

Abstract
The synthesis and in vitro evaluation of four mesoporphyrin IX-peptide conjugates designed to target EGFR, over-expressed in colorectal and other cancers, are reported. Two peptides with known affinity for EGFR, LARLLT (1) and GYHWYGYTPQNVI (2), were conjugated to mesoporphyrin IX (MPIX, 3) via one or both the propionic side chains, directly (4, 5) or with a triethylene glycol spacer (7, 8). The conjugates were characterized using NMR, MS, CD, SPR, UV-vis and fluorescence spectroscopies. Energy minimization and molecular dynamics suggest different conformations for the conjugates. SPR studies show that conjugate 4, bearing two LARLLT with no PEG spacers, has the greatest affinity for binding to EGFR, followed by conjugate 7 with two PEG and two LARLLT sequences. Molecular modeling and docking studies suggest that both conjugates 4 and 7 can bind to monomer and dimer EGFR in open and closed conformations. The cytotoxicity and cellular targeting ability of the conjugates were investigated in human HEp2 cells over-expressing EGFR. All conjugates showed low dark- and photo-toxicities. The cellular uptake was highest for conjugates 4 and 8 and lowest for 7 bearing two LARLLT linked via PEG groups, likely due to decreased hydrophobicity. Among the conjugates investigated 4 is the most efficient EGFR-targeting agent, and therefore the most promising for the detection of cancers that over-express EGFR.
Keywords: porphyrin, peptide, EGFR, photosensitizer, docking
INTRODUCTION
Colorectal cancer (CRC) is one of the most common cancers diagnosed in both men and women, and the third leading cause of cancer-related deaths, expected to cause approximately 50,000 deaths in 2015 in the USA [1]. Since CRC develops overtime in the body, screening methodologies have been developed to detect early stage cancers, with the most cure potential. The detection and removal of polyps and early stage cancer has reduced the incidence of CRC, and has led to the improvement in patient mortality observed in the past 20 years. Two screening methods, chromoendoscopy and confocal laser endomicroscopy (CLE), are particularly suited for the detection of small adenomas (< 5 mm) and flat lesions [2, 3]. These techniques use untargeted contrast dyes, such as methylene blue, fluorescein, acriflavine, and cresyl violet that label both neoplastic and normal mucosal tissue. Targeted agents with fluorescence emissions in the red and near-IR regions could enhance specific delivery to CRC cells with reduced scattering, therefore further improving early detection of small adenomas and further decreasing cancer mortality. One very important target in CRC and several other tumors, such as breast, ovarian and prostate cancers, is the human epidermal growth factor receptor (EGFR), known to be abundantly expressed in both early and advanced CRC [4, 5]. EGFR is a transmembrane glycoprotein with extracellular and intracellular domains, involved in the regulation of signaling pathways that control cell proliferation, differentiation and angiogenesis [6]. Several targeting molecules based on monoclonal antibodies [6, 7], affibody proteins [8, 9], peptides [10], peptomimetics [11, 12] and non-peptidic tyrosine kinase inhibitors [13, 14] have been developed for specific targeting of EGFR. Among these, the use of small peptides as ligands is a particularly attractive approach, because of their easy synthesis and conjugation, and their low immunogenicity. Two peptide ligands, LARLLT and YHWYGYTPQNVI, designed and investigated by Li et al. [15] and Song et al. [16], respectively, have demonstrated high EGFR-targeting ability both in vitro and in vivo. We have recently reported the synthesis of phthalocyanine (Pc) conjugates to one of these peptides, linked to the Pc via a short five-atom or a PEG group [10]. Our studies revealed that Pc conjugates to LARLLT (1) via a PEG spacer, showed enhanced water solubility and targeting ability, accumulating in EGFR over-expressing cells up to 17 times more than unconjugated Pc. However, some Pc conjugates showed very low solubility, in particular those containing the peptide GYHWYGYTPQNVI (2). Herein we investigate conjugates of peptides 1 and 2 to mesoporphyrin IX (MPIX, 3), bearing one or two peptide residues linked via a short three-atom or a PEG linker. MPIX is a derivative of protoporphyrin IX (PPIX) bearing ethyl rather than vinyl groups at the 3,8-positions, and two propionic acids available for conjugation that allow easy introduction of one or two peptide residues. Several conjugates of porphyrins and derivatives to various peptide sequences have been reported [17]. For example, conjugates of PPIX to GnRH-targeting peptides [18] and integrin-targeting peptides, including cycloRGDfk [19], cycloERGDF [20], ATWLPPR [20] and PQRRSARLSA [20] were prepared and shown to have enhanced targeting ability relative to unconjugated PPIX. We have also investigated the use of a low molecular weight polyethylene glycol (PEG) linker between the PPIX and the peptide sequence to enhance the conjugates’ aqueous solubility, reduce intramolecular interactions and favor cellular uptake [20]. In this work, four amphiphilic MPIX conjugates bearing two LARLLT (1) or one GYHWYGYTPQNVI (2) sequences linked directly to the propionic acid group(s) or via low molecular weight PEG spacers, were synthesized and their structures investigated by NMR, MALDI-MS, UV-vis, CD and molecular modeling. Their EGFR-binding ability was studied using SPR, and in cell culture using human HEp2 cells. Based on these experimental studies, a model for binding of conjugates to EGFR protein has been proposed.
RESULTS AND DISCUSSION
Synthesis
The MPIX conjugates 4, 5, 7 and 8 were prepared as shown in Scheme 1. The peptide sequences LARLLT (1) and GYHWYGYTPQNVI (2) were synthesized on Fmoc-Pal-PEG-PS resin using solid phase peptide synthesis [10, 21]. An aminium coupling agent, TBTU, a triazole as electron-poor coupling additive, HOBt, and DIEA as base were used in the solid phase synthesis of 1 and 2 [10, 21–23]. A polar aprotic solvent, DMF, enabled the swelling of the Fmoc-PAL-PEG-PS resin, expanding the active sites and facilitated amino acid coupling to the resin. The C-terminus of the first amino acid was first coupled to the resin followed by the removal of the Fmoc protecting group. The next amino acid was applied to the resin and the procedure repeated until the desired peptide was obtained. In the final step, the protecting group was removed and the peptide was cleaved from the resin and purified by reverse phase HPLC. Peptides 1 and 2 were isolated in 48% and 32%, respectively (Table 1).
Scheme 1.

Synthetic route to porphyrin-peptide conjugates from MPIX 3
Table 1.
Coupling conditions used in the synthesis of compounds 1, 2, 4–8, HPLC retention times (see experimental section for conditions) and isolated yields
| Compound | Rt, min | Conditions | Yield, % |
|---|---|---|---|
| 1 | 18.02 | TBTU/HOBT/DMF/DIEA | 48% |
| 2 | 31.22 | TBTU/HOBT/DMF/DIEA | 32% |
| 4 | 35.01 | TBTU/HOBT/DMF/DIEA/37 °C | 83% |
| 5 | 36.20 | PyAOP/HOBT/DMF/DIEA | 20% |
| DEPBT/HOBT/DMF/DIEA/37 °C | 51% | ||
| 6 | 19.43 | TBTU/HOBT/DMF/DIEA/37 °C | 88% |
| 7 | 19.86 | TBTU/HOBT/DMF/DIEA/37 °C | 16% |
| 8 | 25.86 | DEPBT/HOBT/DMF/DIEA/37 °C | 39% |
MPIX conjugate 4 bearing two LARLLT sequences was prepared by conjugating 1 (2 equiv) to 3 in solution phase, using TBTU, HOBt and DIEA in DMF (Scheme 1). The desired conjugate 4 was isolated in 83% yield after purification by solid phase extraction (SPE). Under similar conditions, MPIX conjugate 5 bearing only one GYHWYGYTPQNVI sequence was also synthesized, as indicated by MALDI-MS, however in low yield (<5%), probably due to higher steric hindrance. Therefore, alternative reaction conditions were investigated for the coupling of 2 to 3, using the organophosphorus DEPBT and the phosphonium salt PyAOP as the coupling agents. DEPBT is normally used to reduce racemization of amino acids, such as tyrosine, serine, threonine, and the imidazole group of histidine [24, 25]. Since peptide 2 contains tyrosine, threonine and histidine residues, DEPBT might be an effective coupling agent for this peptide. On the other hand, PyAOP has been used in challenging coupling reactions, producing cleaner products [26, 27]. Using these reagents, conjugate 5 was isolated in 51% and 20% yields, respectively, following purification by SPE (Table 1).
The conjugation of MPIX to low molecular weight PEG groups bearing a protected carboxylic acid was accomplished using TBTU, HOBt and DIEA in DMF (Scheme 1). After purification by column chromatography, the tert-butyl ester protecting group was cleaved using TFA, giving MPIX-diPEG 6 in 88% yield. Peptides 1 and 2 were conjugated to porphyrin 6 under similar conditions to those described above, producing conjugates 7 and 8 in 16% and 39% yields, respectively (Scheme 1). The retention time, coupling conditions, and yields obtained for peptides 1 and 2 and for porphyrins 4–8 are listed in Table 1.
The structure of the porphyrin conjugates was confirmed by NMR, MALDI-MS and UV-vis spectrophotometry. The spectroscopic properties were investigated in DMF solution and the results obtained are shown in Fig. 1 and summarized in Table 2. The absorption and emission spectra of the porphyrin conjugates were similar to that of MPIX 3, with slight red-shifts observed for the Soret bands for 4, 7 and 8. The conjugates’ fluorescence quantum yields were in the range 0.086–0.13, similar to that reported for porphyrin 3 (ΦF = 0.102) [28]. The quantum yields were slightly larger for the porphyrins conjugated to peptide 2 rather than 1, and were also enhanced for the conjugates bearing PEG spacers, maybe as a result of their higher solubility in DMF.
Fig. 1.

Absorption (a, c) and fluorescence (b, d) spectra of porphyrins 3 (black), 4 (red), 5 (purple), 6 (pink), 7 (green), and 8 (blue) in DMF at room temperature
Table 2.
Spectroscopic data for porphyrins 4, 5, 6, 7 and 8 in DMF at room temperature
| Porphyrin | Absorption (λmax, nm) | Emission (λmax, nm) | Stokes shift, nm | Φfa |
|---|---|---|---|---|
| 4 | 407, 497, 530, 577, 615 | 618, 645, 665, 684 | 220 | 0.089 |
| 5 | 397, 497, 530, 566, 620 | 620, 646, 670, 690 | 223 | 0.124 |
| 6 | 398, 497, 530, 566, 616 | 618, 651, 665, 684 | 220 | 0.086 |
| 7 | 408, 497, 530, 573, 618 | 620, 655, 670, 686 | 212 | 0.124 |
| 8 | 408, 497, 531, 574, 620 | 621, 655, 670, 686 | 213 | 0.132 |
Calculated using porphyrin 3 as standard (0.102) [28].
Circular Dichroism (CD)
The solution conformation of the peptides 1 and 2 and porphyrin conjugates 4 and 7 (bearing two LARLLT sequences linked directly or via PEG spacers to the propionic acid groups of MPIX) were investigated by CD in a membrane-mimicking environment (0.5 mM PBS buffer with 10% TFE). The CD spectra obtained for 1 and 2 up to 1 mM and for 4 and 7 at 15 and 37.5 μM concentrations are shown in Fig. 2, and the molar ellipticity (θ) values are listed in Table 3. For peptide 1, a sharp negative band at ~198 nm and a broad, less intense, negative band at around 225 nm were observed at the highest concentrations (Fig. 2a), suggesting an unordered structure, as expected for a small peptide containing six amino acid residues. On the other hand, peptide 2 showed a negative band at ~198 nm at concentrations <125 μM. This is indicative of a random coil conformation, and two negative bands at 208 and 217 nm at higher concentrations, suggests a mixture of α-helix and β-sheet conformations (Fig. 2b).
Fig. 2.

CD spectra of peptides 1 (a), 2 (b) and porphyrin conjugates 4 (c) and 7 (d) in 0.5 mM PBS buffer with 10% TFE, at different concentrations
Table 3.
Molar ellipticity values for peptides and select conjugates in 0.5 mM PBS with 10% TFE at pH 7.4
| Porphyrin | Concentration, μM | Molar ellipticity θ (deg.cm2.dmol−1) |
|---|---|---|
| 1 | 50 | −59 355 (197 nm) |
| 2 | 50 | −24 097 (198 nm) |
| 4 | 37.5 | −5519 (217 nm) |
| 15 | −4921 (213 nm) and −4223 (224 nm) | |
| 7 | 37.5 | −13 129 (205 nm) and −4860 (227 nm) |
| 15 | −3821 (205 nm) and −2854 (227 nm) |
We have previously observed that peptide sequences conjugated to meso-tetraarylporphyrins via low molecular weight PEG spacers tend to assume extended conformations, in which the peptide retains its original conformation [29–31]. To evaluate the influence of peptide conjugation to the propionic acid groups of MPIX (3), either directly or via two PEG spacers, the CD spectra of conjugates 4 and 7 were obtained at 15 and 37.5 μM concentrations. Porphyrin conjugate 4 exhibited a positive peak at ~200 nm, a negative peak at ~217 nm and a small shoulder at ~222 nm, suggesting a mixture of α-helical and β-sheet conformations in solution (Fig. 2c). On the other hand, the CD spectrum obtained for porphyrin conjugate 7 at 15 μM suggests an α-helix conformation with a positive band at ~193 nm and two negative bands at ~205 and 227 nm. However, at 37.5 μM conjugate 7 appears to adopt a 310-helix conformation [32] with a positive band at 195 nm and two negative bands at 205 and 228 nm (Fig. 2d). A web-based program, K2D2, was used to assess the percentage of secondary structures, α-helix and β-sheet, present in the conformations of conjugates 4 and 7 [33]. Analysis of the results indicated that 4 has 2% (15 μM) — 25% (37.5 μM) α-helix and 48% (15 μM) — 25% (37.5 μM) β-sheet whereas 7 has 3% (15 μM) — 14% (37.5 μM) α-helix and 47% (15 μM) — 27% (37.5 μM) β-sheet [34].
Surface Plasmon Resonance (SPR)
SPR is an optical technique used to evaluate the interaction between an immobilized ligand and an analyte [35, 36]. The EGFR was immobilized onto the gold surface of a chip where the binding and specificity of peptides 1 and 2, and porphyrins 3, 4, 5, 6 and 7 were evaluated. The results obtained are shown in Fig. 3. The relative binding affinity of peptides and conjugates were determined by the maximum response units (RU) reached during association immediately following the injection during the association phase. All conjugates showed higher binding affinity to EGFR than the peptides alone (Fig. 3a) and porphyrin conjugate 4 showed the greatest binding affinity to EGFR of all compounds tested (Fig. 3b). This may in part be due to the hydrophobic nature of the porphyrin macrocycle, which favors binding to the surface of the EGFR protein, as we previously observed with phthalocyanine conjugates [10]. The determined binding affinity (kRU) order is 4 ≫ 7 > 6 ~ 3 > 5 > 2 > 1. Of the two peptides, 2 showed higher binding affinity than 1, probably because 2 binds to the EGF binding pocket [15] while 1 preferentially binds to a pocket away from the EGF binding site in EGFR [16]. Nevertheless, the conjugates bearing two LARLLT sequences, 4 and 7, showed enhanced binding compared with the unconjugated porphyrins 3 and 6, and to conjugate 5 bearing the GYHWYGYTPQNVI sequence. The remarkable strong binding observed for conjugate 4 might in part be due to the +2 charge on the two arginine residues [37, 38]. Dissociation of the bound molecules was observed around 400 s on the sensogram. Conjugates 4, 5 and 7 exhibited slower dissociation rates (Fig. 3) from 400 to 500 s whereas, other conjugates showed relatively rapid dissociation from the EGFR protein.
Fig. 3.

SPR sensorgram of peptides binding to EGFR extracellular domain. (a) 1 (turquoise), 2 (orange) and porphyrins 3 (black), 4 (red), 5 (purple), 6 (pink) and 7 (green) at 100 μM. (b) SPR sensorgrams of 1 to 7 without conjugate 4 (red) is shown for the sake of clarity. Based on the maximum RU reached during association, affinity order is 4 ≫ 7 > 6 ~ 3 > 5 > 2 > 1
Molecular modeling studies
Computational studies were performed to gain further insight on the conformations of the porphyrin conjugates, using energy minimization and molecular dynamics methods in vacuum, and in the presence of water molecules. Results of these studies are shown in Fig. 4. In vacuum, conjugate 4 with two LARLLT sequences (Fig. 4a) showed one of the peptides folded over the porphyrin ring and the other peptide in an extended conformation to minimize steric interactions between the two peptides, while maximizing stabilizing intramolecular interactions with the porphyrin ring. We have previously observed that porphyrin-peptide conjugates bearing short linkers tend to assume folded conformations due to hydrogen bonding and hydrophobic intramolecular interactions, which reduce the conformational entropy compared with the linear conformations [37]. On the other hand, in the presence of water, both peptide chains were extended away from the porphyrin ring suggesting that the interaction of solvent with the peptide chains stabilizes the peptide structure (Fig. 4b). The energy-minimized structure of porphyrin 6, bearing two PEG chains, in vacuum, indicated that the PEG groups are completely folded and interacted with the porphyrin ring, stabilizing the PEG chain structure via hydrophobic interactions (Fig. 4c). Such folded or curled conformations of PEG groups attached to porphyrin derivatives, in the gas phase or water MD simulations have been previously observed [39, 40]. In the presence of water, we observed slight folding of the PEG group positioned away from the porphyrin ring (Fig. 4d). In conjugate 7, bearing two PEG and two LARLLT peptide chains, similar results were observed. In vacuum, the PEG groups were folded onto the porphyrin ring while the peptide extended away from the ring (Fig. 4e). In the presence of water, although the PEG groups were slightly folded towards the porphyrin ring, the overall PEG and peptide chains were extended away from porphyrin ring (Fig. 4f). The energy-minimized conformation of conjugate 5, bearing a single GYHWYGYTPQNVI sequence showed an extended conformation that allows the aromatic amino acids (histidine and tyrosine) to extended away from the peptide backbone (not shown). Conjugate 8 bearing one GYHWYGYTPQNVI sequence linked via a PEG group also adopted an extended conformation (not shown). In agreement with these results, we have previously reported that a PEG spacer tends to minimize folding of the peptide over the porphyrin ring, favoring an extended conformation of the conjugate [29].
Fig. 4.

Energy minimized conformations in a vacuum and in the presence of solvent (layer of water molecules). (a) conjugate 4 in vacuum, (b) conjugate 4 in water, (c) porphyrin 6 in vacuum, (d) porphyrin 6 in water, (e) conjugate 7 in vacuum, and (f) conjugate 7 in water
Since conjugates 4 and 7 exhibited the strongest binding to EGFR by SPR, their mode of interaction with EGFR was modeled using Autodock [41, 42]. For conjugate protein complexes, the models of conjugates generated in solvent molecules were used since these are the most relevant in the presence of the receptor. Furthermore, the models generated in water showed extended structures rather than folded structures, as observed in vacuum (Fig. 4). EGFR exists in both open and closed conformations, and in cancers over-expressing EGFR the majority of the protein exists in the open conformation, which is active for signal transduction processes [43–45]. Based on our previous studies [10], peptide 1 binds to domain I of the extracellular domain and a site on EGFR that is not affected by conformational changes. Therefore, we modeled the interaction of conjugates 4 and 7 with EGFR in both open and closed conformations, as shown in Figs 5a and 5b. The low energy docked structure of peptide 1 to EGFR domain 1 was found to be −5.9 kcal/ mol. Since peptide 1 binds to both open and closed conformations, conjugates 4 and 7 can also bind to both conformations of EGFR. Furthermore, we investigated the hypothesis that conjugates 4 and 7 might bind to two, rather than one, EGFR molecule simultaneously, therefore increasing their binding affinity as observed by SPR. Therefore, the binding of conjugates 4 and 7 to EGFR dimers in open and closed conformations were modeled. Our model suggests that the porphyrin and peptide length in both conjugates 4 and 7 are suitable to bind to two EGFR molecules (Fig. 5). EGFR does not form dimers to generate signaling in closed conformation. Binding of EGF to EGFR or over-expression of receptors on the cell surface induces conformational change from closed to open conformation [43, 45]. However, our SPR data suggests that peptide 1 as well as its conjugates 4 and 7 bind to EGFR domain 1 in the absence of ligand EGF, indicating that the conjugates also bind to EGFR in closed conformation. Such models have been proposed in the literature for binding of bi-specific affibody ligands that target EGFR and HER2 dimers [46], as well as HER2 dimers in open and closed conformations [47]. Hence, our SPR and modeling studies suggest that conjugates 4 and 7 can bind to monomer EGFR, as well as dimer EGFR, in open or closed conformations.
Fig. 5.

Proposed model of conjugates 4 (a, c) and 7 (b, d) binding with two molecules of EGFR in open conformation (a, b) and in closed conformation (c, d). Crystal structure of EGFR (3NJP) was used for modeling the dimers of EGFR
Cellular studies
The cytotoxicity and time-dependent uptake of conjugates 4, 5, 7 and 8, and of porphyrin 3 for comparison purposes, were investigated in human carcinoma HEp2 cells, and the results obtained are shown in Fig. 6 and Table 4. The dark cytotoxicity was evaluated at concentrations up to 400 μM and the phototoxicity (light dose ~1.5 J/cm2) at concentrations up to 200 μM, using the Cell Titer Blue (CTB) assay. All compounds were found to be non-toxic in the dark (IC50 > 400 μM) and only conjugates 4 and 5 showed moderate phototoxicity, with calculated IC50 values of 50 and 150 μM, respectively (Table 4). These results show that the absence of a PEG spacer between the peptide and the porphyrin enhances the phototoxicity of the conjugate. This might be due to the different conformations assumed by the conjugates in the presence and absence of PEG groups, which influences their binding to EGFR and biodistribution.
Fig. 6.
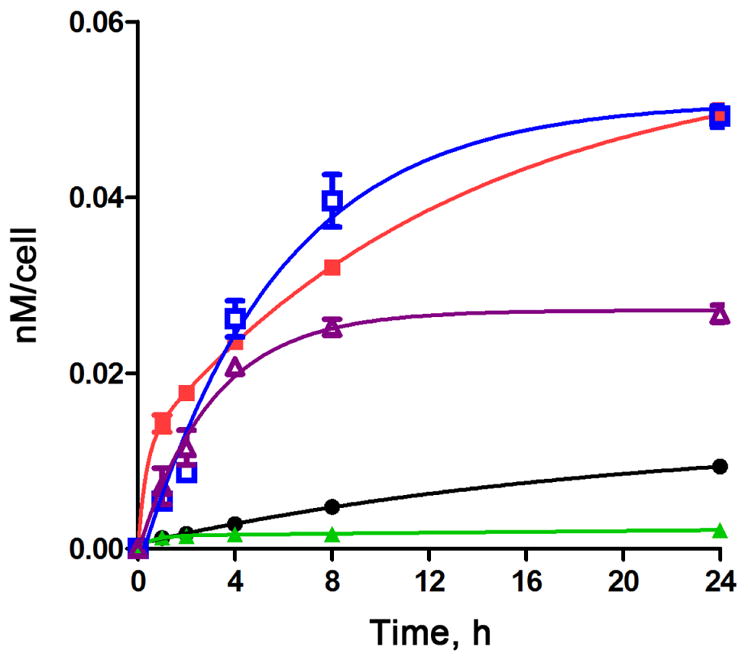
Time-dependent uptake of porphyrins 3 (black), 4 (red), 5 (purple), 7 (green), 8 (blue) at 10 μM by HEp2 cells
Table 4.
Cytotoxicity (CTB assay, light dose ~1.5 J/cm2) for porphyrin conjugates in HEp2 cells
| Porphyrin | Dark toxicity IC50, μM | Phototoxicity IC50, μM |
|---|---|---|
| 3 | > 400 | > 200 |
| 4 | > 400 | 50 |
| 5 | > 400 | 150 |
| 7 | > 400 | > 200 |
| 8 | > 400 | > 200 |
The time-dependent cellular uptake of porphyrin 3 and conjugates 4, 5, 7, and 8 was investigated at 10 μM concentrations over a period of 24 h. Conjugates 4, 5 and 8 accumulated within cells to a much larger extent than MPIX 3 at all time points investigated; however, conjugate 7 accumulated significantly less despite its positive charge, probably as a result of its higher hydrophilicity induced by the two PEG spacers between the porphyrin and the two LARLLT peptides. On the other hand, conjugate 8 bearing two PEG groups and one GYHWYGYTPQNVI sequence steadily accumulated overtime, and after 24 h showed similar uptake as conjugate 4, about 8-fold more than MPIX 3. Conjugate 4 accumulated the most at times < 4 h. These results indicate that the nature of both the peptide and linker affect EGFR targeting and cellular uptake. We have previously observed that in multimeric porphyrin-peptide conjugates, the most amphiphilic molecules tend to show enhanced cellular uptake [31].
Another factor known to influence cellular uptake in peptide conjugates is their polar surface area (PSA), which takes into account the solvent-accessible surface areas of the molecules [48, 49]. The PSA values calculated for peptides 1 and 2, porphyrins 3 and 6 and conjugates 4, 5, 7, and 8 are given in Table 5. In general, low PSA values favor cell permeability and bioavailability. The PSA for peptide 2 is nearly double that for 1, indicating that the latter is significantly more cell permeable, as expected for the smaller sequence containing one cationic residue. Porphyrin 3 has the smallest PSA value, followed by 6. Among the conjugates, those bearing the larger GYHWYGYTPQNVI sequence show larger PSA than the corresponding conjugates bearing two LARLLT sequences, and conjugate 4 has the lowest PSA value of the series, which might contribute to its high uptake. On the other hand, conjugate 8 bearing the largest PSA and molecular weight was also observed to have high cellular uptake, maybe due to its favorable amphiphilicity.
Table 5.
Polar surface area (PSA) of peptides, porphyrins and their conjugates
| Porphyrin | PSA, Å2 |
|---|---|
| 1 | 326 |
| 2 | 613 |
| 3 | 123 |
| 4 | 648 |
| 5 | 702 |
| 6 | 237 |
| 7 | 762 |
| 8 | 816 |
EXPERIMENTAL
Synthesis
General
All reagents and solvents were purchased from commercial suppliers and used directly without further purification. O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU), hydroxybenzotriazole (HOBt), 7-azabenzotriazol-1-yloxy)tripyrrolidnophosphonium hexafluorophosphate (PyAOP), 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT), N-hydroxysuccinimide (NHS), 1-ethyl-3-(3-diimethylaminopropyl) carbodiimide (EDC), 1,8-diazabicycloundec-7-ene (DBU), piperidine, N,N-diisopropylethylamine (DIEA), methanol (MeOH), dichloromethane (DCM), 2,2,2-trifluoroethanol (TFE), ethylacetate, acetone, acetonitrile, liquefied phenol, anhydrous ethyl ether, triisopropylsilane (Tips), sodium acetate, and sodium chloride were purchased from Sigma Aldrich. N,N-dimethylformamide (DMF) peptide sequencing grade, dimethyl sulfoxide (DMSO) cell culture grade, Cremophor EL, trifluoroacetic acid (TFA), malonic acid, oxalic acid, phosphoric acid, and formic acid were purchased from Fisher Scientific. Phosphate buffer solution (PBS) was purchased from VWR International. The peptide sequences were synthesized on an automated peptides synthesizer from Applied Biosystems Pioneer. Analytical thin-layer chromatography (TLC) was carried out using plastic backed TLC plates 254 (precoated, 200 μm) from Sorbent Technologies. Silica gel 60 (230 × 400 mesh, Sorbent Technologies) was used for column chromatography. Prevail C18 reverse phase extract clean was purchased from Grace Davison Discovery Sciences and was used for SPE. NMR spectra were recorded on Liquid AV-400 Bruker spectrometer (400 MHz for 1H, 100 MHz for 13C). The chemical shifts are reported in δ ppm using the following deuterated solvents as internal references: CD3COCD3 2.04 ppm (1H), 29.92 ppm (13C); DMF-d7 8.03 ppm (1H), 163.15 ppm (13C). MALDI-TOF mass spectra were recorded on a Bruker UltrafleXtreme (MALDI-TOF/TOF) instrument using 4-chloro-α-cyanocinnamic acid (CCA) as the matrix. HPLC purifications were carried out on a Waters system including a 2545 quaternary gradient module pump, 2489 UV-visible detector, and a fraction collector III. Analytical HPLC was carried out using a XBridge C18 300 Å, 5 μm, 4.6 × 250 mm (Waters, USA) or an Atlantis C18 300 Å, 5 μm, 4.6 × 250 mm (Waters, USA). Semipreparative HPLC was carried out on XBridge C18 300 Å, 5 μm, 10 × 250 mm (Waters, USA) column. The HPLC solvents contained 0.1% TFA in Millipore water (A) and HPLC grade acetonitrile (B). All tissue culture medium and reagents such as fetal bovine serum (FBS) and penicillin streptomycin (PS) were purchased from Invitrogen. Human carcinoma HEp2 cells were purchased from ATCC. The HEp2 cells were cultured and maintained in 50:50 ATCC formulated Dulbecco’s Modified Eagle’s Medium (DMEM) and advanced MEM (AMEM). A BMG FluoStar Optima micro-plate reader was used for the cell culture assays. Surface Plasmon Resonance was performed using Biacore X100 from GE Health Sciences. Pure recombinant protein EGFR was obtained from Leinco Technologies. The EGF ligand was obtained from Abcam, Inc. 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), ethylenediaminetetraacetic acid (EDTA), and 100 mM glycine at pH 1.5 and 2.5 were purchased from GE Health Science.
Peptides
Peptide sequences LARLLT (1) and GYHWYGYTPQNVI (2) were prepared on an automated peptides synthesizer in a 0.2 mmol scale using standard Fmoc strategy, as we have previously reported [10]. A cleavage cocktail of TFA (88%), Millipore water (5%), liquefied phenol (5%) and Tips (2%) was added to the resin for 3–5 h, then released into cold (−80 °C) anhydrous ethyl ether (5 × 40 mL), and centrifuged. The supernatant was decanted and the residue washed with cold anhydrous ethyl ether. The solid was dissolved in a mixture of Millipore water (A) and acetonitrile (B), freeze-dried and lyophilized. The solvent system for purification of the peptides consisted of Millipore water and HPLC grade acetonitrile with 0.1% TFA. Peptide 1 was obtained as a white solid (145 mg, 48%). HPLC (90% A for 1 min, 90% A to 30% A over 34 min, 30% A to 90% A over 1 min at a flow rate of 1 mL/min) and tR = 18.02 min. The spectroscopic data agrees with that previously reported [15]. Peptide 2 was obtained as a white solid (98 mg, 32%). This peptide was synthesized as per the procedure in literature [16], with an addition of glycine to reduce steric interference from tyrosine. HPLC (90% A for 1 min, 90% A to 10% A over 34 min, 10% A to 90% A over 1 min at a flow rate of 5 mL/min) and tR = 31.22 min. 1H NMR (d-DMSO, 400 MHz): δ, ppm 10.7 (1H, s, ε NH His), 9.18–9.14 (3H, d, J = 18.13 Hz, ι NH Trp, ζ CH His), 8.58–8.49 (2H, m, NH Gly, NH His), 8.32–8.26 (3H, m, NH Tyr), 8.15–8.10 (6H, m, NH Trp, NH Thr, NH Gln, NH Asn, NH Val, NH Ile), 7.56–7.52 (1H, d, ε CH Trp), 7.41 (1H, s, δ CH His), 7.29 (2H, d, J = 8.18 Hz, ζ CH Trp), 7.08–6.97 (12H, m, δ CH Tyr, κ CH Trp, η CH Trp, NH2 Asn, NH2 Gln), 6.75 (2H, s, NH2 Ile), 6.68–6.58 (6H, m, ε CH Tyr), 4.69 (1H, s, α CH Thr), 4.60–4.55 (5H, m, ζ OH Tyr, α CH Trp, α CH His), 4.52–4.48 (1H, m, α CH Pro), 4.45–4.41 (2H, m, α CH2 Gly), 4.39–4.32 (3H, m, α CH Ile, α CH Val, α CH Gln), 4.20–4.12 (4H, m, α CH Tyr, α CH Asn), 4.06 (2H, t, J = 8.18 Hz, α CH2 Gly), 3.88–3.83 (2H, m, δ CH2 Pro), 3.08–2.81 (9H, m, β CH2 Tyr, β CH2 His, β CH Thr), 2.72–2.63 (5H, m, β CH Val, β CH2 Asn, β CH2 Trp), 2.13 (3H, t, J = 7.16 Hz, β CH2 Pro, β CH Ile), 2.06–1.98 (2H, m, γ CH2 Pro), 1.92–1.81 (4H, m, β CH2 Gln, γ CH2 Gln), 1.23 (2H, s, NH2 Gly), 1.10 (6H, s, γ CH2 Ile, γ CH3 Thr), 0.89–0.78 (12H, m, ε CH3 Ile, δ CH3 Ile, γ CH3 Val). MS (MALDI): m/z 1619.80 [M + Na]+, calcd. for C77H101N19O19 1619.74.
MPIX conjugate 4
To a solution of MPIX 3 (19 mg, 0.030 mmol) in DMF (1 mL) was added DIEA (17 mg, 0.13 mmol) under N2 at 37 °C, and the final solution stirred at room temperature for 2 h. TBTU (10 mg, 0.031 mmol), HOBt (5 mg, 0.037 mmol), DMF (2 mL), and DIEA (17 mg, 0.13 mmol) were added and the mixture allowed to stir for 30 min under N2 at 37 °C. Peptide 1 (41 mg, 0.060 mmol) was dissolved in DMF (500 μL) and DIEA (17 mg, 0.128 mmol) and added to the reaction flask. The mixture was stirred for 5 days and dried under N2. A solution consisting of MeOH/acetone/acetonitrile (0.5/6/3.5, 4 mL) was used for SPE. The SPE was conditioned with methanol (10 mL, 5 times), Millipore water (10 mL, 5 times) and MeOH/acetone/ acetonitrile (3 × 10 mL). The crude product was sonicated in MeOH/acetone/acetonitrile (0.5/6/3.5, 5 mL) and loaded onto the SPE cartridge. The cartridge was eluted until the solvent was clear and TFA was added to the solvent system to elute the compound that was stuck on the column. The title conjugate 4 was obtained (30.3 mg) in 83% yield. HPLC (90% A for 1 min, 90% A to 50% A over 5 min, 50% A to 30% A over 55 min, 30% A to 90% A over 1 min at a flow rate of 0.6 mL/min) and tR = 35.01 min. UV-vis (DMF): λmax, nm (log ε) 407 (5.19), 497 (4.06), 530 (4.00), 577 (3.93), 615 (3.63). 1H NMR (d-DMF, 400 MHz): δ, ppm 10.43 (1H, s, MPIX), 10.33 (3H, s, MPIX), 8.51 (2H, s, η NH Arg), 8.41 (2H, s, NH Thr), 8.30 (2H, s, ε NH Arg), 8.00–7.95 (2H, m, NH Ala), 7.93–7.86 (4H, m, NH Leu), 7.58–7.50 (8H, m, NH Leu, NH2 Thr, NH Arg), 7.17 (4H, d, J = 8.42 Hz, ζ NH2 Arg), 4.57–4.45 (4H, m, α CH Ala, α CH Thr), 4.42–4.32 (8H, m, α CH Leu, α CH Arg), 4.29–4.20 (12H, m, β CH Thr, OH Thr, MPIX), 3.78–3.70 (12H, m, MPIX), 3.42–3.34 (8H, m, δ CH2 Arg, MPIX), 1.95–1.85 (10H, m, MPIX, β CH2 Arg), 1.78–1.68 (22H, m, β CH2 Leu, γ CH2 Arg, γ CH Leu), 1.35–1.23 (6H, m, β CH3 Ala), 1.25–1.13 (6H, m, γ CH3 Thr), 0.93–0.78 (36H, m, ε CH3 Leu). MS (MALDI-TOF): m/z 1900.40 [M + 1]+, calcd. for C96H154N24O16 1900.41.
MPIX conjugate 5
To a solution of MPIX 3 (8.32 mg, 0.030 mmol) in DMF (1 mL) was added DIEA (5.6 mg, 0.043 mmol) under inert conditions, and the mixture was stirred at 37 °C for 2 h. DEPBT (8.2 mg, 0.027 mmol), HOBt (12.3 mg, 0.032 mmol), DMF (2 mL), and DIEA (5.6 mg, 0.043 mmol) were added and the final mixture allowed to stir for 30 min under N2 at 37 °C. Peptide 2 (41.5 mg, 0.026 mmol) dissolved in DMF (500 μL) and DIEA (5.6 mg, 0.043 mmol) was added and the mixture allowed to stir at 37 °C for 5 days. The SPE was conditioned with methanol (5 × 10 mL), Millipore water (5 × 10 mL) and MeOH/DCM (3 × 10 mL). Unreacted MPIX 3 was soluble in MeOH/DCM and eluted off the SPE while 5 remained on the SPE. The cartridge was eluted until the solvent was clear and TFA added to the solvent system to elute conjugate 5, obtained (8 mg) in 51% yield. HPLC gradient (90% A for 1 min, 90% A to 50% A over 5 min, 50% A to 10% A over 52 min, 10% A to 90% A over 3 min at a flow rate of 0.7 mL/min) and tR = 36.20 min. UV-vis (DMF): λmax, nm (log ε) 397 (5.19), 497 (4.06), 530 (4.00), 566 (3.93), 620 (3.63). 1H NMR (d-DMF, 400 MHz): δ, ppm 10.80 (1H, s, ε NH His), 10.45 (1H, s, MPIX), 10.34 (3H, s, MPIX), 9.07 (3H, d, J = 11.70 Hz, ι NH Trp, ζ CH His), 8.52–8.25 (6H, m, NH Gly, NH Tyr, NH His), 7.77–7.65 (4H, m, NH Trp, NH Thr, NH Gln, NH Asn), 7.46 (1H, s, NH Val), 7.34 (1H, m, NH Ile), 7.30–7.18 (3H, m, ε CH Trp, δ CH His, η CH Trp), 7.15–6.82 (14H, m, ζ CH Trp, δ CH Tyr, κ CH Trp, NH2 Asn, NH2 Gln, NH Gly), 7.05–6.60 (8H, m, NH2 Ile, ε CH Tyr), 4.78–4.65 (4H, m, MPIX), 4.58–4.43 (5H, m, ζ OH Tyr, α CH Trp, α CH His), 4.35–4.24 (15H, m, α CH Pro, α CH Thr, α CH2 Gly, α CH Ile, MPIX), 3.79–3.66 (19H, m, α CH Asn, α CH Tyr, δ CH2 Pro, α CH Gln, MPIX), 3.46–3.27 (5H, m, MPIX, α CH Val), 3.16–3.09 (2H, m, β CH Thr, OH Thr), 2.44–2.33 (6H, m, β CH2 Tyr), 2.27–2.19 (3H, m, β CH2 His, β CH Val), 2.15–1.95 (2H, m, β CH2 Trp), 1.89–1.75 (12H, m, β CH2 Asn, β CH2 Pro, β CH Ile, MPIX, γ CH2 Pro) 1.30–1.20 (12H, m, β CH2 Gln, γ CH2 Gln, ε CH3 Ile, γ CH3 Thr, γ CH2 Ile), 0.93–0.75 (9H, m, γCH3 Val, δ CH3 Ile). MS (MALDI): m/z 2146.26 [M + 1]+, calcd. for C111H137N23O22 2146.24.
MPIX-diPEG 6
To a solution of MPIX 3 (20.26 mg, 0.032 mmol) in DMF (1 mL) was added DIEA (14 mg, 0.106 mmol) under inert conditions, and the mixture was stirred at 37 °C for 2 h. TBTU (21.4 mg, 0.066 mmol), HOBt (10.7 mg, 0.079 mmol), DMF (2 mL), and DIEA (14 mg, 0.106 mmol) were added and the final mixture stirred for 30 min under N2 at 37 °C. tert-Butyl-12-amino-4,7,10-trioxadodecanoate (17.6 mg, 0.063 mmol) dissolved in DMF (500 μL) and DIEA (14 mg, 0.106 mmol) was added and the mixture allowed to stir at 37 °C for 5 days. The reaction mixture was diluted with water and ethyl acetate (3/7), transferred to a separatory funnel and gently swirled. The organic layer was concentrated under reduced pressure. The crude product was purified by column chromatograph using methanol/dichloromethane (5/95) for elution. 1H NMR (d-DMF, 400 MHz): δ, ppm 10.40 (1H, s, MPIX), 10.29 (3H, s, MPIX), 8.10–8.02 (2H, m, NH PEG), 4.48–4.41 (4H, m, CH2 MPIX), 4.22–4.18 (4H, m, CH2 MPIX), 3.80 (4H, s, OCH2), 3.75–3.70 (14H, m, γ CH2), 3.59 (2H, d, J = 3.23 Hz, γ CH2 PEG), 3.46 (4H, t, J = 6.14 Hz, CH2 MPIX), 3.25–3.17 (12H, m, CH3 MPIX), 3.06–3.01 (8H, t, J = 5.50 Hz, β CH2 PEG, δ CH2 PEG), 2.93–2.87 (2H, m, α CH2 PEG), 2.38–2.30 (4H, m, ε CH2 PEG), 1.95–1.88 (6H, m, CH3 MPIX), 1.40–1.34 (18H, m, tBu). The protected porphyrin was dissolved in TFA (2 mL) and stirred at 0 °C for 4 h. The TFA was evaporated under N2 and purified using HPLC. Conjugate 6 was obtained in 88% yield; HPLC gradient (90% A for 1 min, 90% A to 40% A over 10 min, 40% A to 20% A over 30 min, 20% A to 10% A over 3 min, 10% A to 90% A over 2 min at a flow rate of 0.6 mL/min) and tR = 19.43 min. UV-vis (DMF): λmax, nm (log ε) 398 (5.35), 497 (4.18), 530 (4.18), 566 (4.00), 616 (3.70). 1H NMR (d-DMF, 500 MHz): δ, ppm 10.29 (2H, s, MPIX), 10.23 (2H, s, MPIX), 8.08–8.01 (2H, m, PEG), 4.44–4.38 (4H, m, CH2 MPIX), 4.25–4.18 (4H, m, CH2 MPIX), 3.80 (4H, s, OCH2), 3.72–3.65 (14H, m, γ CH2 PEG), 3.60–3.55 (2H, m, γ CH2 PEG), 3.34–3.25 (4H, m, CH2 MPIX), 3.20–3.15 (12H, m, CH3 MPIX), 3.10–3.02 (4H, m, β CH2 PEG), 2.95–2.86 (8H, m, δ CH2 PEG, α CH2 PEG), 2.46–2.40 (4H, m, ε CH2 PEG), 1.92–1.80 (6H, m, CH3 MPIX). MS (MALDI): m/z 974.50 [M]+ calcd. for C52H72N6O12 974.53.
MPIX conjugate 7
To a solution of MPIX-diPEG 6 (13.94 mg, 0.014 mmol) in DMF (1 mL) was added DIEA (6.20 mg, 0.047 mmol) under inert conditions, and the mixture was stirred at 37 °C for 2 h. TBTU (9.65 mg, 0.030 mmol), HOBt (4.84 mg, 0.036 mmol), DMF (2 mL), and DIEA (6.20 mg, 0.047 mmol) were added and the final mixture allowed to stir for 30 min under N2 at 37 °C. Peptide 1 (19.6 mg, 0.029 mmol) dissolved in DMF (500 μL) and DIEA (8.3 mg, 0.064 mmol) was added and the final mixture was allowed to stir for 5 days. The crude product was dissolved in MeOH/acetone/ acetonitrile (5/60/35) and loaded into a C18 SPE. The SPE was conditioned with methanol (5 × 10 mL), Millipore water (5 × 10 mL) and MeOH/acetone/acetonitrile (3 × 10 mL). When the washes were clear TFA was added to the solvent system to elute the compound, affording conjugate 7 (5.4 mg) in 16% yield. HPLC gradient (90% A for 1 min, 90% A to 50% A over 4 min, 50% A to 10% A over 38 min, 10% A to 90% A over 2 min at a flow rate of 0.6 mL/min) and tR = 19.86 min. UV-vis (DMF): λmax, nm (log ε) 408 (5.19), 497 (4.08), 530 (4.00), 573 (3.90), 618 (3.60). 1H NMR (d-DMF, 400 MHz): δ, ppm 10.46 (1H, s, MPIX), 10.33 (3H, s, MPIX), 8.49–8.42 (6H, m, η NH Arg, NH Thr, ε NH Arg), 7.90 (6H, d, J = 5.83 Hz, NH Leu), 7.81 (2H, d, J = 6.72 Hz, NH Ala), 7.46 (4H, d, J = 8.52 Hz, NH2 Thr), 7.09 (4H, d, ζ NH2 Arg), 4.51–4.46 (8H, m, MPIX), 4.41–4.36 (4H, m, α CH Ala, α CH Thr), 4.30–4.22 (26H, m, α CH Leu, β CH Thr, OH Thr, MPIX), 3.79–3.71 (22H, m, PEG, MPIX), 3.65–3.55 (12H, m, PEG), 2.48–2.44 (4H, m, δ CH2 Arg), 1.90 (16H, t, J = 7.27 Hz, β CH2 Arg, β CH2 Leu), 1.78–1.66 (21H, m, γ CH2 Arg, γ CH2 Leu, MPIX), 1.41–1.34 (6H, m, β CH3 Ala), 1.22–1.15 (7H, m, γ CH3 Thr), 0.89–0.76 (36H, m, δ CH3 Leu). MS (MALDI): m/z 2306.48 [M + 1]+, calcd. for C114H188N26O24 2306.43.
MPIX Conjugate 8
To a solution of MPIX-diPEG 6 (7.29 mg, 0.008 mmol) in DMF (1 mL) was added DIEA (3.25 mg, 0.025 mmol) and the mixture was stirred at 37 °C for 2 h. DEPBT (4.79 mg, 0.016 mmol), HOBt (6.84 mg, 0.018 mmol), DMF (2 mL), and DIEA (3.25 mg, 0.025 mmol) were added and the final mixture allowed to stir for 30 min under N2 at 37 °C. Peptide 2 (28.72 mg, 0.15 mmol) dissolved in DMF (500 μL) and DIEA (4.16 mg, 0.032 mmol) was added and the final mixture was allowed to stir for 5 days. The sample was dissolved in acetone, sonicated, and centrifuged several times to remove the unreacted compound 6. The title bioconjugate 8 was obtained (7.3 mg) in 39% yield. HPLC gradient (90% A for 1 min, 90% A to 50% A over 4 min, 50% A to 10% A over 38 min, 10% A to 90% A over 2 min at a flow rate of 0.6 mL/min) and tR = 25.862 min. UV-vis (DMF): λmax, nm (log ε) 408 (5.24), 497 (4.15), 531 (4.08), 574 (4.00), 620 (3.60). 1H NMR (d-DMF, 400 MHz): δ, ppm 10.30 (1H, s, CH, MPIX), 10.23 (3H, s, CH, MPIX), 9.14 (3H, s, ι NH Trp, ε NH His, ζ CH His), 8.23–8.08 (4H, m, NH Gly, NH Tyr), 7.94–7.78 (4H, m, NH His, NH Trp, NH Thr, NH Gln), 7.68 (6H, m, NH Asn, NH Val, NH Ile, NH PEG, δ CH His), 7.51–7.42 (2H, m, γ CH His, ε CH Trp), 7.33–7.20 (4H, m, ζ CH Trp, η CH Trp, κ CH Trp), 7.05–6.92 (12H, m, δ CH Tyr, NH2 Asn, NH2 Gln, NH2 Ile), 6.61 (6H, d, J = 6.05 Hz, ε CH Tyr), 4.69 (2H, s, α CH Thr, α CH Trp), 4.57 (3H, q, J = 4.03 Hz, OH Tyr), 4.44–4.34 (7H, m, α CH His, α CH Gln, α CH Pro, MPIX), 4.18–4.05 (16H, m, MPIX, α CH Ile, α CH Val, α CH2 Gly, α CH Asn, α CH Tyr, δ CH2 Pro), 3.68–3.54 (26H, m, MPIX, PEG), 3.36–3.25 (16H, m, PEG), 3.20–3.03 (14H, m, β CH2 Tyr, β CH2 His, β CH2 Trp, PEG), 2.72–2.65 (3H, m, β CH Val, β CH2 Asn), 2.15–1.98 (5H, m, β CH Ile, β CH2 Pro, γ CH2 Pro), 1.87–1.65 (17H, m, β CH2 Gln, γ CH2 Gln, MPIX, γ CH2 Ile, PEG), 1.13–1.05 (6H, m, ε CH3 Ile, γ CH3 Thr), 0.87–0.75 (9H, m, δ CH3 Ile, γ CH3 Val). MS (MALDI): m/z 2551.23 [M + 1]+, calcd. for C129H171N25O30 2552.27.
Circular Dichroism
Circular dichroism (CD) studies were performed using a Jasco J-815 spectrometer or on an AVIV 620S circular dichroism spectrometer. The CD measurements were carried out using 1 mm path length quartz cell. Peptide solutions at various concentrations were prepared in PBS/TFE (9/1) with a pH of 7.4. All spectra correspond to an average of three runs and were corrected using the baseline obtained for peptide free solutions.
Spectroscopic properties
Photophysical studies were performed in peptide-sequencing grade DMF solutions. All absorption spectra were measured on a UV-vis Perkin Elmer Lambda 35 spectrophotometer. All experiments were carried out within 3 h of solution preparation at room temperature (23–25 °C) using a 10 mm path length spectrophotometric cell. Emission spectra were recorded on a Fluorolog®-HORIBA JOBINVYON (Model LFI-3751). The fluorescence quantum yields (Φf) were determined using a secondary standard method using MPIX 3 (Φf = 0.102) as the reference [28].
SPR studies
Surface Plasmon Resonance was performed using Biacore X100 (GE Health Sciences) at 25 °C. EGFR was immobilized on a CM5 chip using a standard amine coupling protocol. The carboxyl groups on the sensor chip were activated using 0.2 M EDC and 0.05 M NHS solution in Millipore water (flow rate 5 μL/min). Running buffer consisted of 0.01 M HEPES, 0.15 M NaCl, 3 mM EDTA, 0.005% Tween at pH 7.5 (adjusted using 4 M NaOH). Regeneration buffer for the ligand were 50% acid cocktail (0.15 M malonic acid, 0.15 M oxalic acid, 0.15 phosphoric acid, 0.15 formic acid, adjusted to pH 5 with 4 M NaOH). Regeneration buffer for porphyrin conjugates was 100 mM glycine at pH 2.5 and 100 mM glycine at pH 1.5. For immobilization EGFR was dissolved in NaOAc at pH 4.5 (adjusted using 10% HCl) and injected onto the chips surface at 10 μL/min until 12 000 response units (RU) was obtained. The remaining unreacted activated groups were blocked with 1 M ethanolamine at pH 8.5 (adjusted with 10% HCl). Peptides 1 and 2 were dissolved in DMSO and diluted in running buffer to obtain the desired concentration. Porphyrin conjugates 4, 5 and 7 were dissolved in 10% Cremophor EL in DMSO and diluted in the running buffer to obtain the desired concentration, with maximum DMSO concentration of 1% and Cremophor EL concentration of 0.1%. All solutions were filtered using 0.45 μm filters and the concentration of the peptides was 100 μM.
Computational studies
Three dimensional structures of conjugates 4, 5, 6, 7 and 8 were generated using InsightII (BIOVIA, Sandiego CA) software. These structures were energy minimized by using 100 steps of steepest descent method to remove any short contacts and further minimized for 4000 steps of conjugate gradient method in vacuum. Minimized structures were subjected to 20 ps molecular dynamics simulations at 300 K. Resulting structures were analyzed using time vs. energy graph. From the MD simulations structures were selected at the end of 20 ps and further minimized. These minimized structures were used as representative 3D structures of conjugates. For computational studies in solvent, starting conjugate structures were soaked with a 5 Å layer of water molecules and after equilibration, minimization and dynamics were performed as described above. Final structures were represented using PyMol software (Schrodinger Inc. LLC, Portland OR).
Docking of peptides and conjugates to EGFR was performed using Autodock 4 software [41, 42] using monomer of EGFR structure. A three dimensional structure of conjugate 4 was generated using InsightII (BIOVIA, San Diego, CA). Three dimensional structures of EGFR in the open (3njp) [41] and closed (1nql) [42] conformations were obtained from the protein data bank. The 3D structure of conjugate 4 was generated as described above using molecular modeling and energy minimization. Using the previously published docking results [10], the structure of conjugates 4 and 7 were placed on the binding site of one EGFR molecule in open conformation, via one of the peptide chains. Another EGFR molecule was placed on the other peptide chain of 4 or 7. The two EGFR molecules were rotated and translated so there was no steric clash between the two, however the EGRF binding site for peptide was maintained [10]. A similar procedure was used for generating the binding model of 4 and 7 to EGFR in a closed conformation. The models were energy minimized to remove any possible steric hindrance. Final models were represented using PyMol software (Schrodinger Inc. LLC, Portland OR).
Cell studies
The HEp2 cells (ATCC) were maintained in a 75 cm2 flask (Chemglass) with the medium (DMEM:Advanced, 1:1) containing 10% FBS and 1% antibiotic (Life Technologies). The compound solutions were prepared by dissolving the compound in 100% DMSO at a concentration of 32 mM (stock solution), and a 2 mL of 400 μM compound solution was prepared by adding 25 μL of the 32 mM compound stock solution into 1975 μL growing medium.
Dark cytotoxicity
To investigate the compound dark toxicity, HEp2 cells were placed in a 96-well plate (15,000 cells/well). The cell will be treated with the compound concentration of 200, 100, 50, 25, 12.5, and 0 μM for 20–24 h incubation at 37 °C when each well cell get 100% confluence. To end the treatment, the compound was removed by washing cells with 1X PBS and replaced with the growing media containing 20% CellTiter Blue (Promega). This cell viability assay uses the indicator dye resazurin which is reduced to highly fluorescent resorufin in viable cells, while non-viable cells lose metabolic capacity and are not able to reduce resazurin nor to generate a fluorescent signal. This is a popular, simple, fast, sensitive and accurate method for determination of cell viability that has been demonstrated to correlate well with other methods for measuring cell proliferation and cytotoxicity. The cells were incubated for an additional 4 h at 37 °C, and measured fluorescently at 570/615 nm using a FluoStar Optima micro-plate reader.
Phototoxicity
The concentration range of 100, 50, 25, 12.5, 6.25, 3.125, and 0 μM was used for the phototoxicity experiments. HEp2 cells were placed in 96 well plates as described above, and treated with compound for 24 h at 37 °C. After this treatment the loading media was removed. The cells were washed with 1 X PBS buffer, and then refilled with fresh media. The cells were exposed to a 600 W halogen lamp light source filtered with a water filter (transmits radiation 250–950 nm) and a beam turning mirror (Newport) with 200 nm to 30 μm spectral range, for 20 min. The total light dose was approximately 1.5 J/cm2. After exposed to light, the cells were returned to the incubator for 24 h. After 24 h incubation, the medium was removed and replaced with media containing 20% Cell Titer Blue. The cells were incubated for an additional 4 h. The viable cells is measured fluorescently at 570/615 nm using a FluoStar Optima micro-plate reader.
Time-dependent cellular uptake
The HEp2 cells were plated in a 96-well plate as described above. The cells were treated by adding 100 μL/well of 10 μM working solution at different time periods of 0, 1, 2, 4, 8, and 24 h whenever the cell getting 90–100% confluence monolayer. The 10 μM compound working concentration was made by diluting 400 μM stock solution with growing medium. At the end of the treatments, the cells were washed with 1X PBS, and solubilized by adding 100 μL 0.25% Triton X-100 in 1X PBS per well. A compound standard curve was made by diluting 400 μM compound solution with 0.25% Triton X-100 in 1X PBS to 10, 5, 2.5, 1.25, 0.625, 0.3125, and 0 μM. A cell standard curve was prepared using 10,000, 20,000, 40,000, 60,000, 80,000, and 100,000 cells/well. The cells were quantified by CyQuant Cell Proliferation Assay (Life Technologies). The compound and cell number were determined using a FluoStar Optima micro-plate reader (BMG LRBTEH), with wavelengths 355/615 nm for compounds and 485/520 nm for cells, respectively. Cellular uptake was expressed in terms of nM compound per cell.
CONCLUSION
Two EGFR-targeting peptides with the sequences LARLLT and GYHWYGYTPQNVI were synthesized, characterized, and conjugated to the propionic side chains of mesoporphyrin IX, with or without a triethylene glycol spacer. The conjugations were performed in solution phase at room temperature in DMF, using TBTU/HOBt as the coupling agents for the shorter peptide, and DEPBT/HOBt for the longer peptide. Four conjugates, containing two LARLLT sequences (4 and 7) or one GYHWYGYTPQNVI sequence (5 and 8) were isolated in 16–83% yields.
The structures of the peptides and their conjugates were investigated by NMR, MALDI-MS, CD, UV-vis and fluorescence spectroscopies. SPR, molecular dynamics and docking investigations were used to evaluate their conformations and binding to EGFR. These studies revealed that the conjugates 4 and 7 containing two LARLLT residues have the highest affinity for binding to EGFR, in both open and closed conformations. Furthermore, modeling studies suggested that these two conjugates can also bind to EGFR dimers. The observed enhanced binding of the porphyrin-LARLLT conjugates may be a result of their positive charge and amphiphilic character.
All conjugates were non-toxic in the dark (IC50 > 400 μM), and only conjugate 4 showed moderate phototoxicity (IC50 = 50 μM at 1.5 J/cm2 light dose) toward human HEp2 cells. Conjugates 4 and 8 were the most efficiently taken up by cells, while 7 was the least, due to its lower hydrophobicity. Our results show that amphiphilic mesoporphyrin IX conjugates bearing the LARLLT peptide are very promising for selective EGFR targeting, and hence for the diagnosis of EGFR-over-expressing cancers, such as CRC.
Supplementary Material
Acknowledgments
This study is in memoriam of Emile Fontenot (1928–2010). The research was funded by the National Institutes of Health, grant number R01 CA179902. Computational studies were carried out using the high performance computational facility at LSU, the Louisiana Optical Network Initiative (LONI).
Footnotes
Dedicated to Professor Kevin M. Smith on the occasion of his 70th birthday
Figures S1–S23 and Table S1 are given in the supplementary material. This material is available free of charge via the Internet at http://www.worldscinet.com/jpp/jpp.shtml.
References
- 1.American Cancer Society. Detailed Guide: Colon and Rectum Cancer What Are the Key Statistics for Colorectal Cancer? www.cancer.org/Cancer/ColonandRectumCancer/DetailedGuide/colorectal-cancer-key-statistics.
- 2.Kiesslich R, Goetz M, Vieth M, Galle PR, Neurath MF. Nat Clin Pract Oncol. 2007;4:480–490. doi: 10.1038/ncponc0881. [DOI] [PubMed] [Google Scholar]
- 3.Kantsevoy SV, Adler DG, Conway JD, Diehl DL, Farraye FA, Kaul V, Kethu SR, Kwon RS, Mamula P, Rodriguez SA, Tierney WM, Comm AT. Gastroint Endosc. 2009;70:197–200. doi: 10.1016/j.gie.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 4.Spano JP, Lagorce C, Atlan D, Milano G, Domont J, Bnamouzig R, Attar A, Benichou J, Martin A, Morere JF, Raphael M, Penault-Llorca F, Breau JL, Fagard R, Khayat D, Wind P. Ann Oncol. 2005;16:102–108. doi: 10.1093/annonc/mdi006. [DOI] [PubMed] [Google Scholar]
- 5.Galizia G, Iieto E, Ferraraccio F, De Vita F, Castel-lano P, Orditura M, Imperatore V, La Mura A, La Manna G, Pinto M, Catalano G, Pignatelli C, Ciardiello F. Ann Surg Oncol. 2006;13:823–835. doi: 10.1245/ASO.2006.05.052. [DOI] [PubMed] [Google Scholar]
- 6.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Cancer Cell. 2005;4:301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 7.Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D’Haens G, Pinter T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P. Engl J Med. 2009;360:1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 8.Qi S, Miao Z, Liu H, Xu Y, Feng Y, Cheng Z. Bioconjugate Chem. 2012;23:1149–1156. doi: 10.1021/bc200596a. [DOI] [PubMed] [Google Scholar]
- 9.Ekerljung L, Wallberg H, Sohrabian A, Andersson K, Friedman M, Frejd FY, Stahl S, Gedda L. Bioconjugate Chem. 2012;23:1802–1811. doi: 10.1021/bc3000645. [DOI] [PubMed] [Google Scholar]
- 10.Ongarora BG, Fontenot KR, Hu X, Sehgal I, Satyanarayanajois SD, Vicente MGH. J Med Chem. 2012;55:3725–3738. doi: 10.1021/jm201544y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banappagari S, McCall A, Fontenot KR, Vicente MGH, Gujar A, Satyanarayanajois SD. Eur J Med Chem. 2013;65:60–69. doi: 10.1016/j.ejmech.2013.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanthala S, Gauthier T, Satyanarayanajois SD. Biopolymers. 2014;101:693–702. doi: 10.1002/bip.22441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wheeler DL, Dunn EF, Harari PM. Nat Rev Clin Oncol. 2010;7:49–507. doi: 10.1038/nrclinonc.2010.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meric-Bernstam F, Hung MC. Clin Cancer Res. 2006;12:6326–6330. doi: 10.1158/1078-0432.CCR-06-1732. [DOI] [PubMed] [Google Scholar]
- 15.Li Z, Zhao R, Wu X, Sun Y, Yao M, Li J, Xu Y, Gu J. FASEB J. 2005;19:1978–1985. doi: 10.1096/fj.05-4058com. [DOI] [PubMed] [Google Scholar]
- 16.Song S, Liu D, Peng J, Deng H, Guo Y, Xu LX, Miller AD, Xu Y. FASEB J. 2009;23:1396–1404. doi: 10.1096/fj.08-117002. [DOI] [PubMed] [Google Scholar]
- 17.Williams TM, Sibrian-Vazquez M, Vicente MGH. In: Recent Advancements in PDT. Dougherty T, Pandey R, Kessel D, editors. World Scientific Publishers; Singapore: 2015. in press. [Google Scholar]
- 18.Rahimipour S, Ben-Aroya N, Ziv K, Chen A, Fridkin AM, Koch Y. J Med Chem. 2003;46:3965–3974. doi: 10.1021/jm020535y. [DOI] [PubMed] [Google Scholar]
- 19.Conway CL, Walker I, Bell A, Roberts DJH, Brown SB, Vernon DI. Photochem Photobiol Sci. 2008;7:290–298. doi: 10.1039/b715141a. [DOI] [PubMed] [Google Scholar]
- 20.Sibrian-Vazquez M, Hu X, Jensen TJ, Vicente MGH. J Porphyrins Phthalocyanines. 2012;16:603–615. [Google Scholar]
- 21.Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Oxford University Press; 2004. [Google Scholar]
- 22.Grant GA. Synthetic Peptides: A User’s Guide. Oxford University Press, Inc; New York: 2002. [Google Scholar]
- 23.Benoiton NL. Chemistry of Peptide Synthesis. Taylor & Francis Group: LLC; 2006. [Google Scholar]
- 24.Ye YH, Li HT, Jiang XH. Biopolymers. 2005;80:172–178. doi: 10.1002/bip.20201. [DOI] [PubMed] [Google Scholar]
- 25.Valeur E, Bradley M. Chem Soc Rev. 2009;38:606–631. doi: 10.1039/b701677h. [DOI] [PubMed] [Google Scholar]
- 26.Marder O, Albericio F. Chim Oggi Chem Today. 2003;21:35–40. [Google Scholar]
- 27.Wipf P. Handbook of Reagents for Organic Synthesis: Reagents for High-Throughput Solid-Phase and Solution-Phase Organic Synthesis. John Wiley & Sons; Hoboken: 2005. [Google Scholar]
- 28.Soares ARM, Taniguchi M, Chandrashaker V, Lindsey JS. Chem Sci. 2012;3:1963–1974. [Google Scholar]
- 29.Sibrian-Vazquez M, Jensen TJ, Hammer RP, Vicente MGH. J Med Chem. 2006;49:1364–1372. doi: 10.1021/jm050893b. [DOI] [PubMed] [Google Scholar]
- 30.Sibrian-Vazquez M, Jensen TJ, Vicente MGH. J Med Chem. 2008;51:2915–2923. doi: 10.1021/jm701050j. [DOI] [PubMed] [Google Scholar]
- 31.Sibrian-Vazquez M, Jensen TJ, Vicente MGH. Org Biomol Chem. 2010;8:1160–1172. doi: 10.1039/b917280g. [DOI] [PubMed] [Google Scholar]
- 32.Garnier JR. Structure and the principles of Protein Conformation, The GOR Method for Prediciting Secondary Structures of Proteins. Plenum Press; New York: 1989. [Google Scholar]
- 33.Correa DHAR, Carlos HI. Afr J Biochem Res. 2009;3:164–173. [Google Scholar]
- 34.Perez-Iratxeta C, Andrade-Navarro MA. Bmc Struct Biol. 2008;8:8–25. doi: 10.1186/1472-6807-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komolov KE, Koch KW. In: Methods Mol Biol. De Moi NJ, Fischer MJE, editors. Chapter 17 Springer Science; NY: 2010. [Google Scholar]
- 36.Abdulhalim I, Zourob M, Lakhtakia A. Electromagnetics. 2008;28:214–242. [Google Scholar]
- 37.Sibrian-Vazquez M, Jensen TJ, Fronczek FR, Hammer RP, Vicente MGH. Bioconjugate Chem. 2005;16:852–863. doi: 10.1021/bc050057g. [DOI] [PubMed] [Google Scholar]
- 38.Madeira A, Ohman E, Nilsson A, Sjogren B, Andren PE, Svenningsson P. Nature Protocols. 2009;4:1023–1037. doi: 10.1038/nprot.2009.84. [DOI] [PubMed] [Google Scholar]
- 39.Kimani S, Ghosh G, Ghogare A, Rudshteyn B, Bar-tusik D, Hasan T, Greer A. J Org Chem. 2012;77:10638–10647. doi: 10.1021/jo301889s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li YC, Rissanen S, Stepniewski M, Cramariuc O, Rog T, Mirza S, Xhaard H, Wytrwal M, Kepczyn-ski M, Bunker A. J Phys Chem B. 2012;116:7334–7341. doi: 10.1021/jp300301z. [DOI] [PubMed] [Google Scholar]
- 41.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J Comp Chem. 1998;19:1639–1662. [Google Scholar]
- 42.Huey R, Morris GA, Olson AJ, Goodsell DS. J Comp Chem. 2006;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 43.Burgess AW, Cho HS, Eigenbrot C, Ferguson KM, Garrett TP, Leahy DJ, Lemmon MA, Sliwkowski MX, Ward CW, Yokoyama S. Molecular cell. 2003;12:541–552. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 44.Ferguson KM. Ann Rev Biophys. 2008;37:353–373. doi: 10.1146/annurev.biophys.37.032807.125829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Nat Rev Clin Oncol. 2012;9(1):6–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- 46.Jost C, Schilling J, Tamaskovic R, Schwill M, Honegger A, Pluckthun A. Structure. 2013;21:1979–1991. doi: 10.1016/j.str.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 47.Tan NC, Yu P, Kwon Y-U, Kodadek T. Bioorg Med Chem. 2008;16:5853–5861. doi: 10.1016/j.bmc.2008.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yudin AK. Chem Sci. 2015;6:30–49. doi: 10.1039/c4sc03089c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu C, Mi LZ, Grey MJ, Zhu J, Graef E, Yokoyama S, Springer TA. Mol Cell Biol. 2010;30:5432–5443. doi: 10.1128/MCB.00742-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.