

Abstract
Introduction
Loss of synapses best correlates to cognitive deficits in Alzheimer's disease (AD) in which oligomeric neurotoxic species of amyloid β appears to contribute synaptic pathology. While a number of clinical pathological studies have been performed with limited sample size, there are no systematic studies encompassing large samples. Therefore, we performed a meta-analysis study.
Methods
We identified 417 publications reporting post-mortem synapse and synaptic marker loss from AD patients. Two meta-analyses were performed using a single database of sub-selected publications and calculating the standard mean differences.
Results
Meta-analysis confirmed synaptic loss in selected brain regions is an early event in AD pathogenesis. The second meta-analysis of 57 synaptic markers revealed that pre-synaptic makers were affected more than postsynaptic markers.
Discussion
The present meta-analysis study showed a consistent synaptic loss across brain regions and that molecular machinery including endosomal pathways, vesicular assembly mechanisms, glutamate receptors and axonal transport are often affected.
Keywords: Alzheimer's disease, endosomal/lysomal pathway, meta-analysis, synapse markers, synapse number
1.0 Introduction
Synaptic damage has been extensively studied in AD (reviewed by [1]) since in this neurodegenerative disorder the loss of synapses is the best correlate to the cognitive deficits [2, 3]. Moreover, Aβ oligomers appear to be formed and transported at the synapses and interfere with glutamate receptors [4, 5] and synaptic functioning by interactions with pre- and post-synaptic receptors such as EphA [6], EphB2 [7], PrPc [8], mGluR5 [9], NMDA-R [10], frizzled, insulin-R and NGF-R among others [11].
The loss of synapses in AD and other neurodegenerative disorders is most likely part of a spectrum of alterations and pathogenic molecular cascades which begins with alterations in the synaptic vesicle machinery and glutamate receptors, progressing to mitochondrial dysfunction, reduced axonal flow and loss of neurotrophic support [12]. Together, these alterations might manifest at early stages as synaptic dysfunction that could be reversible, however as the process advances and alterations become irreversible, damage to synapses and spines might occur resulting eventually in synaptic and neuronal loss.
In the very early stages of AD, clinically manifested as amnestic mild cognitive impairment (aMCI) [13], there is sprouting and expansion of pre-synaptic terminals, probably as a compensatory mechanism, that is followed by a 15-25% loss of synapses in the frontal cortex and limbic system [14, 15]. Specifically, a significant reduction in synapse numbers in the CA1 region of the hippocampus and the inferior temporal cortex has been demonstrated by electron microscopy [16, 17]. Moreover, recent studies found a decreased in the dendritic proteins PSD-95 and drebrin in the hippocampus and superior temporal cortex [18-20] while synaptophysin was relatively preserved in these regions but reduced in the dentate gyrus and frontal cortex [15]. In more advanced forms of AD there is a more severe loss of synapses in the neocortex and limbic system varying from 20-40%, depending on the methods to estimate synaptic alterations [15, 21-26] and reviewed by [1].
Over the past 30 years there have been over 400 publications focusing on analyzing synapses and synaptic marker loss in post-mortem tissues from patients with AD and control subjects. To provide a systematic overview of synapse loss and the loss of synaptic markers in AD, twenty-two publications provided data on synapse numbers and 83 publications provided data on synaptic marker levels suitable for meta-analyses. The advantage of employing meta-analysis is that it offers a way to compare a variety of parameters of synaptic pathology with each other without requiring those parameters to use the same scales or units of measurements. To facilitate such comparisons, a database was built by calculating the standard mean difference (SMD) using the reported means and standard deviations for each measured parameter in each study. The present meta-analysis study showed a consistent synaptic loss across brain regions and that the molecular machinery involved endosomal pathways, vesicular assembly mechanisms, glutamate receptors and axonal transport are often affected.
2.0 Methods
2.1. Search strategy and selection criteria
Literature published from 1980 to February, 15th 2015 was systematically screened in the Cochrane Central Register of Controlled Trials, Medline, and Embase electronic databases according to PRISMA guidelines [27] using the following search terms in the title, abstract, or descriptors:
The search resulted in 15217 results that were imported into Endnote. Duplicate references 6268 were automatically removed, followed by manual examination, which retrieved another 658 duplicate references (Fig 1). Conference abstracts (1628) and non-English publications (272) were also excluded from the database (Fig 1). The title and abstract of the remaining 6391 publications were evaluated according to predefined inclusion (AD population; synaptic marker levels, synapse and/or dendritic spine counts) exclusion (non-AD population, non-human data, review/opinion articles) criteria.
Figure 1. Breakdown of publication selection.
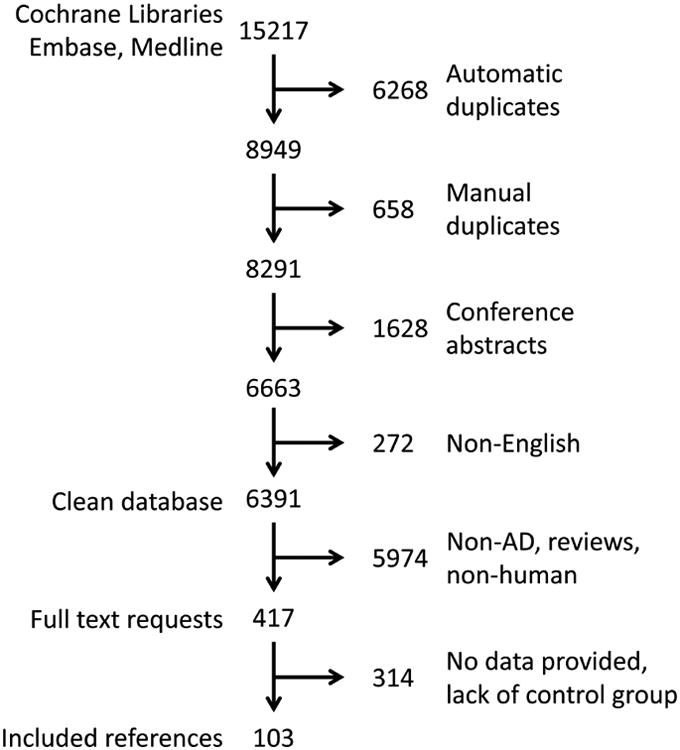
Schematic illustrating the sub-selection of manuscripts for meta-analysis.
We retrieved 417 publications reporting synapse counts or levels of synaptic proteins in patients with AD and cognitively intact elderly, even if not explicitly mentioned in the abstract. The full-text of these publications were analyzed according to the following inclusion criteria: contained AD patient population according to the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer's Disease and Related Disorders Association criteria [28] and/or the Diagnostic and Statistical Manual for Mental Disorders [29] criteria for AD and mentioned the use of a cognitively intact elderly control group, mean and standard deviation or standard error for synapse counts and/or synaptic proteins levels, number of AD patients and controls, and mean age or age range of AD and control groups. The following publications were excluded: those reporting on gene expression, mRNA expression and receptor binding studies (Fig 1).
2.2. Data collection
Out of 417 publications, 103 publications met all inclusion criteria (Fig 1); 20 publications reported counts of synapses [3, 17, 23, 30-46], 81 publications reported synaptic protein levels [2, 19-21, 47-123], and 2 publications reported both [124, 125]. Collectively, the 83 publications [2, 19-21, 47-125] reported 67 different synapse-related proteins in 17 different brain areas. Since, not every possible combination of a synapse protein and brain area has been studied or reported usable data to allow inclusion to the meta-analyses, approximately 35% of these possible combinations were available and provided data suitable for meta-analyses.
Data from the identified publications were extracted on synapse counts and/or synapse protein levels, number of subjects and average age of the AD and control groups. Standard Mean Differences (SMD) were calculated based on the difference between the control and the AD groups took into account the variation within the groups and the number of subjects per group. For publications where more than one measurement was performed, this resulted in more than one SMD, e.g when synaptophysin was measured in hippocampus, temporal cortex and entorhinal cortex three different SMDs were calculated.
2.3. Statistical analysis
All reported comparisons of synapse counts and synapse protein levels in AD patients and controls were integrated and summarized into a final result per brain area-synaptic protein combination, using meta-analysis (regression) methods [126], according to the PRISMA statement [27]. For meta-analysis, a minimum of four publications was required [127, 128]. Comparison across studies did not require conversion to the same unit since our analysis was based on the difference between groups, i.e. not on the absolute value. These data were analyzed using the random-effects meta-analysis model [126] fitted by restricted maximum likelihood using the program metareg from Stata (StataCorp. 2001. Statistical Software: Release 12.1. College Station, TX, USA). As synapse numbers and synaptic proteins were also affected by aging, meta-analyses were conducted with and without a correction by meta-regression for differences in mean age between AD patients and controls.
2.4. Analysis of bias
According to the PRISMA statement [27], the quality of a systematic review depends on the quality of the individual publications and the absence of bias for their inclusion. The quality of the studies was assessed by several inclusion and exclusion criteria (listed in 2.1. Search strategy and selection criteria). Furthermore, results of the meta-analyses were statistically analyzed for possible bias since meta-analyses that are based on small studies reporting larger (smaller) effects may tend to overestimate (underestimate) the actual outcome. Funnel plots, which plot the standard error against the reported mean difference for each publication, can indicate the overestimation or underestimation of the actual difference occurring in the meta-analysis. Therefore, we used Egger's test as implemented in the Stata program meta-bias [129] to test the association between standard error and effect size in the funnel plot. For this analysis, a minimum of eight publications is generally recognized to be required [127, 128].
3.0 Results
Out of 103 references that met all inclusion criterion, 22 references were used to evaluate the extent to which changes in synapse numbers occurred in different brain regions that are affected in AD. A meta analysis of the number of synapses was performed in the hippocampus, frontal cortex, and in the combined regions of the cingulate gyrus, entorhinal cortext, and temporal cortex (C,E,T). Data analysis of patients with AD revealed consistently lower synapse numbers in the hippocampus, the frontal cortex and in the C,E,T (Figure 2) compared to the control group. Synapse numbers were most affected in the hippocampus (SMD -2.12) followed by the C,E,T (SMD -2.55) and the frontal cortex (SMD -1.31) (Figure 2), utilizing the SMD method.
Figure 2. Meta-analyses of synapse numbers in the hippocampus, frontal cortex and C,E,T.
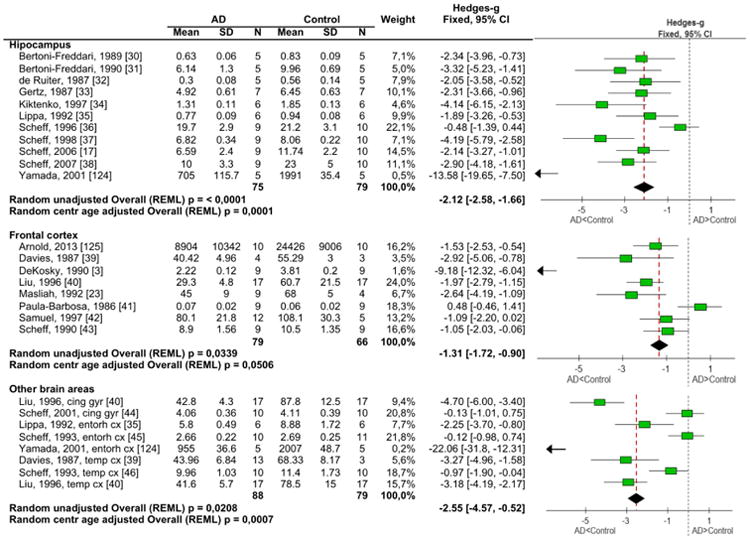
Information extracted from the manuscripts for the meta-analysis of synapse numbers in the different brain regions along with the forest plot of the standard mean differences.
Since synapse numbers were reduced in the hippocampus, C,E,T, and frontal cortex, we performed a second meta-analysis of the effect of AD on 67 pre-and post-synaptic markers to determine if specific molecular pathways in the synapse were selectively affected in AD (Table 1). The most widely-analyzed synaptic marker in the brains of AD patients is the synaptic vesicle protein synaptophysin [1, 12]; however, several other synaptic proteins have been shown to be altered in the brains of patients with AD including: synaptobrevin [19, 72, 77], SNAP 25 [19, 21, 77, 108], synaptotagmin [20, 55, 63, 72, 77, 98], syntaxin [19, 21, 67, 77], Rab3a [55, 72, 77, 104], synapsin I [19, 77, 109-112], and postsynaptic proteins PSD-95 [19, 64, 65, 67], Homer and IRSp53 [89]. The 83 publications reported at least one synaptic marker level and combined provided information on 67 different markers (Table 1). Combining results together in one comparison shows on which markers and brain areas research has focused and which markers and brain areas are underrepresented in the overview (Table 2). Employing the SMD allowed us to pool 67 different synaptic markers into a single overall database for comparison [19-21, 52, 55, 63-65, 67, 72, 77, 78, 80, 85, 86, 88-123]. Irrespective of the brain area these synaptic markers can be divided into 28 pre-synaptic markers in 10 functional categories, 30 postsynaptic markers in 8 functional categories, and 9 markers in 6 functional categories without specific pre- or postsynaptic localization (Table 2).
Table 1. List of individual synaptic markers obtain from the included publications.
| Function | Presynaptic | Postsynaptic | Pre and postsynaptic |
|---|---|---|---|
| Adhesion | Catenin beta | ||
| N-cadherin | |||
| NCAM | |||
|
| |||
| Calcium buffer | Parvalbumin | Calbindin | |
| Calretinin | |||
|
| |||
| Calcium sensor | Synaptotagmin | ||
|
| |||
| Calmodulin-binding protein | Neurogranin | ||
|
| |||
| Cytoskeleton | Septin 5 | Drebrin | Actin |
| Septin 7 | Drebrin/actin | ||
| IRSp53 | |||
| MAP2 | |||
| SAPAP 1/GKAP | |||
| Synaptopodin | |||
|
| |||
| Endocytosis | AP180 | ||
| Dynamin 1 | |||
|
| |||
| ‘Growth’ or ‘plasticity’ protein | GAP43 | ||
|
| |||
| Neuroendocrine secretory proteins | Chromogranin A | ||
| Secretogranin 2 | |||
|
| |||
| Neurotransmitter synthesis | ChAT | ||
|
| |||
| Protein phosphatase | Spinophilin | Calcineurin | |
|
| |||
| Ras GTPase-activating protein SynGAP | synGAP | ||
|
| |||
| Receptor | G-0a | trkA | |
| G-b | |||
| GBR1 GABAB receptor R1 | |||
| G-ia | |||
| G-ia.1 | |||
| G-protein a q/11 | |||
| G-protein b common | |||
| G-sa | |||
| G-sa_s | |||
| Muscarinic M1 | |||
| Muscarinic M4 | |||
| NR1 | |||
| NR2A | |||
| NR2B | |||
|
| |||
| Redox proteins | Thioredoxin | ||
|
| |||
| Signalling | CaMKIIa | ||
| CaMKIIb | |||
| pCaMKII | |||
| PSD95 | |||
| TotalCaMKII | |||
|
| |||
| Small GTPase | Rab3a | ||
| Rab5 | |||
| Rab7 | |||
|
| |||
| SNARE | Complexin 1 | ||
| Complexin 2 | |||
| SNAP25 | |||
| Synaptobrevin | |||
| Synaptobrevin 2 | |||
| Syntaxin | |||
| Syntaxin 1 | |||
| Syntaxin 1A | |||
| Syntaxin 1B | |||
| VAMP2 | |||
| VAMP2/3 | |||
|
| |||
| Tethering | Synapsin-1 | ||
|
| |||
| Transporter | ZnT1 | ||
|
| |||
| Vesicular | SV2 | ||
| Synaptophysin | |||
| VGLUT1 | |||
| VGLUT2 | |||
Table 2. Detailed summary of the standard mean differences of the meta-analysis for pre- and postsynaptic markers by brain region.
| Hippocampus | Frontal cortex | C,E,T | Cingulate gyrus | Entorhinal cortex | Temporal cortex | Amygdala | Basal forebrain | Basal ganglia | Cerebellum | Cortex | Insular | Mesencephalon | Motor cortex | Neocortex | Occipital cortex | Parietal cortex | Thalamus | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Presynaptic | Vesicle related proteins [2, 20, 21, 47-87, 124, 125] | -1.56 | -1.49 | -2.06 | -0.59 | -3.45 | -1.51 | -0.03 | 0.85 | -5.58 | -0.4 | -2.65 | -0.87 | -2.06 | 0.28 | ||||
| Cell adhesion [78, 88, 89] | 0.14 | -2.79 | -1.62 | -1.62 | -4.15 | 0.54 | |||||||||||||
| Calcium buffer [90-97] | -3.74 | -5.11 | -1.84 | -1.84 | -0.43 | ||||||||||||||
| Calcium sensor [20, 55, 63, 72, 77, 98] | -1.07 | -0.46 | -1.45 | -1.19 | -1.7 | -0.12 | -0.26 | 0.08 | |||||||||||
| Cytoskeleton [21, 89, 99] | 1.89 | -0.95 | -0.33 | -0.33 | -0.64 | 0.59 | |||||||||||||
| Endocytosis [52, 78] | -1.2 | -0.44 | -1.39 | -2.2 | -0.74 | -0.33 | |||||||||||||
| Neuroendocrine secretory proteins [63, 80] | -0.28 | 0.22 | 0.22 | ||||||||||||||||
| Neurotransmitter synthesis [97] | -7.27 | ||||||||||||||||||
| Protein phosphatase [100] | 0.5 | -0.24 | |||||||||||||||||
| Receptor [101, 102] | -1.59 | -1.53 | -1.63 | -1.42 | -8.62 | -0.48 | -1.93 | ||||||||||||
| Redox proteins [103] | -1.23 | -0.85 | -1.16 | ||||||||||||||||
| Small GTPase [55, 72, 77, 104-106] | 0.01 | 0.18 | -1.28 | -1.28 | 1.27 | 0.15 | -0.01 | 0.09 | -1.22 | ||||||||||
| SNARE [19, 21, 67, 72, 77, 78, 85, 107, 108] | -0.15 | -1.31 | -1.18 | -0.79 | -1.56 | -0.48 | -0.71 | -0.54 | -0.88 | ||||||||||
| Tethering [19, 77, 109-112] | -0.87 | -0.79 | 0.15 | 0.49 | -0.14 | 0.11 | 0.51 | -0.04 | 0.21 | ||||||||||
| Overall presynaptic markers | -1.21 | -1.35 | -1.62 | -0.58 | -2.47 | -1.34 | -1.16 | -1.46 | 0.02 | 0.12 | -3.32 | -0.71 | -0.04 | -0.4 | -2.65 | -0.55 | -1.57 | 0.28 | |
| postsynaptic | Cell adhesion [78, 88, 89] | 0.14 | -2.79 | -1.62 | -1.62 | -4.15 | 0.54 | ||||||||||||
| Calcium buffer [91, 93-97] | -5.11 | -2.21 | -2.21 | -0.43 | |||||||||||||||
| Calmodulin-binding protein [55, 72] | -0.62 | -0.36 | -0.37 | ||||||||||||||||
| Cytoskeleton [20, 54, 64, 65, 72, 89, 99] | -1.02 | -0.86 | -1.74 | -1.99 | -1.34 | -1.77 | -0.9 | -2.42 | -2.44 | 0.03 | |||||||||
| Growth factor related [55, 72, 113-115] | -1.05 | -0.42 | 0.43 | -0.27 | |||||||||||||||
| Protein phosphatase [64, 65, 100, 116] | -1.66 | -1.08 | -0.44 | -0.44 | 0.5 | -0.24 | |||||||||||||
| Ras GTPase-activating protein SynGAP [89] | -0.35 | ||||||||||||||||||
| Receptor [101, 102, 117-121] | -0.2 | 0.18 | -1.88 | -1.63 | -2.83 | -1.51 | -8.62 | -1.15 | -0.84 | -0.83 | |||||||||
| Redox proteins [103] | -1.23 | -0.85 | -1.16 | ||||||||||||||||
| Signalling [19, 64, 65, 67, 86, 89, 122] | 0.13 | 0.61 | 0.22 | 2.74 | -0.72 | -1.21 | -1.55 | -1.08 | |||||||||||
| Transporter [123] | 4.04 | -3.29 | 3.23 | 3.05 | |||||||||||||||
| Overall postynaptic markers | -0.33 | -1.06 | -1.54 | -0.8 | -1.25 | -1.76 | 1.04 | -3.16 | -1.15 | -0.22 | -2.14 | -1.55 | -0.79 | -0.36 | |||||
| Overall synaptic markers | -1.04 | -1.12 | -1.56 | -0.55 | -2.2 | -1.4 | 3.23 | -1.46 | -0.13 | -0.05 | -2.21 | -0.71 | -0.04 | -0.04 | -2.38 | -0.65 | -1.21 | 0.28 | |
Following the combination of the 67 synaptic markers from all brain regions into one database, we used the same regional division of the brain into hippocampus, frontal cortex and C,E,T. As a result, we evaluated 57 different synaptic markers retrieved from a selection of 70 publications which reported results for the brain regions of interest: hippocampus, frontal cortex and C,E,T (Table 3). These markers can be divided into 24 pre-synaptic markers in 9 functional categories, 25 postsynaptic markers in 7 functional categories, and 7 markers in 5 functional categories without specific pre- or postsynaptic localization (Table 3). Irrespective of the synaptic markers, the hippocampus and the frontal cortex showed equal reduction of synaptic markers with SMDs of -1.04 and -1.12, respectively (Table 3). The C,E,T were affected slightly more with a SMD of -1.56. In the three evaluated brain areas, pre-synaptic makers were affected more than postsynaptic markers; however, this difference was stronger in the hippocampus (pre SMD -1.21 vs post SMD -0.33) than in the frontal cortex (pre SMD -1.35 vs post SMD -1.06) and the C,E,T (pre SMD -1.62 vs post SMD -1.54).
Table 3. further summarization of the standard mean differences for pre- and postsynaptic markers in different brain areas.
| Hippocampus | Frontal cortex | C,E,T | ||
|---|---|---|---|---|
| presynaptic | Calcium regulation [90, 92-96] | -3.74 | -5.11 | -1.84 |
| Cytoskeleton [21, 99] | 1.89 | -0.95 | -0.33 | |
| Vesicular organization [2, 19-21, 47-81, 98, 104, 105, 108-112, 124, 125] | -1.03 | -1.15 | -1.61 | |
| Grand total presynaptic | -1.21 | -1.35 | -1.62 | |
| Cell adhesion [78, 88] | 0.14 | -2.79 | -1.62 | |
| postsynaptic | Calcium regulation [55, 72, 93-96, 122] | -0.62 | -2.39 | -1.67 |
| Cytoskeleton [20, 54, 64, 65, 72, 99, 116] | -1.45 | -0.92 | -1.55 | |
| Intracellular signaling [19, 55, 64, 65, 67, 72, 113, 115] | -1.05 | -0.08 | 1.01 | |
| Neurotransmission [117-121] | -0.2 | 0.63 | -1.96 | |
| Grand total postsynaptic | -0.33 | -1.06 | -1.54 | |
| Overall effect | -1.04 | -1.12 | -1.56 | |
Summarizing the data further showed that some aspects of synaptic organization were affected to a similar extent across brain regions whereas other aspects of synapse function were affected differently (Figure 3). More specifically, calcium homeostasis was negatively affected both pre- and post-synaptically in all the brain regions. Vesicular organization was decreased in the hippocampus (SMD -1.03), frontal cortex (SMD –1.15) and strongest in C,E,T (SMD -1.61). Intracellular signaling was hardly affected in the frontal cortex (SMD -0.08), whereas negatively affected in the hippocampus (SMD -1.05) and C,E,T (SMD -1.96) (Figure 3 and Table 3). Similarly, postsynaptic cytoskeleton organization was decreased in all brain areas (hippocampus -1.45, frontal cortex -0.92 and C,E,T -1.55) whereas pre-synaptic cytoskeleton organization showed a reduced SMD of -0.95 in the frontal cortex and minor changes in the C,E,T (SMD of -0.33), while in the hippocampus there was an increased SMD of 1.89 (Fig 3). Another difference between the hippocampus and the other two brain regions, the frontal cortex, and the C,E,T was the lack of changes in cell adhesion markers (SMD 0.14) in the hippocampus, compared to the decreases in the frontal cortex (SMD -2.79) and the C,E,T (SMD -1.62) (Fig 3).
Figure 3. Pre- and postsynaptic marker changes in different brain areas.
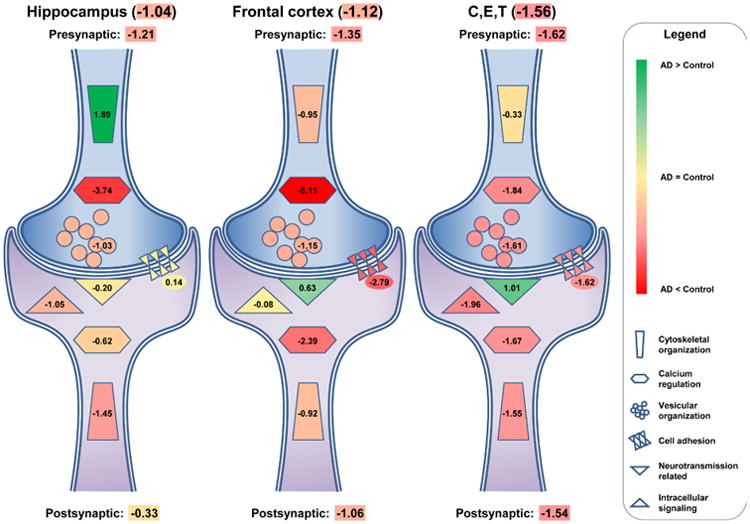
Schematic representation of the standard mean differences (SMD) for pre-and postsynaptic markers in the hippocampus, the frontal cortex, C,E,T (cingulate gyrus, entorhinal cortex and temporal cortex) and the remaining. Presynaptic markers are more affected by AD than the postsynaptic markers in all areas observed. These differences vary by brain area with the hippocampus showing the greatest difference and the C,E,T showing the smallest difference. SMD's are listed in each summarizing structure, for the overall pre- and postsynaptic change and for the overall change per brain area. Green-to-red color change depicts an increase or decrease of synaptic markers in comparison to healthy controls, where more green indicates stronger increase and more red stronger decrease.
4.0 Discussion
The present study reviewed 417 references on synaptic pathology in AD and performed meta-analysis for synapse number (22 publications) and synaptic proteins in a subset of these references (83 publications). The benefits of using meta-analysis are clearly illustrated by the potential of a single database to bring together publications on synaptic pathology, which can grow when data from new publications and existing data (from publications not presenting means plus standard deviations) are added. An additional benefit of this approach is that it allows comparison of the collective results of 83 publications with proteomics studies. The current results are consistent with recent proteomics studies in synaptosomal preparations indicating that proteins such as Rabs, synaptotagmin, annexins, heat shock proteins (HSPs), glutathione (GSH) and others that are involved in regulating energy and calcium metabolism and are dysregulated in AD, such as signal transduction, vesicle transport and antioxidant activity [89, 130, 131].
Interestingly, more recent proteomics studies with PSD preparations from AD patients have shown that brain-specific angiogenesis inhibitor 1-associated protein 2 (IRSp53) was altered. IRSp53 belongs to a family of proteins harboring IRSp53–MIM domain that is associated with both actin and lipids [89]. This cluster of proteins regulates the spine cytoskeleton and membrane trafficking. IRSp53 interacts with PSD scaffold proteins (e.g. PSD-95 and chapsyn-110/PSD-95 and Rabs to modulate dendritic structure [89]. Thus alterations observed in the brains of patients with AD might reflect defects in dendritic spine motility and disorganization of the post-synaptic scaffolds [19].
Although the earliest and most significant alterations in postmortem studies in AD and in APP tg models the earliest and most significant alterations appear to be in proteins located in the pre-synaptic site, it is likely that both the pre- and post-synaptic compartments are affected since the soluble synaptotoxic hydrophobic Aβ oligomers diffuse rapidly between the axonal and dendritic partition [12, 132]. Together these studies suggests that at early stages of AD soluble Aβ oligomers that diffuse from cell to cell might exert their toxic effects by locally affecting in the pre-synaptic site the SNARE machinery components, Rabs, calcium sensors and anti-oxidant molecules and in the post-synaptic site glutamate receptors, PSD scaffold molecules and mitochondria. Moreover these oligomers might engage synaptic receptors that trigger neurotoxic signaling pathways (eg: Fyn, CDK5, GSK3β) that merge in Tau dependent and independent pathways [133-135].
A challenge of the current methodology is that the majority of the studies included in the meta-analysis approach consist of rather small studies. The average study population size is 10 subjects in the AD group versus 10 subjects in the healthy elderly control group. These small sample sizes carry the risk of publication bias, which is observed in the synapse count meta-analyses for hippocampus and frontal cortex. To overcome this problem, future research should aim for larger study populations, which will improve the intrinsic power of the individual study and also the overall power of meta-analysis approaches.
In conclusion the present meta-analysis study showed a consistent synaptic loss across brain regions and that the molecular machinery involved endosomal pathways, vesicular assembly mechanisms, glutamate receptors and axonal transport are often affected. Based on these findings, future research focusing on a set of crucial experiments that are designed to methodically test the hypothesis that synapse loss is due to soluble Aβ oligomers exerting their toxic effects by locally affecting the molecular machinery in the pre-synaptic site including the SNARE machinery components, Rabs, calcium sensors and anti-oxidant molecules and in the post-synaptic site glutamate receptors, PSD scaffold molecules and mitochondria would greatly advance our scientific understanding of synapse loss in AD.
Research in Context.
Systematic review: The authors reviewed the literature on the molecular underpinnings and progression of synapse loss in Alzheimer's disease and found that while several studies have been published but to date no meta-analysis that is inclusive of all the publications has been considered.
Interpretation: Our findings from the meta-analysis of close to 100 of the most important publications showed a consistent synaptic loss across brain regions and that the molecular machinery including endosomal pathways, vesicular assembly mechanisms, glutamate receptors and axonal transport are often affected.
Future directions: The manuscript synthesized data from over 100 papers on synaptic markers and synapse loss; however due to the small average sample sizes for both the control and Alzheimer's disease groups (N=10), future research should aim for larger study populations, which will improve the intrinsic power of the individual study and also the overall power of meta-analysis approaches.
Acknowledgments
Funding Sources: Funding was provided by NIH grants AG5131 and AG18440 (EM).
Footnotes
Conflicts: None of the authors report a conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Scheff SW, Neltner JH, Nelson PT. Is synaptic loss a unique hallmark of Alzheimer's disease? Biochemical pharmacology. 2014;88:517–28. doi: 10.1016/j.bcp.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 3.DeKosky S, Scheff S. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–64. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 4.Mota SI, Ferreira IL, Rego AC. Dysfunctional synapse in Alzheimer's disease -A focus on NMDA receptors. Neuropharmacology. 2014;76 Pt A:16–26. doi: 10.1016/j.neuropharm.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 5.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, et al. APP Processing and Synaptic Function. Neuron. 2003;37:925–37. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 6.Fu AK, Hung KW, Huang H, Gu S, Shen Y, Cheng EY, et al. Blockade of EphA4 signaling ameliorates hippocampal synaptic dysfunctions in mouse models of Alzheimer's disease. Proc Natl Acad Sci U S A. 2014;111:9959–64. doi: 10.1073/pnas.1405803111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cisse M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, et al. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature. 2011;469:47–52. doi: 10.1038/nature09635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, et al. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci. 2012;15:1227–35. doi: 10.1038/nn.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, et al. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66:739–54. doi: 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, et al. Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A. 2013;110:E2518–27. doi: 10.1073/pnas.1306832110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Overk CR, Masliah E. Toward a unified therapeutics approach targeting putative amyloid-beta oligomer receptors. Proc Natl Acad Sci U S A. 2014;111:13680–1. doi: 10.1073/pnas.1414554111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Overk CR, Masliah E. Pathogenesis of synaptic degeneration in Alzheimer's disease and Lewy body disease. Biochemical pharmacology. 2014;88:508–16. doi: 10.1016/j.bcp.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petersen RC. Early diagnosis of Alzheimer's disease: is MCI too late? Current Alzheimer research. 2009;6:324–30. doi: 10.2174/156720509788929237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheff SW, Price DA, Schmitt FA, Scheff MA, Mufson EJ. Synaptic loss in the inferior temporal gyrus in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. 2011;24:547–57. doi: 10.3233/JAD-2011-101782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R. Synaptic and neuritic alterations during the progression of Alzheimer's disease. NeurosciLett. 1994;174:67–72. doi: 10.1016/0304-3940(94)90121-x. [DOI] [PubMed] [Google Scholar]
- 16.Scheff SW, Price DA, Schmitt FA, Roberts KN, Ikonomovic MD, Mufson EJ. Synapse stability in the precuneus early in the progression of Alzheimer's disease. J Alzheimers Dis. 2013;35:599–609. doi: 10.3233/JAD-122353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–84. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 18.Sultana R, Banks WA, Butterfield DA. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer's disease. J Neurosci Res. 2010;88:469–77. doi: 10.1002/jnr.22227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pham E, Crews L, Ubhi K, Hansen L, Adame A, Cartier A, et al. Progressive accumulation of amyloid-beta oligomers in Alzheimer's disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 2010;277:3051–67. doi: 10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Counts SE, He B, Nadeem M, Wuu J, Scheff SW, Mufson EJ. Hippocampal drebrin loss in mild cognitive impairment. Neuro-degenerative diseases. 2012;10:216–9. doi: 10.1159/000333122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wakabayashi K, Honer WG, Masliah E. Synapse alterations in the hippocampal-entorhinal formation in Alzheimer's disease with and without Lewy body disease. Brain Res. 1994;667:24–32. doi: 10.1016/0006-8993(94)91709-4. [DOI] [PubMed] [Google Scholar]
- 22.Masliah E, Terry R, Alford M, DeTeresa R. Quantitative immunohistochemistry of synaptophysin in human neocortex: an alternative method to estimate density of presynaptic terminals in paraffin sections. JHistochemCytochem. 1990;38:837–44. doi: 10.1177/38.6.2110586. [DOI] [PubMed] [Google Scholar]
- 23.Masliah E, Ellisman M, Carragher B, Mallory M, Young S, Hansen L, et al. Three-dimensional analysis of the relationship between synaptic pathology and neuropil threads in Alzheimer disease. JNeuropatholExpNeurol. 1992;51:404–14. doi: 10.1097/00005072-199207000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Masliah E, Terry R, DeTeresa R, Alford M, Hansen L. Morphometric quantification of a synaptic marker in neocortex of Alzheimer and Pick disease. JNeuropatholExpNeurol. 1989;48:333. [Google Scholar]
- 25.Masliah E, Mallory M, Hansen L, DeTeresa R, Terry R. Quantitative synaptic alterations in the human neocortex during normal aging. Neurology. 1993;43:192–7. doi: 10.1212/wnl.43.1_part_1.192. [DOI] [PubMed] [Google Scholar]
- 26.Mitew S, Kirkcaldie MT, Dickson TC, Vickers JC. Altered synapses and gliotransmission in Alzheimer's disease and AD model mice. Neurobiol Aging. 2013;34:2341–51. doi: 10.1016/j.neurobiolaging.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 27.Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. 2009;339:b2535. doi: 10.1136/bmj.b2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan E. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology. 1984;34:939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 29.Association AP. Diagnostic and statistical manual of mental disorders. 3rd. Washington DC: American Psychological Association; 1980. [Google Scholar]
- 30.Bertoni-Freddari C, Fattoretti P, Meier-Ruge W, Ulrich J. Computer-assisted morphometry of synaptic plasticity during aging and dementia. Pathology, research and practice. 1989;185:799–802. doi: 10.1016/S0344-0338(89)80243-2. [DOI] [PubMed] [Google Scholar]
- 31.Bertoni-Freddari C, Fattoretti P, Casoli T, Meier-Ruge W, Ulrich J. Morphological adaptive response of the synaptic junctional zones in the human dentate gyrus during aging and Alzheimer's disease. Brain Res. 1990;517:69–75. doi: 10.1016/0006-8993(90)91009-6. [DOI] [PubMed] [Google Scholar]
- 32.de Ruiter JP, Uylings HB. Morphometric and dendritic analysis of fascia dentata granule cells in human aging and senile dementia. Brain Res. 1987;402:217–29. doi: 10.1016/0006-8993(87)90028-x. [DOI] [PubMed] [Google Scholar]
- 33.Gertz HJ, Cervos-Navarro J, Ewald V. The septo-hippocampal pathway in patients suffering from senile dementia of Alzheimer's type. Evidence for neuronal plasticity? Neurosci Lett. 1987;76:228–32. doi: 10.1016/0304-3940(87)90720-8. [DOI] [PubMed] [Google Scholar]
- 34.Kiktenko AI, Uranova NA, Denisov DV. Quantitative characteristics of changes in synaptic contacts in the hippocampus in Alzheimer's disease. Neurosci Behav Physiol. 1997;27:681–2. doi: 10.1007/BF02461927. [DOI] [PubMed] [Google Scholar]
- 35.Lippa CF, Hamos JE, Pulaski-Salo D, DeGennaro LJ, Drachman DA. Alzheimer's disease and aging: effects on perforant pathway perikarya and synapses. Neurobiol Aging. 1992;13:405–11. doi: 10.1016/0197-4580(92)90115-e. [DOI] [PubMed] [Google Scholar]
- 36.Scheff SW, Sparks DL, Price DA. Quantitative assessment of synaptic density in the outer molecular layer of the hippocampal dentate gyrus in Alzheimer's disease. Dementia. 1996;7:226–32. doi: 10.1159/000106884. [DOI] [PubMed] [Google Scholar]
- 37.Scheff SW, Price DA. Synaptic density in the inner molecular layer of the hippocampal dentate gyrus in Alzheimer disease. J Neuropathol Exp Neurol. 1998;57:1146–53. doi: 10.1097/00005072-199812000-00006. [DOI] [PubMed] [Google Scholar]
- 38.Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–8. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- 39.Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer's disease. J Neurol Sci. 1987;78:151–64. doi: 10.1016/0022-510x(87)90057-8. [DOI] [PubMed] [Google Scholar]
- 40.Liu X, Erikson C, Brun A. Cortical synaptic changes and gliosis in normal aging, Alzheimer's disease and frontal lobe degeneration. Dementia (Basel, Switzerland) 1996;7:128–34. doi: 10.1159/000106867. [DOI] [PubMed] [Google Scholar]
- 41.Paula-Barbosa MM, Saraiva A, Tavares MA, Borges MM, Verwer RW. Alzheimer's disease: maintenance of neuronal and synaptic densities in frontal cortical layers II and III. Acta Neurol Scand. 1986;74:404–8. doi: 10.1111/j.1600-0404.1986.tb03533.x. [DOI] [PubMed] [Google Scholar]
- 42.Samuel W, Alford M, Hofstetter CR, Hansen L. Dementia with Lewy bodies versus pure Alzheimer disease: differences in cognition, neuropathology, cholinergic dysfunction, and synapse density. J Neuropathol Exp Neurol. 1997;56:499–508. doi: 10.1097/00005072-199705000-00006. [DOI] [PubMed] [Google Scholar]
- 43.Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer's disease. Neurobiol Aging. 1990;11:29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- 44.Scheff SW, Price DA. Alzheimer's disease-related synapse loss in the cingulate cortex. J Alzheimers Dis. 2001;3:495–505. doi: 10.3233/jad-2001-3509. [DOI] [PubMed] [Google Scholar]
- 45.Scheff SW, Sparks L, Price DA. Quantitative assessment of synaptic density in the entorhinal cortex in Alzheimer's disease. Ann Neurol. 1993;34:356–61. doi: 10.1002/ana.410340309. [DOI] [PubMed] [Google Scholar]
- 46.Scheff SW, Price DA. Synapse loss in the temporal lobe in Alzheimer's disease. Ann Neurol. 1993;33:190–9. doi: 10.1002/ana.410330209. [DOI] [PubMed] [Google Scholar]
- 47.Alford MF, Masliah E, Hansen LA, Terry RD. A simple dot-immunobinding assay for quantification of synaptophysin-like immunoreactivity in human brain. J Histochem Cytochem. 1994;42:283–7. doi: 10.1177/42.2.8288869. [DOI] [PubMed] [Google Scholar]
- 48.Bigio EH, Vono MB, Satumtira S, Adamson J, Sontag E, Hynan LS, et al. Cortical synapse loss in progressive supranuclear palsy. J Neuropath Exp Neurol. 2001;60:403–10. doi: 10.1093/jnen/60.5.403. [DOI] [PubMed] [Google Scholar]
- 49.Bigio EH, Reisch JS, White CL, 3rd, Satumtira S, Sontag E, Bonte FJ. Synapse loss may be a minor contributor to decreased regional cerebral blood flow in Alzheimer disease. Dementia and geriatric cognitive disorders. 2003;15:72–8. doi: 10.1159/000067970. [DOI] [PubMed] [Google Scholar]
- 50.Brown DF, Risser RC, Bigio EH, Tripp P, Stiegler A, Welch E, et al. Neocortical synapse density and Braak stage in the Lewy body variant of Alzheimer disease: a comparison with classic Alzheimer disease and normal aging. J Neuropath Exp Neurol. 1998;57:955–60. doi: 10.1097/00005072-199810000-00007. [DOI] [PubMed] [Google Scholar]
- 51.Brun A, Liu X, Erikson C. Synapse loss and gliosis in the molecular layer of the cerebral cortex in Alzheimer's disease and in frontal lobe degeneration. Neurodegeneration : a journal for neurodegenerative disorders, neuroprotection, and neuroregeneration. 1995;4:171–7. doi: 10.1006/neur.1995.0021. [DOI] [PubMed] [Google Scholar]
- 52.Cao Y, Xiao Y, Ravid R, Guan ZZ. Changed clathrin regulatory proteins in the brains of Alzheimer's disease patients and animal models. J Alzheimers Dis. 2010;22:329–42. doi: 10.3233/JAD-2010-100162. [DOI] [PubMed] [Google Scholar]
- 53.Corey-Bloom J, Tiraboschi P, Hansen LA, Alford M, Schoos B, Sabbagh MN, et al. E4 allele dosage does not predict cholinergic activity or synapse loss in Alzheimer's disease. Neurology. 2000;54:403–6. doi: 10.1212/wnl.54.2.403. [DOI] [PubMed] [Google Scholar]
- 54.Counts SE, Nadeem M, Lad SP, Wuu J, Mufson EJ. Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J Neuropathol Exp Neurol. 2006;65:592–601. doi: 10.1097/00005072-200606000-00007. [DOI] [PubMed] [Google Scholar]
- 55.Davidsson P, Blennow K. Neurochemical dissection of synaptic pathology in Alzheimer's disease. Int Psychogeriatr. 1998;10:11–23. doi: 10.1017/s1041610298005110. [DOI] [PubMed] [Google Scholar]
- 56.Davidsson P, Bogdanovic N, Lannfelt L, Blennow K. Reduced expression of amyloid precursor protein, presenilin-1 and rab3a in cortical brain regions in Alzheimer's disease. Dement Geriatr Cogn Disord. 2001;12:243–50. doi: 10.1159/000051266. [DOI] [PubMed] [Google Scholar]
- 57.Hansen LA, Daniel SE, Wilcock GK, Love S. Frontal cortical synaptophysin in Lewy body diseases: relation to Alzheimer's disease and dementia. J Neurol Neurosurg Psychiatry. 1998;64:653–6. doi: 10.1136/jnnp.64.5.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heffernan JM, Eastwood SL, Nagy Z, Sanders MW, McDonald B, Harrison PJ. Temporal cortex synaptophysin mRNA is reduced in Alzheimer's disease and is negatively correlated with the severity of dementia. Exp Neurol. 1998;150:235–9. doi: 10.1006/exnr.1997.6772. [DOI] [PubMed] [Google Scholar]
- 59.Heinonen O, Soininen H, Sorvari H, Kosunen O, Paljarvi L, Koivisto E, et al. Loss of synaptophysin-like immunoreactivity in the hippocampal formation is an early phenomenon in Alzheimer's disease. Neuroscience. 1995;64:375–84. doi: 10.1016/0306-4522(94)00422-2. [DOI] [PubMed] [Google Scholar]
- 60.Ho GJ, Hashimoto M, Adame A, Izu M, Alford MF, Thal LJ, et al. Altered p59Fyn kinase expression accompanies disease progression in Alzheimer's disease: implications for its functional role. Neurobiol Aging. 2005;26:625–35. doi: 10.1016/j.neurobiolaging.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 61.Kashani A, Lepicard E, Poirel O, Videau C, David JP, Fallet-Bianco C, et al. Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol Aging. 2008;29:1619–30. doi: 10.1016/j.neurobiolaging.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 62.Kirvell SL, Esiri M, Francis PT. Down-regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer's disease. J Neurochem. 2006;98:939–50. doi: 10.1111/j.1471-4159.2006.03935.x. [DOI] [PubMed] [Google Scholar]
- 63.Lassmann H, Weiler R, Fischer P, Bancher C, Jellinger K, Floor E, et al. Synaptic pathology in Alzheimer's disease: immunological data for markers of synaptic and large dense-core vesicles. Neurosci. 1992;46:1–8. doi: 10.1016/0306-4522(92)90003-k. [DOI] [PubMed] [Google Scholar]
- 64.Leuba G, Savioz A, Vernay A, Carnal B, Kraftsik R, Tardif E, et al. Differential changes in synaptic proteins in the Alzheimer frontal cortex with marked increase in PSD-95 postsynaptic protein. J Alzheimers Dis. 2008;15:139–51. doi: 10.3233/jad-2008-15112. [DOI] [PubMed] [Google Scholar]
- 65.Leuba G, Walzer C, Vernay A, Carnal B, Kraftsik R, Piotton F, et al. Postsynaptic density protein PSD-95 expression in Alzheimer's disease and okadaic acid induced neuritic retraction. Neurobiol Dis. 2008;30:408–19. doi: 10.1016/j.nbd.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 66.Lippa CF. Synaptophysin immunoreactivity in Pick's disease: comparison with Alzheimer's disease and dementia with Lewy bodies. American journal of Alzheimer's disease and other dementias. 2004;19:341–4. doi: 10.1177/153331750401900606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Love S, Siew LK, Dawbarn D, Wilcock GK, Ben-Shlomo Y, Allen SJ. Premorbid effects of APOE on synaptic proteins in human temporal neocortex. Neurobiol Aging. 2006;27:797–803. doi: 10.1016/j.neurobiolaging.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 68.Lue LF, Brachova L, Civin WH, Rogers J. Inflammation, A beta deposition, and neurofibrillary tangle formation as correlates of Alzheimer's disease neurodegeneration. J Neuropathol Exp Neurol. 1996;55:1083–8. [PubMed] [Google Scholar]
- 69.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–62. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Masliah E, Terry RD, Alford M, DeTeresa R, Hansen LA. Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer's disease. Am J Pathol. 1991;138:235–46. [PMC free article] [PubMed] [Google Scholar]
- 71.Mukaetova-Ladinska EB, Abdel-All Z, Mugica ES, Li M, Craggs LJ, Oakley AE, et al. Tau proteins in the temporal and frontal cortices in patients with vascular dementia. J Neuropath Exp Neurol. 2015;74:148–57. doi: 10.1097/NEN.0000000000000157. [DOI] [PubMed] [Google Scholar]
- 72.Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W, Jr, et al. Differential loss of synaptic proteins in Alzheimer's disease: implications for synaptic dysfunction. J Alzheimers Dis. 2005;7:103–17. doi: 10.3233/jad-2005-7203. discussion 73-80. [DOI] [PubMed] [Google Scholar]
- 73.Rei RT, Sabbagh MN, Corey-Bloom J, Tiraboschi P, Thal LJ. Nicotinic receptor losses in dementia with Lewy bodies: comparisons with Alzheimer's disease. Neurobiol Aging. 2000;21:741–6. doi: 10.1016/s0197-4580(00)00168-8. [DOI] [PubMed] [Google Scholar]
- 74.Sabbagh MN, Reid RT, Corey-Bloom J, Rao TS, Hansen LA, Alford M, et al. Correlation of nicotinic binding with neurochemical markers in Alzheimer's disease. J Neural Transm. 1998;105:709–17. doi: 10.1007/s007020050090. [DOI] [PubMed] [Google Scholar]
- 75.Saetre P, Jazin E, Emilsson L. Age-related changes in gene expression are accelerated in Alzheimer's disease. Synapse. 2011;65:971–4. doi: 10.1002/syn.20933. [DOI] [PubMed] [Google Scholar]
- 76.Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. JNeuropatholExpNeurol. 1997;56:933–44. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- 77.Sze CI, Bi H, Kleinschmidt-DeMasters BK, Filley CM, Martin LJ. Selective regional loss of exocytotic presynaptic vesicle proteins in Alzheimer's disease brains. J Neurol Sci. 2000;175:81–90. doi: 10.1016/s0022-510x(00)00285-9. [DOI] [PubMed] [Google Scholar]
- 78.Tannenberg RK, Scott HL, Tannenberg AE, Dodd PR. Selective loss of synaptic proteins in Alzheimer's disease: evidence for an increased severity with APOE varepsilon4. Neurochem Int. 2006;49:631–9. doi: 10.1016/j.neuint.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 79.Tiraboschi P, Hansen LA, Alford M, Masliah E, Thal LJ, Corey-Bloom J. The decline in synapses and cholinergic activity is asynchronous in Alzheimer's disease. Neurology. 2000;55:1278–83. doi: 10.1212/wnl.55.9.1278. [DOI] [PubMed] [Google Scholar]
- 80.Weiler R, Lassmann H, Fischer P, Jellinger K, Winkler H. A high ratio of chromogranin A to synaptin/synaptophysin is a common feature of brains in Alzheimer and Pick disease. FEBS Lett. 1990;263:337–9. doi: 10.1016/0014-5793(90)81408-g. [DOI] [PubMed] [Google Scholar]
- 81.Zhan SS, Beyreuther K, Schmitt HP. Quantitative assessment of the synaptophysin immuno-reactivity of the cortical neuropil in various neurodegenerative disorders with dementia. Dementia. 1993;4:66–74. doi: 10.1159/000107299. [DOI] [PubMed] [Google Scholar]
- 82.Zhan SS, Beyreuther K, Schmitt HP. Vascular dementia in Spatz-Lindenberg's disease (SLD): cortical synaptophysin immunoreactivity as compared with dementia of Alzheimer type and non-demented controls. Acta Neuropathol. 1993;86:259–64. doi: 10.1007/BF00304140. [DOI] [PubMed] [Google Scholar]
- 83.Zhan SS, Beyreuther K, Schmitt HP. Synaptophysin immunoreactivity of the cortical neuropil in vascular dementia of Binswanger type compared with the dementia of Alzheimer type and nondemented controls. Dementia. 1994;5:79–87. doi: 10.1159/000106701. [DOI] [PubMed] [Google Scholar]
- 84.Zhan SS, Sandbrink R, Beyreuther K, Schmitt HP. APP with Kunitz type protease inhibitor domain (KPI) correlates with neuritic plaque density but not with cortical synaptophysin immunoreactivity in Alzheimer's disease and non-demented aged subjects: a multifactorial analysis. Clin Neuropathol. 1995;14:142–9. [PubMed] [Google Scholar]
- 85.Mukaetova-Ladinska EB, Andras A, Milne J, Abdel-All Z, Borr I, Jaros E, et al. Synaptic proteins and choline acetyltransferase loss in visual cortex in dementia with Lewy bodies. J Neuropathol Exp Neurol. 2013;72:53–60. doi: 10.1097/NEN.0b013e31827c5710. [DOI] [PubMed] [Google Scholar]
- 86.Shinohara M, Fujioka S, Murray ME, Wojtas A, Baker M, Rovelet-Lecrux A, et al. Regional distribution of synaptic markers and APP correlate with distinct clinicopathological features in sporadic and familial Alzheimer's disease. Brain. 2014;137:1533–49. doi: 10.1093/brain/awu046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sokolow S, Luu SH, Nandy K, Miller CA, Vinters HV, Poon WW, et al. Preferential accumulation of amyloid-beta in presynaptic glutamatergic terminals (VGluT1 and VGluT2) in Alzheimer's disease cortex. Neurobiol Dis. 2012;45:381–7. doi: 10.1016/j.nbd.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Aisa B, Gil-Bea FJ, Solas M, Garcia-Alloza M, Chen CP, Lai MK, et al. Altered NCAM expression associated with the cholinergic system in Alzheimer's disease. J Alzheimers Dis. 2010;20:659–68. doi: 10.3233/JAD-2010-1398. [DOI] [PubMed] [Google Scholar]
- 89.Zhou J, Jones DR, Duong DM, Levey AI, Lah JJ, Peng J. Proteomic analysis of postsynaptic density in Alzheimer's disease. Clin Chim Acta. 2013;420:62–8. doi: 10.1016/j.cca.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brady DR, Mufson EJ. Parvalbumin-immunoreactive neurons in the hippocampal formation of Alzheimer's diseased brain. Neuroscience. 1997;80:1113–25. doi: 10.1016/s0306-4522(97)00068-7. [DOI] [PubMed] [Google Scholar]
- 91.Chan-Palay V, Hochli M, Savaskan E, Hungerecker G. Calbindin D-28k and monoamine oxidase A immunoreactive neurons in the nucleus basalis of Meynert in senile dementia of the Alzheimer type and Parkinson's disease. Dementia. 1993;4:1–15. doi: 10.1159/000107290. [DOI] [PubMed] [Google Scholar]
- 92.Fonseca M, Soriano E, Ferrer I, Martinez A, Tunon T. Chandelier cell axons identified by parvalbumin-immunoreactivity in the normal human temporal cortex and in Alzheimer's disease. Neuroscience. 1993;55:1107–16. doi: 10.1016/0306-4522(93)90324-9. [DOI] [PubMed] [Google Scholar]
- 93.Fonseca M, Soriano E. Calretinin-immunoreactive neurons in the normal human temporal cortex and in Alzheimer's disease. Brain Res. 1995;691:83–91. doi: 10.1016/0006-8993(95)00622-w. [DOI] [PubMed] [Google Scholar]
- 94.Greene JR, Radenahmad N, Wilcock GK, Neal JW, Pearson RC. Accumulation of calbindin in cortical pyramidal cells with ageing; a putative protective mechanism which fails in Alzheimer's disease. Neuropathol Appl Neurobiol. 2001;27:339–42. doi: 10.1046/j.0305-1846.2001.00351.x. [DOI] [PubMed] [Google Scholar]
- 95.Hof PR, Morrison JH. Neocortical neuronal subpopulations labeled by a monoclonal antibody to calbindin exhibit differential vulnerability in Alzheimer's disease. Exp Neurol. 1991;111:293–301. doi: 10.1016/0014-4886(91)90096-u. [DOI] [PubMed] [Google Scholar]
- 96.Nishiyama E, Ohwada J, Iwamoto N, Arai H. Selective loss of calbindin D28K-immunoreactive neurons in the cortical layer II in brains of Alzheimer's disease: a morphometric study. Neurosci Lett. 1993;163:223–6. doi: 10.1016/0304-3940(93)90388-2. [DOI] [PubMed] [Google Scholar]
- 97.Riascos D, de Leon D, Baker-Nigh A, Nicholas A, Yukhananov R, Bu J, et al. Age-related loss of calcium buffering and selective neuronal vulnerability in Alzheimer's disease. Acta Neuropathol. 2011;122:565–76. doi: 10.1007/s00401-011-0865-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Davidsson P, Jahn R, Bergquist J, Ekman R, Blennow K. Synaptotagmin, a synaptic vesicle protein, is present in human cerebrospinal fluid: a new biochemical marker for synaptic pathology in Alzheimer disease? Mol Chem Neuropathol. 1996;27:195–210. doi: 10.1007/BF02815094. [DOI] [PubMed] [Google Scholar]
- 99.Shim KS, Lubec G. Drebrin, a dendritic spine protein, is manifold decreased in brains of patients with Alzheimer's disease and Down syndrome. Neurosci Lett. 2002;324:209–12. doi: 10.1016/s0304-3940(02)00210-0. [DOI] [PubMed] [Google Scholar]
- 100.Billingsley ML, Ellis C, Kincaid RL, Martin J, Schmidt ML, Lee VM, et al. Calcineurin immunoreactivity in Alzheimer's disease. Exp Neurol. 1994;126:178–84. doi: 10.1006/exnr.1994.1056. [DOI] [PubMed] [Google Scholar]
- 101.Counts SE, Nadeem M, Wuu J, Ginsberg SD, Saragovi HU, Mufson EJ. Reduction of cortical TrkA but not p75(NTR) protein in early-stage Alzheimer's disease. Ann Neurol. 2004;56:520–31. doi: 10.1002/ana.20233. [DOI] [PubMed] [Google Scholar]
- 102.Mufson EJ, Ma SY, Cochran EJ, Bennett DA, Beckett LA, Jaffar S, et al. Loss of nucleus basalis neurons containing trkA immunoreactivity in individuals with mild cognitive impairment and early Alzheimer's disease. J Comp Neurol. 2000;427:19–30. doi: 10.1002/1096-9861(20001106)427:1<19::aid-cne2>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 103.Lovell MA, Xie C, Gabbita SP, Markesbery WR. Decreased thioredoxin and increased thioredoxin reductase levels in Alzheimer's disease brain. Free Radic Biol Med. 2000;28:418–27. doi: 10.1016/s0891-5849(99)00258-0. [DOI] [PubMed] [Google Scholar]
- 104.Blennow K, Bogdanovic N, Alafuzoff I, Ekman R, Davidsson P. Synaptic pathology in Alzheimer's disease: Relation to severity of dementia, but not to senile plaques, neurofibrillary tangles, or the ApoE4 allele. JNeural Transm. 1996;103:603–18. doi: 10.1007/BF01273157. [DOI] [PubMed] [Google Scholar]
- 105.Ginsberg SD, Mufson EJ, Counts SE, Wuu J, Alldred MJ, Nixon RA, et al. Regional selectivity of rab5 and rab7 protein upregulation in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. 2010;22:631–9. doi: 10.3233/JAD-2010-101080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ginsberg SD, Mufson EJ, Alldred MJ, Counts SE, Wuu J, Nixon RA, et al. Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. J Chem Neuroanat. 2011;42:102–10. doi: 10.1016/j.jchemneu.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Brinkmalm A, Brinkmalm G, Honer WG, Moreno JA, Jakobsson J, Mallucci GR, et al. Targeting synaptic pathology with a novel affinity mass spectrometry approach. Molecular & cellular proteomics : MCP. 2014;13:2584–92. doi: 10.1074/mcp.M114.040113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Clinton J, Blackman SE, Royston MC, Roberts GW. Differential synaptic loss in the cortex in Alzheimer's disease: a study using archival material. Neuroreport. 1994;5:497–500. doi: 10.1097/00001756-199401120-00032. [DOI] [PubMed] [Google Scholar]
- 109.Hamos JE, DeGennaro LJ, Drachman DA. Synaptic loss in Alzheimer's disease and other dementias. Neurology. 1989;39:355–61. doi: 10.1212/wnl.39.3.355. [DOI] [PubMed] [Google Scholar]
- 110.Parks KM, Sugar JE, Haroutunian V, Bierer L, Perl D, Wallace WC. Reduced in vitro phosphorylation of synapsin I (site 1) in Alzheimer's disease postmortem tissues. Brain Res Mol Brain Res. 1991;9:125–34. doi: 10.1016/0169-328x(91)90137-m. [DOI] [PubMed] [Google Scholar]
- 111.Perdahl E, Adolfsson R, Alafuzoff I, Albert KA, Nestler EJ, Greengard P, et al. Synapsin I (protein I) in different brain regions in senile dementia of Alzheimer type and in multi-infarct dementia. J Neural Transm. 1984;60:133–41. doi: 10.1007/BF01245030. [DOI] [PubMed] [Google Scholar]
- 112.Qin S, Hu XY, Xu H, Zhou JN. Regional alteration of synapsin I in the hippocampal formation of Alzheimer's disease patients. Acta Neuropathol. 2004;107:209–15. doi: 10.1007/s00401-003-0800-4. [DOI] [PubMed] [Google Scholar]
- 113.Bogdanovic N, Davidsson P, Volkmann I, Winblad B, Blennow K. Growth-associated protein GAP-43 in the frontal cortex and in the hippocampus in Alzheimer's disease: an immunohistochemical and quantitative study. J Neural Transm. 2000;107:463–78. doi: 10.1007/s007020070088. [DOI] [PubMed] [Google Scholar]
- 114.Cheetham JE, Martzen MR, Kazee AM, Coleman PD. Gap-43 message levels in anterior cerebellum in Alzheimer's disease. Brain Res Mol Brain Res. 1996;36:145–51. doi: 10.1016/0169-328x(95)00257-s. [DOI] [PubMed] [Google Scholar]
- 115.Coleman PD, Kazee AM, Lapham L, Eskin T, Rogers K. Reduced GAP-43 message levels are associated with increased neurofibrillary tangle density in the frontal association cortex (area 9) in Alzheimer's disease. Neurobiol Aging. 1992;13:631–9. doi: 10.1016/0197-4580(92)90085-c. [DOI] [PubMed] [Google Scholar]
- 116.Akram A, Christoffel D, Rocher AB, Bouras C, Kovari E, Perl DP, et al. Stereologic estimates of total spinophilin-immunoreactive spine number in area 9 and the CA1 field: relationship with the progression of Alzheimer's disease. Neurobiol Aging. 2008;29:1296–307. doi: 10.1016/j.neurobiolaging.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ferrari-DiLeo G, Mash DC, Flynn DD. Attenuation of muscarinic receptor-G-protein interaction in Alzheimer disease. Mol Chem Neuropathol. 1995;24:69–91. doi: 10.1007/BF03160113. [DOI] [PubMed] [Google Scholar]
- 118.Flynn DD, Ferrari-DiLeo G, Mash DC, Levey AI. Differential regulation of molecular subtypes of muscarinic receptors in Alzheimer's disease. J Neurochem. 1995;64:1888–91. doi: 10.1046/j.1471-4159.1995.64041888.x. [DOI] [PubMed] [Google Scholar]
- 119.Iwakiri M, Mizukami K, Ikonomovic MD, Ishikawa M, Hidaka S, Abrahamson EE, et al. Changes in hippocampal GABABR1 subunit expression in Alzheimer's patients: association with Braak staging. Acta Neuropathol. 2005;109:467–74. doi: 10.1007/s00401-005-0985-9. [DOI] [PubMed] [Google Scholar]
- 120.O'Neill C, Wiehager B, Fowler CJ, Ravid R, Winblad B, Cowburn RF. Regionally selective alterations in G protein subunit levels in the Alzheimer's disease brain. Brain Res. 1994;636:193–201. doi: 10.1016/0006-8993(94)91017-0. [DOI] [PubMed] [Google Scholar]
- 121.Sze C, Bi H, Kleinschmidt-DeMasters BK, Filley CM, Martin LJ. N-Methyl-D-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer's disease. J Neurol Sci. 2001;182:151–9. doi: 10.1016/s0022-510x(00)00467-6. [DOI] [PubMed] [Google Scholar]
- 122.Vallortigara J, Rangarajan S, Whitfield D, Alghamdi A, Howlett D, Hortobagyi T, et al. Dynamin1 concentration in the prefrontal cortex is associated with cognitive impairment in Lewy body dementia. F1000Research. 2014;3:108. doi: 10.12688/f1000research.3786.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lovell MA, Smith JL, Xiong S, Markesbery WR. Alterations in zinc transporter protein-1 (ZnT-1) in the brain of subjects with mild cognitive impairment, early, and late-stage Alzheimer's disease. Neurotox Res. 2005;7:265–71. doi: 10.1007/BF03033884. [DOI] [PubMed] [Google Scholar]
- 124.Yamada M, Itoh Y, Sodeyama N, Suematsu N, Otomo E, Matsushita M, et al. Senile dementia of the neurofibrillary tangle type: a comparison with Alzheimer's disease. Dement Geriatr Cogn Disord. 2001;12:117–26. doi: 10.1159/000051245. [DOI] [PubMed] [Google Scholar]
- 125.Arnold SE, Louneva N, Cao K, Wang LS, Han LY, Wolk DA, et al. Cellular, synaptic, and biochemical features of resilient cognition in Alzheimer's disease. Neurobiol Aging. 2013;34:157–68. doi: 10.1016/j.neurobiolaging.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.van Houwelingen HC, Arends LR, Stijnen T. Advanced methods in meta-analysis: multivariate approach and meta-regression. Stat Med. 2002;21:589–624. doi: 10.1002/sim.1040. [DOI] [PubMed] [Google Scholar]
- 127.Borenstein M, Hedges LV, Higgins JPT, R HR. Introduction to meta-analysis. Hoboken: Wiley; 2009. [Google Scholar]
- 128.Collaboration TC. Cochrane handbook for systematic reviews of interventions. Hoboken: Wiley-Blackwell; 2008. [Google Scholar]
- 129.Rose JB, Crews L, Rockenstein E, Adame A, Mante M, Hersh LB, et al. Neuropeptide Y fragments derived from neprilysin processing are neuroprotective in a transgenic model of Alzheimer's disease. J Neurosci. 2009;29:1115–25. doi: 10.1523/JNEUROSCI.4220-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chang RY, Nouwens AS, Dodd PR, Etheridge N. The synaptic proteome in Alzheimer's disease. Alzheimers Dement. 2013;9:499–511. doi: 10.1016/j.jalz.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 131.Chang RY, Etheridge N, Dodd PR, Nouwens AS. Targeted quantitative analysis of synaptic proteins in Alzheimer's disease brain. Neurochemistry international. 2014;75:66–75. doi: 10.1016/j.neuint.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 132.Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron. 2014;82:756–71. doi: 10.1016/j.neuron.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Proctor DT, Coulson EJ, Dodd PR. Post-synaptic scaffolding protein interactions with glutamate receptors in synaptic dysfunction and Alzheimer's disease. Prog Neurobiol. 2011;93:509–21. doi: 10.1016/j.pneurobio.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 134.Sheng M, Sabatini BL, Sudhof TC. Synapses and Alzheimer's disease. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yu W, Lu B. Synapses and dendritic spines as pathogenic targets in Alzheimer's disease. Neural Plast. 2012;2012:247150. doi: 10.1155/2012/247150. [DOI] [PMC free article] [PubMed] [Google Scholar]