.007; 1FU, p = 0.022; 2FU, p = 0.021). In addition, Aβ1–42 values of persistent MPS participants decreased over time, whereas the control group remained stable over the three time points (p = 0.169). Similar results were found for the ratio Aβ1–42/Aβ1–40. Compared with controls in individuals with persistent MPS, the Aβ1–42/Aβ1–40 ratio tended to be lower at 1FU (p = 0.067) and was significantly decreased at 2FU (p = 0.033). No significant differences were found between controls and individuals with persistent MPS for Aβ1–38 and Aβ1–40 values either cross-sectionally or longitudinally (Fig. 1).
Fig. 1.
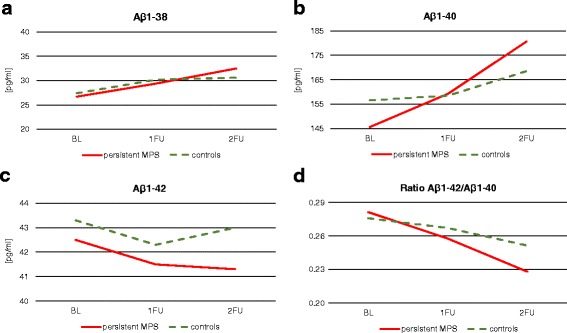
Levels of plasma Aβ1–38, Aβ1–40 and Aβ1–42 in individuals with mild parkinsonian signs (MPS) and controls. a Aβ1–38, b Aβ1–40, c Aβ1–42 and d ratio Aβ1–42/Aβ1–40. Individuals with persistent MPS had significantly lower Aβ1–42 values than controls at all three time points. In addition, Aβ1–42 values and the ratio Aβ1–42/Aβ1–40 of persistent MPS participants decreased over time, whereas the control group remained stable. No significant differences between controls and individuals with persistent MPS were found for Aβ1–38 and Aβ1–40 values either cross-sectionally or longitudinally. Aβ amyloid-beta, BL baseline, 1FU first follow-up, 2FU second follow-up
Further, we found highly significant correlations between the “diagnosis” of MPS and plasma level of Aβ1–42, CERAD total score and TMT (Table 3).
Table 3.
Correlation between persistent MPS and plasma levels of amyloid-beta and cognitive test performance
| Pearson’s r coefficient | |
|---|---|
| Amyloid-beta1–38 | −0.04 |
| Amyloid-beta1–40 | −0.02 |
| Amyloid-beta1–42 | −0.14** |
| CERAD total score | −0.16*** |
| TMT-A | 0.17*** |
| TMT-B | 0.16*** |
| TMT-B – TMT-A | 0.13*** |
**p < 0.01,***p < 0.001
CERAD Consortium to Establish a Registry for Alzheimer’s Disease, MPS mild parkinsonian signs, TMT Trail Making Test
Discussion
The main findings of this prospective longitudinal study are the associations of MPS with both cognitive deficits and low plasma Aβ1–42 levels, as well as the progression of cognitive deterioration and plasma Aβ1–42 reduction in individuals with MPS but not in those without.
In this prospective study of neurologically healthy older adults, the phenomenon of persistent MPS is associated with higher age and lower education but not with other known risk factors for dementia such as ApoE4 positive status, family history for dementia or vascular diseases. This is in line with a large prospective study which found no association of MPS with these risk factors but still found an increased risk for dementia in individuals with baseline MPS [4].
The cognitive profile of participants with persistent MPS was worse than that of the control group. Individuals with persistent MPS were significantly worse in the cognitive tests at all time points investigated (except for ΔTMT-B–TMT-A at baseline) and further showed a non-significant trend for cognitive decrease over time. These results suggest that individuals with persistent MPS might be in a prodromal phase of dementia and might progress to more severe cognitive conditions.
Aβ1–42 is probably the most studied biofluid marker for the prediction of cognitive decline or dementia in neurodegenerative research. We found that individuals with persistent MPS had reduced plasma levels of Aβ1–42 at all study time points and that their levels decrease during the observational period. Taking into account that cerebrospinal fluid (CSF) Aβ1–42 levels are reduced to a level indicative for AD 5–10 years before the onset of dementia [28], with the first changes estimated to occur more than two decades before related cognitive decline [29], one could suggest that the reduced levels of Aβ1–42 in the plasma of individuals with persistent MPS are somehow related to CSF changes of Aβ1–42. Consequently, a reduction of plasma Aβ1–42 may be related to the prodromal phase of AD. In contrast to CSF Aβ1–42, there are only few studies on plasma Aβ1–42 with inconsistent results. In cross-sectional studies of individuals with AD and mild cognitive impairment, lower [30], unchanged [31, 32] or higher [33] levels of Aβ1–42 have been found. A problem of these cross-sectional studies is the relatively low number of participants. The even fewer longitudinal studies measured Aβ in healthy individuals only once at baseline to evaluate the risk of AD, but they did not evaluate the course of Aβ in the prodromal phase. For example, Sundelof et al. found an association of reduced Aβ1–42 and increased risk of AD while Mayeux et al. found an association between high levels of Aβ1–42 and increased risk for AD [34, 35]. Our study is therefore the first reporting repeated measurements of Aβ1–42.
Although plasma Aβ1–42 changes are small, the finding may still be of relevance because probably a (small) subgroup of individuals with persistent MPS will develop AD. It is also possible that the reduction of Aβ1–42 plasma levels in blood occur later than in CSF. Further follow-up of this study and other studies measuring plasma levels of Aβ1–42 would be of great interest because collection of blood is easier, less invasive and can be done in greater cohorts than collection of CSF.
The strength of this study is the prospective longitudinal study design. Moreover, our MPS cohort only included individuals with persistent MPS over the whole observation period, thereby reducing confounders such as individual daily condition (e.g. well rested, mood or nervousness) and inter-rater differences. Most former studies defined their cohort based on one single evaluation but motor signs, especially when they are less distinct, can fluctuate. By taking only individuals with persistent MPS into account, we reduced the amount of false positives and analysed a more specific group at risk for neurodegeneration.
A limitation of this analysis is the relatively short follow-up period of a median of 3.7 years. However, we were able to find significant differences in cognition and Aβ1–42 levels. It is highly probable that these effects become even stronger with a longer follow-up period. Moreover, despite the possible influence of depression on cognitive performance, we did not exclude participants which might be clinically depressed (BDI >18), because the number of those severely depressed individuals was low and equally distributed between the two groups (MPS 9 % and HC 7 %). Therefore, we do not think that the results will be influenced substantially. Additional studies are warranted for further validation.
Conclusion
Taken together, low cognitive performance and plasma Aβ1–42 levels which decrease over time seem to be associated with persistent MPS. This suggests that, at least in a statistically relevant subgroup of individuals with persistent MPS, an underlying neurodegenerative process associated with AD pathology may be present. The potential of the “syndrome” MPS as a predictive marker for future neurodegenerative diseases should therefore be further explored.
Acknowledgements
The authors thank all participants who took part in the TREND study.
The authors acknowledge support of publication by Deutsche Forschungsgemeinschaft and Open Access Publishing Fund of Tuebingen University, Tuebingen, Germany.
Funding
Publication of this article was supported by Deutsche Forschungsgemeinschaft and Open Access Publishing Fund of Tuebingen University, Tuebingen, Germany.
Availability of data and materials
The dataset will not be shared because it contains clinical data which cannot be made public due to privacy rights of the participants and for protection of participants’ identity.
Authors’ contributions
SL substantially contributed to the conception and design of the study, performed the statistical analysis, interpreted the data and drafted the work. KB collected data, helped to draft the manuscript and revised the work critically for important intellectual content. AP, IW, US, MAH, A-KvT and CS collected data and revised the work critically for important intellectual content. ES and HV substantially contributed to the conception and design of the study, and revised the work critically for important intellectual content. VH and BB carried out the immunoassays and revised the work critically for important intellectual content. GWE, FGM, WM and DB substantially contributed to the conception and design of the study, and revised the work critically for important intellectual content. All authors read the manuscript, gave their final approval to version to be published and agreed to be accountable for all aspects of the work.
Authors’ information
Not applicable.
Competing interests
The authors declare that they have no competing interest.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The study was approved by the ethical committee of the Medical Faculty of the University of Tuebingen (Nr. 90/2009BO2), and all subjects gave written informed consent.
Abbreviations
- 1FU
First follow-up
- 2FU
Second follow-up
- AD
Alzheimer’s disease
- ApoE4
Apolipoprotein 4
- Aβ
Amyloid-beta
- BDI-I
Beck Depression Inventory I
- BL
Baseline
- CCA
Common carotid artery
- CERAD
Consortium to Establish a Registry for Alzheimer’s Disease
- CSF
Cerebrospinal fluid
- IMT
Intima-media thickness
- MMSE
Mini Mental State Examination
- MPS
Mild parkinsonian signs
- PD
Parkinson’s disease
- QC
Quality control
- RBD
Rapid eye movement sleep behaviour disorder
- RBDSQ
RBD screening questionnaire
- SD
Standard deviation
- TMT
Trail-Making Test
- TREND
Tübinger Evaluation of Risk Factors for Early Detection of Neurodegenerative Disorders
- UPDRS
Unified Parkinson’s Disease Rating Scale
Additional files
is Table S1 presenting test specifications and characteristics for the methodology. (DOCX 17 kb)
is a figure showing ELISA test run monitoring. Three QC samples were included (single testing) in parallel with the test samples. Only runs with available data points for the three samples were considered. The coloured blocks show the 95 % confidence interval (±2 SD). (PPTX 8306 kb)
is Table S2 presenting surrogate markers of risks and co-morbidities. Participants with persistent MPS did not significantly differ from controls in terms of frequency of vascular co-morbidities. (DOCX 11 kb)
Contributor Information
Stefanie Lerche, Phone: + 49-7071-2980172, Email: Stefanie.lerche@uni-tuebingen.de.
Kathrin Brockmann, Email: Kathrin.Brockmann@uni-tuebingen.de.
Andrea Pilotto, Email: pilottoandreae@gmail.com.
Isabel Wurster, Email: isabel.wurster@uni-tuebingen.de.
Ulrike Sünkel, Email: ulrike.suenkel@uni-tuebingen.de.
Markus A. Hobert, Email: Markus.Hobert@med.uni-tuebingen.de
Anna-Katharina von Thaler, Email: anna-katharina.von-thaler@uni-tuebingen.de.
Claudia Schulte, Email: claudia.schulte@uni-tuebingen.de.
Erik Stoops, Email: erik.stoops@adxneurosciences.com.
Hugo Vanderstichele, Email: hugo.vanderstichele@adxneurosciences.com.
Victor Herbst, Email: v.herbst@euroimmun.de.
Britta Brix, Email: b.brix@euroimmun.de.
Gerhard W. Eschweiler, Email: gerhard.eschweiler@med.uni-tuebingen.de
Florian G. Metzger, Email: florian.metzger@med.uni-tuebingen.de
Walter Maetzler, Email: walter.maetzler@uni-tuebingen.de.
Daniela Berg, Email: Daniela.Berg@uni-tuebingen.de.
References
- 1.Louis ED, Luchsinger JA, Tang MX, Mayeux R. Parkinsonian signs in older people: prevalence and associations with smoking and coffee. Neurology. 2003;61:24–8. doi: 10.1212/01.WNL.0000072330.07328.D6. [DOI] [PubMed] [Google Scholar]
- 2.Bennett DA, Beckett LA, Murray AM, Shannon KM, Goetz CG, Pilgrim DM, et al. Prevalence of parkinsonian signs and associated mortality in a community population of older people. N Engl J Med. 1996;334:71–6. doi: 10.1056/NEJM199601113340202. [DOI] [PubMed] [Google Scholar]
- 3.Berg D, Godau J, Seppi K, Behnke S, Liepelt-Scarfone I, Lerche S, et al. The PRIPS study: screening battery for subjects at risk for Parkinson's disease. Eur J Neurol. 2013;20:102–8. doi: 10.1111/j.1468-1331.2012.03798.x. [DOI] [PubMed] [Google Scholar]
- 4.Louis ED, Tang MX, Schupf N. Mild parkinsonian signs are associated with increased risk of dementia in a prospective, population-based study of elders. Mov Disord. 2010;25:172–8. doi: 10.1002/mds.22943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Louis ED, Schupf N, Marder K, Tang MX. Functional correlates of mild parkinsonian signs in the community-dwelling elderly: poor balance and inability to ambulate independently. Mov Disord. 2006;21:411–6. doi: 10.1002/mds.20735. [DOI] [PubMed] [Google Scholar]
- 6.Wilson RS, Schneider JA, Bienias JL, Evans DA, Bennett DA. Parkinsonianlike signs and risk of incident Alzheimer disease in older persons. Arch Neurol. 2003;60:539–44. doi: 10.1001/archneur.60.4.539. [DOI] [PubMed] [Google Scholar]
- 7.Louis ED, Tang MX, Mayeux R. Parkinsonian signs in older people in a community-based study: risk of incident dementia. Arch Neurol. 2004;61:1273–6. doi: 10.1001/archneur.61.8.1273. [DOI] [PubMed] [Google Scholar]
- 8.Louis ED, Schupf N, Manly J, Marder K, Tang MX, Mayeux R. Association between mild parkinsonian signs and mild cognitive impairment in a community. Neurology. 2005;64:1157–61. doi: 10.1212/01.WNL.0000156157.97411.5E. [DOI] [PubMed] [Google Scholar]
- 9.Rozzini L, Chilovi BV, Bertoletti E, Conti M, Delrio I, Trabucchi M, et al. Mild parkinsonian signs and psycho-behavioral symptoms in subjects with mild cognitive impairment. Int Psychogeriatr. 2008;20:86–95. doi: 10.1017/S1041610207006163. [DOI] [PubMed] [Google Scholar]
- 10.Uemura Y, Wada-Isoe K, Nakashita S, Nakashima K. Depression and cognitive impairment in patients with mild parkinsonian signs. Acta Neurol Scand. 2013;128(3):153–59. [DOI] [PubMed]
- 11.Gaenslen A, Wurster I, Brockmann K, Huber H, Godau J, Faust B, et al. Prodromal features for Parkinson's disease—baseline data from the TREND study. Eur J Neurol. 2014;21:766–72. doi: 10.1111/ene.12382. [DOI] [PubMed] [Google Scholar]
- 12.Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez-Martin P, et al. Movement Disorder Society-sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord. 2008;23:2129–70. doi: 10.1002/mds.22340. [DOI] [PubMed] [Google Scholar]
- 13.Louis ED, Brickman AM, DeCarli C, Small SA, Marder K, Schupf N, et al. Quantitative brain measurements in community-dwelling elderly persons with mild parkinsonian signs. Arch Neurol. 2008;65:1649–54. doi: 10.1001/archneurol.2008.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahlknecht P, Kiechl S, Stockner H, Willeit J, Gasperi A, Poewe W, et al. Predictors for mild parkinsonian signs: a prospective population-based study. Parkinsonism Relat Disord. 2015;21:321–4. doi: 10.1016/j.parkreldis.2014.12.021. [DOI] [PubMed] [Google Scholar]
- 15.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55(3):181–84. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hummel T, Konnerth CG, Rosenheim K, Kobal G. Screening of olfactory function with a four-minute odor identification test: reliability, normative data, and investigations in patients with olfactory loss. Ann Otol Rhinol Laryngol. 2001;110:976–81. doi: 10.1177/000348940111001015. [DOI] [PubMed] [Google Scholar]
- 17.Stiasny-Kolster K, Sixel-Doring F, Trenkwalder C, Heinzel-Gutenbrunner M, Seppi K, Poewe W, et al. Diagnostic value of the REM sleep behavior disorder screening questionnaire in Parkinson's disease. Sleep Med. 2015;16:186–9. doi: 10.1016/j.sleep.2014.08.014. [DOI] [PubMed] [Google Scholar]
- 18.Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatr. 1961;4:561–71. doi: 10.1001/archpsyc.1961.01710120031004. [DOI] [PubMed] [Google Scholar]
- 19.Engelen L, Ferreira I, Stehouwer CD, Boutouyrie P, Laurent S. Reference values for arterial measurements C. Reference intervals for common carotid intima-media thickness measured with echotracking: relation with risk factors. Eur Heart J. 2013;34:2368–80. doi: 10.1093/eurheartj/ehs380. [DOI] [PubMed] [Google Scholar]
- 20.Touboul PJ, Hennerici MG, Meairs S, Adams H, Amarenco P, Bornstein N, et al. Mannheim carotid intima-media thickness and plaque consensus (2004-2006-2011). An update on behalf of the advisory board of the 3rd, 4th and 5th watching the risk symposia, at the 13th, 15th and 20th European Stroke Conferences, Mannheim, Germany, 2004, Brussels, Belgium, 2006, and Hamburg, Germany, 2011. Cerebrovasc Dis. 2012;34:290–6. doi: 10.1159/000343145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stein JH, Korcarz CE, Hurst RT, Lonn E, Kendall CB, Mohler ER, et al. Use of carotid ultrasound to identify subclinical vascular disease and evaluate cardiovascular disease risk: a consensus statement from the American Society of Echocardiography Carotid Intima-Media Thickness Task Force. Endorsed by the Society for Vascular Medicine. J Am Soc Echocardiogr. 2008;21:93–111. doi: 10.1016/j.echo.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 22.Temelkova-Kurktschiev T, Fischer S, Koehler C, Mennicken G, Henkel E, Hanefeld M. Intima-media thickness in healthy probands without risk factors for arteriosclerosis. Deutsch Med Wochenschr. 2001;126:193–7. doi: 10.1055/s-2001-11317. [DOI] [PubMed] [Google Scholar]
- 23.Welsh-Bohmer KA, Mohs RC. Neuropsychological assessment of Alzheimer's disease. Neurology. 1997;49:S11–3. doi: 10.1212/WNL.49.3_Suppl_3.S11. [DOI] [PubMed] [Google Scholar]
- 24.Ble A, Volpato S, Zuliani G, Guralnik JM, Bandinelli S, Lauretani F, et al. Executive function correlates with walking speed in older persons: the InCHIANTI study. J Am Geriatrics Soc. 2005;53:410–5. doi: 10.1111/j.1532-5415.2005.53157.x. [DOI] [PubMed] [Google Scholar]
- 25.Drane DL, Yuspeh RL, Huthwaite JS, Klingler LK. Demographic characteristics and normative observations for derived-trail making test indices. Neuropsychiatry Neuropsychol Behav Neurol. 2002;15:39–43. [PubMed] [Google Scholar]
- 26.Lezak M. Orientation and Attention. New York: Oxford University Press; 1995. pp. 335–84. [Google Scholar]
- 27.Chandler MJ, Lacritz LH, Hynan LS, Barnard HD, Allen G, Deschner M, et al. A total score for the CERAD neuropsychological battery. Neurology. 2005;65:102–6. doi: 10.1212/01.wnl.0000167607.63000.38. [DOI] [PubMed] [Google Scholar]
- 28.Buchhave P, Minthon L, Zetterberg H, Wallin AK, Blennow K, Hansson O. Cerebrospinal fluid levels of beta-amyloid 1–42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatr. 2012;69:98–106. doi: 10.1001/archgenpsychiatry.2011.155. [DOI] [PubMed] [Google Scholar]
- 29.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pesaresi M, Lovati C, Bertora P, Mailland E, Galimberti D, Scarpini E, et al. Plasma levels of beta-amyloid (1–42) in Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2006;27:904–5. doi: 10.1016/j.neurobiolaging.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 31.Tamaoka A, Fukushima T, Sawamura N, Ishikawa K, Oguni E, Komatsuzaki Y, et al. Amyloid beta protein in plasma from patients with sporadic Alzheimer's disease. J Neurol Sci. 1996;141:65–8. doi: 10.1016/0022-510X(96)00143-8. [DOI] [PubMed] [Google Scholar]
- 32.Mehta PD, Pirttila T, Mehta SP, Sersen EA, Aisen PS, Wisniewski HM. Plasma and cerebrospinal fluid levels of amyloid beta proteins 1–40 and 1–42 in Alzheimer disease. Arch Neurol. 2000;57:100–5. doi: 10.1001/archneur.57.1.100. [DOI] [PubMed] [Google Scholar]
- 33.Sobow T, Flirski M, Kloszewska I, Liberski PP. Plasma levels of alpha beta peptides are altered in amnestic mild cognitive impairment but not in sporadic Alzheimer's disease. Acta Neurobiol Exp. 2005;65:117–24. doi: 10.55782/ane-2005-1544. [DOI] [PubMed] [Google Scholar]
- 34.Sundelof J, Giedraitis V, Irizarry MC, Sundstrom J, Ingelsson E, Ronnemaa E, et al. Plasma beta amyloid and the risk of Alzheimer disease and dementia in elderly men: a prospective, population-based cohort study. Arch Neurol. 2008;65:256–63. doi: 10.1001/archneurol.2007.57. [DOI] [PubMed] [Google Scholar]
- 35.Mayeux R, Honig LS, Tang MX, Manly J, Stern Y, Schupf N, et al. Plasma A[beta]40 and A[beta]42 and Alzheimer's disease: relation to age, mortality, and risk. Neurology. 2003;61:1185–90. doi: 10.1212/01.WNL.0000091890.32140.8F. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The dataset will not be shared because it contains clinical data which cannot be made public due to privacy rights of the participants and for protection of participants’ identity.