

ABSTRACT
NEO1 is an essential gene in budding yeast and belongs to a highly conserved subfamily of P-type ATPase genes that encode phospholipid flippases. Inactivation of temperature sensitive neo1ts alleles produces pleiomorphic defects in the secretory and endocytic pathways, including fragmented vacuoles. A screen for multicopy suppressors of neo1-2ts growth defects yielded YPT7, which encodes a Rab7 homolog involved in SNARE-dependent vacuolar fusion. YPT7 suppressed the vacuole fragmentation phenotype of neo1-2, but did not suppress Golgi-associated protein trafficking defects. Neo1 localizes to Golgi and endosomal membranes and was only observed in the vacuole membrane, where Ypt7 localizes, in retromer mutants or when highly overexpressed in wild-type cells. Phosphatidylethanolamine (PE) has been implicated in Ypt7-dependent vacuolar membrane fusion in vitro and is a potential transport substrate of Neo1. Strains deficient in PE synthesis (psd1Δ psd2Δ) displayed fragmented vacuoles and the neo1-2 fragmented vacuole phenotype was also suppressed by overexpression of PSD2, encoding a phosphatidylserine decarboxylase that produces PE at endosomes. In contrast, neo1-2 was not suppressed by overexpression of VPS39, an effector of Ypt7 that forms a membrane contact site potentially involved in PE transfer between vacuoles and mitochondria. These results support the crucial role of PE in vacuole membrane fusion and implicate Neo1 in concentrating PE in the cytosolic leaflet of Golgi and endosomes, and ultimately the vacuole membrane.
KEYWORDS: flippase, membrane asymmetry, P4-ATPase, Rab protein, SNARE, vacuoles, Ypt7
Introduction
Biological membranes are an assembly of protein and lipid molecules organized into a bilayer structure with 2 distinct surfaces. Integral membrane proteins are intrinsically asymmetric and display functionally unique domains on opposite sides of the membrane. The phospholipid composition of the 2 leaflets of a membrane can also be very different and this transverse asymmetry has substantial physiological implications for the membrane system.1,2 Lipid molecules enriched on the cytosolic leaflet of the plasma membrane include phosphatidylserine (PS), phosphatidylethanolamine (PE), phosphatidylinositol and phosphoinositides, whereas sphingolipids, glycosphingolipids and phosphatidylcholine are typically enriched in the extracellular leaflet.3 Phospholipid asymmetry is crucial for recruitment of specific peripheral membrane proteins and the function of numerous transporters and channels within the membrane. Thus, the appropriate asymmetric organization is important for many membrane-centered processes, including signal transduction, cell motility, cell polarity, cell division, vesicular transport and transport of ions and other molecules across the bilayer.1
Phospholipid asymmetry is controlled by flippases, floppases and scramblases; proteins that facilitate interleaflet transport of phospholipid molecules. The type IV P-type ATPases (P4-ATPases) catalyze a unidirectional, ATP-dependent, inward flip of specific phospholipid species from the extracellular leaflet of the plasma membrane to the cytosolic leaflet (flippase activity).4,5 PS and PE are common substrates of P4-ATPases, which appear to be primarily responsible for establishing the asymmetric distribution of these aminophospholipids in the cytosolic leaflet.6 ABC transporters in the multidrug resistance (MDR) family can catalyze outward flop of phospholipids from the cytosolic leaflet to the extracellular leaflet (floppase activity), but often with less headgroup specificity as compared to the flippase activity catalyzed by the P4-ATPases.7 The combined activities of the flippases and floppases set the asymmetric membrane structure, which can be rapidly dissolved by activation of a scramblase.8 TMEM16F and Xkr8 appear to be the scramblases responsible for the regulated exposure of PS in the extracellular leaflet of the plasma membrane required for blood clotting reactions and recognition of apoptotic cells, respectively.9,10
The asymmetric phospholipid organization of the plasma membrane and its physiological significance is becoming clear; however, less is known about how the membrane organization of intracellular organelles influences their function. Eukaryotic cells typically express multiple P4-ATPases and many of these flippases reside within Golgi and endosomal membranes.2 For example, the budding yeast Saccharomyces cerevisiae expresses 5 P4-ATPases; Drs2, Neo1, Dnf1, Dnf2 and Dnf3.11 Only Dnf1 and Dnf2 are present on the plasma membrane of growing cells, although these P4-ATPases also localize to Golgi and endosomal membranes.11 Drs2 and Dnf3 localize primarily to the trans-Golgi network and Neo1 appears to be broadly distributed through the Golgi and endosomal system.11-14 As membrane flows rapidly by vesicular transport between the Golgi, plasma membrane, endosomes and vacuole, the activity of a P4-ATPase in the Golgi should influence the organization of all of these membranes. ATP9A and ATP9B, the mammalian orthologs of Neo1, also localize to Golgi and endosomal membranes.15 In contrast, TAT-5 in Caenorhabditis elegans (Neo1 ortholog) is reported to localize to the plasma membrane and inactivation of TAT-5 leads to the specific exposure of PE in the plasma membrane outer leaflet.16 This result suggests that PE could be the primary substrate of Neo1/TAT-5/ATP9 subgroup of P4-ATPases, although none of these proteins have been purified and assayed in a reconstituted system to clearly define their substrate preferences.
To better understand why NEO1 is an essential gene in budding yeast, several temperature-sensitive (ts) alleles of neo1 have been recovered and used to analyze the cellular consequences of acutely inactivating Neo1 function.12,14 The phenotypes observed are pleiomorphic and include defects in COPI-dependent protein transport from the Golgi to the ER, defects in GGA/clathrin-dependent protein transport from the ER to late endosome, and exposure of both PS and PE in the outer leaflet of the plasma membrane.12,14,17 Loss of PS and PE membrane asymmetry in neo1-1 cells is observed even at a permissive growth temperature (27°C), but a more extensive exposure of PE occurs upon shift to a semipermissive temperature of 30°C where Neo1 is sufficiently inactivated to cause a modest growth defect.17 Golgi glycosylation reactions are also perturbed in neo1-ts cells giving rise to defects in the cell wall.12 Perhaps as a consequence of the cell wall defects, neo1-1 cells grown at permissive temperature are larger and more elongated than normal.12 Inactivation of neo1-1 also causes a V-ATPase-dependent hyperacidification of vacuoles by nearly 1 pH unit, and the vacuoles in neo1-1 cells are fragmented.12,18 This latter phenotype implicates Neo1 in the process of vacuole membrane fusion.
Yeast vacuoles are a large organelle typically present in one to 3 copies per cell, but constantly undergo fission and fusion reactions to maintain this steady state copy number.19 Vacuole fusion is a SNARE-dependent process regulated by the Rab protein Ypt7 and the HOPS tethering complex.20 Ypt7 specifically recruits the multisubunit HOPS complex to the vacuole leading to close apposition of adjacent vacuole membranes along a boundary called the vertex ring.21 Ypt7:GTP bound to HOPS then promotes the formation of a trans-SNARE complex that ultimately drives membrane fusion of the 2 bilayers.20,22,23 A specific set of lipids also play a crucial role in vacuole membrane fusion in vivo, with isolated vacuoles in vitro and in reconstituted liposome fusion assays that recapitulate the role of all of the major protein complexes acting in vacuole fusion.22,24 Ergosterol, diacylglycerol (DAG), phosphatidic acid (PA), phosphoinositides and PE have all been implicated in the vacuole membrane fusion reaction.22,24,25 Strains deficient in production of ergosterol, DAG and phosphoinositides display fragmented vacuoles.26,27 In addition, reagents that deplete or mask these 3 lipids inhibit fusion of vacuoles in vitro and these lipids are concentrated at the site of membrane fusion, the vertex ring, along with the fusion proteins.26,28 The vacuole lipids stimulate a proteoliposome fusion assay that reconstitutes SNARE-, Ypt7- and HOPS-dependent membrane fusion, with phosphatidylcholine (PC), PI3P, PA and PE representing the minimal set of required lipids.22,24 PE has a major influence on the proteoliposome fusion assay,24 but unlike most of the other lipids that facilitate fusion, a role for PE had not emerged from in vivo studies of vacuole fusion. The data presented herein indicates that PE in the cytosolic leaflet of the membrane is an important regulator of the SNARE-mediated vacuole fusion reaction in living cells.
Results
Neo1 is a membrane protein with 10 membrane-spanning segments (Fig. 1A, gray segments), cytosolic N- and C- termini, and large cytosolic loops involved in ATP hydrolysis (Fig. 1A, white segments). Six neo1 ts alleles (neo1-1 to neo1-6) were produced by error-prone PCR for functional studies and each allele bears 6 to 16 mutations.12 The relative positions of neo1-1 and neo1-2 mutations, which are non-overlapping, are shown in Fig. 1A. We had previously screened for multi-copy (2 μ) suppressors of neo1-1 as an approach to identify processes linked to Neo1 function.18 This screen identified several genes encoding subunits of the V-ATPase responsible for acidifying the lumen of vacuoles, endosomes and Golgi compartments. Overexpression of a single subunit perturbs assembly of the V-ATPase complex, thus attenuating V-ATPase activity.29 The results of the suppressor screen led to the discovery that neo1-1 mutants hyperacidify internal organelles and this partially contributes to the growth defects of neo1-1 at nonpermissive temperatures.18 However, we noticed that neo1-2 was not suppressed by overexpression of VMA11 (encoding the V-ATPase c' subunit) nearly as well as neo1-1. Growth of neo1-2 at the semi-permissive temperature of 34°C was marginally improved by VMA11 overexpression (compare 2μ VMA11 to the empty vector control for neo1-2 colony size), whereas neo1-1 growth was significantly improved (Fig. 1). The allele-specific suppression of neo1-1 by VMA11 suggested that a screen for multicopy suppressors of neo1-2 would recover a different set of suppressors.
Figure 1.
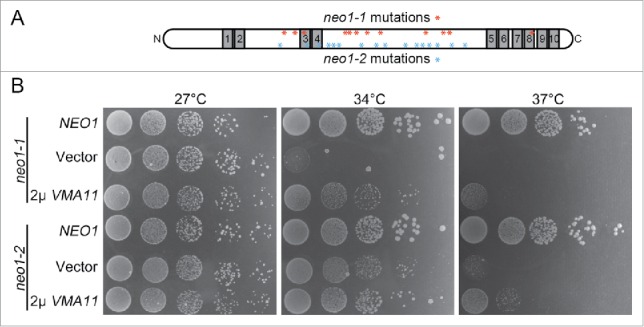
VMA11 differentially suppresses the neo1-1 and neo1-2 growth defects at semi- or non-permissive temperatures. (A) Schematic of Neo1 showing the placement of the 10 transmembrane segments (gray). The N-terminus, C-terminus and large loops between TM2 and 3, and TM4 and 5 are cytosolic. The relative positions of the neo1-1 and neo1-2 mutations are indicated with asterisks (see reference 12 for the specific amino acid substitutions). neo1 ts mutants (YWY160, 161) harboring single copy NEO1 (pRS315-NEO1), empty vector (pRS315) or multi-copy VMA11 (pRS425-VMA11) were spotted on SD-Leu plates with serial dilution and incubated at the indicated temperatures for 3–5 d.
YPT7 overexpression suppresses neo1-2
The neo1-2 strain was transformed with a multicopy genomic library and a clone was isolated that partially suppressed the neo1-2 growth defect at 37°C (Fig. 2A, 2μ clone). This genomic library clone carried a fragment of chromosome XIII containing several genes. Each gene was subcloned and tested for suppression of neo1-2 and only YPT7 was capable of conferring suppression (Fig. 2A, 2μ YPT7 and unpublished observations). Surprisingly, even a single extra copy of YPT7 carried on a centromere-based plasmid suppressed the neo1-2 growth defect at 37°C (Fig. 2A, cen YPT7). In contrast, YPT7 failed to suppress the neo1-1 growth defect at 37°C or at a semipermissive temperature of 30°C (Fig. 2B and C). To further test the allele specificity of suppression, YPT7 or an empty vector was used to transform all 6 independently isolated neo1 ts mutants (neo1-1 – neo1-6,12). Of this group, a single extra copy of YPT7 partially suppressed neo1-2, neo1-3 and neo1-4, but no suppression was observed for neo1-5 and neo1-6 (Fig. 2C). While many of the mutations cluster near each other in the large cytosolic domains, there are no common mutations between neo1-2, neo1-3 and neo1-4 that could provide a simple explanation for the allele-specific suppression by YPT7.12
Figure 2.
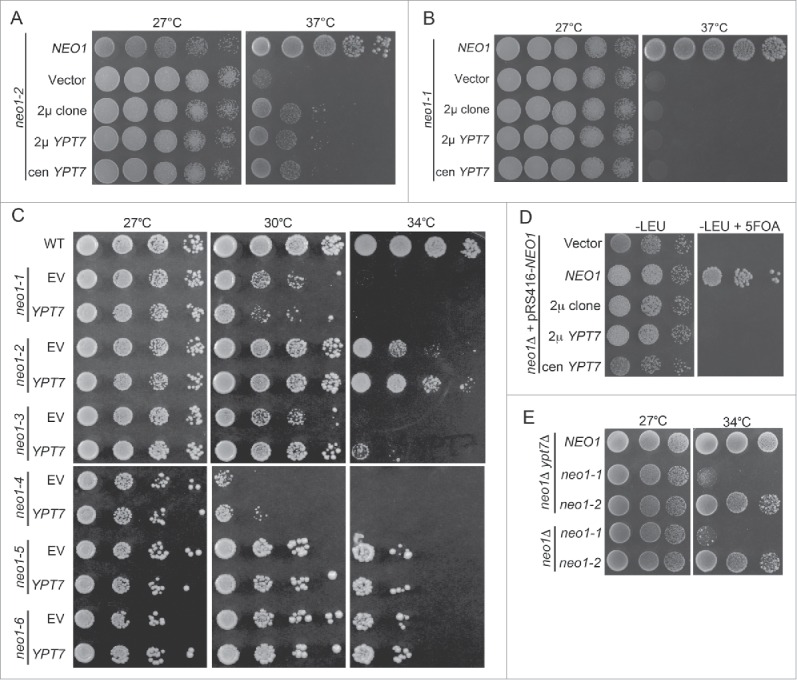
YPT7 suppresses the growth defect of a subset of neo1ts mutants. (A) neo1-2 (YWY161) was transformed with single copy NEO1 (YWY163), empty vector (YWY144), the multicopy genomic DNA suppressor (2 μ clone, YWY157), multicopy (2 μ) YPT7 (YWY158) or single copy (cen) YPT7 (YWY168) respectively. Cells were spotted on SD-Leu plates with serial dilution and incubated at indicated temperatures. (B) A neo1-1 mutant (YWY160) was transformed with the same plasmids and tested as in (A). (C) A neo1Δ pRS416-NEO1 strain (YWY10) was transformed with the same set of constructs and spotted on both SD-Leu and SD-Leu + 5FOA plates at 30°C. (D) The full collection of neo1ts mutants (YWY148-155) carrying either empty vector or single copy YPT7 were spotted on SD-Leu plates at permissive (27°C) or semi-permissive temperatures (30°C, 34°C) to test for suppression. (E) Plasmid shuffling assay with a neo1Δ strain (YWY10) to exchange pRS416-NEO1 for an empty LEU2-marked plasmid (vector), or this LEU2 plasmids carrying carrying NEO1 or YPT7. The medium containing 5-FOA counter-selects the URA3-marked plasmid and only cells that carry complementing or suppressing LEU2 plasmids will grow. (E) Growth of neo1Δypt7Δ double mutants carrying NEO1, neo1-1 or neo1-2 plasmids (YWY129,133,134) was compared to neo1Δ single mutants with neo1-1 and neo1-2 ts alleles on plasmids (YWY160,161).
To test if YPT7 could suppress the lethality caused by deletion of NEO1, we generated haploid neo1Δ strains covered by NEO1 on a URA3-marked plasmid and harboring the same set of YPT7 clones on LEU2-marked plasmids. These strains were serially diluted onto medium containing 5-fluoro-orotic acid (5-FOA) to select for cells capable of losing the NEO1-URA3 plasmid. Only the strain carrying a wild-type NEO1-LEU2 control plasmid was able to grow on 5-FOA, while the strains carrying an empty LEU2 vector or the indicated YPT7 clones failed to grow (Fig. 2D). This result indicates that YPT7 cannot bypass the essential function of NEO1. We then asked if removing YPT7 would have the opposite effect and exacerbate neo1-2 growth defects. For this experiment, we used neo1Δ and neo1Δ ypt7Δ strains expressing NEO1, neo1-1 or neo1-2 from plasmids. However, no difference in growth was observed for neo1-1 or neo1-2 in the presence or absence of YPT7 (Fig. 2E).
Neo1 localization to Golgi and endosomes
The allele-specific suppression of neo1-2 by YPT7 could suggest a direct interaction between the encoded proteins. However, Ypt7 localizes to the vacuole where it regulates membrane fusion reactions and Neo1 has not been reported to localize to the vacuole.12-14,30 However, Neo1 is weakly expressed and localization experiments with functional, epitope- or GFP-tagged forms of Neo1 expressed at endogenous levels reported a broad localization in the ER, Golgi and endosomes, but may have missed a minor fraction of Neo1 in the vacuole membrane. Therefore, we tested if overexpression of a functional, GFP-tagged Neo1 would allow us to detect any Neo1 in the vacuole membrane. GFP-Neo1 (N-terminally tagged) expressed from the moderate-strength alcohol dehydrogenase (ADH) promoter31 co-localized partially with markers of the early Golgi (COP1-mKate), medial Golgi (Aur1-mKate) and late Golgi (Sec7-mKate)(Fig. 3A–C).32 The Pearson's correlation coefficient (r) was slightly higher for the GFP-Neo1 and Aur1-mKate pair perhaps indicating preferential localization of GFP-Neo1 to the medial Golgi at this level of expression.
Figure 3.
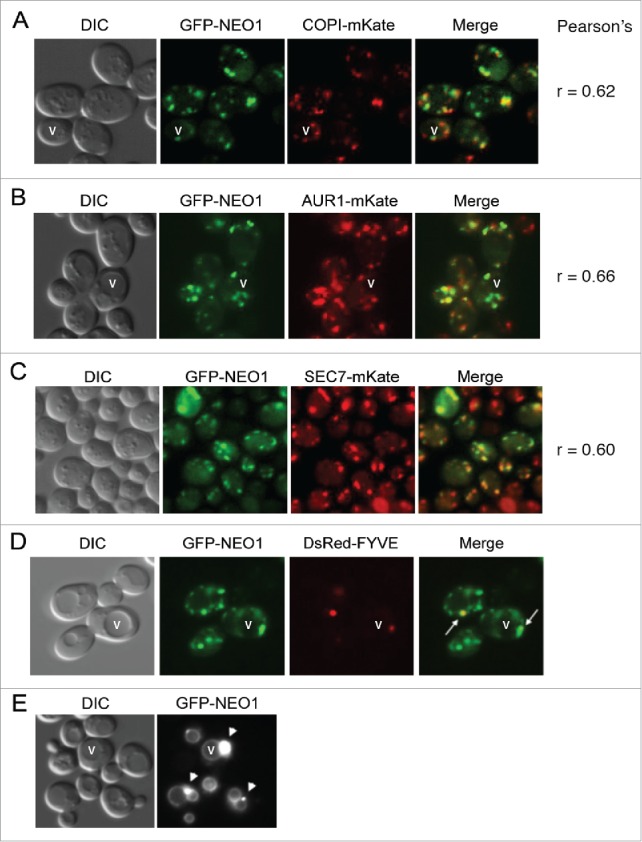
Localization of GFP tagged Neo1p at different levels of expression. (A-C) The construct pRS313-PAdh-GFP-NEO1 was transformed into strains harboring different Golgi markers genomically tagged with mKate in the BY4742 background (YWY208-210) and imaged. The Pearson's correlation coefficient (r) was determined using imageJ's co-localization function. (D) A neo1Δ strain harboring pRS313-PAdh-GFP-NEO1 (which functionally complements neo1Δ to support growth) and pRS425-DsRED-FYVE (late endosomal marker)(YWY128) was imaged. Arrows indicate the overlap between GFP-Neo1 decorated puncta and DsRED-FYVE decorated late endosomes. (E) GFP-NEO1 was highly overexpressed from the GPD promoter in neo1Δ cells (YWY172) and imaged. All strains were cultured overnight and sub-cultured the next morning at 30°C. Cells were harvested at A600 ∼0.6 and imaged.
GFP-Neo1 was also detected in late endosomes marked with DsRed-FYVE (Fig. 3D),33 although only 33% of the cells showed co-localization of GFP-Neo1 with the late endosome marker while every cell showed co-localization with the Golgi markers. A C-terminally epitope tagged form of Neo1 was previously detected in the ER and Golgi,12 but GFP-Neo1 was not detected in the ER even though it was more highly expressed. Importantly, GFP-Neo1 was not detected in the vacuole membrane (marked with a “v” in a few of the cells in Fig. 3) at the moderate expression level driven by the ADH promoter. To drive very high expression, we placed GFP-NEO1 under control of the strong glycerol 3-phosphate dehydrogenase (GPD) promoter31 and expressed the construct from a multicopy plasmid. With these conditions, we can detect vacuole membrane localization of GFP-Neo1 (Fig. 3E, compare the rings of GFP-Neo1 fluorescence to the vacuoles in the DIC image that appear as large depressions in the cells). GFP-Neo1 also localized strongly to a structure adjacent to the vacuoles (arrowheads), which are likely prevacuolar endosomes. Thus, we can only detect GFP-Neo1 in the vacuole membrane when it is expressed at very high, nonphysiological levels.
The observation that GFP-Neo1 mislocalized to the vacuole membrane when highly overexpressed suggested that a saturable retention or retrieval mechanism was acting to maintain a steady-state localization in the Golgi. Because GFP-Neo1 appears to traffic into the late endosome, we tested if retromer was involved in the localization of Neo1. GFP-Neo1 expressed from the ADH promoter was readily detected in the vacuole membrane of vps35Δ cells that are deficient for a retromer subunit (Fig. 4A). In contrast, 2 related P4-ATPases, GFP-Drs2 and GFP-Dnf1,34 were not mislocalized to the vacuole in vps35Δ cells, suggesting that they do not traffic into the late endosome (Fig. 4B). These data suggest that Neo1 is a cargo of retromer and is retrieved from the late endosome to the Golgi to prevent missorting to the vacuole.
Figure 4.
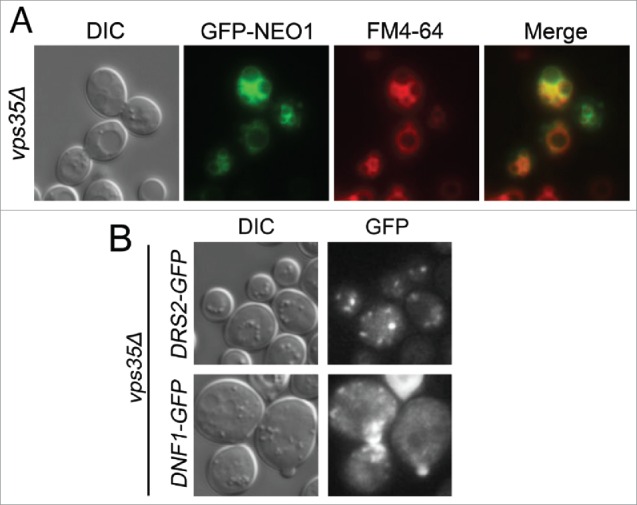
Trafficking of Neo1p is controlled by the retromer complex. (A) A vps35Δ (retromer) mutant expressing pRS413-PAdh-GFP-NEO1 (YWY173) was stained with 32 µM FM4-64 for 20 minutes followed by 1 hour incubation in YPD at 30°C. Cells were rinsed twice with imaging buffer (PBS + 2% glucose) before imaging. (B) Images of a vps35Δ mutant expressing pRS416-PCPY-DRS2-GFP together with pRS425-CDC50 (top, YWY174) or pRS416-PCPY-DNF1-GFP together with pRS425-LEM3 (YWY175).
YPT7 suppresses the fragmented vacuole phenotype of neo1-2
We next examined vacuole morphology in neo1-ts cells and asked if YPT7 overexpression could suppress the fragmented vacuole phenotype of these mutants. WT, neo1-1 and neo1-2 cells expressing YPT7 or an empty vector were grown at the permissive temperature of 30°C and stained with FM4-64 to visualize vacuoles. The WT cells exhibited an average of 2.4 vacuoles per cell while the neo1-1 and neo1-2 exhibited 3.25 and 3.6 vacuoles per cell, respectively (Fig. 5). YPT7 expressed from single-copy (cen) or multicopy (2 μ) vectors suppressed the fragmented vacuole phenotype to WT values in neo1-2 cells (Fig. 5A and B), but not neo1-1 cells (Fig. 5C and D). Therefore, the defect in vacuole fusion caused by neo1-2 can be overcome by modest overexpression of YPT7.
Figure 5.
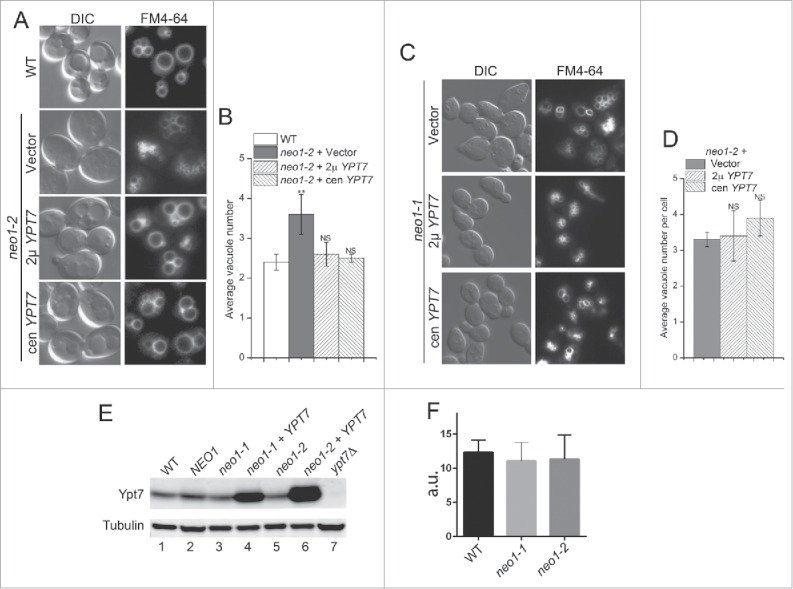
YPT7 suppresses the fragmented vacuole phenotype of a neo1-2 mutant. (A and C) The WT (BY4741), neo1-2 (YWY161), and neo1-1 (YWY160) strains indicated were stained with FM4-64 to mark vacuoles and imaged. (B and D) The average vacuole number per cell was quantified from single image planes of approximately 100 cells, and the average of 3 separate experiments +/− s.e.m.. A student t test was used to compare neo1-ts cells expressing different constructs with WT cells (**, p < 0.01;NS, not significant). (E) An equal number of cells from the indicated strains were lysed in SDS sample buffer and subject to immunoblotting with anti-Ypt7 and anti-tubulin. Ypt7 was overexpressed in neo1-1 and neo1-2 strains from single-copy, centromeric plasmids. (F) Quantitation of Ypt7 band intensities from 3 immunoblots (+/− s.e.m.; a.u., arbitrary units).
Disruption of YPT7 causes highly fragmented vacuoles30 and it is possible that neo1 mutants expressed lower amounts of Ypt7 relative to WT cells, which led to vacuole fragmentation. However, quantitative western blotting showed no significant difference in Ypt7 levels between WT, neo1-1 and neo1-2 strains (Fig. 5E and F). Cells carrying an additional copy of YPT7 on centromere-based plasmids expressed 3-fold more Ypt7 than WT cells, providing an explanation for the dosage suppression by the low copy plasmids. The slightly higher expression of Ypt7 from the plasmids in neo1-2 relative to neo1-1 (compare lane 4 to lane 6) is unlikely to be the cause of the allele-specific suppression because even higher Ypt7 expression from a multicopy plasmid failed to suppress neo1-1 growth defects (Fig. 2).
It is also possible that YPT7 overexpression may somehow protect the mutant Neo1-2 protein from thermal inactivation, thereby conferring suppression. If this is the case, then all phenotypes exhibited by neo1-2 should be suppressed. Inactivation of Neo1 leads to a defect in the COPI-dependent transport of Rer1-GFP from the Golgi to the ER and mislocalization of this reporter to the vacuole.12,35 We examined Rer1-GFP trafficking in neo1-2 cells with or without YPT7 and could not detect any suppression of the Rer1-GFP trafficking defect (Fig. 6). For these data, we found cells in the population with primarily one large vacuole to display to avoid the confounding affect of vacuole morphological changes.
Figure 6.
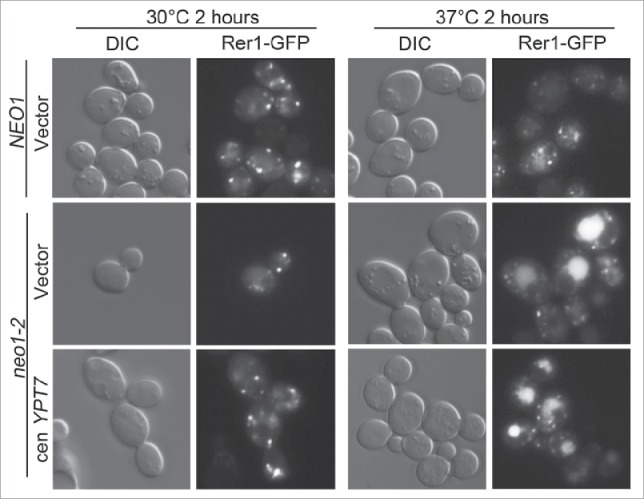
YPT7 does not suppress the neo1-2 Rer1-GFP protein transport defect. neo1-2 strains expressing Rer1-GFP and harboring an empty vector or single copy YPT7 (YWY177-178) was cultured overnight and sub-cultured the next morning at 27°C. After reaching log growth phase, an equal amount of cells were shifted to 30°C or 37°C for 2 hours. A neo1Δ pRS313-NEO1 strain (YWY176) expressing Rer1-GFP was imaged as a control.
PE deficiency cause vacuole fragmentation
Inactivation of Neo1 causes a loss of plasma membrane asymmetry for both PS and PE, although the interleaflet distribution of PE is more strongly affected than PS.17 PE has also been implicated in membrane fusion reactions driven by the vacuolar SNARE proteins.24 Therefore, it seemed likely that the fragmented vacuole phenotype in neo1-ts cells was caused by perturbation of PE distribution. If so, then other means of reducing PE in the vacuole membrane should also lead to fragmented vacuoles. PE is an essential lipid in budding yeast and is synthesized primarily by decarboxylation of PS and by the Kennedy salvage pathway using ethanolamine as a precursor (Fig. 7A).36 To determine if PE deficiency in vivo influences vacuolar morphology, we stained cells deficient for the PS decarboxylases (psd1Δ, psd2Δ and psd1Δ psd2Δ) with FM4-64. As predicted, deletion of PSD1, encoding the mitochondrial PS decarboxylase that synthesizes more than half of the total cellular PE,36 resulted in significantly fragmented vacuoles as compared to WT cells. Deletion of PSD2, encoding a Golgi/endosomal PS decarboxylase with a minor contribution to total PE content (∼5%),36,37 resulted in no significant difference in vacuole number compared to WT cells (Fig. 7B and C). Deletion of PSD1 and PSD2 resulted in highly fragmented vacuoles with ∼90% of the cells displaying 5 or more vacuoles (Fig. 7D and E).
Figure 7.

PE is crucial for vacuole fusion in vivo. (A) Pathways for synthesis of PS, PE and PC in Saccharomyces cerevisiae. WT (SEY6210), psd1Δ and psd2Δ cells (KGS20,21) were stained with FM4-64 to visualize vacuole morphology. (B) Average number of vacuoles per cell was quantified for cells from (A). *, p < 0.05, NS, non-significant. (C) The psd1Δpsd2Δ double mutant (KGS22) was supplemented with 2 mM choline (cho) or 2 mM ethanolamine (etn) and stained with FM4-64. The number of vacuoles per cell (single image plane) was plotted as a percentage of cells in each category. The percentage of cells with 5 or more vacuoles was compared with psd1Δpsd2Δ cells without choline or ethanolamine supplement using Student's t test. **, p < 0.01, ***, p < 0.001.
PE can be further processed through methylation to PC and so psd1Δ psd2Δ diminishes both PE and PC levels in the cells. To distinguish whether a reduction in PE or PC, or an increase in PS, caused vacuole fragmentation in psd1Δ psd2Δ cells, we tested if supplementation of cells with choline (cho) or ethanolamine (etn) to increase flux through the salvage pathways could suppress this phenotype. Choline supplementation modestly decreased the number of vacuoles per psd1Δ psd2Δ cell. In contrast, ethanolamine supplementation to stimulate PE synthesis dramatically decreased vacuole number (Fig. 7D and E). Thus, reduced PE content of the cells rather than reduced PC or increased PS caused the vacuole fragmentation.
Deletion of the PS synthase gene (CHO1) eliminates PS and reduces flux to PE and PC through the de novo synthesis pathway (Fig. 7A). Growth of cho1Δ cells requires supplementation with either choline or ethanolamine and cells grown on choline to support PC synthesis have highly fragmented vacuoles (Fig. 8A and B), comparable to psd1Δ psd2Δ cells. Again, ethanolamine supplementation to increase PE synthesis through the salvage pathway significantly reduced vacuole number in cho1Δ cells, although not as effectively as observed for psd1Δ psd2Δ cells. Together, these data indicate that PE is a crucial membrane lipid for vacuole fusion events in vivo, even in the absence of PS.
Figure 8.
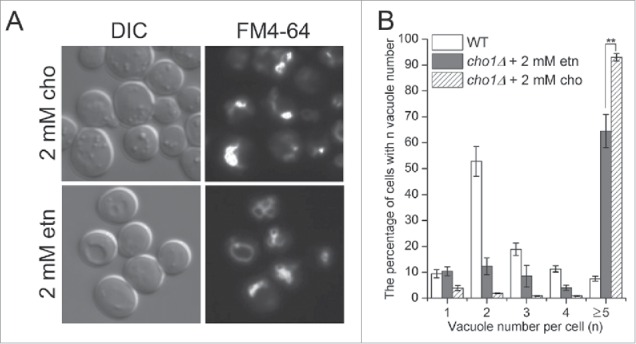
Ethanolamine supplementation reduces vacuole fragmentation in cho1Δ cells. (A) cho1Δ cells (RBY8700) were cultured with 2 mM choline or 2 mM ethanolamine and stained with FM4-64. cho1Δ is inviable in synthetic media without supplement of choline or ethanolamine. (B) The number of vacuoles per cell (single image plane) was plotted as a percentage of cells in each category. **, p < 0.01.
Deletion of PSD1 alone produces a fragmented vacuole phenotype that is similar to neo1-2. Therefore, we tested if overexpression of YPT7 could suppress the fragmented vacuole phenotype of psd1Δ cells. Surprisingly, we found no significant difference in average vacuole number between psd1Δ cells with or without additional copies of YPT7 (Fig. 9). We also tested if ethanolamine supplementation could suppress the neo1-2 vacuole fragmentation phenotype, but surprisingly found no significant suppression (Fig. 10A and B). However, modest overexpression of PSD2 (carried on a single-copy plasmid and expressed from the ADH promoter) was able to significantly suppress the fragmented vacuole phenotype of neo1-2 (Fig. 10A and B), although growth of neo1-2 at 37°C was only marginally improved by PSD2 overexpression (Fig. 10C). These data are consistent with PE being a key lipid in vacuole fusion.
Figure 9.
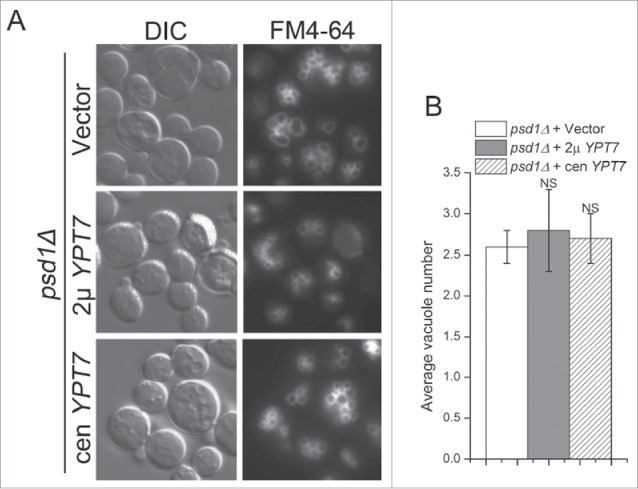
YPT7 does not suppress the fragmented vacuole phenotype of psd1Δ. (A) psd1Δ cells harboring an empty vector, multicopy (2 μ) YPT7 or single copy (cen) YPT7 (YWY140-142) were stained with FM4-64 to visualize vacuole morphology. (B) The average vacuole number of the cells from (A) was quantified. NS, not significant.
Figure 10.
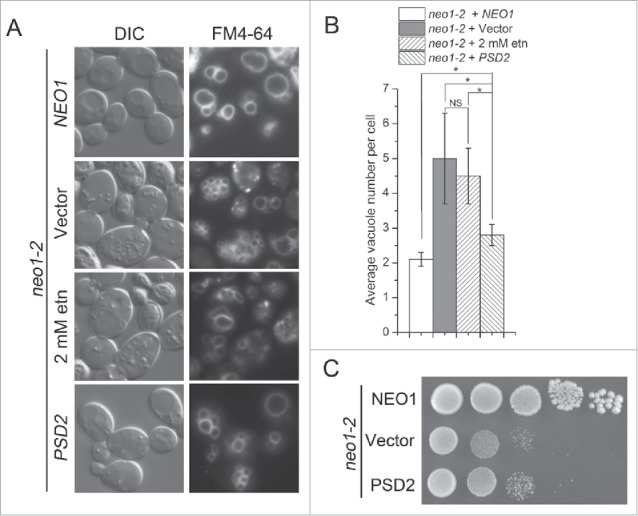
Ethanolamine supplementation and PSD2 partially suppresses the fragmented vacuole phenotype of neo1-2 cells. (A) neo1-2 cells transformed with NEO1, empty vector or PSD2 (YWY179-181), or supplemented with 2 mM ethanolamine during growth at 30°C, were stained with FM4-64. (B) The average vacuole number per cell in strains from (A) was quantified. *, p < 0.05, NS, not significant. (C) Strains from (A) were spotted on SD-Ura plates and incubated at 37°C for 4 d.
In addition to its role in vacuolar fusion, Ypt7 recruits Vps39 to the vacuole membrane to facilitate formation of a membrane contact site between the vacuole and mitochondria called vCLAMP (vacuole and mitochondria patch), a structure that may allow transfer of PE from the mitochondria to the vacuole membrane. Thus, it was possible that YPT7 overexpression suppressed vacuole fragmentation in neo1-2 cells by enhancing the formation of vCLAMP. In this case, we would also expect VPS39 overexpression to suppress neo1-2. However, VPS39 overexpression failed to suppress neo1-2 growth defects. VPS39 also failed to enhance neo1-2 suppression when co-expressed with YPT7 (Fig. 11). Thus, it is unlikely that Ypt7 suppresses neo1-2 by enhancing formation of vCLAMP.
Figure 11.

Overexpression of VPS39 fails to suppress neo1-2 growth defects. neo1-2 strains (from top to bottom: MTY10-39, MTY144-39, MTY158-39 and MTY200-39) expressing the indicated multicopy constructs were grown with serial dilution at the temperatures indicated.
Discussion
PE has been implicated in vacuole membrane fusion based on in vitro liposome-based assays that reconstitutes SNARE-dependent fusion.24 Here we report that PE also plays a crucial role in vacuole membrane fusion in vivo. Deletion of PSD1, encoding a mitochondrial PS decarboxylase responsible for synthesis of ∼50% of the vacuolar PE,37 causes vacuole fragmentation. Even though Psd2 localizes to endosomal membranes, it only makes a modest contribution (∼5%) to vacuole PE levels37 and the psd2Δ mutant does not display fragmented vacuoles. Deletion of both PSD1 and PSD2 causes severe vacuole fragmentation, but supplementing these cells with ethanolamine to stimulate PE synthesis through the Kennedy pathway markedly suppresses the fragmentation phenotype (Fig. 7). Neo1 mutants also cause fragmented vacuoles and a loss of PS and PE asymmetry. The observation that Psd2 overexpression (which should enhance PE levels at the expense of PS) suppresses the vacuole fragmentation in neo1-2 strongly suggests it is a perturbation of PE in the vacuole membrane that initially causes the fragmented vacuole phenotype. These results demonstrate an important role for PE in vacuole fusion and imply that inactivation of Neo1 perturbs PE availability to the fusion machinery in the vacuole membrane.
Budding yeast strains harboring a disruption of NEO1 are inviable in spite of the presence of 4 additional P4-ATPases (Drs2, Dnf1, Dnf2 and Dnf3) that traffic through the Golgi and endosomal system where Neo1 resides. Among the budding yeast P4-ATPases, neo1 mutations uniquely cause vacuole fragmentation and deletion of other P4-ATPases genes, alone or in combination, does not lead to a comparable fragmented vacuolar phenotype.11 Interestingly, both dnf1Δ dnf2Δ and neo1-ts mutants substantially hyperacidify vacuoles, yet the dnf1Δ dnf2Δ cells have 1 – 3 large vacuoles per cell, as do WT cells.18 These observations suggest that Neo1 has a unique substrate that is specifically required for vacuole fusion, most likely PE, but shares a substrate with Dnf1 and Dnf2 that influences vacuolar pH. While Dnf1 and Dnf2 translocate fluorescent derivatives of PE efficiently, these P4-ATPases may preferentially transport lysoPE bearing a single fatty acyl chain and perhaps it is loss of lysoPE asymmetry that influences vacuolar pH.1 Dnf3 is reported to flip PE,38 but this protein is very weakly expressed in budding yeast grown in standard laboratory conditions.11 Drs2 is the major PS flippase in yeast and may also have a weak activity toward PE,39 but Drs2 clearly cannot compensate for the loss of Neo1.17 We speculate that Neo1 is the primary flippase for dually acylated PE in yeast and is needed to concentrate PE on the cytosolic leaflet of Golgi and late endosomes.
None of the P4-ATPases, including Neo1, localize to the vacuole membrane when expressed at endogenous levels or when modestly overexpressed.11,12,14,40,41 Neo1 primarily localizes to the Golgi and we could only detect GFP-Neo1 in the vacuole membrane when expressed from a very strong GPD promoter on a multicopy plasmid, which should generate more than 100-fold more Neo1 than WT cells.31 Mutation of a retromer component involved in late endosome to Golgi retrieval of proteins also led to vacuolar localization at more modest levels of Neo1 expression. Therefore, it seems unlikely that Neo1 is functionally present in the vacuole membrane of WT cells, or that the suppression of neo1-ts by Ypt7 overexpression is mediated by a direct interaction between the Neo1 and the vacuolar localized Ypt7. It is more likely that PE traveling the secretory pathway toward the vacuole is concentrated in the cytosolic leaflet by Neo1 at the level of the Golgi or late endosome prior to arrival at the vacuole. Loss of Neo1 activity could therefore lead to less PE in the cytosolic leaflet of the vacuoles. It is also possible that sorting of SNAREs involved in vacuolar fusion is partially perturbed by neo1ts mutants at the level of the endosomes or Golgi, thereby reducing their concentration in the vacuolar membrane leading to reduced fusion. In this case, increasing Ypt7 or PE concentration on the vacuole may restore wild-type kinetics of fusion with fewer SNAREs.
The observation that only certain neo1ts alleles (neo1-2, neo1-3, and neo1-4) can be suppressed by Ypt7 overexpression may reflect the degree to which PE concentration in the vacuolar membrane cytosolic leaflet is perturbed by the neo1ts mutant. Ypt7 may only suppress neo1ts mutants that reduce PE levels just below a threshold needed for efficient SNARE-mediated fusion. More severely affected mutants, such as neo1-117 or psd1Δ37 may have too little PE in the membranes to support efficient fusion regardless of the Ypt7 concentration. It is also possible that the neo1 mutations that are suppressed by YPT7 alter substrate preference in a unique manner. For example, Neo1 might transport several different phospholipid species differing in either headgroup or acyl chain composition (length and number of double bonds) and the mutations may change these patterns of recognition. Similarly, we find that overexpression of VMA11 suppresses neo1-1 better than it suppresses neo1-2. Clearly these neo1-1 and neo1-2 mutations are differentially affecting Neo1 functions and defining the nature of these differences is an important goal.
The observed suppression of neo1-2 by YPT7 overexpression could imply that cytosolic PE influences Ypt7 activity in vacuole fusion. This could occur through a direct interaction of Ypt7 with PE, or by an influence of PE on the Ypt7 GEF (Mon1-Ccz1 heterodimer) and/or GAP (Gyp1 or Gyp7). However, the vacuolar SNARE-dependent liposome fusion assay requires PE when reconstituted with Ypt7-GTP in the absence of the Ypt7 GEF and GAP, indicating that PE has an important role in vacuole fusion downstream of Ypt7 activation.42 The conically-shaped PE molecule is thought to produce packing defects in the monolayer that facilitate initiation of a hemifusion intermediate driven by zippering of v-SNARE and t-SNAREs in apposing membranes.19,25 One explanation for the suppression of neo1-2 by Ypt7 is that a concentration of PE that is suboptimal to support efficient SNARE catalyzed fusion can be overcome by increasing Ypt7-GTP and HOPS concentration on the vacuolar membrane to enhance tethering and SNARE assembly.
Further tests of the hypotheses described herein will require development of probes that can be used to measure the trans-bilayer distribution of PE in vacuolar membranes of living cells. Alternatively, it would be necessary to reconstitute vacuolar membrane fusion in a proteoliposome system of not only defined composition, but with also an asymmetric distribution of PE between the lumenal and cytosolic leaflet. Because of these technical limitations, we cannot definitively conclude that the fusion defect is caused by the loss of vacuole membrane PE asymmetry in neo1ts mutants. However, we can conclude that perturbations in PE metabolism disrupt vacuole fusion in vivo.
Materials and methods
Media, plasmids and strains
Yeast strains were maintained in complete medium (YP, 1% yeast extract, 2% peptone) or synthetic minimal (SM) medium supplemented with 2% glucose. Sporulation was performed by culturing cells in presporulation media (0.8% yeast extract, 0.3% peptone, 10% glucose) until saturation before shifting to sporulation media (1% KOAc, 0.1% yeast extract, 0.05% glucose) for 3-7 d at 30°C. For random sporulation analysis, sporulated cells were treated with 0.8% glusulase for 15 minutes and then vortexed with equal amount of ether for 30 seconds. The spores were then spread on selective plates.
Plasmids used in this study are listed in Table S1. A NEO1 genomic DNA fragment with 518 bp upstream and 126 bp downstream of the NEO1 open reading frame (ORF) was amplified from yeast genomic DNA using primers containing BamHI and XhoI restriction sites and cloned into pRS416 to generate pRS416-NEO1. The same NEO1 fragment was cloned into pRS315 to generate pRS315-NEO1. The restriction sites used in pRS315 were BamHI and SalI, therefore both XhoI and SalI sites were destroyed when NEO1 was inserted. The NEO1 coding sequence was amplified from pRS416-NEO1 using primers containing BamHI and SalI sites and cloned into pRS413-PNEO1.43 For pRS413-PADH-GFP-NEO1, GFP was amplified from pEGFP-C1 (clontech) using primers containing XbaI and BamHI sites and digested with indicated enzymes, NEO1 coding sequence was removed from pRS413-PNEO1-NEO1 using BamHI and SalI, and pRS413-PADH was digested with XbaI and SalI. These three pieces were then ligated. The GFP-NEO1 fragment was released from pRS413-PADH-GFP-NEO1 using XbaI and SalI, and ligated into pRS423-PGPD digested with SpeI and SalI. All constructs carrying GFP-tagged Neo1 fully supported growth of a neo1Δ strain, indicating that GFP-Neo1 is functional. A YPT7 genomic DNA fragment with 501 bp upstream and 153 bp downstream of coding sequence was amplified from yeast genomic DNA using primers containing BamHI and SalI sites and cloned into pRS315 and pRS425 to generate pRS315-YPT7 and pRS425-YPT7.
The same strategy used to generate pRS413-PADH-GFP-NEO1 was used to generate pRS416-PADH-GFP-YPT7. The GFP fragment was amplified from pYM-N1344 using primers containing BamHI and HindIII sites, the YPT7 coding sequence was amplified from pRS315-YPT7 using primers containing HindIII and XhoI sites, and pRS416-PADH was digested with BamHI and XhoI and 3 pieces were then ligated. The PSD2 coding sequence was amplified from yeast genomic DNA using primers containing EcoRI and SalI sites. The fragment was then digested and cloned into pRS416-PADH. A genomic VPS39 region of 3.95kb was amplified with indicated primers and ligated to BamHI-SalI linearized pRS416 and pRS426 to generate pRS416-PVPS39-VPS39 and pRS426-PVPS39-VPS39 plasmids. All plasmid generated were confirmed by sequencing.
Yeast strains used in this study are listed in Table S2. The plasmid pRS416-NEO1 was transformed into BY4743 neo1Δ and the resulting cells were sporulated. After random sporulation, spore YWY10 (MATa his3 leu2 met15 ura3 lys2 neo1ΔpRS416-NEO1) was selected on SD plates with G418 but lacking uracil. A plasmid-shuffling strategy was used to replace the plasmid pRS416-NEO1 in YWY10 with plasmids containing neo1 ts alleles. pRS413-neo1-1 and pRS413-neo1-2 was transformed into YWY10 and the resulting cells were selected with 5FOA to exclude pRS416-NEO1. The resulting strains were YWY160 and YWY161. Homologous recombination was used for disrupting YPT7 in YWY10. About 50-60 bp flanking homologous sequences immediately outside YPT7 ORF were introduced by PCR primers and the natNT2 cassette was amplified from pFA6a–natNT2.44 The PCR product was transformed into YWY10 cells and colonies harboring successful recombination were selected on clonNAT plates and confirmed by genomic DNA PCR. The resulting strain was designated as YWY129.
Multicopy (2µ) suppressor screen
neo1-2 cells were transformed with 1 µg yeast genomic Tiling collection (GE, Dharmacon),45 plated on SD-Leu plates and incubated at 37°C for 7–10 d. The surviving colonies were picked based on the colony size. The selected colonies were cultured at 30°C in suspension and plasmids were extracted. The mixed plasmids (pRS315-neo1-2 and a 2µ plasmid) from each colony were transformed into E.coli. and screened with kanamycin to select the 2µ plasmid. The 2µ plasmid was then sequenced to identify the inserted genomic DNA fragment. Recovered 2µ plasmids were also transformed back into neo1-2 cells to confirm the suppression of neo1-2 growth defect. The ORFs contained within the genomic DNA fragment with at least 500 bp upstream and 100 bp downstream sequences were amplified and individually cloned into pRS425 or pRS315. The resulting plasmids were transformed into neo1-2 cells to identify YPT7 as the ORF responsible for suppressing neo1-2 growth defects at 37°C.
Vacuole staining
The vacuole staining using FM4-64 was performed as previously described.46 Briefly, cells were shaken with 32 μM of endocytic dye FM4-64 (Life Technologies) for 20 min in the dark in YPD at 30°C, after which cells were harvested at 5,000 rpm for 1 minute, then re-suspended in YPD and incubated with shaking for 60 min at 30°C. Cells were subsequently washed twice in phosphate buffered saline (PBS) containing 2% glucose before visualization. For vacuole quantification in cells with no or moderately fragmented vacuoles, about 100 cells were randomly selected. Total cell number and total recognizable vacuole number inside these cells were counted and the average vacuole number per cell was defined as total vacuole number divided by total cell number. This process was repeated 3 times for each strain. For vacuole quantification in cells with highly fragmented vacuoles, vacuole number per cell in a cell population was classified into 2 categories: cells with 1-4 vacuoles and cells with 5 or more vacuoles. Any cells with distinct FM4-64 staining but too many vacuoles to reliably count were classified into cells with 5 or more vacuoles.
Fluorescence microscopy
Cells were washed twice with PBS containing 2% glucose before imaging. Images were acquired on a Zeiss AxioPlan scope using a 63× Plan-ApoChromat oil DIC objective (NA 1.40). A Cy3 or FITC filter was use for visualizing FM4-64 and DsRED or GFP respectively. Images were analyzed using ImageJ.
Western blots
Lysates from 0.1 OD600 of cells were subjected to SDS-gel electrophoresis. Western blot analysis was performed using Rabbit anti-Ypt7 (1:1000) and mouse anti-α-tubulin (1:4000) antibodies. Secondary antibodies were used at a 1:3000 dilution. Membranes were scanned at 800 nm and 680 nm simultaneously using the Odyssey Infrared Imaging System. Median band intensities were quantified using the Odyssey software (LI-COR Biosciences).
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Bill Wickner for antibodies to Ypt7, and Chris Burd for the mKate strains and the DsRED-FYVE construct. We also thank Aki Nakano for the Rer1-GFP construct and Scott Moye-Rowley for the psd mutants.
Funding
This work was supported by grant GM107978 from the National Institutes of Health to T.R.G..
References
- [1].Hankins HM, Baldridge RD, Xu P, Graham TR. Role of flippases, scramblases and transfer proteins in phosphatidylserine subcellular distribution. Traffic 2015; 16(1):35-47; PMID:25284293; http://dx.doi.org/ 10.1111/tra.12233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Sebastian TT, Baldridge RD, Xu P, Graham TR. Phospholipid flippases: building asymmetric membranes and transport vesicles. Biochim Biophys Acta 2012; 1821(8):1068-77; PMID:22234261; http://dx.doi.org/ 10.1016/j.bbalip.2011.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Schroit AJ, Zwaal RF. Transbilayer movement of phospholipids in red cell and platelet membranes. Biochim Biophys Acta 1991; 1071(3):313-29; PMID:1958692; http://dx.doi.org/ 10.1016/0304-4157(91)90019-S [DOI] [PubMed] [Google Scholar]
- [4].Coleman JA, Kwok MC, Molday RS. Localization, purification, and functional reconstitution of the P4-ATPase Atp8a2, a phosphatidylserine flippase in photoreceptor disc membranes. J Biol Chem 2009; 284(47):32670-9; PMID:19778899; http://dx.doi.org/ 10.1074/jbc.M109.047415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhou X, Graham TR. Reconstitution of phospholipid translocase activity with purified Drs2p, a type-IV P-type ATPase from budding yeast. Proc Natl Acad Sci U S A 2009; 106(39):16586-91; PMID:19805341; http://dx.doi.org/ 10.1073/pnas.0904293106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lopez-Marques RL, Poulsen LR, Bailly A, Geisler M, Pomorski TG, Palmgren MG. Structure and mechanism of ATP-dependent phospholipid transporters. Biochim Biophys Acta 2015; 1850(3):461-75; PMID:24746984; http://dx.doi.org/ 10.1016/j.bbagen.2014.04.008 [DOI] [PubMed] [Google Scholar]
- [7].Coleman JA, Quazi F, Molday RS. Mammalian P4-ATPases and ABC transporters and their role in phospholipid transport. Biochim Biophys Acta 2013; 1831(3):555-74; PMID:23103747; http://dx.doi.org/ 10.1016/j.bbalip.2012.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bevers EM, Williamson PL. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol Rev 2016; 96(2):605-45; PMID:26936867; http://dx.doi.org/ 10.1152/physrev.00020.2015 [DOI] [PubMed] [Google Scholar]
- [9].Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 2013; 341(6144):403-6; PMID:23845944; http://dx.doi.org/ 10.1126/science.1236758 [DOI] [PubMed] [Google Scholar]
- [10].Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 2010; 468(7325):834-8; PMID:21107324; http://dx.doi.org/ 10.1038/nature09583 [DOI] [PubMed] [Google Scholar]
- [11].Hua Z, Fatheddin P, Graham TR. An essential subfamily of Drs2p-related P-type ATPases is required for protein trafficking between Golgi complex and endosomal/vacuolar system. Mol Biol Cell 2002; 13(9):3162-77; PMID:12221123; http://dx.doi.org/ 10.1091/mbc.E02-03-0172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hua Z, Graham TR. Requirement for neo1p in retrograde transport from the Golgi complex to the endoplasmic reticulum. Mol Biol Cell 2003; 14(12):4971-83; PMID:12960419; http://dx.doi.org/ 10.1091/mbc.E03-07-0463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Global analysis of protein localization in budding yeast. Nature 2003; 425(6959):686-91; PMID:14562095; http://dx.doi.org/ 10.1038/nature02026 [DOI] [PubMed] [Google Scholar]
- [14].Wicky S, Schwarz H, Singer-Kruger B. Molecular interactions of yeast Neo1p, an essential member of the Drs2 family of aminophospholipid translocases, and its role in membrane trafficking within the endomembrane system. Mol Cell Biol 2004; 24(17):7402-18; PMID:15314152; http://dx.doi.org/ 10.1128/MCB.24.17.7402-7418.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Takatsu H, Baba K, Shima T, Umino H, Kato U, Umeda M, Nakayama K, Shin HW. ATP9B, a P4-ATPase (a putative aminophospholipid translocase), localizes to the trans-Golgi network in a CDC50 protein-independent manner. J Biol Chem 2011; 286(44):38159-67; PMID:21914794; http://dx.doi.org/ 10.1074/jbc.M111.281006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wehman AM, Poggioli C, Schweinsberg P, Grant BD, Nance J. The P4-ATPase TAT-5 inhibits the budding of extracellular vesicles in C. elegans embryos. Curr Biol 2011; 21(23):1951-9; PMID:22100064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Takar M, Wu Y, Graham TR. The essential Neo1 from budding yeast plays a role in establishing aminophospholipid asymmetry of the plasma membrane. J Biol Chem 2016; 291:15727-39; PMID:27235400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Brett CL, Kallay L, Hua Z, Green R, Chyou A, Zhang Y, Graham TR, Donowitz M, Rao R. Genome-wide analysis reveals the vacuolar pH-stat of Saccharomyces cerevisiae. PloS one 2011; 6(3):e17619; PMID:21423800; http://dx.doi.org/ 10.1371/journal.pone.0017619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wickner W. Membrane fusion: five lipids, four SNAREs, three chaperones, two nucleotides, and a Rab, all dancing in a ring on yeast vacuoles. Ann Rev Cell Dev Biol 2010; 26:115-36; PMID:20521906; http://dx.doi.org/ 10.1146/annurev-cellbio-100109-104131 [DOI] [PubMed] [Google Scholar]
- [20].Hickey CM, Stroupe C, Wickner W. The major role of the Rab Ypt7p in vacuole fusion is supporting HOPS membrane association. J Biol Chem 2009; 284(24):16118-25; PMID:19386605; http://dx.doi.org/ 10.1074/jbc.M109.000737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang L, Seeley ES, Wickner W, Merz AJ. Vacuole fusion at a ring of vertex docking sites leaves membrane fragments within the organelle. Cell 2002; 108(3):357-69; PMID:11853670; http://dx.doi.org/ 10.1016/S0092-8674(02)00632-3 [DOI] [PubMed] [Google Scholar]
- [22].Mima J, Hickey CM, Xu H, Jun Y, Wickner W. Reconstituted membrane fusion requires regulatory lipids, SNAREs and synergistic SNARE chaperones. EMBO J 2008; 27(15):2031-42; PMID:18650938; http://dx.doi.org/ 10.1038/emboj.2008.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Stroupe C, Collins KM, Fratti RA, Wickner W. Purification of active HOPS complex reveals its affinities for phosphoinositides and the SNARE Vam7p. EMBO J 2006; 25(8):1579-89; PMID:16601699; http://dx.doi.org/ 10.1038/sj.emboj.7601051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Mima J, Wickner W. Complex lipid requirements for SNARE- and SNARE chaperone-dependent membrane fusion. J Biol Chem 2009; 284(40):27114-22; PMID:19654322; http://dx.doi.org/ 10.1074/jbc.M109.010223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zick M, Stroupe C, Orr A, Douville D, Wickner WT. Membranes linked by trans-SNARE complexes require lipids prone to non-bilayer structure for progression to fusion. eLife 2014; 3:e01879; PMID:24596153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jun Y, Fratti RA, Wickner W. Diacylglycerol and its formation by phospholipase C regulate Rab- and SNARE-dependent yeast vacuole fusion. J Biol Chem 2004; 279(51):53186-95; PMID:15485855; http://dx.doi.org/ 10.1074/jbc.M411363200 [DOI] [PubMed] [Google Scholar]
- [27].Seeley ES, Kato M, Margolis N, Wickner W, Eitzen G. Genomic analysis of homotypic vacuole fusion. Mol Biol Cell 2002; 13(3):782-94; PMID:11907261; http://dx.doi.org/ 10.1091/mbc.01-10-0512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fratti RA, Jun Y, Merz AJ, Margolis N, Wickner W. Interdependent assembly of specific regulatory lipids and membrane fusion proteins into the vertex ring domain of docked vacuoles. J Cell Biol 2004; 167(6):1087-98; PMID:15611334; http://dx.doi.org/ 10.1083/jcb.200409068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Keenan Curtis K, Kane PM. Novel vacuolar H+-ATPase complexes resulting from overproduction of Vma5p and Vma13p. J Biol Chem 2002; 277(4):2716-24; PMID:11717306; http://dx.doi.org/ 10.1074/jbc.M107777200 [DOI] [PubMed] [Google Scholar]
- [30].Haas A, Scheglmann D, Lazar T, Gallwitz D, Wickner W. The GTPase Ypt7p of Saccharomyces cerevisiae is required on both partner vacuoles for the homotypic fusion step of vacuole inheritance. EMBO J 1995; 14(21):5258-70; PMID:7489715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mumberg D, Muller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 1995; 156(1):119-22; PMID:7737504; http://dx.doi.org/ 10.1016/0378-1119(95)00037-7 [DOI] [PubMed] [Google Scholar]
- [32].Wood CS, Hung CS, Huoh YS, Mousley CJ, Stefan CJ, Bankaitis V, Ferguson KM, Burd CG. Local control of phosphatidylinositol 4-phosphate signaling in the Golgi apparatus by Vps74 and Sac1 phosphoinositide phosphatase. Mol Biol Cell 2012; 23(13):2527-36; PMID:22553352; http://dx.doi.org/ 10.1091/mbc.E12-01-0077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Katzmann DJ, Stefan CJ, Babst M, Emr SD. Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J Cell Biol 2003; 162(3):413-23; PMID:12900393; http://dx.doi.org/ 10.1083/jcb.200302136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Baldridge RD, Graham TR. Identification of residues defining phospholipid flippase substrate specificity of type IV P-type ATPases. Proc Natl Acad Sci U S A 2012; 109(6):E290-8; PMID:22308393; http://dx.doi.org/ 10.1073/pnas.1115725109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sato K, Sato M, Nakano A. Rer1p, a retrieval receptor for endoplasmic reticulum membrane proteins, is dynamically localized to the Golgi apparatus by coatomer. J Cell Biol 2001; 152(5):935-44; PMID:11238450; http://dx.doi.org/ 10.1083/jcb.152.5.935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Birner R, Burgermeister M, Schneiter R, Daum G. Roles of phosphatidylethanolamine and of its several biosynthetic pathways in Saccharomyces cerevisiae. Mol Biol Cell 2001; 12(4):997-1007; PMID:11294902; http://dx.doi.org/ 10.1091/mbc.12.4.997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gulshan K, Shahi P, Moye-Rowley WS. Compartment-specific synthesis of phosphatidylethanolamine is required for normal heavy metal resistance. Mol Biol Cell 2010; 21(3):443-55; PMID:20016005; http://dx.doi.org/ 10.1091/mbc.E09-06-0519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Alder-Baerens N, Lisman Q, Luong L, Pomorski T, Holthuis JC. Loss of P4 ATPases Drs2p and Dnf3p disrupts aminophospholipid transport and asymmetry in yeast post-Golgi secretory vesicles. Mol Biol Cell 2006; 17(4):1632-42; PMID:16452632; http://dx.doi.org/ 10.1091/mbc.E05-10-0912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Natarajan P, Wang J, Hua Z, Graham TR. Drs2p-coupled aminophospholipid translocase activity in yeast Golgi membranes and relationship to in vivo function. Proc Natl Acad Sci U S A 2004; 101(29):10614-9; PMID:15249668; http://dx.doi.org/ 10.1073/pnas.0404146101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu K, Hua Z, Nepute JA, Graham TR. Yeast P4-ATPases Drs2p and Dnf1p are essential cargos of the NPFXD/Sla1p endocytic pathway. Mol Biol Cell 2007; 18(2):487-500; PMID:17122361; http://dx.doi.org/ 10.1091/mbc.E06-07-0592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu K, Surendhran K, Nothwehr SF, Graham TR. P4-ATPase requirement for AP-1/clathrin function in protein transport from the trans-Golgi network and early endosomes. Mol Biol Cell 2008; 19(8):3526-35; PMID:18508916; http://dx.doi.org/ 10.1091/mbc.E08-01-0025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mima J, Wickner W. Phosphoinositides and SNARE chaperones synergistically assemble and remodel SNARE complexes for membrane fusion. Proc Natl Acad Sci U S A 2009; 106(38):16191-6; PMID:19805279; http://dx.doi.org/ 10.1073/pnas.0908694106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hua ZL, Graham TR. Requirement for Neo1p in retrograde transport from the Golgi complex to the endoplasmic reticulum. Mol Biol Cell 2003; 14(12):4971-83; PMID:12960419; http://dx.doi.org/ 10.1091/mbc.E03-07-0463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, et al.. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 2004; 21(11):947-62; PMID:15334558; http://dx.doi.org/ 10.1002/yea.1142 [DOI] [PubMed] [Google Scholar]
- [45].Jones GM, Stalker J, Humphray S, West A, Cox T, Rogers J, Dunham I, Prelich G. A systematic library for comprehensive overexpression screens in Saccharomyces cerevisiae. Nat Methods 2008; 5(3):239-41; PMID:18246075; http://dx.doi.org/ 10.1038/nmeth.1181 [DOI] [PubMed] [Google Scholar]
- [46].Vida TA, Emr SD. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol 1995; 128(5):779-92; PMID:7533169; http://dx.doi.org/ 10.1083/jcb.128.5.779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Katzmann DJ, Stefan CJ, Babst M, Emr SD. Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J Cell Biol 2003; 162(3):413-23; PMID:12900393; http://dx.doi.org/ 10.1083/jcb.200302136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sato K, Sato M, Nakano A. Rer1p, a retrieval receptor for endoplasmic reticulum membrane proteins, is dynamically localized to the Golgi apparatus by coatomer. J Cell Biol 2001; 152(5):935-44; PMID:11238450; http://dx.doi.org/ 10.1083/jcb.152.5.935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wood CS, Hung CS, Huoh YS, Mousley CJ, Stefan CJ, Bankaitis V, Ferguson KM, Burd CG. Local control of phosphatidylinositol 4-phosphate signaling in the Golgi apparatus by Vps74 and Sac1 phosphoinositide phosphatase. Mol Biol Cell 2012; 23(13):2527-36; PMID:22553352; http://dx.doi.org/ 10.1091/mbc.E12-01-0077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gulshan K, Schmidt JA, Shahi P, Moye-Rowley WS. Evidence for the bifunctional nature of mitochondrial phosphatidylserine decarboxylase: role in Pdr3-dependent retrograde regulation of PDR5 expression. Mol Cell Biol 2008; 28(19):5851-64; PMID:18644857; http://dx.doi.org/ 10.1128/MCB.00405-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.