

Abstract
Object
Glioblastoma is the most aggressive and diffusely infiltrative primary brain tumor. Recurrence is expected and is extremely difficult to treat. Over the past decade, the accumulation of knowledge regarding the molecular and genetic profile of glioblastoma has led to numerous molecularly targeted therapies. This article aims to review the literature and highlight the mechanisms and efficacies of molecularly targeted therapies for recurrent glioblastoma.
Methods
A systematic search was performed with the phrase “(name of particular agent) and glioblastoma” as a search term in PubMed to identify all articles published up until 2014 that included this phrase in the title and/or abstract. The references of systematic reviews were also reviewed for additional sources. The review included clinical studies that comprised at least 20 patients and reported results for the treatment of recurrent glioblastoma with molecular targeted therapies.
Results
A total of 42 articles were included in this review. In the treatment of recurrent glioblastoma, various targeted therapies have been tested over the past 10–15 years. The targets of interest include epidermal growth factor receptor, vascular endothelial growth factor receptor, platelet-derived growth factor receptor, Ras pathway, protein kinase C, mammalian target of rapamycin, histone acetylation, and integrins. Unfortunately, the clinical responses to most available targeted therapies are modest at best. Radiographic responses generally range in the realm of 5%–20%. Progression-free survival at 6 months and overall survival were also modest with the majority of studies reporting a 10%–20% 6-month progression-free survival and 5- to 8-month overall survival. There have been several clinical trials evaluating the use of combination therapy for molecularly targeted treatments. In general, the outcomes for combination therapy tend to be superior to single-agent therapy, regardless of the specific agent studied.
Conclusions
Recurrent glioblastoma remains very difficult to treat, even with molecular targeted therapies and anticancer agents. The currently available targeted therapy regimens have poor to modest activity against recurrent glioblastoma. As newer agents are actively being developed, combination regimens have provided the most promising results for improving outcomes. Targeted therapies matched to molecular profiles of individual tumors are predicted to be a critical component necessary for improving efficacy in future trials.
Keywords: combination treatment, glioblastoma, molecular therapy, overall survival, progression-free survival, recurrent
Glioblastoma is the most common primary malignant brain tumor among adults and unfortunately is also the most aggressive. It is a diffusely infiltrative and, almost always, recurrent brain tumor. The prognosis associated with glioblastoma is poor with median survival of 13–16 months even after resection and adjuvant chemotherapy.63,97 While the pathological diagnosis of glioblastoma is characterized by frequent mitosis, palisading nuclei, necrosis, and neovascularization, molecular and genetic studies have revealed glioblastoma to be heterogeneous and highly mutable.15 The features of molecular heterogeneity and mutability contribute to the poor response of glioblastoma to conventional DNA-damaging chemotherapies. In addition, successful treatment of glioblastoma requires anticancer agents to traverse the blood-brain barrier (BBB) and accumulate in tumor tissues at therapeutic levels; without adequate “drug uptake,” treatment will likely be ineffective.
The current landscape of glioblastoma treatment options is notable for only 2 FDA-approved systemically administered chemotherapies: 1) temozolomide, approved for treatment of newly diagnosed glioblastoma, and 2) bevacizumab, which was granted accelerated approval for treatment of recurrent glioblastoma. Before the discovery of temozolomide, maximal resection and adjuvant radiation were the standard therapy, and chemotherapy was controversial due to lack of proven efficacy. Temozolomide, an orally administered alkylating chemotherapeutic agent, has been shown to provide significant benefit in survival.55,97 In the landmark multicenter randomized clinical trial of 573 patients with newly diagnosed glioblastoma performed by Stupp et al., concomitant and adjuvant temozolomide with radiation therapy improved median survival (14.6 vs 12.1 months) and 2-year survival (26.5% vs 10.4%) relative to radiation therapy alone.99 Despite the notable accomplishment that this trial has made, the combination of temozolomide and radiation therapy (also known as the “Stupp protocol”) is unable to eradicate glioblastoma completely. Recurrence of glioblastoma after temozolomide and radiation treatment tends to be the rule and not the exception, with recurrence rates greater than 90%.113 Recurrent glioblastoma tends to be more aggressive and resistant to medical treatment than its primary counterpart.113
Currently, the only approved therapeutic agent for the treatment of recurrent glioblastoma is bevacizumab. Bevacizumab (Avastin) is a humanized monoclonal antibody that binds specifically to vascular endothelial growth factor (VEGF)–A, the most angiogenic isoform of VEGF, which disrupts VEGF binding to its receptor; this prevents the activation of the VEGF/VEGF receptor (VEGFR) axis and angiogenesis.36 In the treatment of recurrent glioblastoma, regimens combining bevacizumab with other chemotherapies like irinotecan were associated with overall survival (OS) of 4.1–9.2 months and survival rates of 77% at 6 months and 31% at 12 months in Phase II clinical trials.10,26,41,48,80–82,108 Other Phase II trials demonstrated comparable efficacy of bevacizumab monotherapy in recurrent glioblastoma treatment.41,61 On the basis of these results, the FDA granted accelerated approval to bevacizumab for the treatment of recurrent glioblastoma as monotherapy or combined with other chemotherapies in 2009. Until a randomized trial of bevacizumab is completed for recurrent glioblastoma, its Phase II results represent a benchmark against which experimental agents and regimens used to treat recurrent glioblastoma can be compared. However, the present lack of overly convincing data for bevacizumab in glioblastoma treatment makes it used less universally in the recurrent setting than temozolomide is used at the time of diagnosis. Therefore, there is an ongoing need for additional treatment regimens for recurrent glioblastoma. Molecularly targeted therapies represent an appealing therapeutic approach worthy of current investigation given the limited number of FDA-approved treatments available for recurrent glioblastoma and the limited efficacy of the treatments that are approved.
To date, multiple mutated genes, including IDH1, PTEN, p16/Arf, EGFRvIII, ERBB2, NF1, and TERT, have been identified in glioblastoma.15 Identification of these mutations and their affected downstream pathways has led to many clinical trials testing drugs designed to target the aberrant genes and pathways, and has created hopes of personalizing therapies for glioblastoma patients.15,77 It is likely that simultaneous targeting of multiple pathways will be needed to develop effective molecularly targeted treatments for glioblastoma. This article aims to review the literature and highlight the mechanisms and efficacies of molecularly targeted therapies for recurrent glioblastoma. Antiangiogenic therapies that target VEGF directly, like bevacizumab and aflibercept, were not reviewed as this distinct mechanism warrants separate consideration.93
Methods
First, a broad search on molecular targeted therapies for glioblastoma was performed. In this search, the phrase “targeted and glioblastoma” was used as a search term in PubMed for all years up to 2014. Based on this search, multiple articles were reviewed for molecular targeted therapies against glioblastoma. The criterion used in the identification of which agents would undergo a systematic search and review was the presence of at least one study evaluating its use against either newly diagnosed or recurrent glioblastoma. After identifying individual molecularly targeted agents for glioblastoma, each agent underwent a formal systematic search. The phrase “(name of agent) and glioblastoma” was used as a search term in PubMed for all years up to 2014 to identify all articles that included this phrase in the title and/or abstract. The references of systematic reviews were also reviewed for additional sources. The abstracts of each article were reviewed for studies that evaluated the efficacy of the agent for the treatment of recurrent glioblastoma. Progression of tumor was also accepted as recurrent disease. The inclusion criteria used for this final review were human studies that included at least 20 patients treated for recurrent glioblastoma. Articles excluded from the review were articles written in languages other than English, those involving cohorts of fewer than 20 patients with recurrent glioblastoma, and those lacking survival or radiographic response data. A total of 42 articles were included in this review.
Results
Receptor Tyrosine Kinase Inhibitors
Receptor tyrosine kinase inhibitors (RTKIs) are bioactive, usually aromatic, small molecules that can bind to the active site of a receptor tyrosine kinase (RTK), thus preventing phosphorylation, and by doing so, inhibit, regulate, or modulate signaling, often with cytostatic activity. Some potent RTKIs will exhibit selectivity for a certain RTK, while others are less selective. Below we classify RTKIs used to treat recurrent glioblastoma into 3 categories based on the primary RTK target: epidermal growth factor receptor (EGFR), VEGFR, and platelet-derived growth factor receptor (PDGFR).
Epidermal Growth Factor Receptor Inhibitors
The EGFR is a TKR known to drive proliferation, transformation, and migration in both normal development and cancer cells. Upon ligand binding, the monomeric cell-surface receptors form homo- and hetero-dimers that autophosphorylate and activate second messenger pathways. The phosphorylated EGFR activates signaling through the PI3K, Ras/MAPK, and STAT3 pathways driving expression of numerous genes that cause proliferation, transformation, and tumor growth. Nearly half of all glioblastomas overexpress wild-type EGFR, and a significant portion also express a mutant variant of EGFR, EGFRvIII.32 The EGFRvIII mutation causes the receptor to be constitutively active, which drives signaling through downstream pathways even in the absence of ligand.68 This observation led to the hypothesis that inhibiting signaling through the EGFR receptor could decrease the growth and proliferative potential of glioblastoma and possibly improve survival. Table 1 highlights outcomes of published studies of using EGFR inhibitors for the treatment of recurrent glioblastoma, with results from each inhibitor summarized below.
TABLE 1.
Outcomes of published studies using EGFR inhibitors for the treatment of recurrent glioblastoma
| Authors & Year | Study Type & Trial Phase | No. of Patients |
Prior Radio- & Chemotherapy Treatment |
Current Intervention & Regimen |
Radiographic Response | PFS | OS* |
|---|---|---|---|---|---|---|---|
| gefitinib | BBB permeability: limited | ||||||
| Rich et al., 2004 | Phase II, open label, single arm |
53 | radiotherapy, chemotherapy, w/ repeat surgery/biopsy documenting progression |
gefitinib | CR (0%), PR (0%), SD (42%) | 13% at 6mos | 9.1 mos |
| Reardon et al., 2006 | Phase I, clinical trial, dose escalation |
34 | radiotherapy, chemotherapy | gefitinib & sirolimus | CR (0%), PR (5%), SD (38%) | 23% at 6mos | not reported |
| Doherty et al., 2006 | retrospective | 28 | radiotherapy, chemotherapy | gefitinb or erlotinib plus sirolimus |
CR(0%), PR (19%), SD(50%) | 25% at 6mos | not reported |
|
| |||||||
| erlotinib | BBB permeability: good | ||||||
| de Groot et al., 2008 | Phase II, clinical trial, single arm |
44 | radiotherapy, chemotherapy | carboplatin & erlotinib | CR (0%), PR (2%), SD (47%) | median 2.1 mos, 14% at 6 mos |
6.9 mos |
| van den Bent et al., 2009 |
Phase II, clinical trial, randomized, controlled |
110 | radiotherapy, chemotherapy | erlotinib vs temozolomide or carmustine (if received prior temozolomide) |
CR (0%); erlotinib arm: PR (3.7%), SD(16.7%); control: PR (9.6%), SD (34.6%) |
erlotinib: 11.4% at 6 mos; control: 24.1% at 6 mos |
erlotinib: 7.7 mos; control: 7.3 mos |
| Raizer et al., 2010 | Phase II, clinical trial, single arm |
38 | radiotherapy, chemotherapy | erlotinib | CR (0%), PR (0%), SD (8%) | median 2 mos, 3% at 6 mos |
6 mos |
| Yung et al., 2010 | Phase II, clinical trial, single arm |
48 | radiotherapy, chemotherapy | erlotinib | CR(2%), PR (4%), SD(16°/c) | 20% at 6 mos | 8.6 mos |
|
| |||||||
| cetuximab | BBB: poor | ||||||
| Neyns et al, 2009 | Phase II, clinical trial, 2 arms, noncomparative |
55 | radiotherapy, chemotherapy | cetuximab | CR(0%), PR (5%), SD (31%), | median 1.9 mos, 7.3% at 6 mos |
5.0 mos, 37.9% at 6 mos |
Values are reported as the median or mean depending on the study.
Gefitinib
Gefitinib, first approved to treat non–small cell lung cancer, was the first EGFR inhibitor to be tested in recurrent glioblastoma. Gefitinib has limited BBB permeability, but increased doses likely accelerate passive permeability.11 In an initial Phase II open-label trial, 53 patients with recurrent glioblastoma with Karnofsky Performance Scale scores greater than 60 were treated with gefinitnib in escalating doses from 500 to 1000 mg for 4-week cycles following repeat resection.89 The median progression-free survival (PFS) was 8.1 weeks, and the median overall survival (OS) was 39 weeks. No patients had radiographic response to treatment, although this finding is tempered by the fact that all patients had resection or biopsy just prior to starting therapy. Of note, 80% of tumors in this series expressed wild-type EGFR and 49% expressed mutant EGFRvIII. Interestingly, many studies of both gefitinib and erlotinib found that longer PFS times correlated with the development of a skin rash, suggesting this particular morbidity to be a potential biomarker of drug response.
Given the relatively limited efficacy of the outcomes from initial studies of EGFR inhibition, it was hypothesized that EGFR inhibition could be augmented by combining inhibitors of downstream second messenger pathways. PI3K, an EGFR target, activates the mTOR pathway, and preclinical data suggested that inhibiting the mTOR pathway could augment antitumor effects of EGFR inhibition. To test the safety of dual-agent therapy for recurrent glioblastoma, a Phase I trial combining sirolimus, an mTOR inhibitor, with gefitinib was conducted.87 The combination was well tolerated, with mucositis, diarrhea, rash, thrombocytopenia, and hypertriglyceridemia being the dose-limiting side effects. No patients had a complete response (CR), but a small subset (6%) had a partial response (PR), and many had stable disease (SD) (38%). Unfortunately, these results were not significantly better than EGFR inhibition alone, and the analysis was limited since less than half of the tumors were analyzed for EGFR expression or the EGFRvIII mutation. A retrospective study assessing response of gefitinib or erlotinib plus sirolimus showed better radiographic responses, but similar 6-month PFS of 25%.28 A Phase II, single-arm study combining gefitinib and everolimus, another mTOR inhibitor, showed a 6-month PFS of only 5% and median PFS of 2.6 months.61
Erlotinib
Erlotinib, like gefitinib, is an orally available EGFR inhibitor. It binds to the EGFR adenosine triphosphate–binding domain, preventing phosphorylation and activation of downstream targets. It was first approved for treatment of non–small cell lung cancer. Erlotinib has been shown to accumulate high concentrations in cerebrospinal fluid, indicating good penetration through the BBB.25 Erlotinib was studied in several single arm Phase II trials. Raizer et al. showed that erlotinib alone had minimal efficacy against recurrent glioblastoma, with 6-month PFS of 3% and an OS of 6 months.79 Yung et al. had slightly better results with a 6-month PFS of 20% and OS of 8.6 months.120 The authors also showed that amplification or mutation status of EGFR did not affect response. The first erlotinib combination trial for recurrent glioblastoma tested erlotinib with carboplatin and found a 6-month PFS of 14%, and median OS of 6.9 months.22 Interestingly, molecular analysis revealed no significant correlation between Akt, PTEN, EGFR, or EGFRvIII expression and survival, although increased EGFR expression had a trend toward longer survival. These studies are consistent with findings from a randomized controlled trial comparing erlotinib to combined temozolomide and carmustine. In that study, van den Bent et al.105 found that median survival and 6-month PFS was worse with erlotinib (1.8 months, 11.4%) compared with combined temozolomide and carmustine (2.4 months, 24.1%). However, there was no difference in OS between the 2 groups (erlotinib: 7.7 months vs temozolomide and carmustine: 7.3 months). Molecular analysis showed that EGFRvIII mutations correlated with poor survival in the erlotinib group, but not in the control arm. Low pAkt expression, albeit in a small number of subjects, trended toward higher PFS in the erlotinib-treated patients, but not in the control arm. This suggested that there may be a particular subset of patients who might best respond to erlotinib and raised the idea that blocking downstream EGFR signaling pathways might sensitize some tumors to EGFR inhibition.
Cetuximab
In addition to small molecule inhibitors, EGFR signaling can also be inhibited by antibodies that bind to the receptor, trigger internalization, and downregulate its expression. Cetuximab is a chimeric monoclonal antibody against EGFR shown to have efficacy in colon cancers with wild-type KRAS. Due to its large size, cetuximab has limited permeability across the BBB.24 In a Phase II clinical trial enrolling 55 patients with recurrent glioblastoma in 2 arms (one included those with EGFR amplification and the other without), median time to progression was 1.9 months.70 Six-month PFS was 7.3%, and the median OS was 5.0 months. Despite a trend toward longer survival in EGFR-amplified subjects, the differences between the groups were not significant. To investigate what molecular factors might be predictive of a good response to cetuximab, further molecular analysis was done on 35 of the 55 subjects in the cetuximab trial.66 The authors found that patients with an EGFRvIII mutation and EGFR amplification had significantly shorter PFS compared with patients with EGFR amplification alone (1.6 vs 3.0 months), and trended toward shorter OS, suggesting that cetuximab may be more effective in tumors that have EGFR amplification without the EGFRvIII mutation. They also found that almost all tumors with the favorable IDH1 mutations did not harbor amplification or mutation of the EGFR pathway.
Vascular Endothelial Growth Factor Receptor Inhibitors
The tumor microenvironment plays a significant role in the pathogenesis of glioblastoma. In particular, angiogenesis is a fundamental process in tumor growth and dissemination, allowing supply of oxygen, nutrients, growth factors, and hormones to tumor cells.38 Extent of angiogenesis and microvascular proliferation has been associated with worse survival and higher rates of recurrence.12 One of the most potent growth factors mediating angiogenesis is VEGF, which is produced mostly by tumor cells and recruits and stimulates the growth of endothelial cells, leading to increased tumor vascularity.38,51 VEGF can be activated by multiple stimuli, including a hypoxic environment via hypoxia-inducible factor-1 (HIF-1), acidosis, growth factors (EGFR, PDGF, and insulin-like growth factor–1), and prostaglandins.33,35,44 The binding of VEGF to the VEGFR leads to activation of PLC-PKC-Raf kinase-MEK-MAPK and PI3K-Akt pathways, which promotes endothelial cell survival, differentiation, migration, and processes aiding in the formation of the tumor micro-vasculature.59 Inhibition of VEGF signaling is felt to cause vascular normalization.5,37,46,54,112 Due to its fundamental role in tumor progression, inhibition of the VEGF and VEGFR pathway has been a major focus of research, drug discovery, and clinical treatment. In the case of glioblastoma, VEGF levels are shown to be high and VEGF overexpression has been associated with poor prognosis.69 In addition, patients with recurrent glioblastomas tend to have higher levels of VEGF, making this a promising target in the treatment of patients with recurrent glioblastoma.91 The VEGF pathway can be targeted by direct VEGF inhibition or by inhibiting its receptor, VEGFR. Table 2 highlights outcomes of published studies using VEGFR inhibitors to treat recurrent glioblastoma, with results using specific inhibitors summarized below.
TABLE 2.
Outcomes of published studies using VEGFR inhibitors for the treatment of recurrent glioblastoma
| Authors & Year | Study Type & Trial Phase |
No. of Patients | Prior Treatment | Current Intervention & Regimen |
Radiographic Response | PFS | OS |
|---|---|---|---|---|---|---|---|
| cediranib | BBB permeability: good but active efflux out via transporter | ||||||
| Batchelor et al, 2010 |
Phase II clinical trial, single arm |
31 | variable | cediranib | PR (57%), minor (20%) | 26% at 6 mos | 7.6 mos |
| Batchelor et al, 2013 |
Phase III clinical trial, randomized, pla- cebo controlled, partially blinded |
325: cediranib (n = 131), cediranib, lomustine (n = 129), lomustine, placebo (n = 65) |
radiotherapy, temozolomide |
cediranib or cedi- ranib + lomustine or lomustine + placebo |
cediranib: CR (1%), PR (14%), SD (64%), PD (9%); cediranib + lomus- tine: CR (2%), PR (16%), SD (55%), PD (16%); lomustine + placebo: CR (0%), PR (9%), SD (41%), PD (41%) |
cediranib: median 92 days; cediranib + lomustine: median 125 days; lomustine + placebo: median 82 days |
cediranib: 8.0 mos; cediranib + lomustine: 9.4 mos; lomustine + placebo 9.8 mos |
Cediranib
Cediranib is a pan-VEGFR TKRIs with additional activity against PDGF β and c-Kit. Cediranib is orally available and dosed daily due to its 22-hour half-life.111 Cediranib is able to traverse the BBB, but active efflux via BBB transporters results in limited cerebrospinal fluid concentrations.110 There are few clinical trials of cediranib for recurrent glioblastoma. In one Phase II clinical trial of 31 patients with recurrent glioblastoma, cediranib was shown to be associated with encouraging proportions of radiographic responses (at least 77% response rate) and 6-month PFS (26%).1 The same group then completed a randomized, Phase II, placebo-controlled, partially blinded clinical trial to determine the efficacy of cediranib in patients with recurrent glioblastoma.2 This 3-arm trial randomized 325 patients to cediranib monotherapy (131 patients), cediranib combined with lomustine (129 patients), and lomustine monotherapy (65 patients). Unfortunately, the trial did not demonstrate a significant benefit in using cediranib alone or in combination with lomustine when compared with lomustine alone. Radiographic response (15% vs 18% vs 9%), median PFS (92 days vs 125 days vs 82 days), and OS (8.0 months vs 9.4 months vs 9.8 months) were all similar. In addition, the combination of cediranib and lomustine increased the incidence of hematological toxicities.
Platelet-Derived Growth Factor Inhibitors
PDGF and PDGF receptors (PDGFRs) are frequently overexpressed in glioblastoma, with certain subtypes of glioblastoma having very high levels.107 The pathways that PDGFR activate are similar to the EGFR axis: PI3K-Akt-mTOR and RAS-RAF-MEK-MAPK. Like EGF signaling, PDGF signaling activates pathways associated with cellular proliferation and growth, but there is also evidence that PDGF is involved in the regulation of angiogenesis.75,76,118 In addition, alterations in PDGF signaling are believed to play a pivotal role in autocrine signaling that promote malignant transformation of normal neural stem cells into glioblastoma.14,20,50,65,94,104 Given the importance of PDGF in glioblastoma pathology, there have been various clinical studies of utilizing molecular agents that target the PDGF axis in the treatment for glioblastoma. Table 3 highlights outcomes of published studies of using PDGFR inhibitors for the treatment of recurrent glioblastoma, which are also summarized below.
TABLE 3.
Outcomes of published studies using PDGFR inhibitors for the treatment of recurrent glioblastoma*
| Authors & Year | Study Type & Trial Phase |
No. of Patients | Prior Treatment | Current Intervention & Regimen |
Radiographic Response |
PFS | OS |
|---|---|---|---|---|---|---|---|
| imatinib | BBB permeability: poor & active efflux out via transporter | ||||||
| Dresemann, 2005 | Phase II clinical trial, single arm |
30 | radiotherapy, temozolo- mide, nitrosurea |
imatinib + hydroxyurea | CR or PR (20%) | 32% at 6 mos, 16% at 24mos |
4.8 mos |
| Reardon et al., 2005 |
Phase II clinical trial, single arm |
33 | at least temozolomide | imatinib + hydroxyurea | CR (3%), PR (6%), SD (42%), PD (48%) |
27% at 6 mos, median 3.5 mos |
12.2 mos |
| Wen et al., 2006 | Phase II clinical trial, single arm |
34 | did not specify | imatinib | PR (6%) | 3% at 6 mos | NA |
| Reardon et al., 2009 |
Phase II clinical trial, parallel arm |
231 | did not specify | imatinib + hydroxyurea | CR or PR (8%) | 11% at 6 mos, median 26.0 mos |
6.3 mos |
| Dresemann et al., 2010 |
Phase III clinical trial, randomized, multi- center, two arm |
240: hydroxyurea (n = 120); imatinib + hydroxyurea (n = 120) |
did not specify | hydroxyurea alone or imatinib + hydroxy- urea |
hydroxyurea: CR or PR (1%); hydroxyurea + imatinib: CR or PR (2%) |
hydroxyurea: 7% at 6 mos; hydroxyurea + imatinib: 5% at 6 mos |
hydroxyurea: 4.8 mos; hydroxyurea + ima- tinib: 5.3 mos |
|
| |||||||
| sorafenib | BBB permeability: good | ||||||
| Reardon et al, 201188 |
Phase II clinical trial, single arm |
32 | variable | sorafenib + temozolo- mide |
CR (0%), PR (3%), SD (47%), PD (50%) |
9% at 6 mos, median 6.4 mos |
10.4 mos |
| Zustovich et al, 2013 |
Phase II clinical trial, single arm |
43 | radiotherapy, temozolo- mide |
sorafenib + temozolo- mide |
CR(0%), PR (12%), SD (48%), PD (48%) |
26% at 6 mos, median 3.2 mos |
7.4 mos |
| Peereboom et al, 2013 |
Phase II clinical trial, single arm |
56 | did not specify | erlotinib + sorafenib | CR (0%), PR (5%), SD (41%), PD (45%) |
14% 6 mos, median 2.5 mos |
5.7 mos |
|
| |||||||
| sunitinib | BBB permeability: good but active efflux out via transporter | ||||||
| Kreisl et al., 2013 |
Phase II clinical trial, parallel arm |
63: bevacizumab resis- tant (n = 31); bevaci- zumab naive (n = 32) |
radiotherapy, temozolo- mide, w/ or w/o beva- cizumab |
sunitinib | bevacizumab resis- tant: CR or PR (0%); bevacizumab naive: CR or PR (10%) |
bevacizumab resis- tant: 0% at 6 mos; bevacizumab naive: 6% at 6 mos |
bevacizumab resistant: 4.4 mos; bevacizum- ab naive: 9.4 mos |
NA = not available.
Imatinib
Imatinib is one of the most well-known molecular targeted therapies because of its efficacy in treating BCR-ABL mutated chronic myelogenous leukemia.52 Imatinib is a small molecule inhibitor of PDGF α and β; it also acts on c-KIT, ABL, BCR-ABL, and arginine tyrosine kinase. There are several clinical trials evaluating the efficacy of imatinib combined with hydroxyurea in the treatment of glioblastoma. It is thought that imatinib works through several possible mechanisms: direct inhibition of the PDGFR, increased capillary-to-interstitium transport leading to enhanced chemotherapy delivery,74 inhibition of angiogenesis,64 and diminish tumor cell DNA repair ability.90 Unfortunately, imatinib has poor penetration through the BBB, and it has been shown that CSF concentrations are 92-fold lower than blood.101
Since 2005, there have been multiple clinical trials examining the efficacy of imatinib in the treatment of recurrent glioblastoma.30,31,84,85,114 Unfortunately, the results of those trials have not been promising. Radiographic responses (CR or PR) range from 1% to 20%. The 6-month PFS ranged from 5% to 32% and OS ranged from 4.8 to 6.3 months. The results of the first Phase II clinical trial by Dresemann et al. were promising with a radiographic response rate (CR and PR) of 20% and 6-month PFS of 32%30 However, subsequent clinical trials were unable to demonstrate similar outcomes. In fact, Dresemann and colleagues conducted a multicenter, randomized, Phase III clinical trial comparing hydroxyurea alone to imatinib with hydroxyurea in 240 patients with recurrent glioblastoma, and there was no significant benefit in adding imatinib with respect to radiographic response, PFS, and OS.31 In that study, patients treated with imatinib and hydroxyurea therapy achieved radiographic response in only 2% of cases. In addition, PFS was only 5% and OS was 5.3 months.
Among the clinical trials reviewed, there was one study that observed a median OS of 12.2 months.85 This median survival was an outlier as other studies consistently demonstrated OS of 4.8–6.3 months.30,31,84,114 Of note, CYP3A4-inducing medications like phenytoin can substantially decrease circulating levels of imatinib. Therefore, if different studies do not carefully adjust or control for this factor, differences in outcomes may be observed. Nonetheless, the data supporting the use of imatinib as a therapy for recurrent glioblastoma are poor. Additional studies on its use as an adjuvant with other agents are ongoing.
Sorafenib
Sorafenib was approved by the FDA in 2005 for the treatment of advanced renal cell carcinoma. Sorafenib is a tyrosine kinase inhibitor that acts on a diverse spectrum of biologically active targets including VEGFR, PDGFR, and Raf kinases such as c-Raf and b-Raf.95,116 Theoretically, such an agent with multiple mechanisms of anticancer activity might be effective in glioblastoma. In addition, sorafenib has good BBB penetration.57 There are a few Phase I studies to date examining sorafenib for the treatment of recurrent glioblastoma, but there are no Phase II clinical trials testing its efficacy as a single agent therapy for glioblastoma.73,88,122 Among the 3 identified studies, 2 studies combined sorafenib and temozolomide as a combination salvage therapy and 1 study combined erlotinib and sorafenib. In the studies of combined sorafenib and temozolomide salvage therapy, the radiographic response rates, PFS, and OS are similar to dose-intense temozolomide salvage therapy, suggesting that the addition of sorafenib has limited activity against recurrent glioblastoma.72,88,106,122 Overall, sorafenib-based regimens were associated with radiographic response rates of only 3%–12%, 6-month PFS of 9%–26%, and OS of 5.7–10.4 months. Some of the studies included patients that had undergone prior antiangiogenic treatment, clouding interpretation of their results.88
Sunitinib
Sunitinib is a nonspecific RTKI initially approved by the FDA for the treatment of renal cell carcinoma and imatinib-resistant gastrointestinal tumors. Sunitinib is a small molecule inhibitor of many receptors, primarily PDGFR and VEGFR, and proteins involved in tumorgenesis.13 Sunitinib also inhibits stem cell–like factor receptor, c-KIT, and colony stimulating factor-1 receptor.13 Because of simultaneous inhibition of multiple targets, sunitinib can induce tumor regression through both reduced tumor vascularization and direct induction of cancer cell death, but at the same time the risk for side effects are higher. While sunitinib is able to readily bypass the BBB, it is actively transported out of the BBB leading to low cerebrospinal fluid concentrations.102 There are 2 studies of sunitinib for glioblastoma.21,62 In vivo studies of human glioblastoma in mice have demonstrated that sunitinib has direct antiproliferative effects (decrease MIB-1 staining) and decreases microvessel density by 74%.21 One clinical trial tested daily sunitinib in the treatment of recurrent glioblastoma. The authors included 63 patients who underwent daily sunitinib treatment.62 Patients were stratified into bevacizumab-resistant and bevacizumab-naive groups. In comparison of the 2 groups, bevacizumab-naive patients consistently had better outcomes than bevacizumab-resistant patients in regard to radiographic response (10% vs 0%), PFS (6% vs 0%), and OS (9.4 months vs 4.4 months). While this study highlights the importance of resistance following antiangiogenic therapy, overall, sunitinib did not offer significant improvement in the measured outcomes.
Ras Pathway Inhibitors
The Ras pathway and its associated proteins are key regulators of normal cell growth and malignant transformation. Upregulation or overexpression of Ras contributes to tumorgenesis in a variety of cancers.29 Ras mutations are found in up to 25% of cancers and up to 90% in lung adenocarcinoma and pancreatic adenocarcinoma. Although Ras mutations are rare in glioblastoma, inhibition of Ras can still be effective because of its role as a second messenger in growth factor signaling cascades.77 Ras is downstream of EGFR, PDGFR, and VEGFR and therefore is intricately involved in the pathogenesis of malignant gliomas. Table 4 highlights outcomes of published studies of using Ras inhibitors for the treatment of recurrent glioblastoma.
TABLE 4.
Outcomes of published studies using Ras inhibitors for the treatment of recurrent glioblastoma
| Authors & Year | Study Type & Trial Phase |
No. of Patients |
Prior Treatment | Current Intervention & Regimen |
Radiographic Response |
PFS | OS |
|---|---|---|---|---|---|---|---|
| Tipifarnib | BBB permeability: unknown | ||||||
| Cloughesy et al., 2006 |
Phase II clinical trial, single arm |
67 | at least radiotherapy, did not specify chemotherapy |
tipifarnib | CR (0%), PR (7%) | 9% at 6 mos | NA |
|
| |||||||
| Lonafarnib | BBB permeability: adequate | ||||||
| Yust-Katz et al., 2013 |
Phase Ib clinical trial |
34 | variable, but not w/ lona- farnib or tipifarnib |
lonafarnib + temo- zolomide |
CR (6%), PR (18%), SD (47%), PD (29%) |
38% at 6 mos, median 3.9 mos |
13.7 mos |
Tipifarnib
Ras is posttranscriptionally modified by farnesyltransferase before being translocated to the cell membrane as an active protein. Therefore, inhibiting farnesyltransferase decreases available activated Ras. Several in vitro studies of glioblastoma have demonstrated that farnesyltransferase inhibitors significantly decrease proliferation, cause cell cycle arrest, and even stimulate apoptosis.34 Tipifarnib is a potent and selective inhibitor of farnesyltransferase. There is only one Phase II clinical trial by Cloughesy et al. that has tested tipifarnib as a treatment for recurrent high-grade gliomas (glioblastoma and anaplastic astrocytoma).16 Among the 67 patients with recurrent glioblastoma, only 7% had radiographic response and 6-month PFS was 9%, a relatively unfavorable result compared with historical controls. The BBB permeability of tipifarnib has yet to be formally investigated.
Lonafarnib
Similar to tipifarnib, lonafarnib is a selective inhibitor of farnesyltransferase, and thereby induces its activity through inhibition of the Ras pathway. The BBB permeability of lonafarnib has yet to be formally studied, but in a Phase I clinical trial of pediatric brain tumors, lonafarnib seemed to demonstrate adequate permeability of the BBB.60 In a Phase Ib clinical trial testing the efficacy of combination lonafarnib and temozolomide for recurrent glioblastoma, outcomes were relatively promising.121 Of 34 patients, CR was achieved in 6% of patients and PR was seen in 18% of patients, a 20% radiographic response rate. The 6-month PFS was 38%. The clinical efficacy of tipifarnib and lonafarnib cannot be directly compared because tipifarnib was studied as a single agent and lonafarnib was combined with temozolomide. However, based on prior studies of temozolomide monotherapy for recurrent glioblastoma, one can extrapolate a potential monotherapy effect with lonafarnib that seems greater than tipifarnib and worthy of further study.106
Protein Kinase C Inhibitors
Protein kinase C (PKC) is a group of cytoplasmic serine/threonine kinases that phosphorylate several target proteins in multiple cellular signaling pathways that regulate cell survival, apoptosis, and stimulation of tumor cell proliferation.77 PKC is also intrinsically involved with the regulation of angiogenesis through VEGF.117 PKC is upregulated in high-grade gliomas when compared with normal astrocytes, and this upregulation correlates with greater tumor proliferation,18 making PKC a candidate for the development of molecularly targeted therapies. Table 5 highlights outcomes of published studies of using PKC inhibitors for the treatment of recurrent glioblastoma.
TABLE 5.
Outcomes of published studies using PKC inhibitors for the treatment of recurrent glioblastoma
| Authors & Year | Study Type & Trial Phase |
No. of Patients | Prior Treatment | Current Intervention & Regimen |
Radiographic Response |
PFS | OS |
|---|---|---|---|---|---|---|---|
| tamoxifen | BBB:good, lipid soluble | ||||||
| Couldwell et al., 1996 | Phase II clinical trial, single arm |
20 | variable | tamoxifen | CR or OR (20%) | NA | 7.2 mos |
| Brandes et al., 1999 | Phase II clinical trial, single arm |
28 | variable | procarbazine, tamoxi- fen |
CR (4%), PR (25%), SD (32%c) |
median 3.3 mos | 6.8 mos |
| Di Cristofori et al., 2013 | retrospective study | 32 | radiotherapy, temo- zolomide |
tamoxifen, temozolo- mide |
CR (0%), PR or SD (41%), PD(59%) |
median 9.5 mos | 17.5 mos |
|
| |||||||
| enzastaurin | BBB: adequate, lipid soluble | ||||||
| Wick et al., 2010 | Phase III clinical trial, comparative trial |
266: enzastaurin (n = 174); lomustine (n = 92) |
did not specify | enzastaurin or lomust- ine |
NA | enzastaurin: 11 % at 6 mos, median 1.5 mos; lomustine: 19% at 6 mos, median 1.6 mos |
enzastaurin: 6.6 mos; lomustine: 7.1 mos |
Tamoxifen
Tamoxifen is a nonsteroidal agent most well known in the treatment of estrogen receptor–positive breast cancer and is highly lipid soluble; thus it readily crosses the BBB. In addition to inhibition of estrogen signaling, tamoxifen also inhibits other pathways, including PKC. In vitro studies have demonstrated that inhibition of PKC by tamoxifen increases cellular apoptosis, and at high dose, can even reverse chemoresistance.67,78 There are only a few studies examining the activity of tamoxifen for recurrent glioblastoma.4,19,27 Two Phase II clinical trials from the 1990s by Couldwell et al. and Brandes et al. tested the efficacy of tamoxifen for recurrent high-grade gliomas. Couldwell et al. tested high-dose tamoxifen alone in 20 patients with recurrent glioblastoma and Brandes et al. tested high-dose tamoxifen with procarbazine in 28 patients with recurrent glioblastoma.4,19 Both studies showed relatively high rates of radiographic responses; combined CR and PR rates were 20%–25%. However, the studies showed OS rates of 6.8 and 7.2 months, and time to progression was 3.3 months. A more recent study demonstrated much more promising results, with a PR and SD rate of 41% and median time to progression of 9.5 months.27 In addition, remarkably, OS was 17.5 months. The study also found no difference in OS and time to progression between glioblastoma patients with methylated vs unmethylated MGMT. Although these results were quite promising, the findings need to be interpreted with caution as the study was retrospective and involved a specific subset of patients. The study had strict eligibility criteria, each of which selected for patients with better prognosis: Karnofsky Performance Scale score higher than 70, high rates of gross-total or subtotal resection (no biopsy), and exclusion of multifocal lesions. Future clinical trials should consider testing tamoxifen more rigorously in a heterogeneous subset of patients.
Enzastaurin
Enzastaurin is small molecular inhibitor of PKCb that has been used for the treatment of a variety of tumors. This agent is lipid soluble and thus has adequate BBB permeability. In an early phase clinical trial, encouraging radiographic response rates were seen in recurrent high-grade gliomas treated with single agent enzastaurin, which led to a follow-up Phase III clinical trial by Wick et al.115 In the Phase III clinical trial, monotherapy enzastaurin was compared with monotherapy lomustine in 266 patients with recurrent glioblastoma. Enrollment was terminated early secondary to the lack of enzastaurin efficacy compared with lomustine. Six-month PFS for enzastaurin was 11% and median PFS was 1.5 months. OS was 6.6 months.
mTOR Inhibitors
The PI3K/AKT pathway is activated through ligand binding to RTK. Downstream signaling activates mammalian target of rapamycin (mTOR), which controls cell growth and cell cycle progression.77,92 Normally the PI3K/ AKT pathway is downregulated by PTEN. PTEN tumor suppressor mutations and deletions lead to unregulated PI3K/AKT activation and increased mTOR activity. PTEN mutations and deletions are found in 30%–40% of glioblastomas.109 The inhibition of mTOR potentially can lead to cell cycle arrest, apoptosis, and overall reduced proliferation.9 Table 6 highlights outcomes of published studies using mTOR inhibitors for the treatment of recurrent glioblastoma, with results from specific inhibitors summarized below.
TABLE 6.
Outcomes of published studies using mTOR inhibitors for the treatment of recurrent glioblastoma
| Authors & Year | Study Type & Trial Phase | Number of Patients |
Prior Treatment | Current Intervention & Regimen |
Radiographic Response | PFS | OS |
|---|---|---|---|---|---|---|---|
| temsirolimus | BBB permeability: unknown | ||||||
| Chang et al., 2005 | Phase II clinical trial, single arm | 43 | radiation | temsirolimus | CR (0%), PR (5%), SD (47%), PD (48%Q |
2% at 6 mos, median 2.3 mos |
NA |
| Galanis et al., 2005 | Phase II clinical trial, single arm | 65 | variable | temsirolimus | CR or PR (0%) | 8% at 6 mos, median 4.4 mos |
4.4 mos |
|
| |||||||
| sirolimus | BBB permeability: adequate | ||||||
| Reardon et al., 2010 | Phase II clinical trial, single arm | 32 | variable | sirolimus + erlotinib | CR (0%), PR (0%), SD (47%), PD (53%) |
3% at 6 mos, median 1.5 mos |
8.5 mos |
|
| |||||||
| everolimus | BBB permeability: good, lipophilic | ||||||
| Kreisl et al., 2009 | Phase II clinical trial, single arm | 22 | did not specify | everolimus + gefitinib | CR (0%), PR (14%c), SD (36%), PD (50%) |
6 mos (5%t), median (2.6 mos) |
5.8 mos |
Temsirolimus
Temsirolimus is an analog of rapamycin (sirolimus). Temsirolimus interacts with mTOR and inhibits mTOR-mediated RNA translation.92 Two independent Phase II clinical trials published in 2005 tested the efficacy of temsirolimus monotherapy for recurrent glioblastoma.9,42 Chang et al.9 enrolled 43 patients, and Galanis et al.42 enrolled 65 patients. Temsirolimus was only able to achieve radiographic responses in 5% and 0% of patients between the 2 studies. Six-month PFS and median PFS were both low at 2%–8% and 2.3–4.4 months, respectively. Based on these available studies, temsirolimus monotherapy seems to have limited activity against recurrent glioblastoma, although there is a lack of large studies examining its efficacy in combination with other agents. Finally, while temsirolimus has been shown to traverse the BBB in rats, its ability to cross the BBB in humans remains unknown and could contribute to its limited efficacy.
Sirolimus
Sirolimus, also known as rapamycin, was first discovered and isolated from a species of Streptomyces and since then has been commonly used as an immunosuppressant for organ transplant. Sirolimus binds to mTOR and prevents its activation. Given its large natural structure, there was initial concern that sirolimus might not be able to traverse the BBB; however, sufficient levels of sirolimus can be found in brain tumors when administered at therapeutic doses.17 There is one Phase II clinical trial conducted by Reardon et al.83 that combined sirolimus and erlotinib for the treatment of recurrent glioblastoma. Radiographic response rate and PFS were discouraging; the CR and PR rates were 0% and 6-month PFS was 3% (median 1.5 months); these results are similar to monotherapy temsirolimus. OS was 8.5 months.
Everolimus
Everolimus is a specific mTOR inhibitor with lipophilic properties that allow it to readily traverse the BBB. The use of everolimus with bevacizumab as a combined modality therapy for newly diagnosed glioblastoma has shown some efficacy; the PFS compared favorably to previous reports of standard radiation and temozolomide therapy.47 However, as a treatment for recurrent glioblastoma, its effects are limited. In a Phase II clinical trial of 22 patients with recurrent glioblastoma, combined everolimus and gefitinib was associated with moderate radiographic response: 14% PR and 36% SD. However, this effect was not durable and the 6-month PFS was 5% and median PFS was 2.6 months. OS was poor as well (5.8 months).
Histone Acetylation Inhibitors
Histone acetylation plays an essential role in epigenetic modification and gene regulation.39 Histone acetylases and histone deacetylases are enzymes that modify lysine residues on histone core proteins. Acetylation of the histone core changes the positive charge to negative, leading to chromatin relaxation and greater levels of transcription.77 Targeting these enzymes results in widespread changes in gene expression profiles involved in cell survival, proliferation, differentiation, cell cycle arrest, and apoptosis.56 Table 7 highlights the outcomes of published studies using histone acetylation inhibitors for the treatment of recurrent glioblastoma.
TABLE 7.
Outcomes of published studies using histone acetylation inhibitors for the treatment of recurrent glioblastoma
| Authors & Year | Study Type & Trial Phase |
No. of Patients |
Prior Treatment | Current Intervention & Regimen |
Radiographic Response | PFS | OS |
|---|---|---|---|---|---|---|---|
| Vorinostat | BBB: adequate in mouse models | ||||||
| Galanis et al., 2009 | Phase II clinical trial, single arm |
66 | did not specify | vorinostat | CR or PR (3%) | 17% at 6 mos, median 1.9 mos |
5.7 mos |
| Friday et al., 2012 | Phase II clinical trial, single arm |
37 | did not specify | vorinostat + bortezomib | NA | 0% at 6 mos, median 1.5 mos |
3.2 mos |
Vorinostat
Vorinostat is a small molecule (composed of a linear suberoylanilide hydroxamic acid) that acts as a specific inhibitor against human Class I and II deacetylases. The inhibition of deacetylase leads to the tight coiling of chromatin and subsequent silencing of gene expression. Several preclinical studies have shown that vorinostat is able to adequately penetrate the BBB and has antitumor properties in glioma models.103,119 Vorinostat has been tested as monotherapy and in combination with other agents for treatment of recurrent glioblastoma.40,43 Galanis et al. reported results of a Phase II clinical trial testing the efficacy of vorinostat in 66 patients with recurrent glioblastoma. Total CR or PR occurred only in 3% of patients. The PFS at 6 months was 17% and the median PFS was 1.9 months. OS was 5.7 months. Friday et al. hypothesized that combination therapy may enhance vorinostat efficacy and performed a Phase II clinical trial to evaluate the treatment effects of combination vorinostat and bortezomib in 37 patients with recurrent glioblastoma.40 The trial was terminated secondary to no patients obtaining 6-month PFS on interim analysis. OS was 3.2 months. In addition, the authors demonstrated that patients who had received prior bevacizumab therapy had a shorter time to progression and shorter OS.
Integrin Inhibitors
Integrins are a large family of cell surface receptors that bind extracellular matrix proteins via an arginine-glycine-aspartic acid peptide. The interaction of integrins with the extracellular matrix activates a number of intracellular signaling pathways involved in cell proliferation, differentiation, motility, and other essential cell functions.100 It has been shown that glioblastoma and its associated vasculature widely express integrins.3 Preclinical studies have demonstrated that integrin inhibition induces apoptosis of human glioma cells and that integrin inhibitors prolong survival of immunodeficient mice carrying glioma xenografts.7,10,53 Table 8 highlights outcomes of published studies of using integrin inhibitors for the treatment of recurrent glioblastoma.
TABLE 8.
Outcomes of published studies using integrin inhibitors for the treatment of recurrent glioblastoma
| Authors & Year | Study Type & Trial Phase |
No. of Patients | Prior Treatment | Current Intervention & Regimen |
Radiographic Response |
PFS | OS |
|---|---|---|---|---|---|---|---|
| cilengitide | BBB permeability: adequate | ||||||
| Reardon et al., 2008 | Phase II clinical trial, randomized, parallel arm |
81: cilengitide high dose (n = 40); cilengitide low dose (n = 41) |
surgery, radiotherapy, no more than 1 chemotherapy |
cilengitide high dose or cilengitide low dose |
cilengitide high dose: CR (0%), PR (13%); cilengitide low dose: CR (0%), PR (5%) |
cilengitide high dose: 15% at 6 mos, median 2.0 mos; cilengitide low dose: 10% at 6 mos, median 2.0 mos |
cilengitide high dose: 9.9 mos; cilengitide low dose: 6.5 mos |
| Gilbert et al., 2012 | Phase II clinical trial, single arm |
30 | did not specify | cilengitide + surgery | NA | 12% at 6 mos, median 2 mos | NA |
Cilengitide
Cilengitide is a cyclic pentapeptide that competitively binds both avb3 and avb5 integrins, inhibiting its function. Compared with historical controls, the addition of concomitant and adjuvant cilengitide to standard chemo-radiation has demonstrated promising activity in glioblastoma patients whose tumors exhibit MGMT promoter methylation.98 In addition, its ability to penetrate the BBB and accumulate at therapeutic levels is adequate.98 Reardon et al.86 performed a randomized Phase II clinical trial of high and low doses of cilengitide for the treatment of recurrent glioblastoma in 81 patients. Modest antitumor activity was observed in the high-dose cilengitide group. PR was seen in 13% of patients. Sixmonth PFS was 15% and median PFS was 2.0 months. OS was encouraging at 9.9 months. The low-dose cilengitide group consistently had significantly worse outcomes. Similarly, Gilbert et al.45 performed a Phase II clinical trial of cilengitide and reoperation for recurrent glioblastoma, showing a similar 6-month PFS of 12% and median PFS of 2 months. Patients were treated prior to surgery, and the authors confirmed that drug delivery and retention in the tumor was adequate.
Discussion
While the prognosis for glioblastoma patients has improved in recent years, OS remains short and there continues to be a dire need for novel therapies. The comprehensive molecular profiling of glioblastoma compiled by The Cancer Genome Atlas (TCGA) has revealed many pharmacological targets and has ushered in the possibility of personalized cancer therapies for glioblastoma based on tumor pathway profiles.6 Unfortunately, the clinical responses to most available molecular targeted therapies are modest at best, especially for recurrent glioblastoma. In the treatment of recurrent glioblastoma, radiographic response rates range from 5% to 20%.2,9,16,19,23,28,30,31,61,62,70,73,84–87,120,122 PFS at 6 months and OS are also quite poor with a majority of studies reporting ranges of 10%–20% and 5–8 months, respectively.
Causes for Failures of Targeted Therapies for Glioblastoma
Based on the results of the clinical trials reviewed in this article, mTOR inhibitors, histone deacetylase inhibitors, and RTK inhibitors have limited activity against recurrent glioblastoma. Additional studies on specific Ras inhibitors are needed, and therefore it is difficult to assess its efficacy and activity against recurrent glioblastoma. There are several potential reasons for these molecular targeting agents to fail. In the case of mTOR inhibitors, molecular screening for potentially high yield subsets of tumors, such as those with PTEN mutations, may be needed to identify optimal tumors to treat. In addition, there is concern that most mTOR inhibitors may not penetrate the BBB. The concern for poor BBB permeability of anticancer compounds is not isolated to only mTOR inhibitors, but applies to most agents that are not lipophilic. However, the BBB is often disrupted near the tumor, and some authors suggest that permeability becomes less of an issue. In the case of histone deacetylation inhibitors, the reasons for failure are not entirely clear, but studies to date have also been limited in scale.
In the case of RTK inhibitors, it was initially believed that the relative nonspecificity of these agents allow them to target multiple RTKs and render them effective as monotherapy. However, this was discouraged by laboratory findings in 2007 that demonstrated concomitant activation of multiple RTKs in primary glioblastoma cells lines, with all such lines having activation of 3 or more RTKs screened.96 In fact, many of the cell lines had considerably more than 3 RTKs activated, several beyond the spectrum of target inhibition achievable with use of a single RTK inhibitor, further suggesting a need for multidrug regimens even when RTK inhibition is the primary therapeutic modality. Consistent with this, a study investigating the phosphorylation status of EGFR in recurrent glioblastoma treated with gefitinib prior to resection found that EGFR was dephosphorylated (inactivated) after gefitinib treatment, suggesting successful targeting. However, gefitinib treatment failed to cause any change in the phosphorylation of downstream targets such as AKT.49 Thus, it is likely that the poor response to EGFR inhibitors is due to other sources of activation of the downstream targets of EGFR in recurrent glioblastoma, underscoring the need for a combined approach when using this RTK inhibitor class.
Molecular Subtypes of Glioblastoma: Steps Toward Personalized Medicine
As mentioned above, TCGA Network full categorization of the molecular alterations in glioblastoma gave rise to a number of potential therapeutic targets. A 2010 study by Verhaak et al. used the TCGA gene expression data to classify glioblastomas into 4 molecular subtypes: proneural, neural, classical, and mesenchymal.107 Aberrations in gene expression of EGFR, NF1, and PDGFRA/ IDH1 each define the classical, mesenchymal, and proneural subtypes, respectively. These findings support the hypothesis that molecularly targeted therapies may best be used against a portion of glioblastomas and suggest a tendency for certain alterations to coassociate in a manner that could make combinations of molecularly targeted agents particularly effective.
Combination Targeted Therapies
Combination anticancer therapy is increasingly recognized as an important treatment strategy against various types of cancer, especially glioblastoma.71 There are several clinical trials that evaluated the use of combination therapy in the treatment of recurrent glioblastoma, and in general, outcomes for combination therapy tend to be superior to monotherapy.4,28,30,41,84,85,87,106,108 A case example demonstrating the efficacy of combination therapy can be seen in Fig. 1. In this case, the patient underwent a gross-total resection of his glioblastoma. He was immediately placed on a clinical trial of adjuvant therapy consisting of combination temozolomide, erlotinib, and bevacizumab for 1 year. He responded well with treatment and was off therapy for the next 2.5 years. This totaled to 3.5 years of PFS. This case emphasizes the clinical efficacy of combination treatment, particularly with agents that have different mechanisms of action.
Fig. 1.
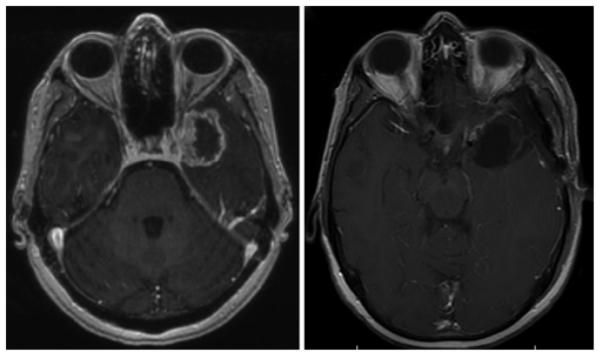
Combination bevacizumab, erlotinib, and temozolomide for glioblastoma. Left: Axial T1-weighted MR image with contrast showing a ring-enhancing lesion in the left temporal lobe, pathology proven glioblastoma. The patient underwent gross-total resection and adjuvant triple therapy with bevacizumab, erlotinib, and temozolomide for 1 year and observation off therapy. Right: Axial T1-weighted MRI with contrast showing no evidence of recurrent disease 3.5 years after initial surgery.
Not all combination regimens led to superior outcomes compared with monotherapy in the treatment of recurrent glioblastoma; rather similar outcomes were observed in certain clinical trials that tested combination regimens. The commonality among these studies is the use of multiple agents that act on the same molecular pathway. As mentioned, glioblastoma often has multiple activated pathways independent of one another. Thus, the effects of utilizing an agent that targets a pathway upstream (e.g., a receptor) are mute when another agent is used to target the same pathway downstream (e.g., regulatory protein). If additive or even synergistic effects are desired, this will likely be best achieved when different pathways or mechanisms are targeted.
Over the past several years, new treatment protocols that combine both traditional anticancer/chemotherapy agents and noncancer drugs have been developed for the treatment of recurrent glioblastoma.58,71 More recently, a group proposed supplementing 9 additional agents (aprepitant, artesunate, auranofin, captopril, copper gluconate, disulfiram, ketoconazole, nelfinavir, and sertraline) to continuous low-dose temozolomide in the treatment of recurrent glioblastoma.58 The authors had a rationale for the addition of each agent; each drug targeted a mechanism or pathway involved with either glioblastoma growth or compensatory mechanisms against temozolomide. However, this regimen has yet to be clinically tested and is limited by the potential for increased morbidity when combining these many targeted therapies in a “one size fits all” approach. A more rational approach could be personalized targeted therapies in which a smaller number of targeted therapies are combined based on the molecular profile of the tumor, using classification such as that of Verhaak et al. described above.107
Conclusions
Currently available molecularly targeted therapies for glioblastoma have had only poor to modest activity in clinical trials of recurrent glioblastoma, and there continues to be a lack of effective treatment regimens. As newer agents are actively being developed, combination regimens are the next step in improving outcomes when utilizing molecularly targeted therapies for recurrent glioblastoma. In addition, stratifying patients based on molecular profiles of their tumor will allow a tailored treatment approach when utilizing molecular targeting agents. In addition, identifying molecular biomarkers of responsiveness will be crucial to allow these treatments to fulfill their therapeutic promise.
Abbreviations used in this paper
- BBB
blood-brain barrier
- CR
complete response
- EGFR
epidermal growth factor receptor
- MGMT
methyguanine DNA methyltransferase
- mTOR
mammalian target of rapamycin
- OS
overall survival
- PD
progressive disease
- PDGFR
platelet-derived growth factor receptor
- PFS
progression-free survival
- PKC
protein kinase C
- PR
partial response
- RTK
receptor tyrosine kinase
- RTKI
RTK inhibitor
- SD
stable disease
- TCGA
The Cancer Genome Atlas
- VEGF
vascular endothelial growth factor
- VEGFR
VEGF receptor
Footnotes
Disclosure
The authors report no conflict of interest concerning the materials or methods used in this study or the findings specified in this paper.
Author contributions to the study and manuscript preparation include the following. Conception and design: Aghi, Lau. Acquisition of data: Lau, Magill. Analysis and interpretation of data: Aghi, Lau. Drafting the article: Lau, Magill. Critically revising the article: Aghi, Lau. Reviewed submitted version of manuscript: all authors. Approved the final version of the manuscript on behalf of all authors: Aghi. Study supervision: Aghi.
References
- 1.Batchelor TT, Duda DG, di Tomaso E, Ancukiewicz M, Plotkin SR, Gerstner E, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. 2010;28:2817–2823. doi: 10.1200/JCO.2009.26.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013;31:3212–3218. doi: 10.1200/JCO.2012.47.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bello L, Francolini M, Marthyn P, Zhang J, Carroll RS, Nikas DC, et al. Alpha(v)beta3 and alpha(v)beta5 integrin expression in glioma periphery. Neurosurgery. 2001;49:380–390. doi: 10.1097/00006123-200108000-00022. [DOI] [PubMed] [Google Scholar]
- 4.Brandes AA, Ermani M, Turazzi S, Scelzi E, Berti F, Amistà P, et al. Procarbazine and high-dose tamoxifen as a secondline regimen in recurrent high-grade gliomas: a phase II study. J Clin Oncol. 1999;17:645–650. doi: 10.1200/JCO.1999.17.2.645. [DOI] [PubMed] [Google Scholar]
- 5.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carbonell WS, DeLay M, Jahangiri A, Park CC, Aghi MK. b1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. 2013;73:3145–3154. doi: 10.1158/0008-5472.CAN-13-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chamberlain MC, Johnston SK. Salvage therapy with single agent bevacizumab for recurrent glioblastoma. J Neurooncol. 2010;96:259–269. doi: 10.1007/s11060-009-9957-6. [DOI] [PubMed] [Google Scholar]
- 9.Chang SM, Wen P, Cloughesy T, Greenberg H, Schiff D, Conrad C, et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23:357–361. doi: 10.1007/s10637-005-1444-0. [DOI] [PubMed] [Google Scholar]
- 10.Chatterjee S, Matsumura A, Schradermeier J, Gillespie GY. Human malignant glioma therapy using anti-alpha(v)beta3 integrin agents. J Neurooncol. 2000;46:135–144. doi: 10.1023/a:1006444300504. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y, Wang M, Zhong W, Zhao J. Pharmacokinetic and pharmacodynamic study of Gefitinib in a mouse model of non-small-cell lung carcinoma with brain metastasis. Lung Cancer. 2013;82:313–318. doi: 10.1016/j.lungcan.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 12.Chi A, Norden AD, Wen PY. Inhibition of angiogenesis and invasion in malignant gliomas. Expert Rev Anticancer Ther. 2007;7:1537–1560. doi: 10.1586/14737140.7.11.1537. [DOI] [PubMed] [Google Scholar]
- 13.Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25:884–896. doi: 10.1200/JCO.2006.06.3602. [DOI] [PubMed] [Google Scholar]
- 14.Clarke ID, Dirks PB. A human brain tumor-derived PDGFR-alpha deletion mutant is transforming. Oncogene. 2003;22:722–733. doi: 10.1038/sj.onc.1206160. [DOI] [PubMed] [Google Scholar]
- 15.Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: from molecular pathology to targeted treatment. Annu Rev Pathol. 2014;9:1–25. doi: 10.1146/annurev-pathol-011110-130324. [DOI] [PubMed] [Google Scholar]
- 16.Cloughesy TF, Wen PY, Robins HI, Chang SM, Groves MD, Fink KL, et al. Phase II trial of tipifarnib in patients with recurrent malignant glioma either receiving or not receiving enzyme-inducing antiepileptic drugs: a North American Brain Tumor Consortium Study. J Clin Oncol. 2006;24:3651–3656. doi: 10.1200/JCO.2006.06.2323. [DOI] [PubMed] [Google Scholar]
- 17.Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5:e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Couldwell WT, Antel JP, Yong VW. Protein kinase C activity correlates with the growth rate of malignant gliomas: Part II. Effects of glioma mitogens and modulators of protein kinase C. Neurosurgery. 1992;31:717–724. doi: 10.1227/00006123-199210000-00015. [DOI] [PubMed] [Google Scholar]
- 19.Couldwell WT, Hinton DR, Surnock AA, DeGiorgio CM, Weiner LP, Apuzzo ML, et al. Treatment of recurrent malignant gliomas with chronic oral high-dose tamoxifen. Clin Cancer Res. 1996;2:619–622. [PubMed] [Google Scholar]
- 20.Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15:1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Boüard S, Herlin P, Christensen JG, Lemoisson E, Gauduchon P, Raymond E, et al. Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro Oncol. 2007;9:412–423. doi: 10.1215/15228517-2007-024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Groot JF, Gilbert MR, Aldape K, Hess KR, Hanna TA, Ictech S, et al. Phase II study of carboplatin and erlotinib (Tarceva, OSI-774) in patients with recurrent glioblastoma. J Neurooncol. 2008;90:89–97. doi: 10.1007/s11060-008-9637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Groot JF, Lamborn KR, Chang SM, Gilbert MR, Cloughesy TF, Aldape K, et al. Phase II study of aflibercept in recurrent malignant glioma: a North American Brain Tumor Consortium study. J Clin Oncol. 2011;29:2689–2695. doi: 10.1200/JCO.2010.34.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deeken JF, Löscher W. The blood-brain barrier and cancer: transporters, treatment, and Trojan horses. Clin Cancer Res. 2007;13:1663–1674. doi: 10.1158/1078-0432.CCR-06-2854. [DOI] [PubMed] [Google Scholar]
- 25.Deng Y, Feng W, Wu J, Chen Z, Tang Y, Zhang H, et al. The concentration of erlotinib in the cerebrospinal fluid of patients with brain metastasis from non-small-cell lung cancer. Mol Clin Oncol. 2014;2:116–120. doi: 10.3892/mco.2013.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Desjardins A, Reardon DA, Coan A, Marcello J, Herndon JE, II, Bailey L, et al. Bevacizumab and daily temozolomide for recurrent glioblastoma. Cancer. 2012;118:1302–1312. doi: 10.1002/cncr.26381. [DOI] [PubMed] [Google Scholar]
- 27.Di Cristofori A, Carrabba G, Lanfranchi G, Menghetti C, Rampini P, Caroli M. Continuous tamoxifen and dose-dense temozolomide in recurrent glioblastoma. Anticancer Res. 2013;33:3383–3389. [PubMed] [Google Scholar]
- 28.Doherty L, Gigas DC, Kesari S, Drappatz J, Kim R, Zimmerman J, et al. Pilot study of the combination of EGFR and mTOR inhibitors in recurrent malignant gliomas. Neurology. 2006;67:156–158. doi: 10.1212/01.wnl.0000223844.77636.29. [DOI] [PubMed] [Google Scholar]
- 29.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 30.Dresemann G. Imatinib and hydroxyurea in pretreated progressive glioblastoma multiforme: a patient series. Ann Oncol. 2005;16:1702–1708. doi: 10.1093/annonc/mdi317. [DOI] [PubMed] [Google Scholar]
- 31.Dresemann G, Weller M, Rosenthal MA, Wedding U, Wagner W, Engel E, et al. Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. J Neurooncol. 2010;96:393–402. doi: 10.1007/s11060-009-9976-3. [DOI] [PubMed] [Google Scholar]
- 32.Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci U S A. 1992;89:4309–4313. doi: 10.1073/pnas.89.10.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eller JL, Longo SL, Hicklin DJ, Canute GW. Activity of anti-epidermal growth factor receptor monoclonal antibody C225 against glioblastoma multiforme. Neurosurgery. 2002;51:1005–1014. doi: 10.1097/00006123-200210000-00028. [DOI] [PubMed] [Google Scholar]
- 34.Feldkamp MM, Lau N, Guha A. Growth inhibition of astrocytoma cells by farnesyl transferase inhibitors is mediated by a combination of anti-proliferative, pro-apoptotic and anti-angiogenic effects. Oncogene. 1999;18:7514–7526. doi: 10.1038/sj.onc.1203105. [DOI] [PubMed] [Google Scholar]
- 35.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 36.Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. 2005;333:328–335. doi: 10.1016/j.bbrc.2005.05.132. [DOI] [PubMed] [Google Scholar]
- 37.Folkins C, Man S, Xu P, Shaked Y, Hicklin DJ, Kerbel RS. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007;67:3560–3564. doi: 10.1158/0008-5472.CAN-06-4238. [DOI] [PubMed] [Google Scholar]
- 38.Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 39.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 40.Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, Geoffroy F, et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. NeuroOncol. 2012;14:215–221. doi: 10.1093/neuonc/nor198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 42.Galanis E, Buckner JC, Maurer MJ, Kreisberg JI, Ballman K, Boni J, et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23:5294–5304. doi: 10.1200/JCO.2005.23.622. [DOI] [PubMed] [Google Scholar]
- 43.Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27:2052–2058. doi: 10.1200/JCO.2008.19.0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gately S. The contributions of cyclooxygenase-2 to tumor angiogenesis. Cancer Metastasis Rev. 2000;19:19–27. doi: 10.1023/a:1026575610124. [DOI] [PubMed] [Google Scholar]
- 45.Gilbert MR, Kuhn J, Lamborn KR, Lieberman F, Wen PY, Mehta M, et al. Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03-02, a phase II trial with measures of treatment delivery. J Neurooncol. 2012;106:147–153. doi: 10.1007/s11060-011-0650-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilbertson RJ, Rich JN. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733–736. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- 47.Hainsworth JD, Shih KC, Shepard GC, Tillinghast GW, Brinker BT, Spigel DR. Phase II study of concurrent radiation therapy, temozolomide, and bevacizumab followed by bevacizumab/everolimus as first-line treatment for patients with glioblastoma. Clin Adv Hematol Oncol. 2012;10:240–246. [PubMed] [Google Scholar]
- 48.Hasselbalch B, Lassen U, Hansen S, Holmberg M, Sørensen M, Kosteljanetz M, et al. Cetuximab, bevacizumab, and irinotecan for patients with primary glioblastoma and progression after radiation therapy and temozolomide: a phase II trial. Neuro Oncol. 2010;12:508–516. doi: 10.1093/neuonc/nop063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hegi ME, Diserens AC, Bady P, Kamoshima Y, Kouwenhoven MC, Delorenzi M, et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib—a phase II trial. Mol Cancer Ther. 2011;10:1102–1112. doi: 10.1158/1535-7163.MCT-11-0048. [DOI] [PubMed] [Google Scholar]
- 50.Hermanson M, Funa K, Hartman M, Claesson-Welsh L, Heldin CH, Westermark B, et al. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992;52:3213–3219. [PubMed] [Google Scholar]
- 51.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011–1027. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 52.Hughes TP, Kaeda J, Branford S, Rudzki Z, Hochhaus A, Hensley ML, et al. Frequency of major molecular responses to imatinib or interferon alfa plus cytarabine in newly diagnosed chronic myeloid leukemia. N Engl J Med. 2003;349:1423–1432. doi: 10.1056/NEJMoa030513. [DOI] [PubMed] [Google Scholar]
- 53.Jahangiri A, Aghi MK, Carbonell WS. b1 integrin: critical path to antiangiogenic therapy resistance and beyond. Cancer Res. 2014;74:3–7. doi: 10.1158/0008-5472.CAN-13-1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7:987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- 55.Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neurooncol. 2012;107:359–364. doi: 10.1007/s11060-011-0749-4. [DOI] [PubMed] [Google Scholar]
- 56.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 57.Kane RC, Farrell AT, Saber H, Tang S, Williams G, Jee JM, et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin Cancer Res. 2006;12:7271–7278. doi: 10.1158/1078-0432.CCR-06-1249. [DOI] [PubMed] [Google Scholar]
- 58.Kast RE, Boockvar JA, Brüning A, Cappello F, Chang WW, Cvek B, et al. A conceptually new treatment approach for relapsed glioblastoma: coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget. 2013;4:502–530. doi: 10.18632/oncotarget.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kieran MW, Packer RJ, Onar A, Blaney SM, Phillips P, Pollack IF, et al. Phase I and pharmacokinetic study of the oral farnesyltransferase inhibitor lonafarnib administered twice daily to pediatric patients with advanced central nervous system tumors using a modified continuous reassessment method: a Pediatric Brain Tumor Consortium Study. J Clin Oncol. 2007;25:3137–3143. doi: 10.1200/JCO.2006.09.4243. [DOI] [PubMed] [Google Scholar]
- 61.Kreisl TN, Lassman AB, Mischel PS, Rosen N, Scher HI, Teruya-Feldstein J, et al. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM) J Neurooncol. 2009;92:99–105. doi: 10.1007/s11060-008-9741-z. [DOI] [PubMed] [Google Scholar]
- 62.Kreisl TN, Smith P, Sul J, Salgado C, Iwamoto FM, Shih JH, et al. Continuous daily sunitinib for recurrent glioblastoma. J Neurooncol. 2013;111:41–48. doi: 10.1007/s11060-012-0988-z. [DOI] [PubMed] [Google Scholar]
- 63.Lacroix M, Abi-Said D, Fourney DR, Gokaslan ZL, Shi W, DeMonte F, et al. A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J Neurosurg. 2001;95:190–198. doi: 10.3171/jns.2001.95.2.0190. [DOI] [PubMed] [Google Scholar]
- 64.Laird AD, Vajkoczy P, Shawver LK, Thurnher A, Liang C, Mohammadi M, et al. SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res. 2000;60:4152–4160. [PubMed] [Google Scholar]
- 65.Lokker NA, Sullivan CM, Hollenbach SJ, Israel MA, Giese NA. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002;62:3729–3735. [PubMed] [Google Scholar]
- 66.Lv S, Teugels E, Sadones J, De Brakeleer S, Duerinck J, Du Four S, et al. Correlation of EGFR, IDH1 and PTEN status with the outcome of patients with recurrent glioblastoma treated in a phase II clinical trial with the EGFR-blocking monoclonal antibody cetuximab. Int J Oncol. 2012;41:1029–1035. doi: 10.3892/ijo.2012.1539. [DOI] [PubMed] [Google Scholar]
- 67.Millward MJ, Cantwell BM, Lien EA, Carmichael J, Harris AL. Intermittent high-dose tamoxifen as a potential modifier of multidrug resistance. Eur J Cancer. 1992;28A:805–810. doi: 10.1016/0959-8049(92)90119-m. [DOI] [PubMed] [Google Scholar]
- 68.Montgomery RB, Moscatello DK, Wong AJ, Cooper JA, Stahl WL. Differential modulation of mitogen-activated protein (MAP) kinase/extracellular signal-related kinase kinase and MAP kinase activities by a mutant epidermal growth factor receptor. J Biol Chem. 1995;270:30562–30566. doi: 10.1074/jbc.270.51.30562. [DOI] [PubMed] [Google Scholar]
- 69.Nam DH, Park K, Suh YL, Kim JH. Expression of VEGF and brain specific angiogenesis inhibitor-1 in glioblastoma: prognostic significance. Oncol Rep. 2004;11:863–869. [PubMed] [Google Scholar]
- 70.Neyns B, Sadones J, Joosens E, Bouttens F, Verbeke L, Baurain JF, et al. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann Oncol. 2009;20:1596–1603. doi: 10.1093/annonc/mdp032. [DOI] [PubMed] [Google Scholar]
- 71.Nonnenmacher L, Westhoff MA, Fulda S, Karpel-Massler G, Halatsch ME, Engelke J, et al. RIST: A potent new combination therapy for glioblastoma. Int J Cancer. 2014 doi: 10.1002/ijc.29138. [epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 72.Norden AD, Lesser GJ, Drappatz J, Ligon KL, Hammond SN, Lee EQ, et al. Phase 2 study of dose-intense temozolomide in recurrent glioblastoma. Neuro Oncol. 2013;15:930–935. doi: 10.1093/neuonc/not040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peereboom DM, Ahluwalia MS, Ye X, Supko JG, Hilderbrand SL, Phuphanich S, et al. NABTT 0502: a phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro Oncol. 2013;15:490–496. doi: 10.1093/neuonc/nos322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pietras K, Ostman A, Sjöquist M, Buchdunger E, Reed RK, Heldin CH, et al. Inhibition of platelet-derived growth factor receptors reduces interstitial hypertension and increases transcapillary transport in tumors. Cancer Res. 2001;61:2929–2934. [PubMed] [Google Scholar]
- 75.Pietras K, Rubin K, Sjöblom T, Buchdunger E, Sjöquist M, Heldin CH, et al. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res. 2002;62:5476–5484. [PubMed] [Google Scholar]
- 76.Plate KH, Breier G, Farrell CL, Risau W. Platelet-derived growth factor receptor-beta is induced during tumor development and upregulated during tumor progression in endothelial cells in human gliomas. Lab Invest. 1992;67:529–534. [PubMed] [Google Scholar]
- 77.Polivka J, Jr, Polivka J, Rohan V, Topolcan O, Ferda J. New molecularly targeted therapies for glioblastoma multiforme. Anticancer Res. 2012;32:2935–2946. [PubMed] [Google Scholar]
- 78.Pollack IF, Randall MS, Kristofik MP, Kelly RH, Selker RG, Vertosick FT., Jr Effect of tamoxifen on DNA synthesis and proliferation of human malignant glioma lines in vitro. Cancer Res. 1990;50:7134–7138. [PubMed] [Google Scholar]
- 79.Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG, et al. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro Oncol. 2010;12:95–103. doi: 10.1093/neuonc/nop015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reardon DA, Desjardins A, Peters K, Gururangan S, Sampson J, Rich JN, et al. Phase II study of metronomic chemotherapy with bevacizumab for recurrent glioblastoma after progression on bevacizumab therapy. J Neurooncol. 2011;103:371–379. doi: 10.1007/s11060-010-0403-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reardon DA, Desjardins A, Peters KB, Gururangan S, Sampson JH, McLendon RE, et al. Phase II study of carboplatin, irinotecan, and bevacizumab for bevacizumab naïve, recurrent glioblastoma. J Neurooncol. 2012;107:155–164. doi: 10.1007/s11060-011-0722-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Reardon DA, Desjardins A, Peters KB, Vredenburgh JJ, Gururangan S, Sampson JH, et al. Phase 2 study of carboplatin, irinotecan, and bevacizumab for recurrent glioblastoma after progression on bevacizumab therapy. Cancer. 2011;117:5351–5358. doi: 10.1002/cncr.26188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reardon DA, Desjardins A, Vredenburgh JJ, Gururangan S, Friedman AH, Herndon JE, II, et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J Neurooncol. 2010;96:219–230. doi: 10.1007/s11060-009-9950-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Reardon DA, Dresemann G, Taillibert S, Campone M, van den Bent M, Clement P, et al. Multicentre phase II studies evaluating imatinib plus hydroxyurea in patients with progressive glioblastoma. Br J Cancer. 2009;101:1995–2004. doi: 10.1038/sj.bjc.6605411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reardon DA, Egorin MJ, Quinn JA, Rich JN, Gururangan S, Vredenburgh JJ, et al. Phase II study of imatinib mesylate plus hydroxyurea in adults with recurrent glioblastoma multiforme. J Clin Oncol. 2005;23:9359–9368. doi: 10.1200/JCO.2005.03.2185. Erratum in J Clin Oncol 24:1224, 2006. [DOI] [PubMed] [Google Scholar]
- 86.Reardon DA, Fink KL, Mikkelsen T, Cloughesy TF, O’Neill A, Plotkin S, et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. J Clin Oncol. 2008;26:5610–5617. doi: 10.1200/JCO.2008.16.7510. [DOI] [PubMed] [Google Scholar]
- 87.Reardon DA, Quinn JA, Vredenburgh JJ, Gururangan S, Friedman AH, Desjardins A, et al. Phase 1 trial of gefitinib plus sirolimus in adults with recurrent malignant glioma. Clin Cancer Res. 2006;12:860–868. doi: 10.1158/1078-0432.CCR-05-2215. [DOI] [PubMed] [Google Scholar]
- 88.Reardon DA, Vredenburgh JJ, Desjardins A, Peters K, Gururangan S, Sampson JH, et al. Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J Neurooncol. 2011;101:57–66. doi: 10.1007/s11060-010-0217-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL, et al. Phase II trial of gefitinib in recurrent glioblastoma. J Clin Oncol. 2004;22:133–142. doi: 10.1200/JCO.2004.08.110. [DOI] [PubMed] [Google Scholar]
- 90.Russell JS, Brady K, Burgan WE, Cerra MA, Oswald KA, Camphausen K, et al. Gleevec-mediated inhibition of Rad51 expression and enhancement of tumor cell radiosensitivity. Cancer Res. 2003;63:7377–7383. [PubMed] [Google Scholar]
- 91.Salmaggi A, Eoli M, Frigerio S, Silvani A, Gelati M, Corsini E, et al. Intracavitary VEGF, bFGF, IL-8, IL-12 levels in primary and recurrent malignant glioma. J Neurooncol. 2003;62:297–303. doi: 10.1023/a:1023367223575. [DOI] [PubMed] [Google Scholar]
- 92.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–262. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 93.Seystahl K, Weller M. Is there a world beyond bevacizumab in targeting angiogenesis in glioblastoma? Expert Opin Investig Drugs. 2012;21:605–617. doi: 10.1517/13543784.2012.670219. [DOI] [PubMed] [Google Scholar]
- 94.Shih AH, Dai C, Hu X, Rosenblum MK, Koutcher JA, Holland EC. Dose-dependent effects of platelet-derived growth factor-B on glial tumorigenesis. Cancer Res. 2004;64:4783–4789. doi: 10.1158/0008-5472.CAN-03-3831. [DOI] [PubMed] [Google Scholar]
- 95.Smalley KS, Xiao M, Villanueva J, Nguyen TK, Flaherty KT, Letrero R, et al. CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations. Oncogene. 2009;28:85–94. doi: 10.1038/onc.2008.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287–290. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- 97.Stupp R, Dietrich PY, Ostermann Kraljevic S, Pica A, Maillard I, Maeder P, et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Oncol. 2002;20:1375–1382. doi: 10.1200/JCO.2002.20.5.1375. [DOI] [PubMed] [Google Scholar]
- 98.Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, Clement PM, et al. Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:2712–2718. doi: 10.1200/JCO.2009.26.6650. [DOI] [PubMed] [Google Scholar]
- 99.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 100.Sun CC, Qu XJ, Gao ZH. Integrins: players in cancer progression and targets in cancer therapy. Anticancer Drugs. 2014;25:1107–1121. doi: 10.1097/CAD.0000000000000145. [DOI] [PubMed] [Google Scholar]
- 101.Takayama N, Sato N, O’Brien SG, Ikeda Y, Okamoto S. Imatinib mesylate has limited activity against the central nervous system involvement of Philadelphia chromosome-positive acute lymphoblastic leukaemia due to poor penetration into cerebrospinal fluid. Br J Haematol. 2002;119:106–108. doi: 10.1046/j.1365-2141.2002.03881.x. [DOI] [PubMed] [Google Scholar]
- 102.Tang SC, Lagas JS, Lankheet NA, Poller B, Hillebrand MJ, Rosing H, et al. Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib coadministration. Int J Cancer. 2012;130:223–233. doi: 10.1002/ijc.26000. [DOI] [PubMed] [Google Scholar]
- 103.Ugur HC, Ramakrishna N, Bello L, Menon LG, Kim SK, Black PM, et al. Continuous intracranial administration of suberoylanilide hydroxamic acid (SAHA) inhibits tumor growth in an orthotopic glioma model. J Neurooncol. 2007;83:267–275. doi: 10.1007/s11060-007-9337-z. [DOI] [PubMed] [Google Scholar]
- 104.Uhrbom L, Hesselager G, Nistér M, Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998;58:5275–5279. [PubMed] [Google Scholar]
- 105.van den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MC, Kros JM, Carpentier AF, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J Clin Oncol. 2009;27:1268–1274. doi: 10.1200/JCO.2008.17.5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.van den Bent MJ, Taal W. Are we done with dose-intense temozolomide in recurrent glioblastoma? Neuro Oncol. 2014;16:1161–1163. doi: 10.1093/neuonc/nou157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vredenburgh JJ, Desjardins A, Herndon JE, II, Marcello J, Reardon DA, Quinn JA, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25:4722–4729. doi: 10.1200/JCO.2007.12.2440. [DOI] [PubMed] [Google Scholar]
- 109.Wang SI, Puc J, Li J, Bruce JN, Cairns P, Sidransky D, et al. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997;57:4183–4186. [PubMed] [Google Scholar]
- 110.Wang T, Agarwal S, Elmquist WF. Brain distribution of cediranib is limited by active efflux at the blood-brain barrier. J Pharmacol Exp Ther. 2012;341:386–395. doi: 10.1124/jpet.111.190488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wedge SR, Kendrew J, Hennequin LF, Valentine PJ, Barry ST, Brave SR, et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389–4400. doi: 10.1158/0008-5472.CAN-04-4409. [DOI] [PubMed] [Google Scholar]
- 112.Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005;437:497–504. doi: 10.1038/nature03987. [DOI] [PubMed] [Google Scholar]
- 113.Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma—are we there yet? Neuro Oncol. 2013;15:4–27. doi: 10.1093/neuonc/nos273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wen PY, Yung WK, Lamborn KR, Dahia PL, Wang Y, Peng B, et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99-08. Clin Cancer Res. 2006;12:4899–4907. doi: 10.1158/1078-0432.CCR-06-0773. [DOI] [PubMed] [Google Scholar]
- 115.Wick W, Puduvalli VK, Chamberlain MC, van den Bent MJ, Carpentier AF, Cher LM, et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol. 2010;28:1168–1174. doi: 10.1200/JCO.2009.23.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7:3129–3140. doi: 10.1158/1535-7163.MCT-08-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xia P, Aiello LP, Ishii H, Jiang ZY, Park DJ, Robinson GS, et al. Characterization of vascular endothelial growth factor’s effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. J Clin Invest. 1996;98:2018–2026. doi: 10.1172/JCI119006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xue Y, Lim S, Yang Y, Wang Z, Jensen LD, Hedlund EM, et al. PDGF-BB modulates hematopoiesis and tumor angiogenesis by inducing erythropoietin production in stromal cells. Nat Med. 2012;18:100–110. doi: 10.1038/nm.2575. [DOI] [PubMed] [Google Scholar]
- 119.Yin D, Ong JM, Hu J, Desmond JC, Kawamata N, Konda BM, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: effects on gene expression and growth of glioma cells in vitro and in vivo. Clin Cancer Res. 2007;13:1045–1052. doi: 10.1158/1078-0432.CCR-06-1261. [DOI] [PubMed] [Google Scholar]
- 120.Yung WK, Vredenburgh JJ, Cloughesy TF, Nghiemphu P, Klencke B, Gilbert MR, et al. Safety and efficacy of erlotinib in first-relapse glioblastoma: a phase II open-label study. Neuro Oncol. 2010;12:1061–1070. doi: 10.1093/neuonc/noq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yust-Katz S, Liu D, Yuan Y, Liu V, Kang S, Groves M, et al. Phase 1/1b study of lonafarnib and temozolomide in patients with recurrent or temozolomide refractory glioblastoma. Cancer. 2013;119:2747–2753. doi: 10.1002/cncr.28031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zustovich F, Landi L, Lombardi G, Porta C, Galli L, Fontana A, et al. Sorafenib plus daily low-dose temozolomide for relapsed glioblastoma: a phase II study. Anticancer Res. 2013;33:3487–3494. [PubMed] [Google Scholar]